

Abstract
Atorvastatin (ATO), one of the most common cholesterol reduction agents, exhibits anti-neoplastic effects in several human cancers. However, the antitumor effects of ATO on cervical cancer have not been extensively reported. Recently, autophagy inhibitors are reported to enhance the efficacy of chemotherapeutics. Here, we showed that ATO reduced cell viability and promoted apoptosis of cervical cancer cells by inducing caspase-3 and PARP activation and upregulating Bim. Treatment of ATO also suppressed tumor growth in vivo. In addition, co-culture with GGPP almost completely reversed the morphological change and apoptosis induced by ATO in cervical cancer cells. Furthermore, ATO induced cellular autophagy in cervical cancer cells, which was confirmed by an increase of LC3-I into LC3-II conversion, downregulation of p62 expression, regulation of AMPK and Akt/mTOR pathways. Moreover, pharmacologic inhibition of autophagy using either Baf-A1 or 3-MA significantly enhanced ATO-mediated apoptosis on cervical cancer cells. In conclusion, combination of ATO with autophagy inhibitors could emerge as a new therapeutic strategy for cervical cancer treatment.
Keywords: Cervical cancer, statins, atorvastatin, autophagy, treatment
Introduction
Cervical cancer, as the fourth cause of cancer-related death among women globally, originates from epithelial neoplastic transformation of uterine cervix [1,2]. According to statistics, the numbers of new cervical cancer cases and deaths in the United States are estimated to reach 13,800 and 4,290, respectively [3]. According to international guidelines, surgery is recommended as the standard treatment for early stage (IA and IB1) cervical cancer patients [4]. But for patients with stage IB2 onwards, concurrent chemoradiation is primarily considered for them especially who are not suitable for radical hysterectomy [4,5]. Nevertheless, both of these therapeutic strategies often induce unfavorable clinical outcomes or serious side effects such as ovarian failure [6] and loss of fertility [7]. Thus, discovering an effective agent with less toxicity is paramount to decreasing cervical cancer related mortality and improving overall survival.
Statins are a well-established type of agents for the treatment of hypercholesterolemia and often are categorized into two classes: hydrophilic (pravastatin and rosuvastatin) and lipophilic (cerivastatin, simvastatin, lovastatin and atorvastatin) [8]. Individual statin, however, displays different pharmacokinetic properties as well as therapeutic dose ranges, which are due to the differing chemical structure [8]. Hydrophilic statins tend to be more hepatoselective whereas lipophilic statins are prone to penetrate extrahepatic cells and hence achieve higher concentration in these tissues [9-11]. Moreover, Kato et al. [12] revealed that cell apoptosis was selectively induced in the presence of lipophilic but not hydrophilic statin in cervical, ovarian and endometrial cancer cells, suggesting a greater anti-cancer effect of lipophilic statins. As a class of drugs, statins are known for causing a few possible adverse effects in which myalgia and rhabdomyolysis are the most important [13]. Notably, several clinical trials show that ATO leads to appreciably lower incidence of myalgia than simvastatin, giving the evidence that ATO is more beneficial than simvastatin in lowering lipid levels and reducing side effects [14,15].
The cholesterol lowering function of statins is dependent on their specific inhibition of the HMG-CoA reductase, the rate-limiting enzyme of the mevalonate pathway [16-18]. Notably, this pathway produces numerous non-steroid isoprenoid metabolites that have been shown to promote tumor progression, including cholesterol, FPP, and GGPP [19,20], indicating that statins may exert antineoplastic functions in human cancers. In fact, various in vitro and in vivo studies have revealed that statins significantly attenuate cell proliferation and metastasis and promote apoptosis in human cervical [21,22], glioma [23], breast [24], ovarian [25], and endometrial cancers [26]. Moreover, epidemiologic researches have unveiled a consistent association between long term statin use and a reduced risk of several human malignancies [27,28]. However, the underlying mechanisms by which statins display anticancer effects in cervical cancer have not been extensively investigated yet. Therefore, we herein selected ATO and investigated its role as a potential therapeutic agent in cervical cancer.
Indeed, autophagy can be activated by statins via the AMPK-mTOR signaling pathway [29], promoting the survival of cancer cells [30]. Dysfunction of autophagy is tightly involved in a series of human diseases including cancer [31,32], whereas downregulation of essential autophagy proteins significantly reduces the survival of cancer cells [33]. Hence, statin-induced autophagy activation may play a potent and important role in reducing the anticancer effect of statins. However, the research focusing on statin use, autophagy, and antitumor effects in cervical cancer has not been conducted yet.
In this study, we explored that the anticancer effect of atorvastatin (ATO), a common statin drug, on the growth and apoptosis of cervical cancer cells and tumor xenografts. Next, the role of intermediates (FPP and GGPP) during the mevalonate pathway in ATO-induced apoptotic function was also investigated. Furthermore, the autophagy induction by ATO in cervical cancer cells was also detected. More importantly, we explored whether ATO-induced cytotoxicity could be enhanced in cervical cancer cells after combination with autophagy inhibitors.
Materials and methods
Antibodies and reagents
Details of primary antibodies used in this study were listed in Table 1. The following secondary antibodies were used: peroxidase-conjugated goat anti-rabbit antibody (1:5000; Biosharp, Cat #BL003A) and anti-mouse antibody (1:5000; Biosharp, Cat #BL001A). Chemical reagents used were as follows: atorvastatin (Sigma, Cat #PHR1422), FPP (Sigma, Cat #F6892), GGPP (Sigma, Cat #G6025), Baf-A1 (MedChemExpress, Cat. #HY-100558), 3-MA (MedChemExpress, Cat #HY-19312), and DMSO (Sigma, Cat #D2650). ATO was dissolved in DMSO and diluted with DMEM to storage concentrations of 5, 10, 20, 40, 80 μM before experiments. FPP and GGPP were both diluted with DMEM and used at 10 μM. Baf-A1 was dissolved in DMSO and used at 10 nM. In addition, 3-MA was dissolved in PBS and used at 5 mM.
Table 1.
