

Abstract
Purpose
Immuno‐MALDI (iMALDI) combines immuno‐enrichment of biomarkers with MALDI‐MS for fast, precise, and specific quantitation, making it a valuable tool for developing clinical assays. iMALDI assays are optimized for the PI3‐kinase signaling pathway members phosphatase and tensin homolog (PTEN) and PI3‐kinase catalytic subunit alpha (p110α), with regard to sensitivity, robustness, and throughput. A standardized template for developing future iMALDI assays, including automation protocols to streamline assay development and translation, is provided.
Experimental Design
Conditions for tryptic digestion and immuno‐enrichment (beads, bead:antibody ratios, incubation times, direct vs. indirect immuno‐enrichment) are rigorously tested. Different strategies for calibration and data readout are compared.
Results
Digestion using 1:2 protein:trypsin (wt:wt) for 1 h yielded high and consistent peptide recoveries. Direct immuno‐enrichment (antibody‐bead coupling prior to antigen‐enrichment) yielded 30% higher peptide recovery with a 1 h shorter incubation time than indirect enrichment. Immuno‐enrichment incubation overnight yielded 1.5‐fold higher sensitivities than 1 h incubation. Quantitation of the endogenous target proteins is not affected by the complexity of the calibration matrix, further simplifying the workflow.
Conclusions and Clinical Relevance
This optimized and automated workflow will facilitate the clinical translation of high‐throughput sensitive iMALDI assays for quantifying cell‐signaling proteins in individual tumor samples, thereby improving patient stratification for targeted treatment.
Keywords: clinical mass spectrometry, formalin‐fixed paraffin‐embedded (FFPE), immunohistochemistry, protein assays, translation
1. Introduction
The most commonly used methods in clinical diagnostics for quantifying protein levels are immunoassays (IAs) such as enzyme‐linked immunosorbent assays (ELISA) and immunohistochemistry (IHC).[ 1 , 2 , 3 ] The general advantages of IAs are a simple workflow and high sensitivity, while IHC additionally provides spatial information about protein concentrations within tissue samples. Despite their widespread use, these assays can suffer from antibody cross‐reactivity and matrix effects. Moreover, IHC is only semi‐quantitative and has limited multiplexing capability.[ 3 , 4 , 5 ]
Mass spectrometry has emerged as a technology that allows these issues to be addressed. Recent advances in untargeted mass spectrometry using liquid chromatography coupled with mass spectrometry (LC‐MS) has enabled the large‐scale detection and quantification of low‐abundance proteins and peptides in complex matrices.[ 6 , 7 , 8 , 9 ] These methods offer high reproducibility and selectivity but have comparatively long analysis times, and require relatively expensive instrumentation and well‐trained operators. Additionally, these methods often lead to only “relative” results (i.e., up or down regulation), whereas, in a clinical context, the precise determination of protein/peptide levels is often required.[ 10 ]
A more appropriate technique for clinical analysis using mass spectrometry is “absolute” quantification using targeted proteomics.[ 10 , 11 ] Combining anti‐peptide antibody‐based immuno‐enrichment of peptides with mass spectrometry as quantitative readout has been shown to be a simple approach for improving selectivity and for achieving high sensitivity and throughput in complex biological samples.[ 12 ]
Clinical Relevance
In precision medicine and particularly precision oncology, there is an urgent need to quantify diagnostic/prognostic/predictive protein biomarkers. Currently, this is mostly done with immuno‐assays which, however, suffer from severe shortcomings—particularly, a lack of standardization, problems with antibody‐specificity, and subjective non‐quantitative data readouts and interpretation. These limitations have led to inconclusive results in biomarker studies, hindering their vital use for better stratification and more consistent treatment decisions, particularly for novel targeted therapeutics. To address the need for more‐standardized and precise assays for protein biomarkers, we present a complete template for developing optimized and automated immuno‐MALDI mass spectrometry assays. Using specific anti‐peptide antibodies, surrogate proteotypic peptides of the proteins‐of‐interest can be immuno‐enriched post‐digestion—together with their stable‐isotope labeled standards (SIS)—from clinical samples, including formalin‐fixed paraffin‐embedded (FFPE) tissues, followed by quantitation using benchtop MALDI‐TOF mass spectrometers which are present in most clinical laboratories. iMALDI synergizes the specificity and sensitivity from immuno‐enrichment with the precision, accuracy, and throughput from MS, and provides highly sensitive and robust data on actual protein expression levels (i.e., fmol of target protein per µg of total lysate protein). Here, we apply our standardized universal template to the quantitation of two cancer proteins, PTEN and PI3K.
In immuno‐matrix assisted laser desorption/ionization time of flight (iMALDI) mass spectrometry (MS), endogenous, or proteolytically derived peptides are enriched using antibodies that are coupled to magnetic beads, which then are directly spotted onto a MALDI plate, eluted by the matrix solvent, and analyzed using MALDI‐time‐of‐flight (TOF) MS (Figure 1 ).[ 13 ] The low complexity of the immuno‐enriched samples circumvents the need for elaborate and expensive LC‐MS instrumentation, while allowing the use of comparatively low‐cost MALDI‐TOF MS instrumentation for the readout. The presence of MALDI‐MS instruments in many clinical laboratories for microbial identification makes this technology especially well‐suited for clinical translation.[ 14 , 15 ]
Figure 1.
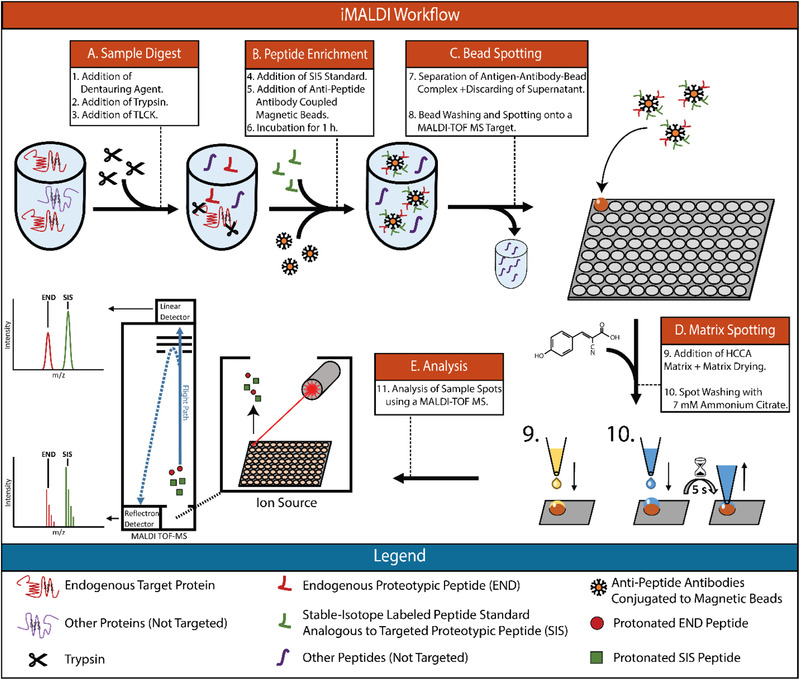
Immuno‐MALDI Mass Spectrometry Workflow. END—Endogenous Peptide. SIS—Stable‐isotope labelled standard. HCCA—hydroxy‐Cyanocinammic acid. Nα‐Tosyl‐l‐lysine chloromethyl ketone hydrochloride.
One iMALDI application of particular interest is the quantification of cell signaling proteins in patient tumor samples, which should be helpful for stratifying patients for targeted cancer treatment.[ 2 , 16 , 17 , 18 ]
In this study, we focused on the PI3K pathway which controls cell proliferation, survival, and apoptosis. It is commonly altered in many cancers, including breast cancer and colorectal cancer, and is a major drug target. PI3K p110α, and PTEN are commonly dysregulated on both the genetic and protein levels, and are indicators of PI3K pathway activity. [ 19 , 20 , 21 , 22 ] Quantifying these proteins in cancer tumors might, therefore, improve patient stratification for treatments targeting the PI3K pathway.
Since iMALDI is typically conducted using anti‐peptide antibodies specific to proteotypic peptides of the target protein, the use of synthetic stable‐isotope labelled internal standard (SIS) peptides allows the precise determination of protein concentrations.
