

Abstract
Persistent measurable residual disease (MRD) is an increasingly important prognostic marker in acute myeloid leukemia (AML). Currently, MRD is determined by multi‐parameter flow cytometry (MFC) or PCR‐based methods detecting leukemia‐specific fusion transcripts and mutations. However, while MFC is highly operator‐dependent and difficult to standardize, PCR‐based methods are only available for a minority of AML patients. Here we describe a novel, highly sensitive and broadly applicable method for MRD detection by combining MFC‐based leukemic cell enrichment using an optimized combinatorial antibody panel targeting CLL‐1, TIM‐3, CD123 and CD117, followed by mutational analysis of recurrently mutated genes in AML. In dilution experiments this method showed a sensitivity of 10−4 to 10−5 for residual disease detection. In prospectively collected remission samples this marker combination allowed for a median 67‐fold cell enrichment with sufficient DNA quality for mutational analysis using next generation sequencing (NGS) or digital PCR in 39 out of 41 patients. Twenty‐one samples (53.8%) tested MRD positive, whereas 18 (46.2%) were negative. With a median follow‐up of 559 days, 71.4% of MRD positive (15/21) and 27.8% (5/18) of MRD negative patients relapsed (P = .007). The cumulative incidence of relapse (CIR) was higher for MRD positive patients (5‐year CIR: 90.5% vs 28%, P < .001). In multivariate analysis, MRD positivity was a prominent factor for CIR. Thus, MFC‐based leukemic cell enrichment using antibodies against CLL‐1, TIM‐3, CD123 and CD117 followed by mutational analysis allows high sensitive MRD detection and is informative on relapse risk in the majority of AML patients.
1. INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous group of clonal hematopoietic stem‐cell and progenitor‐cell (HSPC) disorders with a variable response to therapy. Although the majority of patients achieve morphologic complete remission (CR) after induction chemotherapy, relapse rates are high due to the persistence of trace amounts of chemoresistant leukemic cells. 1 Nevertheless, the choice of post‐remission treatment is still based on risk stratification at the time of diagnosis. Distinct chromosomal and molecular aberrations assign patient risk and guide post‐remission therapy conceivably including allogeneic hematopoietic stem cell transplantation. 2 In recent years the detection of residual leukemic cells beyond CR, termed as measurable residual disease (MRD), using molecular methods 3 , 4 , 5 , 6 or multiparameter flow cytometry (MFC) 7 , 8 has been shown to provide additional independent prognostic information, with MRD negative patients having a better clinical outcome than MRD positive ones. Thus, current guidelines from the European Leukemia Net propose the separation of CR into CR‐MRD positive and CR‐MRD negative subgroups, with the former carrying a higher risk of relapse. 9 , 10
Quantitative polymerase chain reaction (qPCR) techniques detecting fusion transcripts and mutated genes are the best established methods for measuring MRD in AML with sensitivities ranging from 10−2 to 10−6. However, qPCR is applicable only in approximately 40% of patients. 10 , 11 , 12 In contrast, MRD detection by MFC employing leukemia‐associated immunophenotypes (LAIPs), such as lack of antigen expression, cross‐lineage expression, over‐expression, and asynchronous expression of surface markers can be applied to the majority of AML patients. 10 , 13 , 14 , 15 However, the sensitivities are lower ranging only from 10−2 to 10−4. In addition, MFC is laborious, difficult to standardize, and immunophenotypic shifts resulting in false negative MRD detection results can frequently occur during the course of disease. 16 , 17 Next generation sequencing approaches are increasingly used in clinical trials, but are not yet generally recommended in clinical practice. 10 Thus, although MFC and molecular methods have proven their value in predicting relapse‐free (RFS) and overall survival (OS), there is no current standard to detect MRD, which is applicable to virtually all AML patients.
In the present study, we aimed to establish a novel method for monitoring MRD in AML with a broader applicability by combining MFC‐based leukemic cell enrichment followed by mutational profiling of recurrently mutated genes in AML using next generation sequencing (NGS) or digital PCR. After having optimized an antibody panel for leukemic cell enrichment, this novel method of MRD detection was validated in a prospective pilot trial. Indeed, it was applicable in >90% of AML patients with a high sensitivity to detect residual leukemic cells and was informative on relapse risk with a significantly shorter RFS in patients with a positive MRD status.
2. PATIENTS AND METHODS
2.1. Clinical samples
To establish surface markers for leukemic cell enrichment a retrospective cohort comprising 150 adults diagnosed with AML according to WHO criteria at the Division of Hematology, Medical University of Graz, Austria were included in this study. The cohort also included 25 cases, in which relapse material was available. To assess the normal CD34+ HSPC compartments, bone marrow samples (NBM) were obtained from 12 lymphoma patients without any evidence of disease in the BM. To validate the MRD assay, a prospective cohort comprising of 41 patients who achieved CR after induction chemotherapy was included. The BM or peripheral blood (PB) samples collected from patients were processed as described. 18 Informed consent was obtained from all patients and the study was approved by the Institutional Review Board of Medical University Graz, Austria (protocol 26‐050 ex 13/14 and 29‐499 ex 16/17).
2.2. MFC analysis and cell sorting
Multi‐parameter flow cytometry was performed using a Fortessa cytometer (Becton Dickinson; BD; San Jose, CA, USA) with strictly harmonized baseline settings as described. 19 In brief, cryopreserved cells were thawed, washed with phosphate‐buffered saline and stained with the appropriate antibodies (Table S1). At least 200 000 events were recorded and data were analyzed using either Kaluza software (Beckman Coulter, USA) or by merging all panels using the Infinicyt software (Cytognos, Salamanca, Spain).
For enrichment of residual cells of remission samples, phycoerythrin (PE)‐labeled antibodies targeting CLL‐1, TIM‐3, CD117, and CD123, along with the backbone markers CD45, CD34 and CD38 were used (Table S2). After excluding CD14+ monocytes and CD203c+ basophils, the mononuclear cells were sorted into PE positive and PE negative fractions using a FACS Aria II (Becton Dickinson). Purity of the sorted fractions was >95%.