Lists of primary antibodies used in this study
| Primary antibodies | Source | Catalog no. | Western blot |
|---|---|---|---|
| Rabbit monoclonal anti-PARP | Cell Signaling Technology | 9532 | 1:1000 |
| Rabbit monoclonal anti-cleaved PARP | Cell Signaling Technology | 5625 | 1:1000 |
| Rabbit monoclonal anti-Cleaved Caspase-3 | Cell Signaling Technology | 9664 | 1:1000 |
| Rabbit monoclonal anti-Bim | Cell Signaling Technology | 2933 | 1:1000 |
| Rabbit monoclonal anti-AMPK | Cell Signaling Technology | 5831 | 1:1000 |
| Rabbit monoclonal anti-p-AMPK (Thr172) | Cell Signaling Technology | 50081 | 1:1000 |
| Rabbit monoclonal anti-Akt | Cell Signaling Technology | 4685 | 1:1000 |
| Rabbit monoclonal anti-p-Akt (Ser473) | Cell Signaling Technology | 4060 | 1:1000 |
| Rabbit monoclonal anti-LC3B | Cell Signaling Technology | 3868 | 1:1000 |
| Rabbit monoclonal anti-p62 (Ser349) | Cell Signaling Technology | 16177 | 1:1000 |
| Rabbit polyclonal anti-mTOR | Abways Technology | CY3456 | 1:1000 |
| Rabbit monoclonal anti-p-mTOR (S2481) | Abways Technology | CY5996 | 1:1000 |
| Mouse monoclonal anti-Caspase-3 | Abcam | Ab208161 | 1:1000 |
| Mouse monoclonal anti-GAPDH | Beyotime | AF0006 | 1:2000 |
| Mouse monoclonal anti-Tubulin | Beyotime | AT819 | 1:2000 |
Cell culture
Human cervical cancer cell lines SiHa and Caski were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All cells were cultured in DMEM (Gibco, Cat #11965092) supplemented with 10% FBS (Gibco, Cat #26010074) and 1% penicillin-streptomycin solution (Solarbio, Cat #P1400) at 37°C in a humidified atmosphere containing 5% CO2. The cells were split at approximately 80% confluence by using 0.25% trypsin with EDTA (Gibco, Cat #25200072).
Cell viability assay
Viability of cervical cancer cells was measured by using CCK-8 according to the manufacturer’s protocol (Dojindo, Cat #FH783). In brief, SiHa and Caski cells (3000 cells/well) were seeded into 96-well plates with a volume of 100 μl/well and cultured under different treatment conditions. After incubation for the indicated times, 10 μl CCK-8 solution in 90 μl DMEM was added into each well, followed by incubation at 37°C for 2 h. Then, the absorbance of each well at 450 nm was measured under a Micro-plate Reader (Bio-Rad, USA). After correction by subtracting the optical density value of wells that did not contain cells, cell viability was calculated as a percentage of control. Each sample was detected in triplicate.
Cell apoptosis assay
Cell apoptosis was determined using Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, Cat #556547). After relevant treatment, cells in each group were collected with 0.25% trypsin, washed with ice-cold PBS for twice, resuspended in 100 μl 1 × binding buffer, and stained using kit solution at room temperature for 15 min in the dark. After incubation, 400 μl of 1 × binding buffer was added to the mixture, percentage of apoptotic cells was analyzed by flow cytometry (Beckman Coulter, USA). All experiments were repeated three times.
Western blotting analysis
Western blotting for measuring the expression of target proteins were performed using the standard approach as described previously [34]. At the indicated time points, total proteins from cells were isolated using RIPA lysis buffer (Beyotime, Cat no. P0013B) on ice. Protein concentration was determined by the BCA protein assay kit (Beyotime, Cat #P0012). Equal amounts of proteins (40 μg) were separated by 8~15% SDS-PAGE, and then transferred onto PVDF membranes (Millipore, Cat #ISEQ00010). The membranes were blocked with 5% skim milk and incubated with primary antibodies overnight at 4°C. Subsequently, the membranes were washed in Tris-buffered saline (Cell Signaling Technology, Cat #9997) with 1% Tween-20 (Sigma, Cat #P1379) solution, followed by incubation with appropriate peroxidase-conjugated secondary antibodies for 1 h at room temperature. The epitopes were detected and visualized with ECL kit (Invitrogen, Cat #WP20005). The intensities of the blots were quantified by densitometry and normalized by corresponding levels of internal loading controls (GAPDH or Tubulin). Image Lab 3.0 software (Bio-Rad, USA) was employed to analyze intensities of bands. Each experiment was repeated three times.
Transmission electron microscopy
In brief, SiHa cells were treated with or without 20 μM ATO for 48 h. Then, the cells were washed with PBS for 3 times and collected by centrifugation at 800 × g for 5 min. Subsequently, 2% preheated agarose was dripped to the cell pellet and mixed uniformly. After that, the agar was cut into 1 mm3 blocks and placed into a 2.5% glutaraldehyde/0.1 M PBS at 4°C overnight for fixation. Then, the sections were washed in PBS for 3 times and post fixed in 1% osmium tetroxide for 1 h at 4°C. After dehydration in a graded series of ethanol solutions, samples were embedded into araldite and cut into semi-thin sections. Finally, ultra-thin sections were stained with 1% uranyl acetate and observed under a transmission electron microscope (Hitachi, Japan).