An iMALDI assay comprises a number of sample preparation steps that can affect the overall outcome: i) digestion of protein lysates (both digestion time and relative trypsin amount used) to release endogenous target peptides (Figure 1), ii) coupling of the antibodies to magnetic beads, iii) peptide enrichment using anti‐peptide antibodies (Figure 1B), iv) separation of the antibody‐antigen‐bead complex from the sample, v) transfer of the beads to a MALDI target plate (Figure 1C), vi) matrix addition and target spot washing (Figure 1D), and vii) data acquisition and analysis (Figure 1E). Optimization and automation of these steps in order to improve handling time, robustness, and sensitivity is essential for translation of the developed assay into the clinic. For instance, digestion conditions need to be optimized to achieve reproducible release of the target peptide(s) as quickly as possible.[ 23 ] Similarly, antibody enrichment conditions need to be optimized, which can be challenging since various antibodies can exhibit very different binding kinetics.[ 24 , 25 ] The surface chemistry of magnetic beads used to bind anti‐peptide antibodies plays a crucial role as well, as it is important to minimize non‐specific binding of background peptides to the antibody‐coupled magnetic beads. This is of major importance for iMALDI, as it does not include liquid chromatography to reduce sample complexity, and is therefore more susceptible to interferences from the sample and dynamic range issues than other MS‐based assays. While this lack of liquid chromatography is a major advantage for the ease of translation, it requires a higher level of specificity of the actual enrichment in order to minimize and avoid interferences from the matrix.
Another challenge is to find suitable calibration matrices. Calibration using standard addition, that is, by using the sample itself as the matrix, is often impractical since large amounts of sample would be required for the complete workflow. Since, after successful and efficient immuno‐enrichment, both the complexity and the dynamic range of the matrix are limited, the use of surrogate matrices for generating calibration curves and determining the parameters of the assay is a valid strategy.[ 26 ]
In this study, we systematically evaluated and optimized critical steps of the iMALDI workflow in order to streamline the procedure and to further improve the robustness and sensitivity of iMALDI for quantifying cell signaling proteins. This study provides a template that can be followed to optimize future iMALDI assays.
2. Experimental Section
2.1. Materials
Reagents were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise specified. LC‐MS grade water and LC‐MS grade acetonitrile were purchased from Thermo Fisher (Waltham, MA). Unlabeled (NAT) and stable‐isotope labelled peptides (SIS) analogues of the PTEN peptide 148AQEALDFYGEVR159, as well as a double‐isotope labelled standard (dSIS) analogue of the AKT2 peptide 468THFPQFSYSASIRE481, were synthesized, purified, and quantified in‐house at the University of Victoria‐Genome BC Proteomics Centre (Victoria, Canada).[ 27 ] PTEN dSIS, as well as unlabeled, mono‐and‐double isotope labelled standards analogues of the p110α peptide 503EAGFSYSHAGLSNR516 were obtained from SynPeptide (Shanghai, China). Peptide concentration and purity were determined by amino‐acid analysis and capillary zone electrophoresis at the University of Victoria‐Genome BC Proteomics Centre.[ 27 ] Polyclonal rabbit anti‐peptide antibodies (pAbs) were ordered from Signatope (Reutlingen, Germany). Antibodies were generated and purified as described previously, but using the peptides AQEALDFYGEVR for PTEN and EAGFSYSHAGLSNR for p110α as antigens.[ 28 ]
Protein G Dynabeads and M280 Tosylactivated Dynabeads were obtained from Invitrogen (Carlsbad, CA). MagReSyn Protein G beads were purchased from Resyn Biosciences (Gauteng, South Africa). Trypsin (TLCK treated) was purchased from Worthington (Lakewood, USA). A Bravo 96 LT liquid handling robot (Agilent Technologies), equipped with a tip wash station and a plate shaker, was used for assay automation. Samples were analyzed using a Microflex LRT MALDI‐TOF MS (Bruker Daltonics, Bremen, Germany). µFocus MALDI target plates were purchased from Hudson Surface Technologies (Suwon, S. Korea).
2.2. General iMALDI Method
2.2.1. Preparation of Cell Lysates
E. coli BL21 DE3 cells were grown overnight in lysogeny broth (10 g L−1 Tryptone, 10 g L−1 NaCl, 5 g L−1 yeast extract) at 37 °C. Cells were pelleted and resuspended in PBS (pH 7.4). Protein extraction was performed using T‐PER buffer (Thermo Fisher). Protein concentration was determined using a Bicinchoninic acid assay (BCA) assay (Thermo Fisher).
MDA‐MB 231 cell lysates were prepared as described previously.[ 13 ]
2.2.2. Antibody‐Bead Coupling
Protein G Dynabeads were washed 7× with 25:75 acetonitrile: PBSC (i.e., PBS+0.015% CHAPS, w:w) and 3× PBSC buffer, using 1:10 bead‐slurry: buffer (v:v). This step was automated using a Bravo 96 LT liquid handling robot (Figure S2A, Supporting Information). Rabbit polyclonal anti‐peptide antibodies (1 µg µL−1 in PBS+0.05% sodium azide) were added (0.2 µg antibody per 30 µg beads) and incubated while being rotated at room temperature for 1 h. Prior to use, the antibody‐coupled beads were washed 3× with PBSC and reconstituted in PBSC to give a final concentration of 1.5 µg beads µL−1 (0.01 µg antibody µL−1).
2.2.3. Tryptic Digest
E. coli and MDA‐MB 231 lysates were diluted to a concentration of 0.1 µg protein µL−1 using cold (4 °C) 20 mm TRIS HCl at pH 8 supplemented with 0.015% CHAPS (TRIS+C). Each sample was aliquoted in 100‐µL aliquots (10 µg total protein, each). Using a Bravo 96LT liquid handling robot (Figure S2B, Supporting Information), 10 µL of 10% sodium deoxycholate (to give a final concentration of 0.9%) were added to each aliquot, and samples were incubated for 30 min at 60 °C. 10 µL of trypsin solution (0.2 µg trypsin µL−1 in 1 mm HCl, 20 µg total trypsin per replicate) were added and samples were incubated at 37 °C for 1 h. Ten µL of 170 µm Nα‐Tosyl‐l‐lysine chloromethyl ketone hydrochloride (TLCK) solution were added to stop the digestion (Figure 1A).
2.2.4. Peptide Enrichment
The following liquid handling steps were performed using a Bravo 96 LT liquid handling robot (Figures S1 and S2, Supporting Information). Internal standard (SIS or dSIS) and, where applicable, NAT were added to the digested samples prior to enrichment, the precise amounts are specified in the according sections below. Twenty µL of antibody‐bead slurry (1.5 µg beads µL−1, 10 ng antibody µL−1) was added to the sample and incubated for 1 h at room temperature, while shaking at 1000 RPM (Microplate Vortex 120 V ADV, Thermo Fisher). The antigen‐antibody‐bead complex was separated, washed 1× using 70 µL of PBSC, 3× using 80 µL of 5 mm ammonium bicarbonate (AmBic). After resuspension in 10 µL of AmBic, the beads were subsequently spotted onto a 2600 µm µFocus MALDI target plate. After the spots were dried, 1.5 µL of matrix (3 mg mL−1 α‐cyano‐4‐hydroxycinnamic acid, 7 mm ammonium citrate dibasic in 70% acetonitrile (ACN)/0.1% trifluoroacetic acid (TFA, Thermo Fisher)) were added. After drying, spots were washed three times as follows: 5 µL of 7 mm ammonium citrate dibasic (AmCit, pH≈5) were added on top of each spot, and removed after 5 s (Figure 1B–E).