2.3. Detection of the NPM1 W288fs*12 mutation by digital PCR (dPCR)
Sorted cells from patients positive for NPM1 mutation W288fs*12 were analyzed by dPCR as described. 20 In brief, after DNA extraction using the QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) dPCR was performed in duplicates using QuantStudio 3D Master Mix v2 and a QuantStudio 3D Digital PCR System (Applied Biosystems). A control sample with a known NPM1 W288fs*12 mutation along with a no template control was added to each run. Chips were imaged in the QuantStudio 3D Chip Reader and raw data were analyzed using the QuantStudio 3D Analysis Suite Software (Applied Biosystems).
2.4. Mutational analysis by NGS
Sequencing of other mutations was done using an Ion Torrent platform as described. 20 In brief, after isolation of DNA from sorted cells with an Ion AmpliSeq Direct FFPE DNA Kit (Thermofisher Scientific, Waltham, USA), NGS libraries were prepared using the AmpliSeq library kit 2.0 (Thermo Fisher Scientific) and one of the following Ion AmpliSeq Custom Next‐Generation Sequencing DNA Panels: either the AMLv2‐Panel covering the whole coding sequence of CEBPA, DNMT3A, GATA2, TET2 and TP53 as well as hotspot mutations in ASXL1, BRAF, CBL, FLT3 (D835), IDH1, IDH2, JAK2, KIT, KRAS, NPM1, NRAS, PTPN11, RUNX1 and WT1 or the MN Panel covering the whole coding sequence of CEBPA, BCOR, DDX41, DNMT3A, ETV6, GATA2, NF1, PHF6, SF3B2, SFRP1, SRP72, STAG2, TP53, ZRSR2, and hotspot mutations in NPM1, ASXL1, BRAF, CALR, CBL, CSF3R, ETNK1, EZH2, FLT3, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NRAS, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, STAT3, TET2, U2AF1 and WT1. Sequencing was performed in duplicate using an Ion Proton benchtop sequencer (Thermo Fisher Scientific). On average, 1 × 106 reads were obtained for each sample with more than 90% of bases above AQ20 and 87% to 93% reads on‐target. For data analysis the Ion Torrent Suite Software Plug‐ins (Thermo Fisher Scientific, open source, GPL, https://github.com/iontorrent/) was used. Called variants were annotated using open source software ANNOVAR and SnpEff. All coding, nonsynonymous mutations were further evaluated and visually inspected in IGV (http://www.broadinstitute.org/igv/) and variant calls resulting from technical read errors or sequence effects were excluded from the analysis.
2.5. Statistical analysis
Differences in patient characteristics were calculated using a two‐sided Fisher´s exact or Mann‐Whitney tests. Comparison of cell populations concerning the percentage of cells positive for distinct surface markers was done using the Mann‐Whitney test. The package survival 2.44‐1.1 of R 3.6.1 (www.r-project.org) was used to calculate relapse‐free survival and cumulative incidences of relapse with death as competing risk. 9 The Fine and Gray model was used for univariate and multivariate assessment of risk factors for relapse using cmprisk 2.2‐8. Sensitivity and specificity to predict relapse was calculated by the package timeROC 0.3 with a method that is appropriate for cumulative incidence settings. All remaining statistical analyses were performed using GraphPad Prism software version 8.0 (GraphPad Software, La Jolla, CA, USA).
3. RESULTS
3.1. MFC analysis of cell surface markers for residual leukemic cell enrichment
An ideal cell surface marker for MFC‐based leukemic cell enrichment should display exclusive expression on the vast majority (>90%) of leukemic cells in all AML patients, but not on normal hematopoietic cells. We therefore analyzed the expression of 24 surface markers on bulk leukemic cells in 150 diagnostic AML samples by MFC. For each marker the number of AML cases expressing them on more than 90%, 50%‐90%, 10%‐50% and less than 10% of blasts was determined (Figure 1A). By these criteria, CD44 and CD305 were expressed on >90% of bulk leukemic cells in the majority of evaluated AML cases. When including samples with a surface expression on 50%‐90% of cells, the markers CD47, CD33, CLL1, CD45RA, CD117, CD123, GPR56, TIM‐3 and CD99 were prevalent in a substantial number of evaluated AML cases. The rest of markers were hardly expressed on 30% of the evaluated AML cases. We then analyzed the expression of these markers on normal CD34+38− HSPCs counterparts. Both CD44 and GPR56 were expressed on >90% of CD34+38− HSPCs, while the markers CD117, CD305, CD47 and CD33 were prevalent in substantial numbers of normal HSPCs with a surface expression on 50%‐90% of cells, comparably to what has been reported. 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 The remaining markers were hardly expressed on CD34+38− HSPCs (Figure 1B).
FIGURE 1.