Confocal microscope
A tandem mRFP-GFP-LC3 adenovirus was purchased from Hanheng Biotechnology (Shanghai, China, Cat #HH20180703DHP-AP01). The GFP fluorescence (green) could be quenched under the acidic condition (pH between 4 and 5) or in the lysosome lumen, whereas the mRFP fluorescent (red) is stable. Adenoviral infection was performed according to the manufacturer’s instructions. Cells were seeded in the 35 mm confocal culture dishes (Wuxi NEST Biotechnology, China) at a density of 1 × 104 cells until cells reached 30% to 40% confluence, and then transfected with mRFP-GFP-LC3 adenovirus (multiplicity of infection = 100) for 2 h at 37°C in 10% serum-containing DMEM. After DMEM were removed, cells were washed with PBS twice and treated with or without 20 μM ATO for 48 h at 37°C in 5% CO2. Finally, LC3 punctas were observed and captured using the confocal laser scanning microscope (Leica, Germany). In brief, mRFP fluorescence without GFP indicated that autophagosomes have been fused with lysosome. The yellow punctas (colocalization of both GFP and mRFP fluorescence) are shown in phagophores or autophagosomes. The increase of both yellow and red punctas indicated the increase of autophagic flux in cells, while the only elevation on yellow punctas without red punctas alteration, or the decrease of both yellow and red punctas suggested the blockade of autophagic flux.
Animal experiments
Five-week-old female BALB/c nude mice were purchased from Beijing Vital River Laboratory Animal Technology (Beijing, China), and were housed under SPF condition in Wenzhou Medical University for in vivo animal experiments. All procedures were performed under sterile precautions. To generate a xenograft model, 5 × 106 SiHa cells in 100 μL PBS were injected subcutaneously into the right flank of each mouse. Two weeks after inoculation, mice were divided into two groups and gavaged daily either PBS (control) or ATO (50 mg/kg) for six weeks. The length (a) and width (b) of tumor tissue were monitored with a caliper once a week, with the following formula to calculate the tumor volume: V (mm3) = 0.53*ab2. All mice were sacrificed after tumor inoculation for a total of eight weeks. After that, the xenograft tumors were removed, weighed and imaged with the camera. Tissues including liver and kidney were collected, formalin-fixed, embedded in paraffin and stained with hematoxylin and eosin (HE) to examine the morphology. All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health. Approval for all animal experiments was obtained from the Ethics Review Committee of the Second Affiliated Hospital of Wenzhou Medical University.
TUNEL staining
The TUNEL staining analysis was conducted according to the instructions of the TUNEL assay kit (Roche, Cat #6432344001). Briefly, the paraffin-embedded sections were cut and dewaxed in xylene, and the placed in proteinase K (Servicebio, Cat #G1205) solution for antigen retrieval. Tumor cell apoptosis in xenograft tissues was confirmed by terminal deoxynucleotidyl transferase dUTP nick end labeling technique for 30 min at 37°C in a humidified atmosphere in dark. Subsequently, the slides were washed with PBS for 3 times, and incubated in the dark for 10 s after a circle of DAPI attaining solution was slowly added. Subsequently, the slides were placed in PBS to remove excess stain. The TUNEL positive (shining green) cells were apoptotic cells and the nuclei was stained with DAPI (blue). Imaging and cell counting were conducted under the Olympus light microscope (Olympus, Japan).
Statistical analysis
Statistical analyses were performed with SPSS 22.0 software (Chicago, USA). Continuous variables were presented as the means ± standard deviation (SD). Two-tailed independent Student’s t-test was applied for comparison of two experimental groups. Statistical differences among the more than two groups were assessed by one-way ANOVA and LSD post-hoc test. P value < 0.05 was defined as statistical significance.
Results
ATO induces morphological changes and reduces the viability of cervical cancer cells
The effects of ATO on cell morphological changes were examined in SiHa and Caski cervical cancer cell lines. Both cell lines were seeded into the 6-well plate and exposed to different doses of ATO (0, 5 and 20 μM) for 24 h. As shown in Figure 1, ATO exposure dose-dependently changed the morphological appearance in both SiHa (Figure 1A) and Caski (Figure 1B) cells. Both cell lines after ATO treatment showed a characteristic shrinkage of cytoplasm as well as plasma membrane budding (Figure 1A, 1B). Subsequently, a time course and concentration curve of ATO was also carried out to detect the effect of ATO on viability of SiHa and Caski cells. Efficacy of ATO treatment with 10, 20, 40, or 80 μM was evaluated at 24, 48, and 72 h of incubation time. Results showed that ATO significantly reduced cell viability in a dose- and time-dependent way in both cell lines (Figure 1C, 1D).
Figure 1.

ATO inhibited the viability of cervical cancer cells in vitro. A, B. Morphological changes in SiHa and Caski cells in response to different concentrations of ATO (0, 5 and 20 μM) for 24 h (magnification × 100). C, D. Cell viability of SiHa and Caski cells were determined by CCK-8 assay after treatment of ATO with increasing concentrations (0, 10, 20, 40 and 80 μM) for 0, 24, 48 and 72 h. Data were presented as mean ± SD; *P < 0.05 vs. control group. Each experiment was performed three times.
ATO induces cell apoptosis of cervical cancer cells
Annexin V-FITC/PI double staining was performed to exam the ability of ATO to induce apoptosis in cervical cancer cells. SiHa and Caski cells were treated with ATO with different concentrations (0, 20, 40, and 80 μM) for 48 h, and then followed by flow cytometry. As shown in Figure 2, an increase of apoptosis was detected in both SiHa (P < 0.05; Figure 2A) and Caski (P < 0.05; Figure 2B) cells. Additional western blotting analysis firmly established the apoptotic induction by ATO in both cells, accompanying with a significant elevation of pro-apoptotic proteins including cleaved-caspase-3, cleaved-PARP, and Bim in a dose-dependent manner (P < 0.05; Figure 2C, 2D).
Figure 2.

ATO promoted the apoptosis of cervical cancer cells in vitro. A, B. Apoptosis of SiHa and Caski cells after treatment of ATO (0, 20, 40 and 80 μM) for 48 h was evaluated by Annexin V-FITC/PI double staining using a flow cytometry, and percent of apoptotic cells was quantified. Numbers represent the percentage of the frequency in each quadrant. C, D. Western blot for the detection of cell apoptosis markers (PARP, Caspase-3, and BIMs) in SiHa and Caski cells after treatment of ATO (0, 5, 10, 20 and 40 μM) for 48 h. The bar graphs represented the ratios of Cleaved PARP/PARP, Cleaved Caspase-3/Caspase-3, and BIMs/Tubulin. Data were presented as mean ± SD. *P < 0.05 vs. 0 μM ATO group. Each experiment was performed three times.
