

Abstract
Background & Aims
Molecular mechanisms by which hypoxia might contribute to hepatosteatosis, the earliest stage in non‐alcoholic fatty liver disease (NAFLD) pathogenesis, remain still to be elucidated. We aimed to assess the impact of hypoxia‐inducible factor 2α (HIF2α) on the fatty acid translocase CD36 expression and function in vivo and in vitro.
Methods
CD36 expression and intracellular lipid content were determined in hypoxic hepatocytes, and in hypoxic CD36‐ or HIF2α ‐silenced human liver cells. Histological analysis, and HIF2α and CD36 expression were evaluated in livers from animals in which von Hippel‐Lindau (Vhl) gene is inactivated (Vhlf/f‐deficient mice), or both Vhl and Hif2a are simultaneously inactivated (Vhlf/fHif2α/f‐deficient mice), and from 33 biopsy‐proven NAFLD patients and 18 subjects with histologically normal liver.
Results
In hypoxic hepatocytes, CD36 expression and intracellular lipid content were augmented. Noteworthy, CD36 knockdown significantly reduced lipid accumulation, and HIF2A gene silencing markedly reverted both hypoxia‐induced events in hypoxic liver cells. Moreover livers from Vhlf/f‐deficient mice showed histologic characteristics of non‐alcoholic steatohepatitis (NASH) and increased CD36 mRNA and protein amounts, whereas both significantly decreased and NASH features markedly ameliorated in Vhlf/fHif2αf/f‐deficient mice. In addition, both HIF2α and CD36 were significantly overexpressed within the liver of NAFLD patients and, interestingly, a significant positive correlation between hepatic transcript levels of CD36 and erythropoietin (EPO), a HIF2α ‐dependent gene target, was observed in NAFLD patients.
Conclusions
This study provides evidence that HIF2α drives lipid accumulation in human hepatocytes by upregulating CD36 expression and function, and could contribute to hepatosteatosis setup.
Keywords: CD36, HIF2α, hypoxia, NAFLD, steatosis
KEY POINTS.
CD36 knockdown attenuates hypoxia‐induced lipid accumulation in hepatocytes. HIF2α silencing reverts both lipid and CD36 content accumulation in hypoxic hepatocytes and in livers from a murine experimental model which displays NAFLD features due to an overexpression of HIF1 and HIF2. Expression of HIF2α and CD36 is increased in the liver of NAFLD patients.
1. INTRODUCTION
Overnutrition is a major contributor to the development of non‐alcoholic fatty liver disease (NAFLD) because a high consumption of saturated fatty acids, cholesterol and fructose along with a low intake of polyunsaturated fatty acids, featuring NAFLD patients, alters hepatic lipid metabolism homeostasis leading to an excessive fat accumulation within the liver which activates inflammation, hepatocellular damage and fibrogenesis. 1 , 2 There is extensive clinical and experimental evidence indicating that chronic intermittent hypoxia, featuring a respiratory disorder of growing prevalence worldwide termed obstructive sleep apnoea, could contribute to the progression of NAFLD from simple steatosis, also termed non‐alcoholic fatty liver (NAFL) or hepatosteatosis, to non‐alcoholic steatohepatitis (NASH), 3 , 4 , 5 , 6 the most clinically relevant form of NAFLD with a significant risk to progress into cirrhosis and hepatocellular carcinoma, 7 , 8 as well as increasing the cardiovascular morbidity and mortality and the incidence of extrahepatic cancers. 9 , 10
Hypoxia‐inducible factors (HIFs), particularly HIF1α and HIF2α, are master regulators of hypoxia‐induced cellular adaptive responses elicited to restore cell metabolism and survival. 11 HIFs are implicated in numerous physiological and pathological conditions, and it has been reported that HIF2α promotes NASH in mice, 12 , 13 and dysregulates lipid metabolism in HepG2 cells. 14 Since lipotoxicity due to free fatty acids (FFAs) overload within hepatocytes plays a central role in NAFLD pathophysiology, 15 and that this process is largely regulated by membrane‐bound FFA transporters, it is conceivable that HIFs might contribute to NAFLD pathogenesis by upregulating the expression and function of FFA transporters in the membrane of hepatocytes.
Among membrane‐bound FFA transporters, the fatty acid translocase CD36 (CD36) is the best characterized. 16 CD36 functions as a high affinity receptor for long‐chain FFAs contributing under excessive fat supply to lipid accumulation and metabolic dysfunction. 17 This FFA receptor is involved in several aspects of lipid metabolism including fat taste perception, fat intestinal absorption and FFA utilization by muscle, adipose tissues and liver. 18 Regarding the latter, hepatic CD36 expression is normally weak but it increases by a number of different stimuli such as cytokines or insulin. 18 , 19 Noteworthy, experimental studies have demonstrated that CD36 plays a key role in the hepatosteatosis setup in rodents 20 , 21 and, more interestingly, NAFLD patients present high hepatic CD36 mRNA levels, 22 and this FFA transporter is largely overexpressed in the plasma membrane of hepatocytes. 23 While there are evidences that HIF1α upregulates CD36 expression and function in retinal epithelial cells and macrophages, 24 , 25 whether HIF2α is able to regulate CD36 gene expression in hepatocytes still remains to be elucidated.
Therefore, the aim of the present study was to determine the impact of HIF2α on CD36 expression and function as well as on lipid content in hepatocytes submitted to hypoxic conditions, in livers from genetically‐modified mice in which von Hippel‐Lindau (Vhl) gene is inactivated (Vhlf/f‐deficient mice), a murine experimental model which displays NAFLD features due to an overexpression of HIF1 and HIF2, in livers from mice in which both Vhl and Hif2a are simultaneously inactivated (Vhlf/fHif2αf/f‐deficient mice), and in livers from patients with biopsy‐proven NAFLD.
2. MATERIALS AND METHODS
2.1. Cell culture and treatment
Human hepatoma Huh7 cells and AML12 (alpha mouse liver 12) hepatocyte cell line were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Huh7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing high glucose, penicillin/streptomycin and 10% fetal bovine serum, and AML12 hepatocytes were cultured in DMEM:F12 medium containing high glucose, penicillin/streptomycin and 10% fetal bovine serum, supplemented with 10 µg/mL insulin, 5.5 µg/mL transferrin, 5 ng/mL selenium and 40 ng/mL dexamethasone. The cells were submitted to normoxic (21% O2) or hypoxic conditions (1% O2) in a hypoxic chamber InvivO2 200 (Ruskinn Technology Ltd.) for 36 hours.
2.2. Short hairpin CD36 or HIF2α knockdown
Human scrambled (shControl, shC), CD36 shRNA or HIF2α shRNA lentiviral particles (Dharmacon, Madrid, Spain) were used to produce stable CD36 or HIF2α knockdown in Huh7 cells. Proliferating cells were co‐incubated with lentiviral transducing particles in culture media containing polybrene (Santa Cruz Biotechnology Inc, Heidelberg, Germany) for 24 hours, and then cultured with 2‐5 µg/mL of puromycin (Santa Cruz Biotechnology, Inc). Resistant cells were expanded and examined for CD36 or HIF2A mRNA levels respectively.
2.3. Nile Red staining
The cells were fixed with paraformaldehyde 4% for 30 minutes at 4ºC, and resuspended in Nile Red working solution to 0.4 µL/mL (Sigma‐Aldrich Inc). The fluorescence was determined using a flow cytometer Cytomics FC500 MPLTM (Beckman‐Coulter Inc).