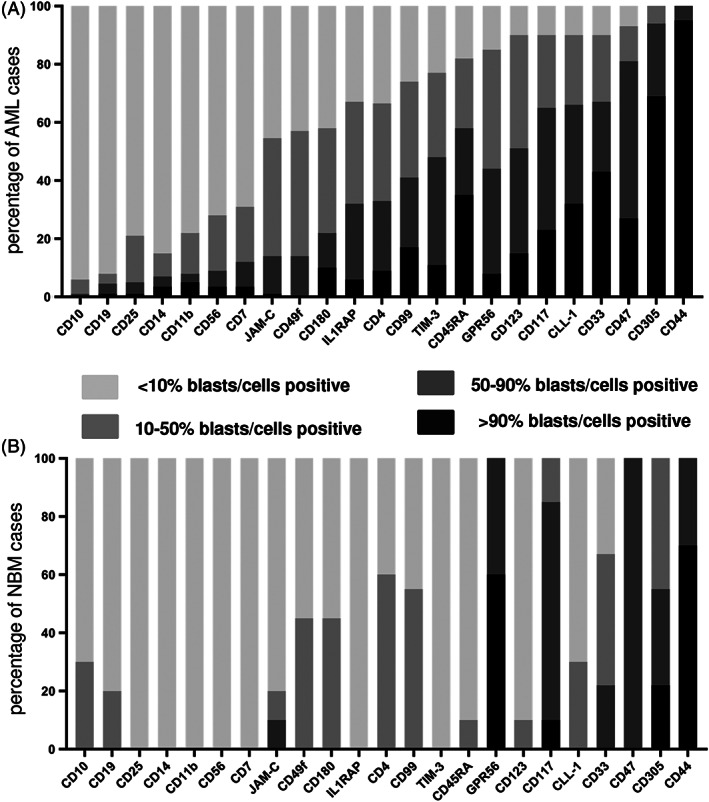
Expression of putative enrichment markers on AML bulk cells and normal HSPCs. A, The percentages of AML samples or B, CD34 + 38‐ HSPC NBM samples of which cells were either <10%, 10%–50%, 50%–90% or > 90% positive for a distinct marker, are given
Since no single marker showed an ideal expression profile for leukemic cell enrichment, we aimed to determine the best marker combination enabling enrichment of residual leukemic cells in a maximum number of AML cases. Although CD47, GPR56, CD33, CD305 and CD45RA were expressed on >90% blasts of the majority of AML cases evaluated, these markers were not considered further for enrichment, because they were also widely expressed on normal hematopoietic cell subtypes including CD34+38− HSPCs 21 , 22 , 23 , 24 , 25 (Figure 1B and Figure S2). From that perspective CLL‐1, TIM‐3, CD117 and CD123 were the most promising markers, since they were only expressed on either a distinct progenitor population or on small subtypes of mature leukocytes. 26 , 27 , 28 The remaining markers were expressed at levels too low to allow reliable enrichment and therefore were not considered further.
3.2. Establishment and validation of an MFC leukemic cell enrichment panel
To establish the numbers of AML samples covered sufficiently by our enrichment panel, the performance of the possible enrichment markers CLL‐1, TIM‐3, CD117 and CD123 was reanalyzed. First, the number of AML cases where at least one of the enrichment markers was expressed on >90% of leukemic blasts was determined. Among the 150 AML cases analyzed, 58% of the AML cases (86/150) showed expression of at least one marker on >90% blasts in our initial analysis, which was done for each marker using a separate fluorochrome for each antibody (Figure 2A). Sixty of the remaining 64 AML samples (in four samples no additional material was available), which did not have any single enrichment marker expressed on >90% of cells were further analyzed using an antibody cocktail targeting all four enrichment markers labeled with the same fluorochrome (PE). In 48 samples >90% of blasts were identified within the PE (marker) positive gate (Figure 2A) indicating that the cocktail including antibodies against CLL‐1, TIM‐3, CD117 and CD123 sufficiently labeled >90% of AML cells. Thus, in total 134 out of 146 AML samples (91.7%) showed adequate labelling (>90% of cells) using this enrichment panel.
FIGURE 2.
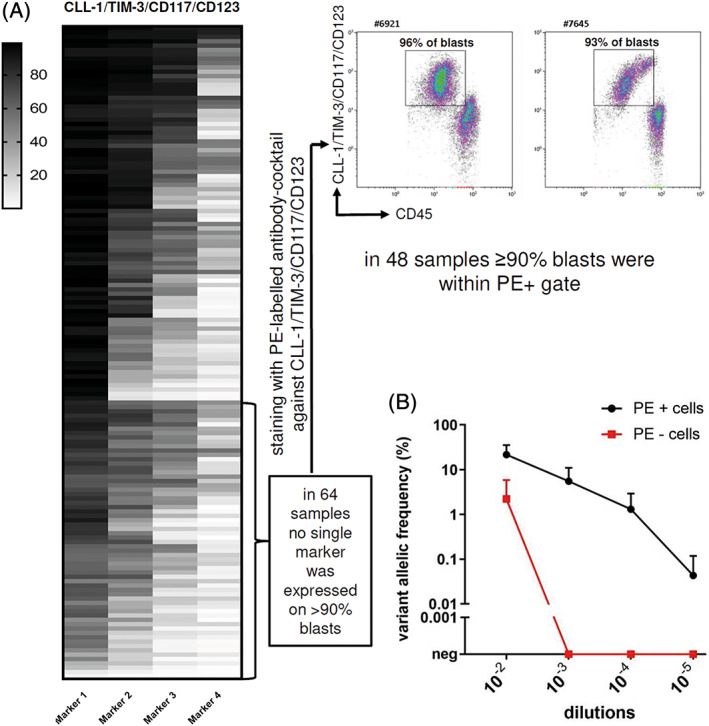
Expression of enrichment markers in 150 AML samples and dilution experiment to determine the sensitivity for detecting a leukemic cell in NBM. A, Expression of enrichment markers in 150 diagnostic AML samples. The makers are arranged in descending order with the marker displaying the highest percentage of cells at the left while the marker displaying the lowest percentage of cells positive is on the right. Each row represents one patient sample. In 64 samples no single marker was expressed on >90% of blasts. Thus, these samples were analyzed with an PE‐labeled antibody cocktail targeting all four enrichment markers. Two representative examples of primary AML samples are shown. In 48 samples the majority of blasts (>90%) were then identified within the PE (marker) positive gate. B, NPM1 variant allelic frequency (VAF) of sorted PE positive (black) and PE negative cells (red) of various dilutions of three NPM1 W288fs*12 mutated leukemic samples mixed with normal BM cells (mean ± SE). neg, negative; NBM, normal bone marrow [Color figure can be viewed at wileyonlinelibrary.com]
Next, we compared expression levels of the selected enrichment markers between leukemic cells and their normal HSPC counterparts. While TIM‐3, CD123 and CLL‐1 were hardly expressed on normal HSPCs (P < .001; Figure S3), CD117 was present on a subset of normal HSPCs. We thus speculated whether CD117 could be omitted due to its expression on a significant fraction of normal HSPCs. However, a combination of CLL‐1, CD123 and TIM‐3 allowed coverage of >90% leukemic cells in less than 75% of AML samples. Therefore, in order to be able to target the majority of AML samples, CD117 was necessary to include along with the other selected markers. Based on these results the markers CLL‐1, TIM‐3, CD117 and CD123 were eventually assigned for enrichment of residual leukemic cells in a maximum number of AML patients. Although CD117 has been used as a backbone marker for detection of MRD in previous studies, 29 , 30 the fact that the enrichment markers, especially CD117, are also expressed on a fraction of normal HSPCs, excludes the use of this marker combination for MRD detection based on MFC alone.