ATO exhibits anti-cancer effects in vivo
To characterize whether ATO could inhibit tumor growth in vivo, subcutaneous cervical cancer xenografts in nude mice injected with SiHa cells were generated. Two weeks after inoculation, the nude mice bearing SiHa tumor xenografts were gavaged with 50 mg/kg/d ATO (the treatment group) or PBS (the control group) for 6 weeks. The size of implanted tumors was measured weekly by calipers. Animals in both groups stayed alive till sacrifice. After sacrifice, the tumor weight was measured. Results showed that tumor xenograft growth was markedly suppressed by ATO (Figure 3). As graphically presented in Figure 3A, tumor volumes were significantly smaller in the treatment group compared to controls from week 2 onwards (P < 0.05). As compared to the control group, the mean wet tumor weight in ATO-treated group was approximately 30% lighter (P < 0.05; Figure 3B, 3C). Figure 3D showed that ATO treatment did not lead to morphological and histological changes in the mouse liver and kidney under macroscopic examination, indicating that ATO use may be less toxic to organs or tissues. TUNEL assay results showed that the number of apoptotic cells in tumor xenografts was significantly increased in ATO treatment group compared to that in control group (Figure 3E).
Figure 3.

ATO inhibited SiHa tumor growth and induced apoptosis in vivo. Groups of seven female BALB/c nude mice were subcutaneously transplanted with 5 × 106 SiHa cells. Two weeks after inoculation, the mice were treated with 50 mg/kg/d ATO p.o. for 6 weeks consecutively. Control groups received PBS only. Then, all mice were sacrificed and xenograft tumors were isolated. Tissues including liver and kidney were also collected. A. Tumor volume (mm3) from ATO treatment and control groups were measured weekly by calipers. Data were presented as mean ± SD. *P < 0.05 vs. 0 weeks group. B. Macroscopic photos of tumor isolated from individual mice after sacrifice. C. Tumor weight (g) from ATO treatment and control groups were measured immediately after sacrifice. Data were presented as mean ± SD. *P < 0.05 vs. control group. D. Representative hematoxylin and ecosin (HE) staining images of liver and kidney tissues from control and ATO-treated mice (magnification × 100). E. Representative photomicrographs of TUNEL staining in tumor tissues sections from control and ATO-treated mice under a fluorescent microscopy (magnification × 100).
ATO-induced apoptosis in cervical cancer cells is reversed by mevalonate metabolites
We explored whether mevalonate metabolites could rescue the ATO-induced apoptosis in cervical cancer cells. The intermediates during the mevalonate pathway, FPP (10 μM) and GGPP (10 μM), were added into the 6-well plate alone or together with ATO (20 μM) at the same time. After incubation for 24 h, the morphological appearance of cells was observed under microscope. Furthermore, cell viability of each group was detected after 72 h incubation. DMSO was used as the control group. As shown in Figure 4, FPP or GGPP alone in culture medium showed no effect on the morphological changes and viability of SiHa (Figure 4A, 4B) and Caski (Figure 4C, 4D) cells compared to the control group. Notably, supplementation of GGPP almost completely rescued the morphological changes and viability of both SiHa (P < 0.05; Figure 4B) and Caski (P < 0.05; Figure 4D) cells after ATO treatment, while a minor effect was observed with FPP exposure concomitantly.
Figure 4.

FPP and GGPP reversed ATO-mediated viability-inhibiting and apoptosis-promoting effects in cervical cancer cells in vitro. A, C. Morphological changes in SiHa and Caski cells in response to DMSO, 10 μM FPP, 10 μM GGPP, 20 μM ATO, 20 μM ATO plus 10 μM FPP, and 20 μM ATO plus 10 μM GGPP for 72 h (magnification × 100). B, D. Cell viability of SiHa and Caski cells was determined by CCK-8 assay after treatment of DMSO, 10 μM FPP, 10 μM GGPP, 20 μM ATO, 20 μM ATO plus 10 μM FPP, and 20 μM ATO plus 10 μM GGPP for 72 h. E, F. Western blot for the detection of cell apoptosis markers (PARP and Caspase-3) in SiHa and Caski cells after treatment of DMSO, 10 μM FPP, 10 μM GGPP, 20 μM ATO, 20 μM ATO plus 10 μM FPP, and 20 μM ATO plus 10 μM GGPP for 72 h. The bar graphs represented the ratios of Cleaved PARP/PARP and Cleaved Caspase-3/Caspase-3. Data were presented as mean ± SD. *P < 0.05 vs. DMSO group; #P < 0.05 vs. ATO group. Each experiment was performed three times.
Consistently, GGPP or FPP alone also did not alter apoptosis-related proteins expression levels in both cell lines (Figure 4E, 4F). However, downregulation of cleaved-PARP and cleaved-caspase-3 was detected in ATO with co-treatment of GGPP in SiHa (P < 0.05; Figure 4E) and Caski (P < 0.05; Figure 4F) cells, while addition of FPP had a little influence on atorvastatin-induced cleavage of PARP and caspase-3. These results indicated that ATO may induce apoptosis of cervical cancer through negatively control of the mevalonate pathway.
ATO induces cellular autophagy in cervical cancer cells
Transmission electron microscopy images showed an increased number of the double-membrane and autophagosome-like structures upon ATO stimulation in SiHa cells (Figure 5A). To detect the status of autophagic flux, the tandem mRFP-GFP-LC3 adenovirus was used. Compared to control group, more numbers of red puncta (GFP-mRFP-) and yellow puncta (GFP-mRFP-) were observed in ATO treatment SiHa cells (Figure 5B).
Figure 5.