2.4. Animals
All animal experimentation was conducted in accordance with Spanish and European legislation and approved by the research ethics committee at the Universidad Autónoma de Madrid (UAM) (CEI55‐1002‐A049). Mice used in this study were maintained in light/dark (12h light/12 hours dark), temperature (22°C) and humidity‐controlled rooms, fed with standard chow diet (LASQCdiet®Rod14, LASvendi, Germany) ad libitum, and had free access to drinking water at the animal facilities of the UAM. Vhlfloxed‐Ubc‐Cre‐ERT2 (Vhlf/f‐deficient mice), VhlfloxedHIF2αfloxed‐Ubc‐Cre‐ERT2 (Vhlf/fHif2αf/f‐deficient mice) mice and their corresponding controls (Vhlfloxed lacking UBC‐Cre‐ERT2 or VhlfloxedHIF2αfloxed lacking UBC‐Cre‐ERT2) were generated as described. 26 Male mice at 4‐5 months of age were used in this study. For gene inactivation, mice were fed ad libitum with Teckland CRD TAM400/CreER tamoxifen pellets (Harland‐Teklad, Valencia, Spain) for 10‐15 days, including control mice, and were later returned to standard chow diet for 15 days. After that, mice were sacrificed and livers were harvested.
2.5. Patients
This study included 33 patients with a clinical diagnosis of NAFLD (18 NAFL and 15 NASH), and 18 subjects with normal liver (NL) who underwent a liver biopsy by a percutaneous route during programmed cholecystectomy. Characteristics of the study population are detailed in Table 1. All NAFLD patients and NL subjects studied drank less than 20 g/day of alcohol, were not having potentially hepatotoxic drugs, had no analytical evidence of iron overload, and were seronegative for autoantibodies and for hepatitis B virus, hepatitis C virus and human immunodeficiency virus. This study was performed in agreement with the Declaration of Helsinki, and with local and national laws. The Human Ethics Committee of the Hospital Universitario Santa Cristina approved the study procedures (PI‐688A), and all participants signed an informed written consent before inclusion in the study.
Table 1.
Characteristics of the study population
| Feature | NL (n = 18) | NAFL (n = 18) | NASH (n = 15) |
|---|---|---|---|
| Age (years) | 48.2 ± 12.9 | 54.3 ± 14.9 | 47.4 ± 11.7 |
| Body mass index (kg/m2) | 27.1 ± 4.2 | 29.9 ± 4* | 29.4 ± 2.7 |
| Glucose (mg/dL) | 91.4 ± 11.1 | 96.1 ± 9.6 | 98.6 ± 15.6 |
| Insulin (µU/L) | 7.6 ± 6.6 | 9.6 ± 4.1* | 11.5 ± 5.3* |
| HOMA‐IR | 1.7 ± 0.9 | 2.3 ± 1.1* | 2.8 ± 1.4* |
| Triglycerides (mg/dL) | 110 ± 38.8 | 135.3 ± 39.8* | 138.9 ± 40.4* |
| HDL‐cholesterol (mg/dL) | 48.3 ± 10.8 | 49.5 ± 8.9 | 46.3 ± 8.9 |
| ALT (IU/L) | 17.2 ± 5.9 | 24.6 ± 9.3** | 42.8 ± 18.6*** |
| AST (IU/L) | 17.3 ± 3.8 | 20.3 ± 5.1* | 27 ± 11.9*** |
| GGT (IU/L) | 26.2 ± 16.6 | 39.4 ± 29.6* | 52.6 ± 35.2*** |
| Steatosis (%) | |||
| Grade 0 | 100% | ||
| Grade 1 | 66.6% | ||
| Grade 2 | 33.3% | 50% | |
| Grade 3 | 50% | ||
| Lobular inflammation (%) | |||
| Grade 0 | 100% | 100% | |
| Grade 1 | 80% | ||
| Grade 2 | 10% | ||
| Grade 3 | 10% | ||
| Ballooning (%) | |||
| Grade 0 | 100% | 100% | |
| Grade 1 | 70% | ||
| Grade 2 | 30% | ||
| Grade 3 | |||
| Fibrosis (%) | |||
| Stage 0 | 100% | 100% | 30% |
| Stage 1 | 70% | ||
| Stage 2 | |||
| Stage 3 |
Data are presented as mean ± standard deviation or as number of cases (%). Study population: Normal liver (NL) individuals (n = 18), NAFL patients (n = 18) and NASH patients (n = 15). HOMA‐IR, homeostatic model assessment‐insulin resistance; HDL, high‐density lipoprotein; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma‐glutamyltransferase. *P < .05, **P < .01 and ***P < .005, NAFL or NASH vs NL.
2.6. Gene expression analysis by real‐time quantitative PCR
Total RNA from cells or liver samples was extracted using TRIzol reagent (Vitro, Sevilla, Spain) and was reverse transcribed using a reverse transcription system (Promega Inc) in a T100TM Thermal Cycler (BioRad Inc, Madrid, Spain) following manufacturer's indications. Quantitative real‐time polymerase chain reaction (qPCR) was performed with StepOnePlusTM Real Time PCR System sequence detector (Thermo Fisher Scientific, Inc, Madrid, Spain) using the SYBR Green method and d(N)6 random hexamer with primers purchased from Metabion (Steinkirchen, Germany). Each sample was run in duplicated and normalized to 36B4 (mouse and human samples) or HPRT (cell samples) gene expression. Fold changes were determined using the ΔΔCt method. Primer sequences are detailed in Table 2.
Table 2.
Primer sequences for RT‐qPCR
| Gene | Forward (5’→3’) | Reverse (5’→3’) |
|---|---|---|
| m‐Cd36 | AGATGACGTGGCAAAGAACAG | CCTTGGCTAGATAACGAACTCTG |
| m‐Epo | CAAAGTCAACTTCTATGCTTGGAAAA | CAGGCCTTGCCAAACTTCTATG |
| m‐Hif2a | GAGGAAGGAGAAATCCCGTGA | TATGTGTCCGAAGGAAGCTGA |
| m‐Pgk1 | CAGTTGCTGCTGAACTCAAATCTC | CCCACACAATCCTTCAAGAACA |
| m‐Vhl | ATCCCTGAAGAGCCAAAGATGA | TCAGCCCTACCCGATCTTACC |
| m‐36b4 | AGATGCAGCAGATCCGCAT | GTTCTTGCCATCAGCACC |
| h‐CD36 | ATGTGTGTGGAGAGCGTCAACC | TGAGCAGAGTCTTCAGAGACAGCC |
| h‐EPO | TTCGCAGCCTCACCACTCT | GAGATGGCTTCCTTCTGGGC |
| h‐HIF2A | CTCATCCCTGCGACCATGA | TTCCCAAAACCAGAGCCATT |
| h‐PGK1 | TGGCTTCTGGCATACCTGCT | GCTGCTTTCAGGACCACAGCT |
| h‐PHD3 | TGCATCACCTGCATCTACTATCTG | CGCAGGATCCCACCATGTA |
| h‐VHL | CGCCGCATCCACAGCTA | TGTGTCCCTGCATCTCTGAAGA |
| h‐36B4 | CAGGCGTCCTCGTGGAAGTGAC | CCAGGTCGCCCTGTCTTCCCT |
| h‐HPRT | ATTGTAATGACCAGTCAACAGGG | GCATTGTTTTGCCAGTGTCAA |
2.7. Preparation of total protein extracts
Liver biopsy samples were homogenized in 16 volumes (w/v) of cold lysis buffer (50 mM Tris‐HCl, 1% Triton X‐100, 2 mM EGTA, 10 mM EDTA acid, 100 mM NaF, 1 mM Na4P2O7, 2 mM Na3VO4, 100 µg/ml phenylmethylsulphonyl fluoride and protease inhibitors). To obtain total cell lysates, at the end of the experiment, attached cells were scraped off and incubated for 10 minutes on ice with RIPA buffer (50 mM Tris HCl, pH 7.4, 1% Triton X‐100, 0.2% sodium dodecyl sulfate (SDS), 1 mM EDTA, 1 mM PMSF and 5 µg/ml leupeptin). Protein extracts were stored at −80ºC after centrifugation.