Analysis of paired diagnostic and relapse samples (n = 25) indicated that the expression of all the four enrichment markers was conserved throughout the disease course with either an increase or no significant change between diagnosis and relapse gate (Figure S4A). Most importantly, among the 25 cases studied, 17 cases had at least one of the selected enrichment markers positive and expressed on >90% of cells. In the remaining eight samples ≥90% of blasts were identified within the PE (marker) positive gate using an PE‐labeled antibody cocktail targeting all four enrichment markers (Figure S4B). Thus, the antibody cocktail targeting CLL‐1, TIM‐3, CD117 and CD123 sufficiently labeled >90% of AML cells both at diagnosis and at relapse in all 25 patients studied ensuring that enrichment was not hampered by immunophenotypic shifts. In addition, analysis of matched remission and relapse samples revealed that the majority of blasts from the relapse sample were within the same PE (marker) positive gate as in remission sample (Figure S5) suggesting that this enrichment panel was accurate.
Thus, for flow cytometry based enrichment of residual leukemic cells in remission BM samples, a single tube was designed by combining antibodies targeting CLL‐1, TIM‐3, CD117 and CD123 in the PE‐fluorescence channel along with the backbone markers CD45, CD14, CD34 and CD38. Furthermore, an antibody against CD203c, a basophil marker, was included to improve enrichment for leukemic cells by exclusion of CD123+ basophils (Table S2). The cells were sorted into PE (marker) positive and PE (marker) negative fractions according to the sorting strategy described in the methods section (Figure 3A).
FIGURE 3.
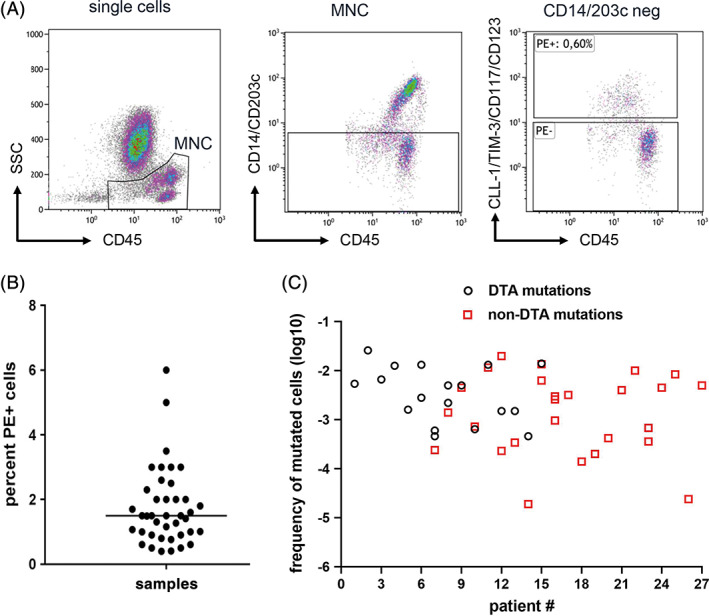
Leukemic cell enrichment using MFC‐based sorting followed by parallel sequencing for MRD detection. A, Sorting strategy for enrichment of residual leukemic cells. The MNCs were identified using SSC low and CD45 low. The monocytes and basophils were excluded using CD14/CD203c. The CD14/CD203c negative cells were gated on marker cocktail and the marker positive and marker negative fractions were sorted. B, Percentage of sorted PE (marker) positive cells calculated as percentage of total nucleated BM cells obtained after FACS based cell sorting in 41 remission samples. C, Calculated frequencies of mutated cells detected during complete remission using our two‐step MRD detection method. Black circles denote mutated cell frequencies as detected by DTA mutations and red squares display mutated cell frequencies as detected by non‐DTA mutations. MFC, multi‐parameter flow cytometry; MRD, measurable residual disease; MNC, mononuclear cells; DTA, DNMT3A, TET2 or ASXL1 [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Sensitivity for detection of residual leukemic cells after MFC‐based cell enrichment and mutational analysis
In order to determine the sensitivity of this newly developed two‐step MRD assay to detect residual leukemic cells, serial dilutions experiments were performed. The NPM1‐W288fs*12 mutated leukemic cells from three different patients were diluted with normal BM cells at ratios from 1:102 to 1:105. Sorting of these normal BM leukemic cell mixtures was then performed by applying the above described enrichment panel and NPM1 mutational analysis of sorted cells was done using dPCR. The NPM1 mutation was clearly enriched in all sorted PE (marker)‐positive samples up to the dilution of 1:104. In two out of three samples tested, the NPM1 mutation could even be traced in PE positive cells in dilutions 1:105. In contrast, NPM1‐mutations were not detected in PE (marker) negative cells in dilutions higher than 1:102 (Figure 2B). Thus, the sensitivity to detect a NPM1‐mutated AML cell among normal BM cells with this approach was 10−4 to 10−5.