ATO induced cellular autophagy in cervical cancer cells in vitro. A. Transmission electron microscopic ultrastructures of SiHa cells treated with 10 μM ATO for 48 h (magnification × 15000, × 30000). Arrowhead represented autophagosomes. B. Representative images of mRFP-GFP-LC3 transfected SiHa cells treated with 10 μM ATO for 48 h under fluorescence microscopy (Bar = 30 μm). C, D. Western blot for the detection of cell autophagic markers (LC3 and p62) in SiHa and Caski cells after treatment of ATO (0, 5, 10, 20, and 40 μM) for 48 h. The bar graphs represented the ratios of LC3-II/LC3-I and p62/GAPDH. E, F. Western blot for the detection of autophagy-related pathway markers (AMPK, Akt, and mTOR) in SiHa and Caski cells after treatment of ATO (0, 5, 10, 20, and 40 μM) for 48 h. The bar graphs represented the ratios of p-AMPK/AMPK, p-Akt/Akt, and p-mTOR/mTOR. Data were presented as mean ± SD. *P < 0.05 vs. 0 μM ATO group. Each experiment was performed three times.
Cellular autophagy activation was also confirmed by conversion of the cytosolic LC3-I into the autophagosome-associated LC3-II [35]. Therefore, we employed the ratio of LC3-II to LC3-I to determine the LC3 conversion during autophagy in this study. To further determine the induction of autophagy by ATO in cervical cancer cells, the expression patterns of endogenous LC3 and p62 were evaluated by western blotting analysis after ATO treatments with varying doses (0, 5, 10, 20, and 40 μM) for 48 h. As shown in Figure 5C, 5D, treatment of both SiHa and Caski cells with ATO led to a dramatic elevation of LC3-II:LC3-I ratio (P < 0.05) and a sharp decline of p62 level (P < 0.05).
mTOR is a core regulator of autophagic process and the autophagic process is induced by suppression of mTOR signaling [36]. It is widely accepted that AMPK and Akt are important upstream regulator of mTOR signaling in mammalian cells. Specifically, AMPK blocks mTOR activity thereby promoting the occurrence of autophagy [37] whereas Akt activates mTOR signaling and induces autophagy inhibition [38]. To further confirm if ATO could influence the expression of AMPK and Akt/mTOR signaling pathways that were critically involved in autophagy in SiHa and Caski cells, the expression of related proteins (AMPK, p-AMPK, Akt, p-Akt, mTOR, p-mTOR) was measured by western blotting assay under ATO exposure at different concentrations (0, 5, 10, 20, and 40 μM) for 48 h. As shown in Figure 5E, there was no significant change on the total protein of AMPK after ATO administration, while ATO use significantly increased the level of p-AMPK in SiHa cells than that in control (P < 0.05). In addition, ATO showed no effect on the total level of Akt and mTOR, while the level of phosphorylation of Akt and mTOR were suppressed in SiHa cells (P < 0.05). Similar results were also found in Caski cells (Figure 5F). Taken together, these findings suggested that ATO induced cellular autophagy in cervical cancer cells, which might be associated with the AMPK and Akt/mTOR signaling pathways.
Autophagy inhibition increases anticancer effects of ATO in cervical cancer cells
To investigate whether the anticancer effects of ATO could be enhanced by autophagy inhibition, both SiHa and Caski cells were treated with 40 μM ATO alone or in presence of chemical autophagy inhibitors (10 nM Baf-A1 or 5 mM 3-MA) for 48 h. Cell viability was evaluated using CCK-8 assay and apoptotic-related proteins were detected by western blotting, respectively. Data showed that ATO suppressed the growth of SiHa (P < 0.05; Figure 6A) and Caski (P < 0.05; Figure 6B) cells, and this inhibitory effect was significantly enhanced after supplementation with Baf-A1 (P < 0.05), as confirmed by enhancement of cleaved-caspase-3 and cleaved-PARP (P < 0.05; Figure 6C, 6D). Similar results were also obtained when ATO was co-treated with 3-MA in both two cell lines (P < 0.05; Figure 6E, 6H). These observations suggested that autophagy inhibitors inhibited the autophagic process and markedly improved the anti-cancer effects of ATO on cervical cancer cells.
Figure 6.

Autophagy inhibitors Baf-A1 or 3-MA increased anticancer effects of ATO in cervical cancer cells in vitro. A, B. Cell viability of SiHa and Caski cells was determined by CCK-8 assay after treatment of 10 nM Baf-A1, 40 μM ATO, and 10 nM Baf-A1 plus 40 μM ATO for 48 h. C, D. Western blot for the detection of cell apoptosis markers (PARP and Caspase-3) in SiHa and Caski cells after treatment of 10 nM Baf-A1, 40 μM ATO, and 10 nM Baf-A1 plus 40 μM ATO for 48 h. The bar graphs represented the ratios of Cleaved PARP/PARP and Cleaved Caspase-3/Caspase-3. E, F. Cell viablity of SiHa and Caski cells was determined by CCK-8 assay after treatment of 5 mM 3-MA, 40 μM ATO, and 5 mM 3-MA plus 40 μM ATO for 48 h. G, H. Western blot for the detection of cell apoptosis markers (PARP and Caspase-3) in SiHa and Caski cells after treatment of 5 mM 3-MA, 40 μM ATO, and 5 mM 3-MA plus 40 μM ATO for 48 h. The bar graphs represented the ratios of Cleaved PARP/PARP and Cleaved Caspase-3/Caspase-3. Data were presented as mean ± SD. *P < 0.05 vs. control group; #P < 0.05 vs. ATO group. Each experiment was performed three times.