2.8. Extraction of nuclear protein liver extracts
Liver biopsy samples were homogenized in 4 volumes (w/v) of cold buffer A (0.3 M Sucrose solution with protease inhibitors). Samples were centrifuged and the supernatant containing the cytosolic fraction was stored at −80ºC. The pellet containing the nuclear fraction was washed for a total of three times with buffer A. Samples were incubated in rotation for 1 hour with cold buffer B (1% Trion x100, 1% Sodium Deoxycholate, 0.1% SDS, 5 mM EDTA, 200 nM NaCl, 20 mM Tris HCl pH 8 with protease inhibitors). After sonication, cellular debris was removed by centrifugation and the supernatant fraction containing the nuclear fraction was stored at −80ºC.
2.9. Western blot analysis
After protein content determination with Bradford reagent, 50 µg of total protein or100 µg of nuclear protein was boiled in Laemmli sample buffer and submitted to 8% SDS‐PAGE gels. Proteins were transferred to Inmunoblot nitrocellulose membrane (BioRad Inc) and, after blocking with 5% non‐fat dry milk, incubated overnight with different antibodies as indicated: anti‐HIF1α (1:1000, 10006421, Cayman Chemical, Hamburg, Germany), anti‐HIF2α (1:1000, ab199, Abcam, Cambridge, UK) and anti‐CD36 (1:1000, NB400‐144, Novus, Abingdon, UK). Immunoreactive bands were visualized using the enhanced chemiluminescence (ECL) Western blotting protocol (BioRad Inc). The anti‐βactin (1:5000, A‐5441, Sigma‐Aldrich Inc) and the anti‐lamin B (1:1000, ab65986, Abcam) antibodies were used as loading control for total and nuclear protein respectively. Densitometric analysis of the bands was performed using Image J software (NIH, Bethesda, MD).
2.10. Histopathology assessment
Paraffin‐embedded liver biopsy sections (4 µm thick) were stained with haematoxylin/eosin and evaluated by a single‐blinded hepatopathologist. Steatosis was determined grading percentage involvement by steatotic hepatocytes as follows: grade 0, 0%‐5%; grade 1, >5%‐33%; grade 2, >33%‐66%; and grade 3, >66%, as described by Kleiner et al. 27 In addition, this scoring system was used to evaluate the degree of lobular inflammation and hepatocellular ballooning. NAFLD activity score was calculated for each liver biopsy studied as described elsewhere. 27 Histologic diagnosis of liver biopsies from NAFLD patients was classified into two groups: simple steatosis without hepatocellular ballooning nor lobular inflammation, also termed NAFL, and NASH. Minimal criteria for NASH included the combined presence of grade 1 steatosis, lobular inflammation and hepatocellular ballooning with or without fibrosis. As we want to study early stages of NAFLD, only NASH patients with either mild fibrosis (F1) or without fibrosis (F0) were studied and included in the same group. Representative images were taken using an optical microscope Nikon Eclipse E400 (Nikon, Tokyo, Japan).
2.11. Oil Red O staining
Cryoprotected liver biopsy sections (5 µm) were stained with an Oil red O (ORO, Sigma‐Aldrich Inc) working solution (60% ORO/isopropanol w:v) and counter‐stained with haematoxylin. Red staining was quantified from the images taken using an optical microscope Nikon Eclipse E400 (Nikon). Image analysis procedures were performed with the FIJI software (NIH, Bethesda, MD). Values were obtained in six different lobular areas where hepatocytes are the predominant cell type. The average value was considered as ORO staining index for each liver biopsy sample, and expressed as arbitrary units which reflect the intensity of the staining.
2.12. HIF2α and CD36 immunohistochemistry
Paraffin‐embedded liver biopsy sections (4 µm thick) were immunostained with a primary rabbit antibody against HIF2α (ab199, Abcam) 28 or CD36 (NB400‐144, Novus) diluted to 1:50 and 1:200 respectively, using the DAKO EnVision™+ System (DAKO, Glostrup, Denmark) as described by the manufacturer. Liver tissue area occupied by nuclear HIF2α or CD36‐positive cells was quantified from the images taken using an optical microscope Nikon Eclipse E400 (Nikon). Image analysis procedures were performed with the FIJI software (NIH). Values were obtained in six different lobular areas where hepatocytes are the predominant cell type. The average value was considered as nuclear HIF2α or CD36 expression index for each liver biopsy sample, and expressed as percentage of positive nuclei or arbitrary units respectively, which reflects the intensity of the immunostaining.
2.13. CD36 and N‐cadherin immunofluorescence
Paraffin‐embedded liver biopsy sections (4 μm thick) were co‐incubated with an anti‐CD36 antibody (NB400‐144, Novus) and anti‐N‐cadherin antibody (BP1‐48309, Novus), diluted to 1:200 and 1:25 respectively, following with the appropriated conjugated secondary antibodies Alexa Fluor® 568 goat anti‐rabbit IgG (A11011, Life Technologies) or Alexa Fluor® 488 goat anti‐mouse IgG (A11029, Life Technologies). The immunofluorescence‐mounting medium used was Fluoromont G® (BioNova cientifica). Representative images were taken using a confocal microscope Leica TCS SP5 X (Leica, Barcelona, Spain).
2.14. Statistical analysis
Data from qPCR and western blot experiments are expressed as percentage, and presented as mean ± standard error of mean (SEM) relative to control condition (100%); data from immunohistochemistry are expressed as percentage or arbitrary units, and presented as mean ± SEM; data from flow cytometry experiments are expressed as fold increase, and presented as mean ± SEM relative to control condition (1). Data were compared using one‐way analysis of variance (ANOVA) followed by Bonferroni test. All statistical analyses were performed using the GraphPad Prism 6.0 software (GraphPad Software Inc, San Diego, CA, USA) and the IBM SPSS Statistics 24.0 (SPSS Inc, IBM, Armonk, NY) software with two‐sided tests, with a P‐value of < .05 considered as statistically significant.
3. RESULTS
3.1. Hypoxia induces lipid accumulation and CD36 expression in both human and murine hepatocytes
To explore the molecular mechanisms involved in the regulation of CD36 by hypoxia, Huh7 human liver cells were maintained under normoxic conditions (Nx, 21%O2) or submitted to hypoxia (Hp, 1%O2) for 36h. To assure that hypoxia was achieved in Huh7 cell cultures, we assessed the expression of HIFα protein subunits by western blot. As Figure 1A shows, HIF1α and HIF2α expression was markedly induced by hypoxia. Accordingly, PHD3 upregulation, a well‐recognized HIF responsive gene, was found in hypoxia‐exposed Huh7 liver cells (Figure 1B).
Figure 1.