3.4. Validation of the combination of MFC‐based cell enrichment and mutational analysis for MRD detection in a prospective AML cohort
For validation of the combination of MFC‐based cell enrichment and mutational analysis for MRD detection we performed a pilot study including 41 patients with AML in complete remission after induction chemotherapy (for patient characteristics see Table S3). In total, 93 mutations were identified at diagnosis in these patients with at least one informative mutation per patient, which could serve as a marker for residual disease (Table S4). Remission BM samples were subjected to MFC‐based cell enrichment. A median of 6499 PE (marker) positive cells (range 2066‐38 700 cells) were sorted from CD45+ cells (median 499.600, range 190.400‐1.760.000 cells) resulting in 0.4% ‐ 6% PE (marker) positive cells (median of 1.5% cells). This indicated an enrichment of ≈15‐250‐fold with a median enrichment of 67‐fold (Figure 3B). Since subsequent mutational profiling of sorted PE (marker) positive cells was possible in all but two patient samples, data from 39 patients were included for further analysis. In both samples with unsuccessful sequencing cell numbers were low (2070 and 1780 cells, respectively). We previously reported that the sensitivity of detecting a mutated variant using our sequencing approach was 0.5%. 20 However, insertions and deletions, as found for example with NPM1 or RUNX1 mutations were reliably detected with a sensitivity of 0.1% as also reported by others. 31 , 32 In addition, our dPCR approach to detect the NPM1 W288fs mutation showed also a sensitivity of 0.1%. 20 Therefore, we defined persisting residual disease as a detected VAF of ≥0.5% while in the case of insertions we used ≥0.1% as cut‐off. By applying these cut‐off values, 44 of the 93 mutations (47.3%) still persisted in 27 patients (69%) at remission. The VAF ranged from 0.1% to 47%. Mutations in DNMT3A and TET2 followed by NPM1 were the most common detectable mutations at remission (Table S4). Because DNMT3A, TET2 and ASXL1 (DTA) mutations were shown to persist in regenerating BM despite clearance of AML blasts, 20 , 31 , 32 , 33 DTA mutations were excluded for analysis of residual disease. Interestingly, in six of 27 (22%) patients, who had both DTA mutations and non‐DTA mutations, DTA mutations still persisted after induction therapy while the non‐DTA mutations were cleared. These observations were consistent with the idea that residual cells bearing DTA mutations represent pre‐leukemic clones. 34 , 35 Persistence of non‐DTA mutations within the PE (marker)‐positive fraction was found in twenty‐one out of 39 samples (21/39) with a median VAF of 5% (mean 11.5%, range: 0.14%–45%) indicating MRD positivity in 53.8% of our analyzed patients. The calculated frequencies of mutated leukemic cells in these MRD positive remission BM samples ranged between 1.98 × 10−2‐1.9 × 10−5 (Figure 3C, Table S4). In 19 of the 39 samples we also did mutational analysis of the PE (marker)‐negative cell fraction. Notably, leukemia‐specific mutations were absent in the PE (marker)‐negative cells of all samples tested. Since nine out of these 19 samples were MRD positive in the PE (marker)‐positive cells, these results clearly suggested a leukemic cell enrichment using our enrichment panel (Table S5).
3.5. MRD positivity as detected by this two‐step assay serves as a reliable biomarker for relapsing disease
Next, we analyzed RFS and OS according to the MRD status of the patients. Median follow‐up time was 559 days. Among the 21 MRD positive patients, 15 patients relapsed (71.4%), while only five of 18 MRD negative patients (27.8%) relapsed (P = .007). After 3 years the calculated sensitivity and specificity of our two‐step method to predict relapse was 0.75 and 0.71, respectively. Patients with a positive MRD status had a shorter RFS than MRD negative patients (median RFS 283 vs not reached, P = .003) (Figure 4A). Accordingly, the cumulative incidence of relapse (CIR) was significantly higher for MRD positive patients (5‐year CIR: 90.5% vs 28%, P < .001, Figure 4B). However, no prognostic significance of MRD status on overall survival was seen in our cohort (P = .085, Figure S6).
FIGURE 4.
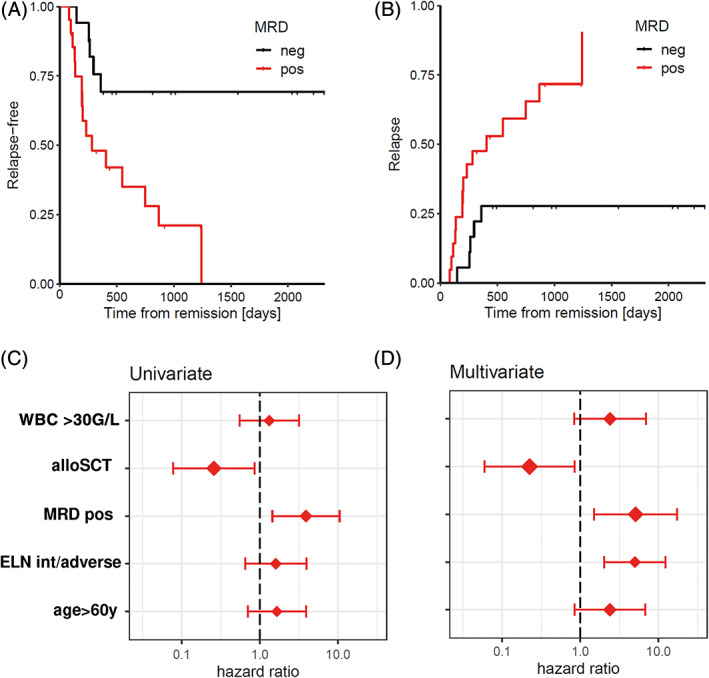
Relapse‐free survival (RFS) and cumulative incidence of relapse (CIR) in AML patients receiving intensive chemotherapy according to MRD status. A, RFS of AML patients according to MRD status (n = 39). Patients with a positive MRD status as measured by our two‐step MRD assay had a significantly shorter duration of RFS (P = .0031). B, Competing risk analysis for CIR in AML patients according to their MRD status. C, Box plot displaying univariate analysis of hazard ratios of risk factors for CIR. D, Box plot displaying multivariate analysis of hazard ratios of risk factors for CIR. alloSCT, allogeneic stem cell transplantation; MRD pos, measureable residual disease positive; ELN int/adverse, European Leukemia Net intermediate/adverse risk [Color figure can be viewed at wileyonlinelibrary.com]
Univariate analysis including also age, ELN risk category and leukocyte counts, MRD positivity was the only significant risk factor for CIR (HR, 3.9; 95% confidence interval [CI], 1.4‐10.5; P = .00724, Figure 4C), while allogeneic stem cell transplantation as consolidation reduced CIR (HR, 0.26; 95% CI, 0.08‐0.86; P = .0266). When tested in a multivariate analysis, MRD positivity (HR, 5. 1; 95% CI, 1.5‐17.3; P = .00902), as well as intermediate/adverse ELN risk groups (HR, 5.0; 95% CI, 2.0‐12.3; P = .00046) and stem cell transplantation (HR, 0.22; 95% CI, 0.06‐0.84; P = .0271) were factors significantly affecting CIR (Figure 4D).