Discussion
ATO has been previously reported as an authentic agent in inhibiting the proliferation and inducing cell death of human cervical cancer cells [21], which is also confirmed by a recent study [22]. Consistently, the current study indicated that ATO displayed antineoplastic activity in cervical cancer cells via suppression of viability and induction of apoptosis. It has been known that caspases are both the initiators and the executors of cell death that initially trigger the cellular apoptosis, among which the caspase-3 is the well-characterized [39]. After being activated by auto-proteolysis, the executor caspase-3 is cleaved off by the initiator caspases and further promotes the cleavage of specific cellular substrates [40,41]. During this process, several important apoptotic hallmarks are observed including chromatin condensation, plasma membrane asymmetry and cellular blebbing [41], which eventually induce the typical morphological change of cells [42]. In line with this notion, the elevation of cleaved-caspase-3 accounted for the morphological change of both SiHa and Caski cells after ATO administration. Notably, caspases family has also participated in the regulation of non-apoptotic processes such as cell differentiation [43], strongly indicating the existence of other apoptotic cascade in cells. In fact, numerous signaling molecules apart from caspases have been reported to be involved in cell apoptotic pathway, such as PARP [44] and Bim proteins [45]. Western blotting experiments in our study also demonstrated that treatment of ATO, in a concentration-dependent way, induced the apoptosis of cervical cancer cells through enhancing the expression of cleaved-PARP and Bim. Furthermore, the anticancer effect of ATO in cervical cancer was also explored in vivo. Results showed that ATO induced a remarkable inhibition of tumor growth, as evidenced by smaller volume and lighter weight of xenograft tumors in atorvastatin treatment group compared with control group (PBS). Additional TUNEL assay study confirmed that ATO significantly enhanced the number of apoptotic cervical cancer cells.
The mevalonate metabolic pathway is responsible for the biosynthesis of several isoprenoid products during lipid metabolism [46]. Among these metabolites, FPP and GGPP are often dysregulated during this pathway thereby leading to tumorigenesis [19]. It has been widely accepted that ATO exerts its lipid-lowering functions via specifically blocking the mevalonate pathway and reducing the level of FPP and GGPP [17]. In this study, we added either FPP or GGPP as supplements and investigated whether it could reverse the anticancer effects of ATO in these cells. Results showed that either FPP or GGPP alone had no significant cytotoxicity in cervical cancer cells. Although only a subtle effect was detected in FPP supplemented with ATO treatment group, the ATO-induced morphological transformation and viability reduction were almost completely reversed after administration of GGPP and ATO simultaneously in cervical cancer cells. Similarly, supplementation of GGPP resulted in a sharp decline of the cleaved-PARP and cleaved-caspase-3 levels after ATO treatment, while a little effect of FPP was observed. These findings indicated that ATO might induce cervical cancer cells apoptosis through suppressing the mevalonate pathway and biosynthesis of the isoprenoid intermediates.
Autophagy is a cellular process responsible for cytoplasmic components degradation during periods of stress related to nutrient starvation, infection and apoptosis [47]. In this process, a number of intracellular macromolecules and organelles are engulfed by double membrane-bound autophagosomes and delivered to lysosomes or vacuoles for subsequent degradation [48]. Accumulated evidences have demonstrated that ATO could induce autophagy in various human cancers [29]. In this study, the double-membrane and autophagosome-like structures were observed in SiHa cells after ATO stimulation using transmission electron microscopy. Moreover, ATO treatment significant elevated the expression of LC3-II and downregulated p62 proteins, which are considered as the two classic autophagy-related markers [49].
Previous study has reported that AMPK and Akt/mTOR signaling pathways are critically involved in autophagy regulation [50]. In our study, western blotting results showed a significant accumulation of p-AMPK as well as a dramatic reduction of Akt and mTOR phosphorylation. These results together suggested the autophagic process was activated by ATO in cervical cancer cells. Notably, multiple cancer treatments can also induce the autophagic process, which provides a survival advantage for cancer cells under exposure to cancer treatments such as chemotherapies [51]. Therefore, interference with autophagy represents a rational therapeutic strategy to enhance the effect of anti-cancer therapeutics for human cancers. Herein, inhibition of autophagy by either Baf-A1 or 3-MA indeed suppressed proliferation and promoted apoptosis induced by ATO in cervical cancer cells. These findings suggested that suppression of autophagy strengthened the anticancer effect of ATO in cervical cancer cells, which was consistent with a previous study focusing on hepatocellular carcinoma and colorectal cancer [16].
In conclusion (Figure 7), this study showed that ATO inhibited tumor growth and promoted apoptosis of cervical cancer both in vitro and in vivo, which might be associated with the suppression of the mevalonate pathway. Furthermore, ATO could also induce autophagy in cervical cancer cells. More importantly, pharmacologic inhibition of autophagy significantly enhanced ATO-induced cytotoxicity in cervical cancer. Therefore, our results recommended that combination of autophagy inhibitors and ATO could be a promising therapeutic approach for cervical cancer.
Figure 7.
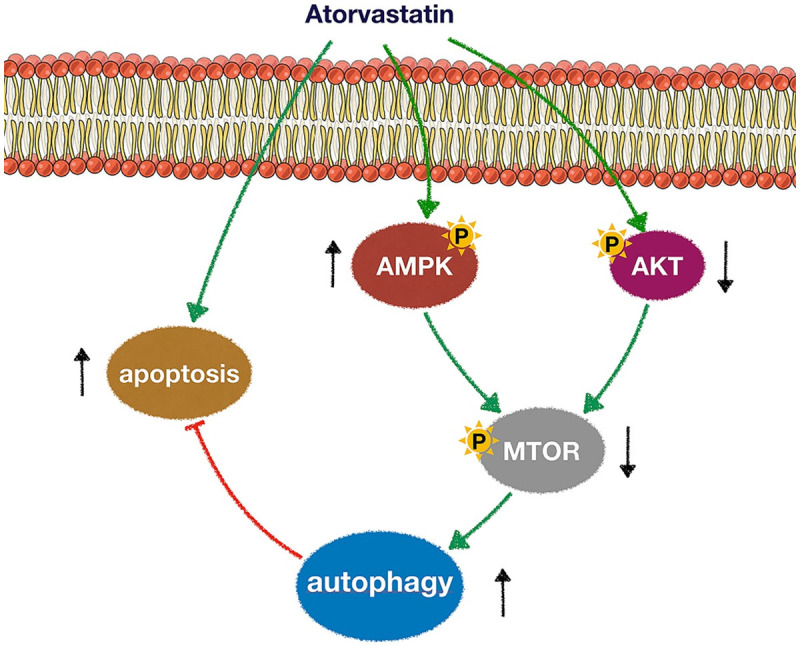
A schematic diagram depicting the potential molecular mechanisms underlying anticancer effects of ATO in cervical cancer cells. ATO-mediated blockade of HMG-CoA activity led to activation of AMPK and inhibition of Akt. mTOR, a major AMPK/Akt downstream target and negative autophagy regulator, was also down-regulated by ATO. Pharmacologic inhibition of autophagy significantly enhanced ATO-mediated apoptosis.