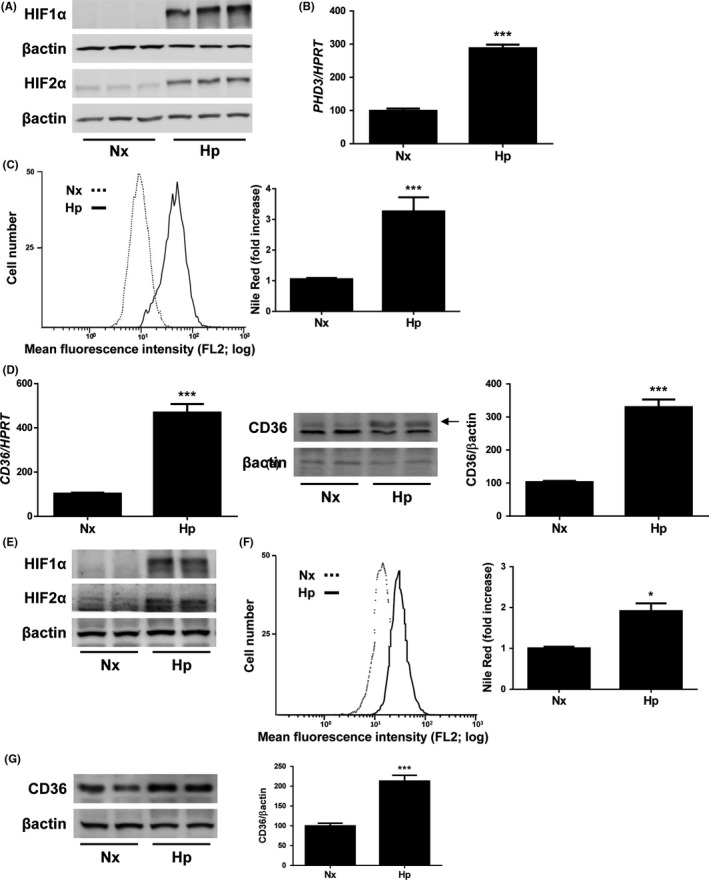
Hypoxia induces lipid accumulation and CD36 expression in hepatocytes. Huh7 cells maintained under normoxic (Nx, 21% O2), or hypoxic conditions (Hp, 1% O2) in a hypoxia chamber for 36h. A, Representative blots with the indicated antibodies. B, PHD3 mRNA levels. C, (left panel) Representative experiment of Nile Red fluorescence intensity. (right panel) Analysis of intracellular lipid content by Nile Red staining. D, (left panel) CD36 mRNA. (right panel) Representative blots with the indicated antibodies and densitometric analysis from all blots. ***P < .005, Hp vs Nx (n = 4 independent experiments performed by triplicate). AML12 cells maintained under normoxic (Nx, 21% O2), or hypoxic conditions (Hp, 1% O2) in a hypoxia chamber for 36 hours. E, Representative blots with the indicated antibodies. F, (left panel) Representative experiment of Nile Red fluorescence intensity. (right panel) Analysis of intracellular lipid content by Nile Red staining. G, Representative blots with the indicated antibodies and densitometric analysis from all blots. *P < .005 and ***P < .005, Hp vs Nx (n = 3 independent experiments performed by duplicate)
Next, we investigated whether Huh7 cells submitted to hypoxic conditions would increase their intracellular lipid content performing Nile Red staining experiments. After flow cytometry analysis, we observed a significant increase in the lipid content of Huh7 cells submitted to hypoxia, compared to those maintained under normoxic conditions (Figure 1C). Interestingly, a parallel increase in CD36 mRNA and protein levels was found in hypoxic Huh7 cells (Figure 1D). Similar findings were observed in mouse hepatocytes (AML12 cell line) submitted to hypoxia in which both intracellular lipid content and CD36 expression were significantly augmented when compared to normoxic cells (Figure 1E‐G).
3.2. Silencing of CD36 attenuates hypoxia‐induced lipid accumulation in liver cells
To elucidate whether CD36 is involved in hypoxia‐induced lipid accumulation in hepatic cells, we infected Huh7 cells with scrambled (control, shC) or CD36 shRNA (shCD36) lentiviral particles. With this approach, we obtained an average of 70% decrease of mRNA and protein levels of CD36 (Figure 2A,B). As depicted in Figure 2F, silencing of CD36 significantly reduced lipid accumulation in Huh7 submitted to hypoxia without altering the induction of hypoxia markers (Figure 2C,D) and partly blocking hypoxia‐induced CD36 increase (Figure 2E).
Figure 2.
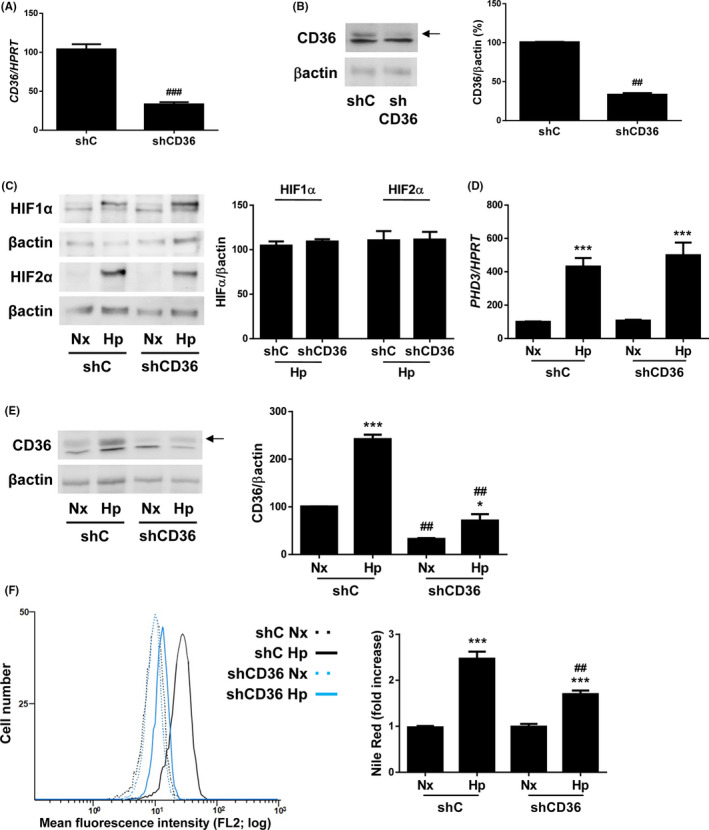
CD36 knockdown attenuates hypoxia‐induced lipid accumulation in human liver cells. Huh7 cells infected with scrambled (shC) or CD36 shRNA (shCD36) lentiviral particles maintained under normoxic (Nx, 21% O2), or hypoxic conditions (Hp, 1% O2) in a hypoxia chamber for 36h. A, CD36 mRNA in normoxia. B, C, and E, Representative blots with the indicated antibodies and densitometric analysis from all blots. D, PHD3 mRNA levels. F. (left panel) Representative experiment of Nile Red fluorescence intensity. (right panel) Analysis of intracellular lipid content by Nile Red staining. * P < .005 and ***P < .005, Hp vs Nx; ## P < .01 and ### P < .005, shCD36 vs shC (n = 4 independent experiments performed by duplicate)
These data propose that CD36 might play a major role in the onset of hepatosteatosis under hypoxic conditions.
3.3. HIF2A silencing markedly reduces both lipid accumulation and CD36 upregulation in hypoxic human hepatocytes
We next wanted to determine whether HIF2α might be linked to hypoxia‐induced CD36 upregulation. To this end, we infected Huh7 cells with scrambled (control, shC) or HIF2α shRNA (shHIF2) lentiviral particles achieving a 75% decrease in HIF2A mRNA levels (Figure 3A). Under hypoxia conditions, it was observed a reduced HIF2α protein stabilization (Figure 3B), as well as PHD3 and EPO mRNA levels, while the hypoxia‐induced increased expression of PGK1, a major HIF1α target gene, remained unchanged (Figure 3C). Noteworthy, the reduction of HIF2α significantly decreased both lipid accumulation and CD36 upregulation observed in Huh7 cells submitted to hypoxia for 36 hours (Figure 3D,E). Taken together these data suggest that HIF2α is the responsible for the hypoxia‐induced CD36 upregulation and, ultimately, for the increase of lipid content in hypoxic hepatocytes.
Figure 3.
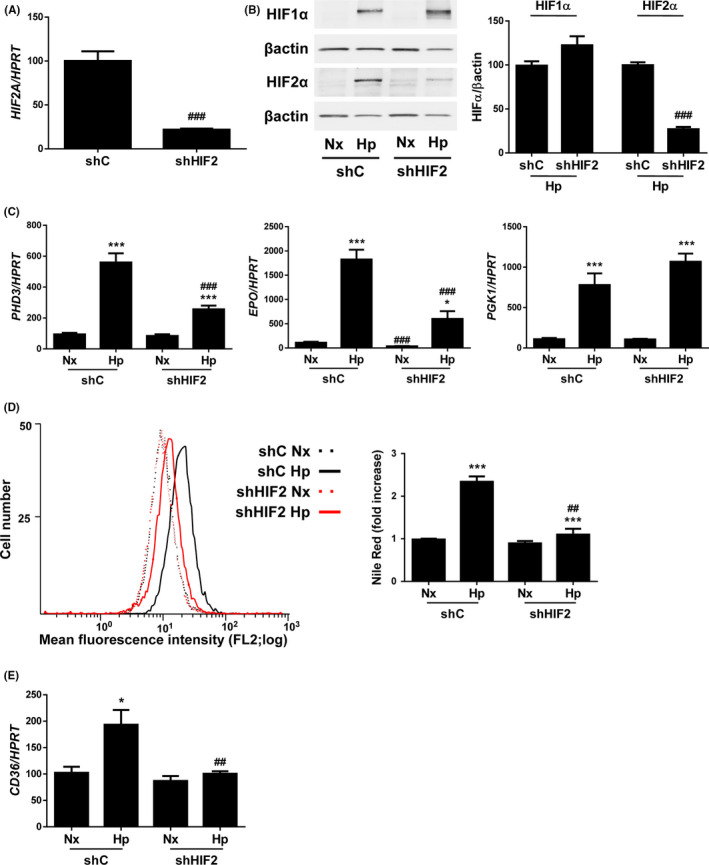
HIF2α silencing reduces both lipid accumulation and induction of CD36 expression in human liver cells submitted to hypoxic conditions. Huh7 cells infected with scrambled (shC) or HIF2α shRNA (shHIF2) lentiviral particles maintained under normoxic (Nx, 21% O2), or hypoxic conditions (Hp, 1% O2) in a hypoxia chamber for 36h. A, HIF2A mRNA levels in normoxia. B, Representative blots with the indicated antibodies and densitometric analysis from all blots. C, PHD3, EPO and PGK1 mRNA levels. D, (left panel) Representative experiment of Nile Red fluorescence intensity. (right panel) Analysis of intracellular lipid content by Nile Red staining. E, CD36 mRNA levels. *P < .05 and ***P < .005, Hp vs Nx; ## P < .01 and ### P < .005, shHIF2 vs shC (n = 4 independent experiments performed by duplicate)
3.4. Lack of HIF2α ameliorates NASH features and decreases CD36 content in livers from Vhlf/f‐deficient mice
To investigate the role of HIF2α ‐CD36 pathways in the hepatic lipid homeostasis in vivo, we used adult Vhlfloxed‐Ubc‐Cre‐ERT2 mice (Vhlf/f‐deficient mice) in which Vhl gene inactivation leads to an elevated expression of HIFα protein subunits, and VhlfloxedHif2αfloxed‐UBC‐Cre‐ERT2 mice (Vhlf/fHif2αf/f‐deficient mice) in which both Vhl and Hif2a are simultaneously inactivated (Figure 4A).
Figure 4.
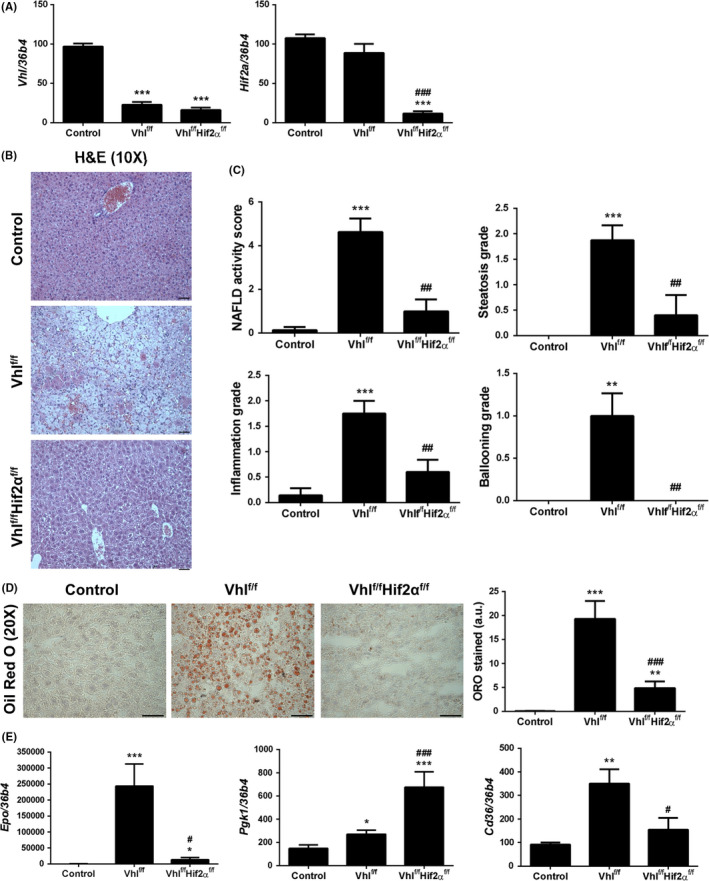
Lack of HIF2α reverts Vhl inactivation‐induced NASH. A, Hepatic Vhl and Hif2a mRNA levels. B, Representative 10X images of haematoxylin/eosin (H&E) staining. Scale bar 100 µm. C, NAFLD activity score, steatosis grade, lobular inflammation and hepatocellular ballooning grade. D, Representative 20X images of Oil Red O (ORO) staining, and its quantification. Scale bar 100 µm. E, Hepatic Epo, Pgk1 and Cd36 mRNA levels. Experimental groups: Control, Vhlf/f‐deficient mice and Vhlf/fHif2αf/f‐deficient mice (n = 6‐8 animals/group). *P < .05, **P < .01 and ***P < .005, Vhlf/f or Vhlf/fHif2αf/f‐deficient vs Control mice; # P < .05, ## P < .01 and ### P < .005, Vhlf/fHif2αf/f vs Vhlf/f‐deficient mice
Histological examination of liver biopsies revealed that control mice displayed a normal histology while Vhlf/f‐deficient mice exhibited borderline or definite NASH due to the combined presence of steatosis, inflammation and hepatocyte ballooning. Interestingly, these NASH features were markedly attenuated in livers from Vhlf/fHif2αf/f‐deficient mice (Figure 4B,C). Accordingly, an Oil Red O staining revealed that lipid accumulation observed in liver sections from Vhlf/f‐deficient mice was higher than in control animals, and was reverted in livers from Vhlf/fHif2αf/f‐deficient mice (Figure 4D).
Interestingly, hepatic Cd36 mRNA levels were significantly higher in Vhlf/f‐deficient mice than in control mice, as well as Epo and Pgk1 mRNA levels which are HIF2α and HIF1α‐dependent genes respectively. Noteworthy, in Vhlf/fHif2αf/f‐deficient mice, hepatic Cd36 mRNA levels were significantly lower than in Vhlf/f‐deficient mice, in parallel to Epo mRNA levels, whereas Pgk1 gene expression was not repressed but rather it was over‐induced (Figure 4E).
Moreover HIF2α protein expression determined by western blot (Figure 5A) and by immunostaining (Figure 5B) was elevated in the livers of Vhlf/f‐deficient mice. As expected, this increase in HIF2α content was not observed in mice with both Vhl and Hif2a inactivated (Figure 5A,B). Regarding HIF2α immunostaining, nuclear staining was lost in livers from Vhlf/fHif2αf/f‐deficient mice with respect to those from Vhlf/f‐deficient mice, suggesting that the nuclear signal observed correspond to endogenous HIF2α (Figure 5B). Indeed, we also analysed HIF2α protein content in nuclear extracts by western blot, and we found similar results which indicate that hepatic HIF2α translocation into the nucleus was nearly blocked in Vhlf/fHif2αf/f‐deficient mice (Figure S1A).
Figure 5.
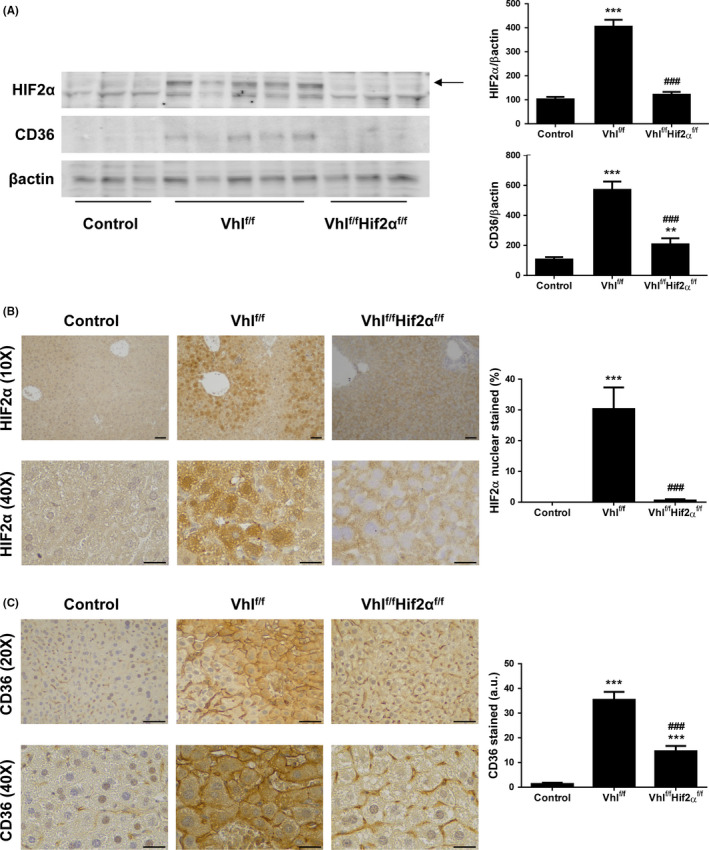
HIF2α deficiency attenuates Vhl inactivation‐induced hepatic CD36 overexpression. A, Representative blots with the indicated antibodies and densitometric analysis from all blots. B, Representative 10X and 40X images of HIF2α immunostaining, and quantification of nuclear HIF2α‐expressing cells. Scale bar 100 and 50 µm respectively. C, Representative 20× and 40× images of CD36 immunostaining, and quantification of CD36‐expressing cells. Scale bar 100 and 50 µm respectively. Experimental groups: Control, Vhlf/f‐deficient mice and Vhlf/fHif2αf/f‐deficient mice (n = 6‐8 animals/group). **P < .01 and ***P < .005, Vhlf/f or Vhlf/fHif2αf/f‐deficient vs Control mice; ### P < .005, Vhlf/fHif2αf/f vs Vhlf/f‐deficient mice
In parallel, an increase in CD36 protein expression was observed in the livers of Vhlf/f‐deficient mice detected by western blot (Figure 5A) and by immunostaining (Figure 5C). In these animals, the intensification of CD36 immunostaining was also enhanced in the plasma membrane of hepatocytes. Indeed, a co‐localization between CD36 and N‐cadherin, a well‐characterized hepatocyte plasma membrane marker, 29 was observed (Figure S1B). Noteworthy, hepatic CD36 expression was also significantly lower in Vhlf/fHif2αf/f‐deficient mice than in Vhlf/f‐deficient mice (Figure 5A,C).
Taken together, these findings strongly suggest that HIF2α could play an important role on hepatic lipid homeostasis by regulating the expression and function of the fatty acid receptor CD36 in hepatocytes.
3.5. Expression of HIF2α and CD36 is increased within the liver of NAFLD patients
Finally, we wanted to explore whether this link between HIF2α and CD36 exists in human liver as well. Representative haematoxylin/eosin staining liver pictures and the mean of the NAFLD activity score from the study patients are shown in Figure 6A,B. Furthermore, we estimated the hepatic protein content of nuclear HIF2α and total CD36 assessing their expression by immunohistochemistry. A higher expression of HIF2α was observed in NAFLD patients, such in NAFL as in NASH cases, than in NL individuals (Figure 6C). Interestingly, HIF2α immunostaining was also more intense in the nucleus of hepatocytes in NAFLD patients, which is in line with nuclear HIF2 expression upon Vhl inactivation in mouse liver shown in Figure 5B. Moreover in agreement with previous results reported by our group, 23 CD36 was weakly expressed in liver biopsies from NL subjects, while is markedly expressed at the plasma membrane and cytoplasm of numerous hepatocytes in NAFL and NASH patients (Figure 6D). Further immunofluorescence staining revealed that CD36 co‐localized with N‐cadherin in NAFLD human samples (Figure S1C), which was also found in Vhlf/f‐deficient mice (Figure S1B). Finally, we measured hepatic mRNA levels of CD36 and EPO, being the latter one of the best‐characterized HIF2α‐dependent gene targets. Therefore, we used EPO mRNA content as a surrogate marker of HIF2α activation. We found that both CD36 and EPO mRNA expression was elevated in NAFLD patients and, interestingly, a significant positive correlation between the mRNA levels of these genes was observed in the entire study population (Figure 6E,F).
Figure 6.