4. DISCUSSION
In this study we established a novel method for monitoring MRD in AML by combining flow cytometry‐based leukemic cell enrichment using antibodies targeting CLL‐1, TIM‐3, CD117 and CD123 followed by mutational analysis using NGS or dPCR. Monitoring MRD by this method proved to be feasible with clinically relevant sensitivity to detect one leukemic cell in at least 10 000 normal BM cells or even more. This two‐step MRD method may therefore overcome some of the limiting issues of current MRD detection making its use attractive for further clinical development: First, this approach can be used successfully in the vast majority of AML patients. Using a combination of these four markers we were able to enrich leukemic cells in 91.7% percent of diagnostic AML samples and in our prospectively collected remission samples DNA quality of sorted cells was sufficient for mutational analysis in 39 out of 41 samples (95%). In all samples tested an informative leukemia‐specific mutation was present. Second, there is no need for specific reagents for distinct samples involving multiple antibody or sequencing primer combinations as used by others 36 , 37 allowing standardization and even automation of this method more easily. Third, by using a combination of four enrichment antibodies in one fluorescence channel with our approach, an immunophenotypic shift in one marker might affect MRD measurement only minimally. This is in contrast to MRD detection by MFC alone, where an immunophenotypic shift of one surface marker during relapse may result in false negative reports due to the loss of the LAIP. 17 , 38 In addition, the documentation of the presence of a leukemia‐specific mutation by parallel sequencing of enriched cells may completely overcome this problem.
Although we are not aware of any study performing more than one technique for MRD detection in a combinatorial manner as described here, several studies have explored using more than one MRD detection method in parallel. Simultaneous use of MFC and qPCR for detection of fusion transcripts in CBF AMLs was equally effective in predicting CIR. 39 Note, MFC in addition to WT1‐based molecular assessment of pre‐transplant MRD could predict the risk of post‐transplant relapse in AML. 40 , 41 A study comparing MFC with multigene sequencing to measure MRD in AML patients before HSCT found that the presence of MRD as detected by either method prior to HSCT was predictive for relapse risk. 42 Interestingly, in a recent study using NGS as well as MFC concomitantly, the highest relapse risk was observed in patients, who were positive by both assays. 32 Patients with a discordant status had an intermediate risk, while patients negative in both assays had the lowest relapse risk. These data show that the combinatorial use of more than one MRD detection technique may improve prognostication. Our combined approach takes advantage of both techniques and therefore may display improved sensitivity for the detection of residual leukemic cells. Indeed, in our remission samples, where we performed mutational profiling in both populations, leukemia‐specific mutations were present within sorted PE (marker) positive cells, but absent in PE (marker) negative cells.
Our pilot study also has some limitations. While in >90% of AML patients MRD can be detected using the selected antibodies, still a proportion of patient samples lacks sufficient expression of the enrichment markers. Interestingly, subsequent MFC analysis of frozen diagnostic material of the five patients who tested MRD negative but relapsed, three samples fell in the latter category and lacked sufficient expression of CLL‐1, TIM‐3, CD117 and/or CD123. Another relapsing patient tested MRD negative showed cytogenetic evolution at relapse suggesting that or enrichment strategy may miss persisting low‐level subclones at remission. In addition, our sorting strategy makes MRD detection in patients with CD14+ monocytic/monoblastic AMLs challenging, because in these cases leukemic cells may significantly overlap with normal/regenerating monocytes. However, despite these limitations of the enrichment process, the calculated sensitivity of the two‐step MRD assay with 0.75 was absolutely comparable with other assays 5 , 6 , 7 , 8 , 43 , 44 , 45 and only 28% of MRD‐negative patients relapsed. Previous studies exploring the prognostic significance of either mutation‐based 3 or MFC‐based 7 , 8 MRD detection reported higher relapse rates, with nearly 40% of MRD negative patients relapsing. Furthermore, expression of CD117 on normal HSPC may hamper sufficient enrichment of residual leukemic cells for subsequent sequencing, and therefore reduce the sensitivity to detect a leukemic cell in remission BM. To improve enrichment the use of newly identified markers with better specificity for leukemic cells may be useful. The markers CD191, CD70 and LILRB2 were recently found to be expressed on more than 75% of leukemic cells in almost all AML cases with low expression on normal hematopoietic cells, making them good candidates for enrichment of residual leukemic cells. 46 Furthermore, the background error rate in calling mutations in our NGS approach limited the detection of already known mutations to a sensitivity of 0.1% to 0.5%. 20 , 47 Recent studies using error‐corrected sequencing approaches have shown increased sensitivities to detect leukemic cells 48 , 49 and were found to be highly predictive for relapse risk. 50 Application of such molecular barcoding in future studies might help to increase the sensitivity as well as specificity of our two‐step method.