2.2.5. Data Acquisition and Analysis
The MALDI plates were analyzed on a Bruker Microflex LRT in both linear positive (LP) and reflectron positive (RP) mode. One thousand shots were accumulated per spot in 25‐shot intervals using a “random walk” pattern. The data was analyzed using FlexAnalysis (Bruker, v3.4, Build 70). Linear mode spectra were smoothed using the Savitzky Golay algorithm (10 cycles with a 1‐Da width and TopHat baseline subtraction). Peaks were detected using centroid mode (Peak width = 1 Da, height = 80%). Reflectron mode mass spectra were smoothed using Savitzky Golay (1 cycle, Peak width = 0.2 Da and TopHat baseline subtraction). Peaks were detected using SNAP (SNAP average composition = Averagine[ 29 ]). Mass lists were exported and analyzed using R.[ 30 ]
2.3. Selection of Proteotypic Peptides for Antibody Development
Recombinant PTEN protein was purchased from Abcam (85% purity). Recombinant PI3K p85α p110α was gifted by Dr. John Burke. Candidate proteotypic peptides for antibody development were identified using Peptide Picker.[ 31 ] Candidate peptides were experimentally confirmed using tryptic digests of the respective recombinant proteins analyzed by MALDI‐TOF MS.
One microgram of recombinant protein was denaturated in 15 µL of 2 m Urea, reduced with 2 µL 20 mm dithiotreitol (DTT) for 30 min at 37 °C, and alkylated with 2 µL of 80 mm iodoacetamide for 30 min at 37 °C. Alkylation was quenched using 1 µL 80 mm of DTT, followed by digestion using 1 µL trypsin solution (0.1 µg µL−1 in 1 mm HCl) for 1 h at 37 °C. The digests were desalted using C18 ZipTips (Millipore, Burlington, MA) and eluted in 20 µL of 50% ACN, 0.1% TFA, 2 µL (0.05 µg digested protein) of which were used for analysis. Matrix addition and spot washing were done as described above. The samples were analyzed using an Ultraflex III (Bruker).
Polyclonal antibodies (pAbs) were raised against the most intense proteotypic peptides identified in the respective protein digests (PTEN: 148AQEALDFYGEVR159, p110α: 503EAGFSYSHAGLSNR516). Two pAbs were generated for each target peptide.
Corresponding synthetic peptide standards with and without isotopic labeling were generated as described above.
To compare the pAb performance, the E. coli lysate digest (10 µg total protein per replicate) was spiked with 2.5 fmol per replicate of PTEN and p110α NAT peptides. Using the spiked E. coli digest as the sample, PTEN and p110α NAT peptides were enriched using the different pAbs coupled to Protein G Dynabeads, as described above (N = 4 per pAb). Additionally, the MALDI matrix was spiked with AKT2 dSIS peptide as external standard to a concentration of 0.67 fmol µL−1, resulting in 1 fmol peptide per sample spot using 1.5 µL of matrix. Antibody‐enrichment efficiencies of the different antibodies were compared using the NAT:AKT2 dSIS ratios and two‐sided t‐tests with a confidence level of 0.99.
2.4. Comparison of Manual versus Automated Wash
Ten 10‐µg aliquots of E. coli cell lysate were each spiked with 2.5 fmol PTEN (AQEAL(+7)DFYGEVR(+10)) and p110α (EAGFSYSHAGL(+7)SNR(+10)) double‐SIS (dSIS) peptide. iMALDI assays were done according to the procedure described above. For five samples, the antigen‐antibody‐magnetic bead complexes were washed and spotted manually after incubation with the sample; the other five were washed and spotted using the automated bead washing + spotting protocol (Figure S1, Supporting Information). AKT2 dSIS (THF(+10)PQFSYSASIR(+10)E) peptide was added to the matrix to a concentration of 0.67 fmol µL−1 as an external standard, resulting in 1 fmol AKT dSIS peptide per sample spot. Automated and manual bead washing were compared based on the ratio between PTEN and p110α dSIS to the AKT2 dSIS intensities, respectively by two‐sided t‐tests with a confidence level of 0.99.
2.5. Optimization of Tryptic Digestion
Protein:trypsin ratios of 10:1 and 1:2 as well as digestion times of 0.5, 1, 2, and 4 h were compared. Using MDA‐MB 231 cell lysate (0.1 µg total protein µL−1), thirty two 100‐µL aliquots were prepared (10 µg of total protein each). Sixteen aliquots each were digested using a protein:trypsin ratio of 10:1 (w:w) and 1:2 (w:w), respectively. For both tested ratios, four replicates each were digested for 0.5, 1, 2, and 4 h at 37 °C. Afterwards, 2.5 fmol PTEN and p110α SIS and dSIS peptides were added as internal standards. iMALDI assays, with AKT2 dSIS peptide added as internal standard to the matrix, were done as described above. Peptide recoveries were calculated using the ratio of the peak intensities of the released endogenous peptide to the spiked‐in dSIS peptide. The release of endogenous proteotypic peptides was compared for the different incubation times and for both protein:trypsin ratios. The results were evaluated using a two‐sided t‐tests with a confidence level of 0.99.
2.6. Optimization of Calibration Strategies
PBSC, BSA digest (0.1 µg µL−1 in TRIS+C buffer), and E. coli digest (0.1 µg µL−1 in TRIS+C buffer) were tested as possible calibration matrices. PTEN (AQEALDFYGEVR) and p110α (EAGFSYSHAGLSNR) NAT peptide standards (c = 1.000, 0.500, 0.250, 0.125, 0.062, 0.030, 0.000 fmol µL−1) were prepared manually.
Calibration curves were prepared by adding 20 µL of the respective standards to either PBSC, BSA digest (10 µg total protein per replicate) or E. coli digest (10 µg total protein per replicate), yielding amounts from 0 to 20 fmol per replicate. Additionally, four replicates of MDA‐MB 231 digest (100 µL of 0.1 µg total protein µL−1) were prepared to quantify endogenous PTEN and p110α using the different calibration strategies. A solution containing 2.5 fmol PTEN and p110α SIS and dSIS peptides was added as internal standards to each sample. iMALDI assays were conducted as described above using automated wash protocols.
To determine whether the peak parameters used for generating the calibration curve would affect the quantification, calibration curves were generated using NAT:dSIS ratios based on either the peak intensities, the peak areas, or the S/N ratios of the NAT and dSIS peak heights, generating a total of three calibration curves per matrix. For the MDA‐MB 231 samples, the NAT:dSIS ratios were determined the same way: PTEN and p110α were quantified using the three different calibration curve strategies for each matrix. The results were compared using two‐sided t‐tests with a confidence level of 0.99.
2.7. Optimization of Immuno‐Enrichment
E. coli cell lysate digest was used as sample matrix for the experiments described below. Prior to digestion as described above, the lysate was reduced and alkylated as described previously.[ 13 ]
2.8. Optimization of Bead Types and MALDI Plate Spot Sizes
Three different types of magnetic beads were tested for antibody coupling: Protein G Dynabeads, M280 Tosylactivated Dynabeads, and Protein G MagReSyn microspheres. Additionally, two types of MALDI plates were tested for each bead type: 2600 µm µFocus MALDI plates and 700 µm MFX µFocus MALDI plates (Hudson).
2.8.1. Bead Preparation
Protein G Dynabeads were prepared as described above (bead suspension “PG #1”). The bead suspension was diluted 10‐fold using PBSC (3 µg beads (0.02 µg antibody) per 10 µL, bead suspension “PG #1_1/10”).
M280 Tosylactivated Dynabeads were prepared by washing 0.625 mg of beads with 200 µL of 0.1 m sodium phosphate buffer (pH 7.5), resuspending them in 6.25 µL of PTEN and 6.25 µL p110α antibody (c = 1 mg mL−1 each) in 1 m ammonium sulfate, and incubating them for 20 h at 37 °C with rotation. The supernatant was removed, 0.2 mL of 20 mm TRIS+0.015% CHAPS buffer (buffer C) were added for quenching, and samples were incubated for 1 h at 37 °C with rotation. The beads were washed twice with 0.2 mL of buffer C and resuspended in 20.6 µL of buffer C (30 µg beads (0.6 µg antibody µL−1)). From this stock suspension, two dilutions were prepared using PBSC: “C #1” (30 µg beads, 0.6 µg total antibody per 20 µL), and “C #1_1/10” (3 µg beads, 0.06 µg total antibody per 20 µL).
Protein G MagReSyn beads (bead suspension “PG #2”) were prepared by resuspending 40.8 µg beads in 600 µL PBSC and sonicating them with a sonication probe (Sonic Dismembrator Model 100, Thermo Fisher) using short bursts (1 s using intensity level 1) until the suspension was homogenous and free of clumps. The beads were then placed on a magnet to remove the supernatant and the beads were resuspended in 20 µL PBSC. The beads were washed 7x with 25:75 ACN:PBSC and 3× with PBSC, as described for the in Antibody‐bead Coupling section. After washing, the beads were resuspended in 50 µL of PBSC and split into two aliquots. A solution of 4.3 µL of PTEN and p110α antibody solution were added, respectively, and incubated for 1 h with rotation. The conjugated beads were stored at 4 °C with rotation, until used. Prior to use, PTEN and p110α aliquots were combined, the supernatant was removed, and the beads were washed three times using 300 µL of PBSC. Finally, the beads were resuspended in 263 µL of PBSC (final concentration = 0.155 µg beads, 0.025 µg antibody per 1 µL).
2.8.2. Assay Preparation
E. coli digest (0.083 µg total protein µL−1) was used as sample matrix, from which thirty two 120‐µL aliquots were prepared. Additionally, eight 100‐µL PBSC aliquots were prepared. PTEN+p110α NAT+SIS+dSIS peptides (1.25 fmol) were added to each sample. To 4 replicates of E. coli digest and one replicate of PBS+CHAPS, 20 µL of bead suspensions “PG #1”, “PG 1_1/10”, “PG #2”, or “C” #1’ and “C #1_1_1/10” were added, respectively. To eight replicates of E. coli digest and two replicates of PBSC, 20 µL of bead suspension “PG #2” were added.
The assays were performed as described above. The antigen‐antibody‐bead complexes were spotted onto two different MALDI Plates: “PG #1”, “C #1”, and “PG #2” (half of the prepared replicates) were spotted onto a 2600 µm µFocus MALDI plate: “PG #1_1/10”, “C #1_1/10”, and “PG #2” (second half of the prepared replicates) were spotted onto a 700 µm MFX µFocus MALDI plate. Matrix spotting for the 700 µm MFX µFocus MALDI plate was performed by manually adding 0.2 µL of matrix solution.
2.9. Direct versus Indirect Immuno‐Enrichment
Twelve aliquots of E.coli digest (150 µL, 10 µg of total protein), spiked with 1 fmol PTEN NAT and dSIS standards, were prepared in a 1.1‐mL Deep Well plate. Six samples were enriched using direct immuno‐enrichment; the other six were enriched using indirect immuno‐enrichment.
Indirect enrichment was performed as follows: Ten µL of 0.02 µg µL−1 PTEN anti‐peptide antibody solution were added to each of the six aliquots and incubating for 1 h at 1000 RPM at room temperature. A 30‐µg aliquot of Protein G Dynabeads was added to the sample solution to bind the peptide‐antibody complex. Bead washing, spotting, and analysis were done as described before. After immuno‐enrichment, the matrix (1.5 µL) was spiked to 0.67 fmol µL−1 AKT2 dSIS peptide, resulting in 1 fmol of external standard per sample spot. Two‐sided t‐tests with a confidence level of 0.99 were performed for comparing the NAT: AKT2 dSIS peptide ratios between the two conditions tested.
2.10. Incubation Times
Three PCR plates were prepared containing 10 aliquots of E. coli digest (150 µL, 10 µg of total protein) spiked with 1.25 fmol (20 µL 0.0625 fmol peptide µL−1) of PTEN and p110α NAT standard (Plates A, B, C). Additionally, six more aliquots were prepared in the same way in a 1.1‐mL U‐bottom deep well plate (Plate D). Plates A, B, and C were incubated rotating (8 RPM), Plate D was incubated shaking at 1000 RPM. Plates A, B, and D were incubated at room temperature, while Plate C was incubated at 4 °C. Plates A and D were incubated for 1 h, while plates B and C were incubated for 22 h overnight. Plate incubation was timed so that all plates would finish the incubation step at the same time. iMALDI assays were conducted as described above, but the matrix was spiked with AKT2 dSIS peptide as external standard to a concentration of 0.67 fmol µL−1, resulting in 1 fmol peptide per sample spot using 1.5 µL matrix. Two‐sided t‐tests with a confidence level of 0.99 were performed to compare the NAT:AKT2 dSIS peptide ratios of plate A to plates B, C, and D, as well as plate B to plate C.
3. Results and Discussion
3.1. Antibody Generation
Potential proteotypic peptide targets for PTEN and p110α were identified in silico using our Peptide Picker software.[ 31 ] Peptides with a length of 7–20 amino acids following Keil rules, present in all isoforms and without Tryptophan or strings of Proline and Serine were considered. Candidate peptides were experimentally confirmed by analyzing tryptic digests of recombinant PTEN and p110α using MALDI‐TOF MS (Figure S3A,B, Table S1, Supporting Information). The most intense proteotypic peptides were selected for antibody generation, namely the PTEN peptide 148AQEALDFYGEVR159 (Figure S3A, Supporting Information) and the p110α peptide 503EAGFSYSHAGLSNR516 (Figure S3B, Supporting Information). Having identified the proteotypic peptides with the highest sensitivity using MALDI‐TOF MS, rabbit polyclonal antibodies were generated against these target peptides and used for the experiments described below.
3.2. Comparison of Automated and Manual Wash
All liquid handling steps were automated using a Bravo liquid handling robot (Agilent Technologies). The automated separation of the antigen‐antibody‐bead complex from the sample solution, and the washing and spotting onto a MALDI plate can lead to losses of target peptide at various stages. Loss of antigen‐antibody‐bead complex may occur during liquid transfer and bead washing steps, and bead washing steps may be less effective due to less efficient removal of supernatants, leading to higher non‐specific background levels. Due to the complexity of the procedure and the potential for numerous pitfalls during automation, automated bead washing and spotting were also compared to the manual procedure (Figure 2 , Table S2, Supporting Information).
Figure 2.
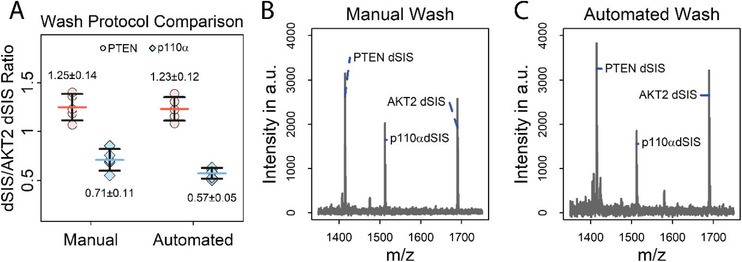
Comparison of automated versus manual washing of the antigen‐antibody‐bead complexes. A) Comparison of the efficiency of manual versus automated bead washing for enriching PTEN and p110α dSIS peptide (2.5 fmol) spiked into E. coli digest. The signal of the stable isotope labelled AKT2 peptide (THF(+10)PQFSYSASIR(+10)E) spiked into the MALDI matrix (1 fmol per spot) was used for normalization. Error bars represent standard deviation, horizontal bars indicate means. Values above and below the data points represent the mean and absolute standard deviation; N = 5. B,C) Mass spectra of antigen‐antibody‐bead complex using B) manual and C) automated bead washes, with data recorded in the reflectron mode, showing similar non‐specific backgrounds in both bead‐washing methods.
A comparison of manual and automated washing and spotting revealed no significant difference (p > 0.01) in enrichment efficiency, as measured by the ratio of PTEN and p110α dSIS to AKT2 dSIS (PTENautomated/manual = 0.98; p110αautomated/manual = 0.80), but particularly for p110α automation improved the CV considerably from 15% to 9%. (Figure 2A). Additionally, mass spectra of iMALDI assays with automated and manual bead washing and spotting show similar backgrounds, demonstrating that both are equally effective (Figure 2B). However, hands‐on time is reduced by approximately a factor of six using automation—manual bead washing and spotting using a full 96‐well plate took approximately 90 min, compared to 15 min using the liquid handling system. Manual matrix spotting and spot washing of 96 spots took approximately 60–80 min, whereas the automated protocols required 30 min with little hands‐on time. Thus, automation allows the preparation of hundreds of samples per day, without compromising precision (PTENManual/Automated CV = 11/10%, p110αManual/Automated CV = 15/9%).
3.3. Optimization of Tryptic Digestion
One of the characteristics of our previously reported protein iMALDI assays is the use of protease inhibitors during lysis and consequently the use of a relatively high amount of trypsin, compared to standard proteomics workflows.[ 23 , 32 ] In this paper, we report the optimization of the iMALDI digestion conditions for the PTEN and p110α assays in which different protein:trypsin ratios were tested. Specifically, we compared the results of using a 1:2 protein:trypsin ratio, which had previously been used for an AKT iMALDI assay, to a 10:1 protein:trypsin ratio which is more commonly used in typical proteomics experiments, to reduce the occurrence of potential none‐tryptic cleavages derived from residual contamination with chymotrypsin and to reduce the abundance of tryptic autoproteolysis products.[ 13 ] In addition, different incubation times (from 0.5 to 4 h) were tested (Figure 3 ). Ten µg of MDA‐MB 231 lysate was used as sample, 2.5 fmol of PTEN and p110α dSIS were spiked in.
Figure 3.
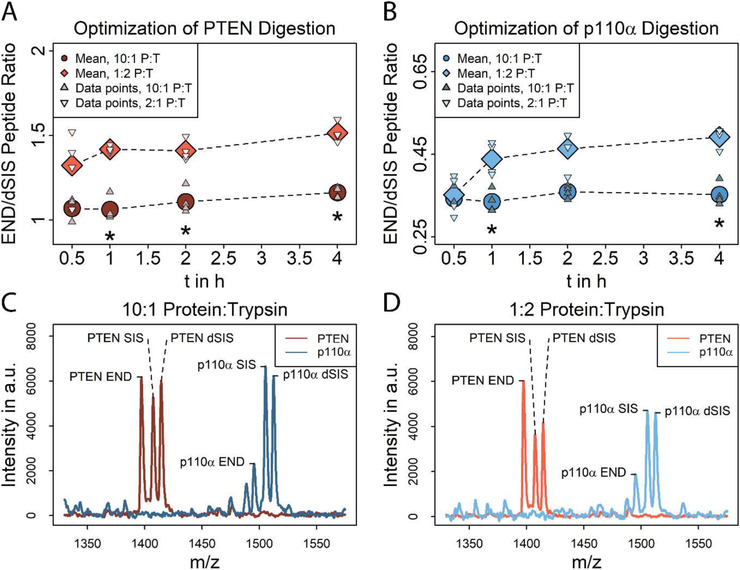
Optimization of tryptic digestion using MDA‐MB 231 lysate spiked with 2.5 fmol of both SIS and dSIS peptide. A) PTEN (148AQEALDFYGEVR159) peptide recoveries at different incubation times (0.5–4 h) and at protein:trypsin (P:T) ratios of 1:10 (dark red) and 2:1 (light red). (*) indicates a significant difference (p < 0.01) between two protein:trypsin ratios at the same incubation time. N = 4 per tested incubation time. B) p110α (503EAGFSYSHAGLSNR516) recoveries at different incubation times (0.5–4 h) at protein:trypsin ratios of 1:10 (light blue) and 2:1 (dark blue). (*) indicates significant difference (p < 0.01) between two protein:trypsin ratios at the same incubation time. N = 4 per tested incubation time. C,D) Overlaid mass spectra of enriched PTEN and p110α peptides after 1 h digest using C) 10:1 and D) 1:2 protein:trypsin, recorded in the linear mode. Spectra show similar background for both tested protein:trypsin ratios and no peaks are interfering with the target peptide peaks.
For both target peptides and the two protein:trypsin ratios tested, the release of the endogenous target peptide as determined by the END:dSIS ratio, did not significantly improve with incubation times greater than 1 h. For example, no significant difference (p > 0.01) between END:dSIS ratios was observed between a 1 and 4 ‐h incubation using 10:1 protein:trypsin, for either target peptide (PTEN1 h:4 h = 0.91, p110α 1 h:4 h = 0.97). Peptides were readily released, and the END:dSIS ratios observed indicate that chymotryptic side‐activity is unlikely, even when very high amounts of trypsin were used, as chymotryptic cleavage of the target peptides would have been more pronounced in the samples with higher trypsin concentration, leading to reduced END levels that would have been reflected by lower END:dSIS ratios.
Overall, peptide recoveries were approximately 30% higher when using a 1:2 protein:trypsin ratio (Figure 3A,B, Table S3, Supporting Information).
For both proteins, the protein:trypsin ratios and the digestion times tested did not change the non‐specific background. No background peaks interfered with either PTEN or p110α END peptides, or their respective SIS and dSIS standards at either digestion ratio (Figure 3C,D). CVs were consistently below 10% for both peptides, with the exception of 0.5 h incubation using 1:2 protein:trypsin (PTEN/p110α CV = 15/14%).
For further experiments, we chose to use a 1 h incubation time using a 1:2 protein:trypsin ratio, since incubation times of greater than 1 h did not improve the release of endogenous target peptide and it was desirable to keep turnaround times short. Using a high ratio of trypsin to protein ensures high digestion efficiency, and after immuno‐enrichment, we found no background peaks which interfered with the peaks from the enriched peptides.
3.4. Optimization of Calibration Strategies
Different methods of creating a calibration curve were evaluated by quantifying endogenous PTEN and p110α in MDA‐MB 231 cell lysate. This was done to test the influence of the calibration matrix and the peak parameters used for quantification. Three external calibration curves were generated using matrices of increasing complexity: PBSC buffer, BSA digest (10 µg total protein/replicate), and E. coli digest (10 µg total protein per replicate). The target peptides were measured using both the linear and reflectron modes to determine whether the difference in resolution and signal intensity would affect the quantitation.
NAT:dSIS ratios of samples and calibrators were calculated using either i) peak intensities, ii) S/N ratios, or iii) peak areas of the respective peaks, in both linear and reflectron modes, resulting in six calibration curves for each matrix. The amounts of PTEN and p110α in the individual samples were calculated independently using the different calibration matrices and NAT:dSIS readouts (intensity, S/N ratio, peak area) (Figure 4A–D, Table S4–S6, Supporting Information).
Figure 4.
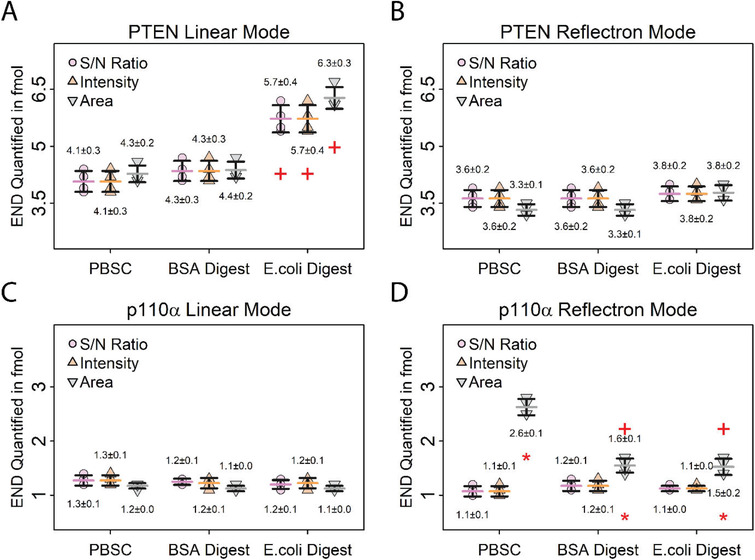
Evaluation of different calibration strategies by quantifying PTEN and p110α in the same sample using different calibration matrices and peak parameters. Error bars represent standard deviations, horizontal bars indicate means. Values above and below the data points represent the means and absolute standard deviations. A,B) Quantification of endogenous PTEN in 10 µg MDA‐MB 231 digest using different calibration matrices and peak parameters. Data recorded in the linear mode A) shows differences in PTEN quantification using E. coli digest as matrix, whereas data recorded in the B) reflectron mode shows no difference between the matrices. (+) indicates a significant difference (p < 0.01) between E. coli digest and PBS+CHAPS matrices for PTEN quantification using the same peak parameters for calculating the calibration. This difference is due to an interference from the calibration matrix with the internal standard for PTEN. C,D) Quantification of endogenous p110α in 10 µg MDA‐MB 231 digest using different calibration matrices and peak parameters. Data recorded in the C) linear mode shows no difference in p110α quantification using different matrices and peak parameters, while the D) reflectron mode data shows differences when peak area is used for quantification, likely due to less accurate SNAP peak modelling for low‐intensity peaks. (+) indicates a significant difference (p < 0.01) between E. coli digest and BSA to PBS+CHAPS matrices using the same peak parameter for calculating the calibration. (*) indicates a significant difference (p < 0.01) between peak area and S/N ratio for p110α quantification. N = 4 per calibration matrix.
Comparing the amounts of quantified PTEN peptide in the samples using the different peak properties for generating the calibration curve showed no significant difference using either S/N, peak intensities, or peak areas in the linear and reflectron modes. For example, using PBSC as calibrator matrix, the mean quantified peptide amounts in the linear mode were 4.1 fmol using S/N (CV = 7%), 4.1 fmol using intensities (CV = 7%), and 4.3 fmol using peak areas (CV = 5%). No significant difference was found between using S/N and intensities (p = 1.00), S/N and peak areas (p = 0.38), or Intensities and peak areas (p = 0.38). Furthermore, no significant differences were observed between using S/N ratio, peak area or intensity when using BSA or E. coli digests as the matrix (Figure 4A,B).
Notably, a significant difference was observed when quantifying p110α in the reflectron mode based on peak areas. For example, when PBS+CHAPS was used as the calibrator matrix, mean p110α amounts of 1.1 fmol (CV = 9%) were found using both S/N and peak intensities, compared to 2.6 fmol (CV = 6%, p < 0.01) calculated using peak areas (Figure 4C,D). This is likely due to inaccurate peak fitting during the reflectron‐mode peak analysis. Particularly for low‐intensity peaks, such as the endogenous p110α peaks observed here, the peak modelling using SNAP may be less accurate for projected isotope patterns, which affects peak areas more than S/N ratios.
A comparison between the calibrator matrices showed no significant differences for the quantification of p110α (Figure 4C,D).
For PTEN, no significant differences between calibration matrices were observed in the reflectron mode. Using the linear mode, the quantified amounts were higher using an E. coli digest as the matrix. This can be explained by interference from a background peak with the internal standard peak in the linear mode, which was avoided in the reflectron mode because of the increased resolution (Figure 4A, Figure S4, Supporting Information). These results indicate that evaluating different matrices in both linear and reflectron modes is recommended when developing a new iMALDI assay.
In conclusion, we found that the calibration matrix has little impact on the quantification, other than potential interferences from background peaks in the mass spectrum. This, however, can be a major concern, especially in MALDI‐TOF MS, since — without LC separation — there are fewer options for resolving such interferences compared to LC‐MS. Thus, the use of a BSA digest (10 µg per replicate) seems to be the most appropriate compromise. Peak intensity is the preferred quantification strategy, as we have found that peak area is more dependent on the particular peak fitting method used and therefore is more prone to errors, for example, in the case of p110α in the reflectron mode. In our hands, S/N ratios were slightly more prone to errors when calculated by the FlexAnalysis software, so again peak intensity appears to be the parameter that is the least influenced by spectrum processing. The lower limit of quantitation was 0.7 fmol on the MALDI target spot for both peptides in the linear mode, or 0.9 fmol for p110α and 1.4 fmol for PTEN in the reflectron mode. Notably, both the linear and reflectron modes were found to be suitable for peptide quantification, and it is feasible to measure all samples in both modes as an internal quality control measure.
3.5. Optimization of Immuno‐Enrichment
3.5.1. Optimization of Bead Types and MALDI Plate Spot Sizes
Different types of magnetic beads were compared for antibody coupling (Figure 5A,B, Table S7, Supporting Information): two types of protein G coupled beads (one a solid bead (PG #1) and the other a porous microsphere (PG #2)), as well as tosyl‐activated magnetic beads (solid spheres) which covalently bind antibodies (C #1). In addition to different bead types, different MALDI plates with different spot sizes were tested: 2600 µm µFocus and 700 µm µFocus plates. Concentrating the same amount of analyte on a much smaller surface area could potentially lead to higher sensitivities.[ 33 ]
Figure 5.
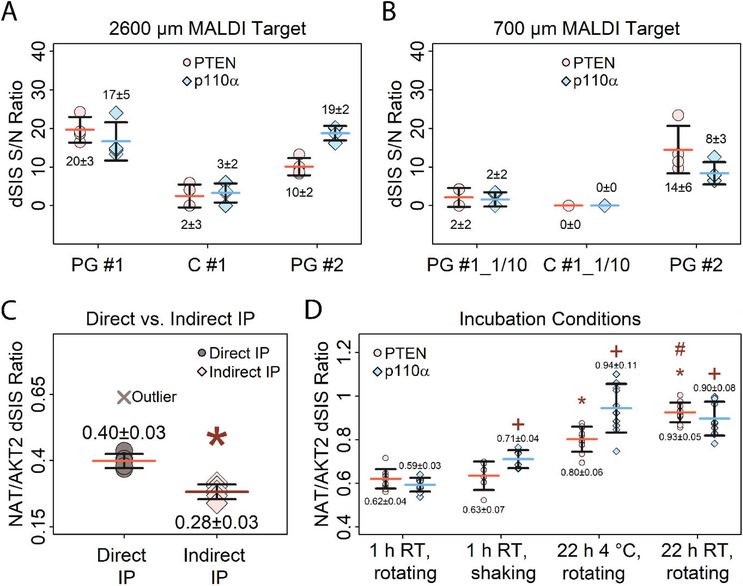
Optimization of immuno‐enrichment. Error bars represent standard deviation, horizontal bars indicate means. Values above and below the data points represent the mean and absolute standard deviation A) Comparison of different bead types for enriching 2.5 fmol PTEN and p110α dSIS peptides spiked into 10 µg E. coli digest, using a 2600 µm MALDI target plate. Protein G Dynabeads (PG #1), Protein G MagReSyn beads (PG #2) and M280 tosylactivated Dynabeads (C #1) were tested. Peak S/N ratios of the enriched dSIS peptides were used for comparison. B) Comparison of different beads for enriching 2.5 fmol PTEN and p110α dSIS peptides spiked into 10 µg E. coli digest, using a 700 µm MALDI target plate. Peak S/N ratios of the enriched dSIS peptides are used for comparison. The same antibody‐coupled beads as in (A) were tested, using 1/10 of the bead amount (PG #1_1/10, C #1_1/10), except for PG #2 (same as in (A)). N = 4 per bead type and MALDI target plate. Data for (A) and (B) were recorded in the reflectron mode. C) Comparison of direct and indirect immuno‐enrichment for PTEN NAT peptide (1 fmol) spiked into 10 µg E. coli digest, showing better recovery using direct IP. Outlier (>Q3+3x Interquartile Range) was excluded due to poor quality of the mass spectrum. MeanDirect IP (outlier included) = 0.44 ± 0.1. The signal of AKT2 dSIS standard spiked into the MALDI matrix (1 fmol per spot) was used for normalization. (*) indicates significant difference (p < 0.01) to direct IP. Data was recorded in the linear mode. D) Testing different incubation times, temperatures and mixing conditions for enriching PTEN and p110α NAT peptide (1.25 fmol) spiked into 10 µg of E. coli digest. The signal of the double‐stable‐isotope‐labelled AKT2 peptide (THF(+10)PQFSYSASIR(+10)E) spiked into the MALDI matrix (1 fmol per spot) was used for normalization. (*) indicates significant difference (p < 0.01) of PTEN enrichment after 22 h incubation compared to 1 h incubation, (+) significant difference (p < 0.01) between p110α enrichment compared to 1 h incubation, and (#) indicates significant difference between PTEN enrichment after 22 h at room temperature (RT) and 22 h at 4 °C. N = 10 for each condition, except 1 h shaking (N = 6). Data was recorded in the linear mode.
A comparison of different types and amounts of beads showed that the covalently coupled antibody‐beads, in general, did not perform as well as antibodies non‐covalently bound to protein G beads. This was indicated by a mean p110α dSIS S/N of three for the covalently coupled beads, compared to 17 for the protein G‐coupled beads (PG #1). Both protein‐G beads performed similarly, giving mean p110α S/N ratios of 17 for PG #1 compared to 19 for PG #2, and mean PTEN S/N ratios of 20 versus 10 for PG #1 and #2. The porous microspheric PG #2 beads have an approximately 20× higher binding capacity than PG #1, which is why only 3 µg of PG #2 beads were used per replicate, compared to 30 µg for PG #1 (note that using <3 µg of PG #2 proved impractical). Still, using 3 µg PG #2 resulted in 0.5 µg of antibody available per replicate, twice as much as the 0.2 µg of antibody using 30 µg PG #1. However, neither the lower amount of beads nor the higher amount of available antibody using PG #2 seemed to improve enrichment. CVs of S/N ratios between PG #1 and PG #2 were comparable for PTEN, while CVs for p110α peptides were slightly lower using PG #2 (PTENPG#1/PG#2 CV = 0.8, p110αPG#1/PG#2 CV = 3).
For evaluating the 700 µm µFocus plates, PG #1 and C #1 bead amounts had to be reduced by a factor of 10 in order to avoid overloading the MALDI plate (PG #1_1/10 and C #1_1/10). PG #2 already required a very small amount of beads for the 2600 µm µFocus plates (3 µg) and did not need downscaling.
Using a 700 µm µFocus plate, very low S/N ratios (S/N = 2) were observed for both PTEN and p110α using PG #1_1/10. Using C #1_1/10, no analytes were detected. In contrast, S/N ratios of 14 for PTEN and 8 for p110α were observed using PG #2, which are closer to the observed S/N ratios of ten for PTEN and 19 for p110α on a 2600 µm µFocus plate. Thus, using PG #2 beads yielded comparable results on both MALDI target spot sizes tested. However, no increase in sensitivity was achieved. Handling the extremely small volumes (as low as 0.2 µL) and bead amounts (3 µg and lower) necessary for using small anchor plates also posed major challenges to the liquid handling system used in this study.
In conclusion, both types of protein G beads were found to be suitable for iMALDI. However, in our experience, bead performance may vary substantially depending on the antibody and sample matrix used. We found no improvement using smaller MALDI target spots for iMALDI, which may be related to the additional challenges posed by the automation of a miniaturized system.
3.5.2. Direct versus Indirect Immuno‐Enrichment
Both direct immuno‐enrichment (i.e., adding antibodies immobilized to magnetic beads to the sample) and indirect immuno‐enrichment (i.e., adding unbound antibody to the sample, then enriching the antigen‐antibody complex using magnetic beads) of the target peptides were compared using E. coli digest spiked with PTEN NAT and dSIS peptides as the samples (Figure 5C, Table S8, Supporting Information). Samples were immuno‐enriched on antibody‐immobilized beads for 1 h (direct) or by using free antibody for 1 h, followed by a 1 h incubation with magnetic protein G coupled beads (indirect). AKT2 dSIS peptide was added to the MALDI matrix as an internal standard to allow comparison of the two methods.
Our data demonstrate that the direct approach increases the recovery of the target peptide (NATDirect/Indirect = 1.4, p < 0.01). Low CVs were achieved using either approach (CVDirect = 7%, CVIndirect = 10%). Although direct enrichment performed slightly better, indirect enrichment is still feasible with iMALDI.
3.5.3. Incubation Times
To optimize incubation conditions, the influence of i) incubation time of the sample with the antibody‐coupled beads, ii) sample mixing during enrichment, and iii) enrichment temperature were evaluated (Figure 5D, Table S9, Supporting Information). An E. coli digest spiked with PTEN and p110α NAT and dSIS peptides was used as the sample. AKT2 dSIS peptide was added to the MALDI matrix as internal standard, and enrichment efficiencies were compared based on the ratios of PTEN NAT/AKT2 dSIS, and p110α dSIS.
To test the impact of incubation time on peptide yield, samples were incubated at room temperature either overnight or for 1 h, while rotating. Longer incubation time slightly improved the enrichment (PTEN1h_rotating/Overnight_RT_rotating = 0.67, p110α1h_rotating/Overnight_RT_rotating = 0.66).
Immuno‐enrichment for 1 h at room temperature either using end‐over‐end rotation or shaking at 1000 RPM, yielded comparable results for PTEN (PTEN1h_rotating/1h_shaking = 0.98) and slightly better enrichment for p110α (p < 0.01, although the observed increase was small, with p110α1h_rotating/1h_shaking = 0.83).
The effect of incubation temperature (RT vs. 4 °C) was tested by incubating samples overnight while rotating.
Both incubation temperatures showed comparable enrichment (PTENOvernight_4 °C_rotating/Overnight_RT_rotating = 0.86, p110αOvernight_4 °C_rotating/Overnight_RT_rotating = 1.04). Low CVs between 5–12% were achieved regardless of incubation time or sample mixing methods.
In conclusion, increasing incubation times clearly improved enrichment efficiency, while neither the incubation temperature nor the mixing method led to significant changes in assay performance. Indeed, this is an important finding for clinical translation: the general iMALDI workflow yielded reproducible results even when conducted under slightly varying conditions, a situation which cannot be avoided even within a given hospital setting and even more so across different sites.
4. Conclusions
The iMALDI workflow was optimized for the efficient and automated quantification of cell signaling proteins from cell lysates. The essential steps inherent in every iMALDI procedure, including assay automation, tryptic digest, calibration, antibody‐sample incubation times, temperatures as well as using different enrichment strategies and different types of antibody‐coupled magnetic beads, were systematically evaluated (Table 1 ).
Table 1.
Summary of optimized iMALDI method parameters
| Parameter | Optimal condition |
|---|---|
| Peptide length for antibody development and MALDI‐TOF MS analysis | 7–20 Amino acids |
| Tryptic digest | 1:2 protein:trypsin (w:w) for 1 h at 37°C |
| Bead type | Protein G Dynabeads (MagReSyn Protein G beads perform similarly but with different background peaks observed in the mass spectrum) |
| Enrichment | Direct enrichment (antibodies coupled to beads prior to enrichment). |
| Incubation condition | 1 h to overnight, shaking (1000 RPM, too rapid shaking may cause loss of antigen) or rotating. |
| Incubation temperature | Room temperature (1 h incubation) or 4°C (overnight incubation) |
| Wash protocol | 1 × 70 µL PBS +0.015% (w:w) CHAPS, 3 × 80 µL 5 mm ammonium bicarbonate (automated) |
| MALDI matrix | 3 mg mL−1 HCCA+ 7 mm ammonium citrate dibasic in 70:29.9:0.1 ACN:H2O:TFA (1.5 µL matrix used per spot) |
| Spot wash procedure | 3 × 7 mm ammonium citrate dibasic (pH = 5, 10 µL wash buffer with 5 s incubation time on spot) |
| Calibration surrogate matrix | BSA Digest (0.1 µg µL−1 in TRIS+ 0.015% (w:w) CHAPS buffer) |
| Peak parameter used for peak calculations | Peak intensity |
The liquid handling steps were successfully automated. In particular, the labor‐intensive and comparatively complicated washing and spotting of the antibody‐antigen bead complex were automated without compromising sensitivity, while the turnaround time for 96 samples was reduced from approximately 90 min using manual preparation to 15 min using automation (see Supporting Information).
Regarding the sample preparation, it was found that digestion using 1:2 protein:trypsin was the ratio‐of‐choice, as it combined high digestion efficiency with short incubation times. In contrast to conventional proteomics experiments, most potential interferences are removed during anti‐peptide immuno‐enrichment after the proteolytic digestion. For the enrichment step, direct enrichment using protein G coupled beads was found to be the most sensitive method for cell lysate digests. Importantly, we found that different pAbs from the same manufacturer (Signatope) provided the same results. Given that only minimal amounts of antibody are required to conduct iMALDI assays with high sensitivity — even for proteins of low abundance — this is an important result, as 10 000s of assays can be conducted using a single pAb preparation, theoretically reducing the need for the generation of monoclonal antibodies.
In addition, different matrices were evaluated, as well as alternative peak parameters (S/N, intensity, area) for quantifying peptides in cell digests. For generating an external calibration, the complexity of the matrix was found to have little influence on the amount of target peptide measured in the sample and a BSA digest was found to be completely acceptable as a generic sample matrix.
In summary, this study provides a comprehensive template on how to set up, optimize, and evaluate future iMALDI assays. This will facilitate the transfer of this technology into the clinic, where protein‐based assays are becoming increasingly important for patient stratification.[ 10 , 34 ]
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
The authors are grateful to Genome Canada and Genome British Columbia for financial support through the Genomics Innovation Network (project codes 204PRO for operations and 214PRO for technology development). They are also grateful for financial support from Genome Canada, Genome British Columbia, and Genome Quebec through the Genomics Applications Partnership Program (GAPP: 183AKT), from Genome Canada and Genome Quebec through the Genomics Applications Partnership Program (GAPP: PD‐L1), and from the Terry Fox Research Institute. C.H.B. is also grateful for support from the Leading Edge Endowment Fund (University of Victoria), for support from the Segal McGill Chair in Molecular Oncology at McGill University (Montreal, Quebec, Canada), and for support from the Warren Y. Soper Charitable Trust and the Alvin Segal Family Foundation to the Jewish General Hospital (Montreal, Quebec, Canada). The study was supported by the MegaGrant of the Ministry of Science and Higher Education of the Russian Federation (Agreement with Skolkovo Institute of Science and Technology, No. 075‐10‐2019‐083). The authors are grateful to Dr. John Burke for providing the recombinant p110α/p85α protein.
Froehlich B. C., Popp R., Sobsey C. A., Ibrahim S., LeBlanc A. M., Mohammed Y., Aguilar‐Mahecha A., Poetz O., Chen M. X., Spatz A., Basik M., Batist G., Zahedi R. P., Borchers C. H., Systematic Optimization of the iMALDI Workflow for the Robust and Straightforward Quantification of Signaling Proteins in Cancer Cells. Prot. Clin. Appl. 2020, 14, 2000034 10.1002/prca.202000034
References
- 1. Cross T. G., Hornshaw M. P., J. Appl. Bioanal. 2016, 2, 108. [Google Scholar]
- 2. de Gramont A., Watson S., Ellis L. M., Rodón J., Tabernero J., de Gramont A., Hamilton S. R., Nat. Rev. Clin. Oncol. 2015, 12, 197. [DOI] [PubMed] [Google Scholar]
- 3. De Matos L. L., Trufelli D. C., De Matos M. G. L., Da Silva Pinhal M. A., Biomarker Insights 2010, 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoofnagle A. N., Wener M. H., J. Immunol. Methods 2009, 347, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Hurley G., Sjöstedt E., Rahman A., Li B., Kampf C., Pontén F., Gallagher W. M., Lindskog C., Mol. Oncol. 2014, 8, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meier F., Geyer P. E., Virreira Winter S., Cox J., Mann M., Nat. Methods 2018, 15, 440. [DOI] [PubMed] [Google Scholar]
- 7. Ludwig C., Gillet L., Rosenberger G., Amon S., Collins B. C., Aebersold R., Mol. Sys. Biol. 2018, 14, e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pfammatter S., Bonneil E., McManus F. P., Prasad S., Bailey D. J., Belford M., Dunyach J.‐J., Thibault P., Mol. Cell. Proteomics 2018, 17, 2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parker C. E., Borchers C. H., Mol. Oncol. 2014, 8, 840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sobsey C. A., Ibrahim S., Richard V. R., Gaspar V., Mitsa G., Lacasse V., Zahedi R. P., Batist G., Borchers C. H., Proteomics 2020, 20, 1900029. [DOI] [PubMed] [Google Scholar]
- 11. Faria S. S., Morris C. F. M., Silva A. R., Fonseca M. P., Forget P., Castro M. S., Fontes W., Front. Oncol. 2017, 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anderson N. L., Anderson N. G., Haines L. R., Hardie D. B., Olafson R. W., Pearson T. W., J. Proteome Res. 2004, 3, 235. [DOI] [PubMed] [Google Scholar]
- 13. Popp R., Li H., LeBlanc A., Mohammed Y., Aguilar‐Mahecha A., Chambers A. G., Lan C., Poetz O., Basik M., Batist G., Borchers C. H., Anal. Chem. 2017, 89, 10592. [DOI] [PubMed] [Google Scholar]
- 14. Lévesque S., Dufresne P. J., Soualhine H., Domingo M.‐C., Bekal S., Lefebvre B., Tremblay C., PLoS One 2015, 10, e0144878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel R., Clin. Chem. 2015, 61, 100. [DOI] [PubMed] [Google Scholar]
- 16. Popp R., Basik M., Spatz A., Batist G., Zahedi R. P., Borchers C. H., Analyst 2018, 143, 2197. [DOI] [PubMed] [Google Scholar]
- 17. Li L., Wei Y., To C., Zhu C.‐Q., Tong J., Pham N.‐A., Taylor P., Ignatchenko V., Ignatchenko A., Zhang W., Wang D., Yanagawa N., Li M., Pintilie M., Liu G., Muthuswamy L., Shepherd F. A., Tsao M. S., Kislinger T., Moran M. F., Nat. Commun. 2014, 5, 5469. [DOI] [PubMed] [Google Scholar]
- 18. Lapek J. D., Greninger P., Morris R., Amzallag A., Pruteanu‐Malinici I., Benes C. H., Haas W., Nat. Biotechnol. 2017, 35, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Janku F., Yap T. A., Meric‐Bernstam F., Nat. Rev. Clin. Oncol. 2018, 15, 273. [DOI] [PubMed] [Google Scholar]
- 20. Danielsen S. A., Eide P. W., Nesbakken A., Guren T., Leithe E., Lothe R. A., Biochim. Biophys. Acta, Rev. Cancer 2015, 1855, 104. [DOI] [PubMed] [Google Scholar]
- 21. Lee J. S., Lee H. W., Lee E. H., Park M., Lee J. S., Kim M.‐S., Kim T. G., Nam H.‐Y., Hwang S. W., Park J. H., Int. J. Clin. Exp. Pathol. 2018, 11, 1554. [PMC free article] [PubMed] [Google Scholar]
- 22. Li S., Shen Y., Wang M., Yang J., Lv M., Li P., Chen Z., Yang J., Oncotarget 2017, 8, 32043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Proc J. L., Kuzyk M. A., Hardie D. B., Yang J., Smith D. S., Jackson A. M., Parker C. E., Borchers C. H., J. Proteome Res. 2010, 9, 5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Landry J. P., Ke Y., Yu G.‐L., Zhu X. D., J. Immunol. Methods 2015, 417, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Landry J. P., Fei Y., Zhu X., Assay Drug Dev. Technol. 2012, 10, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neubert H., Shuford C. M., Olah T. V., Garofolo F., Schultz G. A., Jones B. R., Amaravadi L., Laterza O. F., Xu K., Ackermann B. L., Clin. Chem. 2020, 66, 282. [DOI] [PubMed] [Google Scholar]
- 27. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H., Mol. Cell. Proteomics 2009, 8, 1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoeppe S., Schreiber T. D., Planatscher H., Zell A., Templin M. F., Stoll D., Mol. Cell. Proteomics 2010, 10, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Senko M. W., Beu S. C., McLaffertycor F. W., J. Am. Soc. Mass Spectrom. 1995, 6, 229. [DOI] [PubMed] [Google Scholar]
- 30. Core Team R, R: A Language and Environment for Statistical Computing, R Foundation For Statistical Computing, Vienna, Austria: 2019. [Google Scholar]
- 31. Mohammed Y., Domański D., Jackson A. M., Smith D. S., Deelder A. M., Palmblad M., Borchers C. H., J. Proteomics 2014, 106, 151. [DOI] [PubMed] [Google Scholar]
- 32. Burkhart J. M., Schumbrutzki C., Wortelkamp S., Sickmann A., Zahedi R. P., J. Proteomics 2012, 75, 1454. [DOI] [PubMed] [Google Scholar]
- 33. Tu T., Gross M. L., TrAC Trends Anal. Chem. 2009, 28, 833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blank‐Landeshammer B., Richard V. R., Mitsa G., Marques M., LeBlanc A., Kollipara L., Feldmann I., Couetoux du Tertre M., Gambaro K., McNamara S., Spatz A., Zahedi R. P., Sickmann A., Batist G., Borchers C. H., Cancers 2019, 11, 1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information