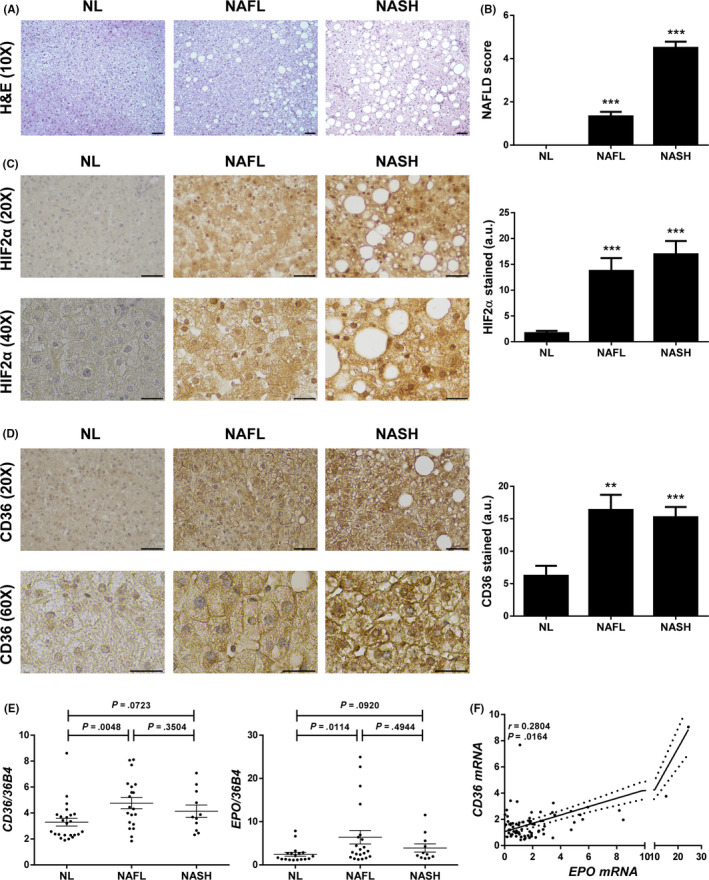
Expression of HIF2α and CD36 is increased within the liver of NAFLD patients. A, Representative 10X images of haematoxylin/eosin (H&E) staining. Scale bar 100 µm. B, NAFLD activity score. C, Representative 20× and 40× images of HIF2α immunostaining, and quantification of nuclear HIF2α‐expressing cells. Scale bar 100 and 50 µm respectively. D, Representative 20× and 60× images of CD36 immunostaining, and quantification of CD36‐expressing cells. Scale bar 100 and 50 µm respectively. E, CD36 and EPO mRNA levels. F, Correlation in the study population of matched mRNA expression levels. Study population: Normal liver (NL) individuals (n = 18), NAFL patients (n = 18) and NASH patients (n = 15). **P < .01 and ***P < .005, NAFL or NASH vs NL
4. DISCUSSION
This study not only demonstrates for the first time that HIF2α upregulates CD36 expression and function contributing to hepatosteatosis in hepatocytes, but it also provides evidence suggesting that a HIF2α‐induced CD36 upregulation could be operative in vivo and play a relevant role in NAFLD pathophysiology in humans, as we found a significant increase of both HIF2α and CD36 protein content, and an elevated mRNA levels of CD36 and EPO, being the latter a well‐known surrogate marker of HIF2α activation, as well as a marked positive correlation between these genes, in the livers of NAFLD patients. It is important to note that although hepatic mRNA levels of both CD36 and EPO were higher in NAFL than in NASH patients, the differences between these groups were not significant. Likewise, hepatic protein amounts of both CD36 and HIF2α were similar in NAFL and NASH patients indicating that its hepatic expression remains largely stable during histological progression from NAFL to NASH. Taken together, our data strongly suggest that the HIF2α/CD36 pathway could have a pathogenic role in the phase of hepatosteatosis, but further investigation is needed for understanding in depth its impact on NASH progression.
There is a growing evidence indicating that hypoxia contributes to NAFLD development and progression to NASH likely by the effects that HIFs exert on target genes regulating glucose and lipid homeostasis in the adipose tissue, small intestine and liver. 30 , 31 , 32 Regarding the latter, HIF2α‐dependent effects in the liver appear to be linked to its activation level, thus mild HIF2α activation will enhance insulin signalling and fatty acid oxidation whereas potent HIF2α activation will lead to liver dysfunction and steatosis. 33 In our study, 2 weeks after transgene excision, Vhlf/f‐deficient mice showed mild to moderate NASH features with a median NAFLD activity score of 4.5, similar findings to that reported by Qu et al 13 In particular, these authors observed that livers of mice overexpressing HIFs after 2 weeks of disrupting the Vhl gene in hepatocytes displayed NASH features along with a marked increase of Cd36 mRNA levels. More interestingly, in agreement with our findings showed herein, they also found that the combined inactivation of Vhl and Hif2a genes in hepatocytes reverted hepatic steatosis and inflammation observed in Vhlf/f‐deficient mice, indicating that HIF2α is a direct regulator of lipid homeostasis in the liver, but whether HIF2α exerts its steatogenic effect by upregulating genes important for FFA uptake in hepatocytes, such as CD36, is still unknown. Shedding light on this issue, our findings provide a robust experimental evidence demonstrating that HIF2α not only upregulates CD36 expression in human liver cells but also its function as FFA transporter because we found a marked decrease of hypoxia‐induced lipid content and CD36 mRNA levels when we knocked down HIF2α in hepatocytes. Further experimental studies, however, are needed to prove whether the induction of CD36 by HIF2α occurs directly or indirectly, but it is conceivable that HIF2α could activate CD36 gene expression in human hepatocytes at the transcriptional level as it has been reported at least one putative hypoxia response element consensus site at human CD36 gene promoter. 24
To the best of our knowledge, this is the first study demonstrating that hypoxia upregulates CD36 expression and function in hepatocytes, and that CD36 gene knockdown markedly reduces the increased FFA uptake in these hypoxic hepatocytes. In addition, another novel finding of this study is the concomitant increase in HIF2α and CD36 protein content in the liver of NAFLD patients. In particular, noteworthy, such in NAFLD patients as in Vhlf/f‐deficient mice, the increase in HIF2α expression was markedly observed in the nuclei of hepatocytes and that of CD36 was also detected at the plasma membrane of hepatocytes. More interestingly, we observed that histological features of NASH significantly ameliorated in Vhlf/fHif2αf/f‐deficient mice along with a marked decrease of both mRNA and protein hepatic CD36 levels, supporting our assumption that HIF2α may contribute to NAFLD onset by upregulating CD36 expression and function in hepatocytes.
The subcellular distribution of CD36 is critical for the regulation of its functional activity as FFA transporter facilitating the uptake and influx of FFA to the cells, remaining functionally inactive at intracellular storage pools and active when translocated to the plasma membrane. 34 In line with our present findings and reinforcing others we previously reported, 23 Zhao et al 35 have recently confirmed that CD36 is largely located in the plasma membrane of hepatocytes such in NASH patients as in mice with histological features of NASH, providing further evidence indicating the key role of palmitoylation in regulating CD36 translocation to the plasma membrane of hepatocytes. Protein palmitoylation is mediated by a family of palmitoyl acyltransferases (PAT). 36 In humans, 23 genes encoding PAT have been described so far, whose expression and function could be regulated from transcriptional to post‐translational level. 37 It is tempting to speculate that HIF2α might induce CD36 expression and its translocation to the plasma membrane of human hepatocytes by upregulating PAT gene expression, thus increasing CD36 palmitoylation which facilitates incorporation of CD36 into plasma membranes, but this hypothesis still remains to be addressed.
In conclusion, this study provides novel evidence indicating that HIF2α upregulates CD36 expression and function in hepatocytes, and could contribute to the onset of hepatosteatosis. Unveiling the molecular mechanisms underlying the HIF2α‐dependent CD36 upregulation in human hepatocytes may have notable implications for the development of new pharmacological therapies inhibiting the HIF2α/CD36 pathway trying to attenuate the excessive fat accumulation within the liver and its detrimental effects on the outcome of NAFLD.
5. CONFLICT OF INTEREST STATEMENT
Authors have not conflict of interest.
Supporting information
Fig S1
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Esther Fuertes Yebra for helpful technical assistance.
Rey E, Meléndez‐Rodríguez F, Marañón P, et al. Hypoxia‐inducible factor 2α drives hepatosteatosis through the fatty acid translocase CD36. Liver Int. 2020;40:2553–2567. 10.1111/liv.14519
Esther Rey and Florinda Meléndez‐Rodríguez are contributed equally to this work.
Carmelo García‐Monzón and Águeda González‐Rodríguez share correspondence and senior authorship.
Handling editor: Isabelle Leclercq
Funding information
This work was supported by PI13/01299, PI17/00535 and CIBEREHD from Instituto de Salud Carlos III (ISCIII/FEDER, Spain) to CGM; CP14/00181, PI16/00823 and PI19/00123 (ISCIII/FEDER, Spain), and Beca Eduardo Gallego 2016 (Fundación Francisco Cobos, Spain) to AGR; SAF2016‐76815 (Ministerio de Economía y Competitividad/FEDER, Spain), 534/C/2016 (TV3 Marató, Spain) and CIBERCV (ISCIII/FEDER, Spain) to JA.
Contributor Information
Carmelo García‐Monzón, Email: garciamonzon@hotmail.com, Email: aguedagr.phd@gmail.com.
Águeda González‐Rodríguez, Email: garciamonzon@hotmail.com, Email: aguedagr.phd@gmail.com.
REFERENCES
- 1. Dongiovanni P, Valenti L. A nutrigenomic approach to non‐alcoholic fatty liver disease. Int J Mol Sci. 2017;18(7):1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Musso G, Gambino R, De Michieli F, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37(4):909‐916. [DOI] [PubMed] [Google Scholar]
- 3. Aron‐Wisnewsky J, Clement K, Pepin JL. Nonalcoholic fatty liver disease and obstructive sleep apnea. Metabolism. 2016;65(8):1124‐1135. [DOI] [PubMed] [Google Scholar]
- 4. Aron‐Wisnewsky J, Minville C, Tordjman J, et al. Chronic intermittent hypoxia is a major trigger for non‐alcoholic fatty liver disease in morbid obese. J Hepatol. 2012;56(1):225‐233. [DOI] [PubMed] [Google Scholar]
- 5. Lefere S, Van Steenkiste C, Verhelst X, Van Vlierberghe H, Devisscher L, Geerts A. Hypoxia‐regulated mechanisms in the pathogenesis of obesity and non‐alcoholic fatty liver disease. Cell Mol Life Sci. 2016;73(18):3419‐3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Musso G, Cassader M, Olivetti C, Rosina F, Carbone G, Gambino R. Association of obstructive sleep apnoea with the presence and severity of non‐alcoholic fatty liver disease. A systematic review and meta‐analysis. Obesity reviews : an official journal of the International Association for the Study of . Obesity. 2013;14(5):417‐431. [DOI] [PubMed] [Google Scholar]
- 7. European Association for the Study of the L, European Association for the Study of D, European Association for the Study of O . EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388‐1402. [DOI] [PubMed] [Google Scholar]
- 8. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐Meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73‐84. [DOI] [PubMed] [Google Scholar]
- 9. Allen AM, Hicks SB, Mara KC, Larson JJ, Therneau TM. The risk of incident extrahepatic cancers is higher in non‐alcoholic fatty liver disease than obesity ‐ A longitudinal cohort study. J Hepatol. 2019;71(6):1229‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim D, Adejumo AC, Yoo ER, et al. Trends in mortality from extrahepatic complications in patients with chronic liver disease, From 2007 Through 2017. Gastroenterology. 2019;157(4):1055‐1066.e11. [DOI] [PubMed] [Google Scholar]
- 11. Aragones J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009;9(1):11‐22. [DOI] [PubMed] [Google Scholar]
- 12. Morello E, Sutti S, Foglia B, et al. Hypoxia‐inducible factor 2alpha drives nonalcoholic fatty liver progression by triggering hepatocyte release of histidine‐rich glycoprotein. Hepatology. 2018;67(6):2196‐2214. [DOI] [PubMed] [Google Scholar]
- 13. Qu A, Taylor M, Xue X, et al. Hypoxia‐inducible transcription factor 2alpha promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology. 2011;54(2):472‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cao R, Zhao X, Li S, et al. Hypoxia induces dysregulation of lipid metabolism in HepG2 cells via activation of HIF‐2alpha. Cell Physiol Biochem. 2014;34(5):1427‐1441. [DOI] [PubMed] [Google Scholar]
- 15. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52(2):774‐788. [DOI] [PubMed] [Google Scholar]
- 16. Su X, Abumrad NA. Cellular fatty acid uptake: a pathway under construction. Trends Endocrinol Metab. 2009;20(2):72‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure‐function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. 2014;34:281‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonen A, Chabowski A, Luiken JJ, Glatz JF. Is membrane transport of FFA mediated by lipid, protein, or both? Mechanisms and regulation of protein‐mediated cellular fatty acid uptake: molecular, biochemical, and physiological evidence. Physiology. 2007;22:15‐29. [DOI] [PubMed] [Google Scholar]
- 19. Buque X, Cano A, Miquilena‐Colina ME, Garcia‐Monzon C, Ochoa B, Aspichueta P. High insulin levels are required for FAT/CD36 plasma membrane translocation and enhanced fatty acid uptake in obese Zucker rat hepatocytes. Am J Physiol Endocrinol Metab. 2012;303(4):E504‐514. [DOI] [PubMed] [Google Scholar]
- 20. Koonen DP, Jacobs RL, Febbraio M, et al. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet‐induced obesity. Diabetes. 2007;56(12):2863‐2871. [DOI] [PubMed] [Google Scholar]
- 21. Zhou J, Febbraio M, Wada T, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134(2):556‐567. [DOI] [PubMed] [Google Scholar]
- 22. Greco D, Kotronen A, Westerbacka J, et al. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol. 2008;294(5):G1281‐1287. [DOI] [PubMed] [Google Scholar]
- 23. Miquilena‐Colina ME, Lima‐Cabello E, Sanchez‐Campos S, et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non‐alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60(10):1394‐1402. [DOI] [PubMed] [Google Scholar]
- 24. Mwaikambo BR, Yang C, Chemtob S, Hardy P. Hypoxia up‐regulates CD36 expression and function via hypoxia‐inducible factor‐1‐ and phosphatidylinositol 3‐kinase‐dependent mechanisms. J Biol Chem. 2009;284(39):26695‐26707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ortiz‐Masià D, Díez I, Calatayud S, et al. Induction of CD36 and thrombospondin‐1 in macrophages by hypoxia‐inducible factor 1 and its relevance in the inflammatory process. PLoS One. 2012;7(10):e48535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elorza A, Soro‐Arnaiz I, Melendez‐Rodriguez F, et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell. 2012;48(5):681‐691. [DOI] [PubMed] [Google Scholar]
- 27. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313‐1321. [DOI] [PubMed] [Google Scholar]
- 28. Torres‐Capelli M, Marsboom G, Li QO, et al. Role Of Hif2alpha Oxygen Sensing Pathway In Bronchial Epithelial Club Cell Proliferation. Sci Rep. 2016;6:25357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hempel M, Schmitz A, Winkler S, et al. Pathological implications of cadherin zonation in mouse liver. Cell Mol Life Sci. 2015;72(13):2599‐2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee KY, Gesta S, Boucher J, Wang XL, Kahn CR. The differential role of Hif1beta/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 2011;14(4):491‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taniguchi CM, Finger EC, Krieg AJ, et al. Cross‐talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med. 2013;19(10):1325‐1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie C, Yagai T, Luo Y, et al. Activation of intestinal hypoxia‐inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat Med. 2017;23(11):1298‐1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. LaGory EL, Giaccia AJ. Long‐range hypoxia signaling in NAFLD. Nat Med. 2017;23(11):1251‐1252. [DOI] [PubMed] [Google Scholar]
- 34. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase‐ and CD36‐mediated pathways. J Lipid Res. 2009;50(Suppl):S86‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao L, Zhang C, Luo X, et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non‐alcoholic steatohepatitis. J Hepatol. 2018;69(3):705‐717. [DOI] [PubMed] [Google Scholar]
- 36. Rocks O, Gerauer M, Vartak N, et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010;141(3):458‐471. [DOI] [PubMed] [Google Scholar]
- 37. Chavda B, Arnott JA, Planey SL. Targeting protein palmitoylation: selective inhibitors and implications in disease. Expert Opin Drug Discov. 2014;9(9):1005‐1019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Supplementary Material