Irrespective of the technique used, a strong association between MRD status and relapse risk has been shown in many studies. 4 , 5 , 6 , 10 , 32 , 43 , 44 In this pilot study, patients with a positive MRD status as assessed by our two‐step MRD assay had an about 5‐fold higher risk of relapse, therefore confirming and extending data from previous studies. In contrast to previous studies, we could not detect a significant impact of the MRD status on overall survival, which is possibly attributable to the rather low number of patients in our pilot study. Thus, although a high sensitivity to detect residual leukemic cells and clinical applicability of the assay could be established with this pilot study, a proper prospective evaluation in larger cohorts is required. Moreover, a comparison of our two‐step MRD assay with the current standard methods of MFC‐MRD and qPCR as well as novel NGS methods, such as error‐corrected sequencing, is warranted to further solidify its performance. In conclusion, our two‐step MRD assay is predictive of impending relapse and allows for faster simpler MRD detection in more than 90% of the AML patients with high sensitivity making this novel test highly attractive for further clinical development.
CONTRIBUTIONS
S.D., A.Ro., I.H., R.G., N.K., and B.P. performed experiments; S.D., K.K., E.H., F.Q., G.H., A.Re., A.Z., H.S, and A.W. analyzed results; S.D. and A.W. designed the research and wrote the paper. All other authors contributed to and approved the final version of the manuscript.
CONFLICT OF INTERESTS
B.P. and A.W. report research support from Becton Dickinson BioSciences. All other authors declare no conflict of interest.
Supporting information
Appendix S1 Supporting information
ACKNOWLEDGMENTS
This study was supported by the Austrian Research Promotion Agency (FFG) within the COMET program (CBmed) and funds of the Oesterreichische Nationalbank (Anniversary Fund, project number: 15689 to A.W.). A.W. was also supported by an unrestricted grant from the Ingrid‐Shaker‐Nessmann Krebsforschungsvereinigung and Leukämiehilfe Steiermark. Furthermore, we acknowledge the contributions to this project by Noel Warner, Jim Keenan and Gert Boschman, all from Becton Dickinson BioSciences.
Daga S, Rosenberger A, Kashofer K, et al. Sensitive and broadly applicable residual disease detection in acute myeloid leukemia using flow cytometry‐based leukemic cell enrichment followed by mutational profiling. Am J Hematol. 2020;95:1148–1157. 10.1002/ajh.25918
Funding information Oesterreichische Nationalbank, Grant/Award Number: 15689; Austrian Research Promotion Agency, Grant/Award Number: CBmed
REFERENCES
- 1. Bachas C, Schuurhuis GJ, Assaraf YG, et al. The role of minor subpopulations within the leukemic blast compartment of AML patients at initial diagnosis in the development of relapse. Leukemia. 2012;26:1313‐1320. [DOI] [PubMed] [Google Scholar]
- 2. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136‐1152. [DOI] [PubMed] [Google Scholar]
- 3. Yin JA, O'Brien MA, Hills RK, Daly SB, Wheatley K, Burnett AK. Minimal residual disease monitoring by quantitative RT‐PCR in core binding factor AML allows risk stratification and predicts relapse: results of the United Kingdom MRC AML‐15 trial. Blood. 2012;120:2826‐2835. [DOI] [PubMed] [Google Scholar]
- 4. Rücker FG, Agrawal M, Corbacioglu A, et al. Measurable residual disease monitoring in acute myeloid leukemia with t(8;21)(q22;q22.1): results from the AML Study Group. Blood. 2019;134:1608‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ivey A, Hills RK, Simpson MA, et al. Assessment of minimal residual disease in standard‐risk AML. N Engl J Med. 2016;374(5):422‐433. [DOI] [PubMed] [Google Scholar]
- 6. Krönke J, Schlenk RF, Jensen KO, et al. Monitoring of minimal residual disease in NPM1‐mutated acute myeloid leukemia: a study from the German‐Austrian acute myeloid leukemia study group. J Clin Oncol. 2011;29:2709‐2716. [DOI] [PubMed] [Google Scholar]
- 7. Freeman SD, Virgo P, Couzens S, et al. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol. 2013;31:4123‐4131. [DOI] [PubMed] [Google Scholar]
- 8. Terwijn M, van Putten WL, Kelder A, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol. 2013;31:3889‐3897. [DOI] [PubMed] [Google Scholar]
- 9. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukemia: which platforms are ready for “prime time”? Hematology. 2014;2014:222‐233. [DOI] [PubMed] [Google Scholar]
- 12. Hokland P, Ommen HB, Nyvold CG, Roug AS. Sensitivity of minimal residual disease in acute myeloid leukaemia in first remission–methodologies in relation to their clinical situation. Br J Haematol. 2012;158:569‐580. [DOI] [PubMed] [Google Scholar]
- 13. Al‐Mawali A, Gillis D, Lewis I. The role of multiparameter flow cytometry for detection of minimal residual disease in acute myeloid leukemia. Am J Clin Pathol. 2009;131:16‐26. [DOI] [PubMed] [Google Scholar]
- 14. Buccisano F, Maurillo L, Del Principe MI, et al. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood. 2012;119:332‐341. [DOI] [PubMed] [Google Scholar]
- 15. Ossenkoppele GJ, van de Loosdrecht AA, Schuurhuis GJ. Review of the relevance of aberrant antigen expression by flow cytometry in myeloid neoplasms. Br J Haematol. 2011;153:421‐436. [DOI] [PubMed] [Google Scholar]
- 16. DiNardo CD, Luger SM. Beyond morphology: minimal residual disease detection in acute myeloid leukemia. Curr Opin Hematol. 2012;19:82‐88. [DOI] [PubMed] [Google Scholar]
- 17. Kern W, Bacher U, Haferlach C, Schnittger S, Haferlach T. The role of multiparameter flow cytometry for disease monitoring in AML. Best Pract Res Clin Haematol. 2010;23:379‐390. [DOI] [PubMed] [Google Scholar]
- 18. Wölfler A, Erkeland SJ, Bodner C, et al. A functional single‐nucleotide polymorphism of the G‐CSF receptor gene predisposes individuals to high‐risk myelodysplastic syndrome. Blood. 2005;105:3731‐3736. [DOI] [PubMed] [Google Scholar]