Acknowledgements
This work was supported by grants to XZ from Center for Uterine Cancer Diagnosis & Therapy Research of Zhejiang Province and Project of Wenzhou science and Technology Bureau (No. ZS2017006).
Disclosure of conflict of interest
None.
Abbreviations
- 3-MA
3-methyladenine
- Akt
protein kinase B
- AMPK
adenosine 5’-monophosphate (AMP)-activated protein kinase
- ANOVA
analysis of variance
- ATO
atorvastatin
- Baf-A1
bafilomycin-A1
- BCA
bicinchoninic acid
- DAPI
4, 6-diamino-2-phenyl indole
- DMEM
dulbecco’s modifed eagle medium
- DMSO
dimethyl sulfoxide
- ECL
enhanced chemiluminescence
- EDTA
ethylene diamine tetraacetic acid
- FBS
fetal bovine serum
- FPP
farnesylpyrophosphate
- GFP
green fluorescent protein
- GGPP
geranylgeranylpyrophosphate
- HE
hematoxylin and eosin
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- LC3
light chain 3
- LSD
least significance difference
- RFP
red fluorescent protein
- mTOR
mammalian target of rapamycin
- p-AMPK
phosphorylated-AMPK
- p-Akt
phosphorylated-Akt
- PARP
poly ADP-ribose polymerase
- PBS
phosphate-buffered saline
- p-mTOR
phosphorylated-mTOR
- PVDF
polyvinylidene fluoride
- RIPA
radioimmunoprecipitation assay
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SPF
specific pathogen free
References
- 1.Koh WJ, Abu-Rustum NR, Bean S, Bradley K, Campos SM, Cho KR, Chon HS, Chu C, Clark R, Cohn D, Crispens MA, Damast S, Dorigo O, Eifel PJ, Fisher CM, Frederick P, Gaffney DK, Han E, Huh WK, Lurain JR 3rd, Mariani A, Mutch D, Nagel C, Nekhlyudov L, Fader AN, Remmenga SW, Reynolds RK, Tillmanns T, Ueda S, Wyse E, Yashar CM, McMillian NR, Scavone JL. Cervical cancer, version 3.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2019;17:64–84. doi: 10.6004/jnccn.2019.0001. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 4.Tomao F, Santangelo G, Musacchio L, Di Donato V, Fischetti M, Giancotti A, Perniola G, Petrella MC, Monti M, Palaia I, Muzii L, Benedetti Panici P. Targeting cervical cancer: is there a role for poly (ADP-ribose) polymerase inhibition? J Cell Physiol. 2020;235:5050–5058. doi: 10.1002/jcp.29440. [DOI] [PubMed] [Google Scholar]
- 5.Kumar L, Harish P, Malik PS, Khurana S. Chemotherapy and targeted therapy in the management of cervical cancer. Curr Probl Cancer. 2018;42:120–128. doi: 10.1016/j.currproblcancer.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Wo JY, Viswanathan AN. Impact of radiotherapy on fertility, pregnancy, and neonatal outcomes in female cancer patients. Int J Radiat Oncol Biol Phys. 2009;73:1304–1312. doi: 10.1016/j.ijrobp.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, Li W, Kanis MJ, Qi G, Li M, Yang X, Kong B. Oncologic and obstetrical outcomes with fertility-sparing treatment of cervical cancer: a systematic review and meta-analysis. Oncotarget. 2017;8:46580–46592. doi: 10.18632/oncotarget.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19:117–125. doi: 10.1111/j.1472-8206.2004.00299.x. [DOI] [PubMed] [Google Scholar]
- 9.Germershausen JI, Hunt VM, Bostedor RG, Bailey PJ, Karkas JD, Alberts AW. Tissue selectivity of the cholesterol-lowering agents lovastatin, simvastatin and pravastatin in rats in vivo. Biochem Biophys Res Commun. 1989;158:667–675. doi: 10.1016/0006-291x(89)92773-3. [DOI] [PubMed] [Google Scholar]
- 10.McKenney JM. Pharmacologic characteristics of statins. Clin Cardiol. 2003;26:III32–38. doi: 10.1002/clc.4960261507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Q, Liao JK. Pleiotropic effects of statins. - Basic research and clinical perspectives. Circ J. 2010;74:818–826. doi: 10.1253/circj.cj-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kato S, Smalley S, Sadarangani A, Chen-Lin K, Oliva B, Branes J, Carvajal J, Gejman R, Owen GI, Cuello M. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J Cell Mol Med. 2010;14:1180–1193. doi: 10.1111/j.1582-4934.2009.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simic I, Reiner Z. Adverse effects of statins - myths and reality. Curr Pharm Des. 2015;21:1220–1226. doi: 10.2174/1381612820666141013134447. [DOI] [PubMed] [Google Scholar]
- 14.Stoekenbroek RM, Boekholdt SM, Fayyad R, Laskey R, Tikkanen MJ, Pedersen TR, Hovingh GK Incremental Decrease in End Points Through Aggressive Lipid Lowering Study Group. High-dose atorvastatin is superior to moderate-dose simvastatin in preventing peripheral arterial disease. Heart. 2015;101:356–362. doi: 10.1136/heartjnl-2014-306906. [DOI] [PubMed] [Google Scholar]
- 15.Bakker-Arkema RG, Best J, Fayyad R, Heinonen TM, Marais AD, Nawrocki JW, Black DM. A brief review paper of the efficacy and safety of atorvastatin in early clinical trials. Atherosclerosis. 1997;131:17–23. doi: 10.1016/s0021-9150(97)06066-8. [DOI] [PubMed] [Google Scholar]
- 16.Auer J, Berent R, Weber T, Eber B. Clinical significance of pleiotropic effects of statins: lipid reduction and beyond. Curr Med Chem. 2002;9:1831–1850. doi: 10.2174/0929867023369024. [DOI] [PubMed] [Google Scholar]
- 17.Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–1900. doi: 10.1056/NEJMoa1201832. [DOI] [PubMed] [Google Scholar]
- 18.Reboulleau A, Robert V, Vedie B, Doublet A, Grynberg A, Paul JL, Fournier N. Involvement of cholesterol efflux pathway in the control of cardiomyocytes cholesterol homeostasis. J Mol Cell Cardiol. 2012;53:196–205. doi: 10.1016/j.yjmcc.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016;16:718–731. doi: 10.1038/nrc.2016.76. [DOI] [PubMed] [Google Scholar]
- 20.Dimitroulakos J, Lorimer IA, Goss G. Strategies to enhance epidermal growth factor inhibition: targeting the mevalonate pathway. Clin Cancer Res. 2006;12:4426s–4431s. doi: 10.1158/1078-0432.CCR-06-0089. [DOI] [PubMed] [Google Scholar]
- 21.Crescencio ME, Rodriguez E, Paez A, Masso FA, Montano LF, Lopez-Marure R. Statins inhibit the proliferation and induce cell death of human papilloma virus positive and negative cervical cancer cells. Int J Biomed Sci. 2009;5:411–420. [PMC free article] [PubMed] [Google Scholar]
- 22.AlKhalil M, Al-Hiari Y, Kasabri V, Arabiyat S, Al-Zweiri M, Mamdooh N, Telfah A. Selected pharmacotherapy agents as antiproliferative and anti-inflammatory compounds. Drug Dev Res. 2020;81:470–490. doi: 10.1002/ddr.21640. [DOI] [PubMed] [Google Scholar]
- 23.Oliveira KA, Dal-Cim T, Lopes FG, Ludka FK, Nedel CB, Tasca CI. Atorvastatin promotes cytotoxicity and reduces migration and proliferation of human A172 glioma cells. Mol Neurobiol. 2018;55:1509–1523. doi: 10.1007/s12035-017-0423-8. [DOI] [PubMed] [Google Scholar]
- 24.Kotamraju S, Williams CL, Kalyanaraman B. Statin-induced breast cancer cell death: role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 2007;67:7386–7394. doi: 10.1158/0008-5472.CAN-07-0993. [DOI] [PubMed] [Google Scholar]
- 25.Jones HM, Fang Z, Sun W, Clark LH, Stine JE, Tran AQ, Sullivan SA, Gilliam TP, Zhou C, Bae-Jump VL. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am J Cancer Res. 2017;7:2478–2490. [PMC free article] [PubMed] [Google Scholar]
- 26.Schointuch MN, Gilliam TP, Stine JE, Han X, Zhou C, Gehrig PA, Kim K, Bae-Jump VL. Simvastatin, an HMG-CoA reductase inhibitor, exhibits anti-metastatic and anti-tumorigenic effects in endometrial cancer. Gynecol Oncol. 2014;134:346–355. doi: 10.1016/j.ygyno.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Platz EA, Leitzmann MF, Visvanathan K, Rimm EB, Stampfer MJ, Willett WC, Giovannucci E. Statin drugs and risk of advanced prostate cancer. J Natl Cancer Inst. 2006;98:1819–1825. doi: 10.1093/jnci/djj499. [DOI] [PubMed] [Google Scholar]
- 28.Elmore RG, Ioffe Y, Scoles DR, Karlan BY, Li AJ. Impact of statin therapy on survival in epithelial ovarian cancer. Gynecol Oncol. 2008;111:102–105. doi: 10.1016/j.ygyno.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Yang Z, Xie L, Xu L, Xu D, Liu X. Statins, autophagy and cancer metastasis. Int J Biochem Cell Biol. 2013;45:745–752. doi: 10.1016/j.biocel.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Liu B, Wen X, Cheng Y. Survival or death: disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013;4:e892. doi: 10.1038/cddis.2013.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 33.Xu HM, Liang Y, Chen Q, Wu QN, Guo YM, Shen GP, Zhang RH, He ZW, Zeng YX, Xie FY, Kang TB. Correlation of Skp2 overexpression to prognosis of patients with nasopharyngeal carcinoma from South China. Chin J Cancer. 2011;30:204–212. doi: 10.5732/cjc.010.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen Z, Li S, Sheng B, Shen Q, Sun LZ, Zhu H, Zhu X. The role of atorvastatin in suppressing tumor growth of uterine fibroids. J Transl Med. 2018;16:53. doi: 10.1186/s12967-018-1430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 36.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma X, Hu Y. Targeting PI3K/Akt/mTOR cascade: the medicinal potential, updated research highlights and challenges ahead. Curr Med Chem. 2013;20:2991–3010. doi: 10.2174/09298673113209990124. [DOI] [PubMed] [Google Scholar]
- 39.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 40.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 41.Stennicke HR, Salvesen GS. Caspases - controlling intracellular signals by protease zymogen activation. Biochim Biophys Acta. 2000;1477:299–306. doi: 10.1016/s0167-4838(99)00281-2. [DOI] [PubMed] [Google Scholar]
- 42.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 43.Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–539. doi: 10.1038/cdd.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Virag L, Robaszkiewicz A, Rodriguez-Vargas JM, Oliver FJ. Poly(ADP-ribose) signaling in cell death. Mol Aspects Med. 2013;34:1153–1167. doi: 10.1016/j.mam.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 45.Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI, Wong KK, Letai A. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67:11867–11875. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 46.Liao P, Hemmerlin A, Bach TJ, Chye ML. The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnol Adv. 2016;34:697–713. doi: 10.1016/j.biotechadv.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagelkerke A, Bussink J, Geurts-Moespot A, Sweep FC, Span PN. Therapeutic targeting of autophagy in cancer. Part II: pharmacological modulation of treatment-induced autophagy. Semin Cancer Biol. 2015;31:99–105. doi: 10.1016/j.semcancer.2014.06.001. [DOI] [PubMed] [Google Scholar]