- 19. Daga S, Rosenberger A, Quehenberger F, et al. High GPR56 surface expression correlates with a leukemic stem cell gene signature in CD34‐positive AML. Cancer Med. 2019;8:1771‐1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaksch L, Kashofer K, Heitzer E, et al. Residual disease detection using targeted parallel sequencing predicts relapse in cytogenetically normal acute myeloid leukemia. Am J Hematol. 2018;93:23‐30. [DOI] [PubMed] [Google Scholar]
- 21. Kenderian SS, Ruella M, Shestova O, et al. CD33‐specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29:1637‐1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kersten B, Valkering M, Wouters R, et al. CD45RA, a specific marker for leukaemia stem cell sub‐populations in acute myeloid leukaemia. Br J Haematol. 2016;173:219‐235. [DOI] [PubMed] [Google Scholar]
- 24. Pabst C, Bergeron A, Lavallée VP, et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood. 2016;127:2018‐2027. [DOI] [PubMed] [Google Scholar]
- 25. Haubner S, Perna F, Köhnke T, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019;33:64‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Rhenen A, van Dongen GA, Kelder A, et al. The novel AML stem cell–associated antigen CLL‐1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110:2659‐2666. [DOI] [PubMed] [Google Scholar]
- 27. Kikushige Y, Shima T, Takayanagi S, et al. TIM‐3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7:708‐717. [DOI] [PubMed] [Google Scholar]
- 28. Testa U, Pelosi E, Frankel A. CD123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coustan‐Smith E, Song G, Shurtleff S, et al. Universal monitoring of minimal residual disease in acute myeloid leukemia. JCI Insight. 2018;3:98561 10.1172/jci.insight.98561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roug AS, Larsen HØ, Nederby L, et al. hMICL and CD123 in combination with a CD45/CD34/CD117 backbone–a universal marker combination for the detection of minimal residual disease in acute myeloid leukaemia. Br J Haematol. 2014;164:212‐222. [DOI] [PubMed] [Google Scholar]
- 31. Onecha E, Linares M, Rapado I, et al. A novel deep targeted sequencing method for minimal residual disease monitoring in acute myeloid leukemia. Haematologica. 2019;104:288‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jongen‐Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378:1189‐1199. [DOI] [PubMed] [Google Scholar]
- 33. Gaidzik VI, Weber D, Paschka P, et al. DNMT3A mutant transcript levels persist in remission and do not predict outcome in patients with acute myeloid leukemia. Leukemia. 2018;32:30‐37. [DOI] [PubMed] [Google Scholar]
- 34. Corces‐Zimmerman MR, Majeti R. Pre‐leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28:2276‐2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Metzeler KH, Herold T, Rothenberg‐Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128:686‐698. [DOI] [PubMed] [Google Scholar]
- 36. Kern W, Haferlach C, Schnittger S, Haferlach T. Clinical utility of multiparameter flow cytometry in the diagnosis of 1013 patients with suspected myelodysplastic syndrome: correlation to cytomorphology, cytogenetics, and clinical data. Cancer. 2010;116:4549‐4563. [DOI] [PubMed] [Google Scholar]
- 37. Béné MC, Nebe T, Bettelheim P, et al. Immunophenotyping of acute leukemia and lymphoproliferative disorders: a consensus proposal of the European Leukemia Net Work Package 10. Leukemia. 2011;25:567‐574. [DOI] [PubMed] [Google Scholar]
- 38. Oelschlägel U, Nowak R, Schaub A, et al. Shift of aberrant antigen expression at relapse or at treatment failure in acute leukemia. Cytometry. 2000;42:247‐253. [DOI] [PubMed] [Google Scholar]
- 39. Perea G, Lasa A, Aventín A, et al. Prognostic value of minimal residual disease (MRD) in acute myeloid leukemia (AML) with favorable cytogenetics [t (8; 21) and inv (16)]. Leukemia. 2006;20:87‐94. [DOI] [PubMed] [Google Scholar]
- 40. Marani C, Clavio M, Grasso R, et al. Integrating post induction WT1 quantification and flow‐cytometry results improves minimal residual disease stratification in acute myeloid leukemia. Leuk Res. 2013;37:1606‐1611. [DOI] [PubMed] [Google Scholar]
- 41. Zhao XS, Yan CH, Liu DH, et al. Combined use of WT1 and flow cytometry monitoring can promote sensitivity of predicting relapse after allogeneic HSCT without affecting specificity. Ann Hematol. 2013;92:1111‐1119. [DOI] [PubMed] [Google Scholar]
- 42. Getta BM, Devlin SM, Levine RL, et al. Multicolor flow cytometry and multigene next‐generation sequencing are complementary and highly predictive for relapse in acute myeloid leukemia after allogeneic transplantation. Biol Blood Marrow Transplant. 2017;23:1064‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jourdan E, Boissel N, Chevret S, et al. French AML Intergroup. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood. 2013;121:2213‐2223. [DOI] [PubMed] [Google Scholar]
- 44. Balsat M, Renneville A, Thomas X, et al. Post induction minimal residual disease predicts outcome and benefit from allogeneic stem cell transplantation in acute myeloid leukemia with NPM1 mutation: a study by the Acute Leukemia French Association Group. J Clin Oncol. 2017;35:185‐193. [DOI] [PubMed] [Google Scholar]
- 45. Buccisano F, Dillon R, Freeman SD, Venditti A. Role of minimal (measurable) residual disease assessment in older patients with acute myeloid leukemia. Cancer. 2018;10:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perna F, Berman SH, Soni RK, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. 2017;32:506‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra‐rare mutations by next‐generation sequencing. Proc Natl Acad Sci U S A. 2012;109:14508‐14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Young AL, Wong TN, Hughes AE, et al. Quantifying ultra‐rare pre‐leukemic clones via targeted error‐corrected sequencing. Leukemia. 2015;29:1608‐1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thol F, Gabdoulline R, Liebich A, et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood. 2018;132:1703‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patkar N, Kodgule R, Kakirde C, et al. Clinical impact of measurable residual disease monitoring by ultradeep next generation sequencing in NPM1 mutated acute myeloid leukemia. Oncotarget. 2018;9:36613‐36624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting information