

Abstract
Olmesartan (OMST) is a new angiotensin II receptor antagonist recently approved by the FDA to treat cardiovascular diseases. We investigated the molecular mechanisms by which OMST regulates vascular senescence. In the present study, bleomycin (BLM) was used to induce senescence in vascular smooth muscle cells (VSMCs); after which, the cells were treated with OMST. The effects of OMST on BLM-mediated cell senescence were evaluated using cell adhesion, NAD+/NADH, and Annevin V/PI double staining assays, as well as by immunofluorescence staining of γH2AX, Edu flow cytometry, and evaluations of senescence-associated β-gal activity. Differentially expressed microRNAs (DEMs) were identified by miRNA microarray assays, and subsequently validated by quantitative real time PCR. Bisulfite sequencing PCR (BSP) was used to detect the methylation status of the miR-665 promoter. The target genes of miR-665 were predicted and confirmed using luciferase reporter assays. We found that miR-665 was upregulated in VSMCs in response to BLM-induced cellular senescence. BSP studies revealed that CpG sites in the promoter region of the miR-665 gene underwent extensive demethylation during BLM-induced cellular senescence, and there was a concomitant up-regulation of miR-665 expression. SDC1 mRNA was identified as a direct target of miR-665. Either miR-665 overexpression or SDC1 knockdown significantly reversed the effects of OMST on BLM-induced VSMC senescence. Moreover, SDC1 overexpression partially reversed the changes that occurred in cells with BLM-induced senescence caused by miR-665 overexpression. Our findings suggest that the miR-665/SDC1 axis functions as a vital modulator of VSMC senescence, and may represent a novel biological target for treating atherosclerosis.
Keywords: Atherosclerosis, vascular smooth muscle cell senescence, olmesartan, MiR-665, SDC1
Introduction
Atherosclerosis is the major cause of coronary heart disease, cerebral infarction, and peripheral vascular disease [1,2]. It is well recognized that vascular senescence plays an important role in age-associated cardiovascular diseases, including atherosclerosis [3]. Most arterial walls [4] and human vascular smooth muscle cells (VSMCs) entering senescence exhibit a significant loss of proliferative capability and self-repair capacity [5], and have increased levels of certain senescence markers, such as senescence-associated β-galactosidase (SA-β-gal), p21, and p16 [6,7]. Bleomycin (BLM) is one of the most commonly used sclerosants, and produces several biochemical effects, including induction of cell cycle G2/M phase arrest and apoptosis, and cellular RNA degradation [8]. BLM has been used to induce devastating aging-associated diseases such as pulmonary fibrosis [9] and dermal fibrosis [10], and has also been reported to diminish epithelial regenerative capacity and induce alveolar epithelial cell senescence [11]. Those reports suggest that BLM-induced VSMC senescence could be used as a cellular senescence model in which to explore the biochemical mechanisms underlying atherosclerosis and identify potential molecular targets for its treatment.
Olmesartan (OMST) is the pharmacologically active metabolite of olmesartanmedoxomil, which was found to reduce inflammation responses and increase microvascular neogenesis [12,13]. Recently, OMST has attracted attention as an effective drug for treating hypertension, albuminuria, and glomerular hypertrophy [14,15]. In particular, OMST has been approved by the FDA as a new angiotensin II receptor antagonist for use in treating certain cardiovascular diseases [16]. OMST exerts its therapeutic effects by directly inhibiting hub kinases or indirectly modulating protein kinase signaling pathways involved in the pathogenesis of atherosclerosis. Furthermore, OMST was shown to exert anti-atherosclerotic effects [16-19]. However, the molecular mechanisms by which OMST regulates vascular senescence remain unclear.
MicroRNAs (miRNAs) are small non-coding RNAs with a length of 19-25 nucleotides. MiRNAs play a vital role in regulating gene expression by binding to the 3’-untranslated region (3’UTR) of their target mRNA molecules [20]. Recent studies have focused on the roles played by miRNAs in vascular diseases, including atherosclerosis. For example, miRNA-30e was shown to regulate TGF-β-mediated NADPH oxidase 4-dependent oxidative stress in atherosclerosis via Snai1 [21]. However, the changes in miRNA expression that allow OMST to regulate vascular smooth muscle cell senescence have not been thoroughly investigated.
In this study, we systematically evaluated the levels of miRNA expression in senescent vascular smooth muscle cells after treatment with OMST. The key differentially expressed miRNA was identified, and its potential target gene was investigated. Our study provides an experimental-based rationale for investigating the mechanism by which OMST regulates BLM-mediated vascular smooth muscle cell senescence.
Materials and methods
Cell culture and treatment
Human VMSCs (No: CRL-1999) were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) containing 10% FBS (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a 5% CO2 atmosphere. For senescence induction, cells at 90% confluence were further cultured in growth medium containing 50 μM bleomycin (BLM group) or vehicle (PBS-non-senescent control) for 3 h in atmospheric oxygen. Next, an aliquot of cells in the senescence group was treated with 50 nM olmesartan (the BLM+OMST group).
Cell adhesion assay
Cell adhesion was measured with a VybrantTM Cell Adhesion Assay Kit (V-13181, Molecular Probes, Inc., Eugene, OR, USA) according to the manufacturer’s instructions. Briefly, VMSCs were washed with PBS, re-suspended in FBS-free medium at a density of 5 × 106 cells per mL, and subsequently incubated with a calcein-labeled cell suspension for 60 h at 37°C. Next, cellular fluorescence was detected at an emission maximum of 517 nm.
NAD+/NADH assay
The cellular content of NAD+ and NADH was determined using a NAD+/NADH Assay Kit with WST-8 (S0175, Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. In brief, ~1 × 106 VMSCs were plated into each well of six-well plates and incubated with 200 μL of NAD+/NADH extraction solution. The NAD+/NADH ratio was calculated by using standard curves for NAD+ and NADH.
Immunofluorescence
Approximately 5 × 105 VMSCs were inoculated into each well of a six-well plate that had been preset with sterilized coverslips; the cells were then cultured overnight. The next day, the cells were fixed with 4% paraformaldehyde for 10 min and then permeabilized with 0.2% Triton X-100 for 5 min. After blocking in 4% FBS for 30 min, the cells were incubated with a red fluorescent antibody against γH2AX, followed by incubation with an Alexa Fluor 594 dye-conjugated secondary antibody; after which, the cellular nuclei were counterstained with DAPI. Finally, the slides were photographed under an Olympus confocal microscope (FV1000MPE, Olympus, Tokyo, Japan).
Senescence-associated β-gal activity assay
Cellular senescence was analyzed using a senescence-associated β-galactosidase (SA-β-gal) Staining Kit (Beyotime Biotechnology) according tothe manufacturer’s protocol. Briefly, VMSCs were fixed with β-galactosidase fixation solution (2% formaldehyde/0.2% glutaraldehyde in PBS) for 5 min, washed with PBS, and then incubated overnight with SA-β-gal staining solution at 37°C. The next day, the cells were observed under a light microscope, and the percentage of cells that stained positive for SA-β-gal was calculated as previously described [22].
Edu flow cytometry assay
Proliferating VMSCs were detected by using a Cell Light™ EdU Apollo®488 In Vitro Imaging Kit (Ribobio Co., Ltd., Guangzhou, China) according to the manufacturer’s instructions. Briefly, VMSCs were incubated with 50 μmol of EdU labeling medium for 2 h at 37°C; after which, they were washed three times with PBS and then stained with 200 μL of 1 × Apollo solution for 30 min at 37°C in the dark. Finally, the percentage of Edu-positive cells was analyzed using a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Cell apoptosis assay
Cell apoptosis was measured using an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s protocol. VMSCs were collected and fixed with 75% ethanol for 20 min. The VMSCs were then washed with PBS, resuspended in 500 μL of Annexin-V-FITC for 10 min at room temperature, and then incubated with 5 μL of PI for 60 min at 4°C in the dark. Cell apoptosis was detected using a flow cytometer (FACSCalibur, BD Biosciences) equipped with FlowJo software.
RNA extraction
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and further purified using an RNeasy Mini Spin Column Kit (Qiagen, Inc., Valencia, CA, USA. The total RNA was then quantified via spectrophotometry.
MiRNA microarray
After being checked for quality, the RNA samples were labeled using reagents in a miRCURY™ Power Labeling Kit (Exiqon, Denmark) according to the manufacturer’s instructions. The labeled RNA samples were hybridized onto a miRCURYTM LNA Array (Exiqon, Denmark), washed with a Wash Buffer Kit (Exiqon, Denmark), and then scanned with a GenePix 4000B microarray scanner (Axon Molecular Devices, San Jose, CA, USA).
Microarray data analysis
Raw data extraction was performed with GeneChip Command Console software (version 4.0; Affymetrix, Inc., Santa Clara, CA, USA). After normalization, miRNAs that were expressed at significantly different levels in the two groups were identified by using specific cut-off criteria (a p-value < 0.05 and a fold-change > 2.0). Hierarchical clustering was performed to display the differentially expressed miRNAs. A VENN analysis was performed to screen the overlapping significantly differentially expressed miRNAs (DEMs). Subsequently, the target genes of the overlapping DEMs were predicted by the TargetScan database (http://targetscan.org/). These putative target genes were then subjected to Gene Ontology (GO) classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. The significant GO and KEGG pathway terms were identified by using a cut-off p-value < 0.05 and a count > 2.
Quantitative real time PCR
First, cDNA was synthesized from the same RNA samples used in the prior microarray analysis. Next, PCR analysis was performed on an ABI Prism 7900 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) using SYBR-Green PCR Master Mix (Applied Biosystems).The expression levels of miRNAs and SDC1 were normalized to those of U6 and GAPDH, respectively, using the 2-ΔΔCT method. The primer sequences used in this study are listed in Table 1.
Table 1.
Primers used for quantitative real time PCR analysis
| Gene | Primer sequence 5’-3’ |
|---|---|
| miR-665 | F: ACACTCCAGCTGGGACCAGGAGGCTGAGG |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAGGGGCCT | |
| miR-1267 | F: ACACTCCAGCTGGGCCTGTTGAAGTGTAAT |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGGGGATT | |
| miR-4742-3p | F: ACACTCCAGCTGGGTCTGTATTCTCCTTTGC |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCTGCAGGCA | |
| miR-193a-3p | F: ACACTCCAGCTGGGAACTGGCCTACAAAGTC |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACTGGGAC | |
| miR-5579-3p | F: ACACTCCAGCTGGGTTAGCTTAAGGAGTACC |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGATCTGGT | |
| miR-3133 | F: ACACTCCAGCTGGGTAAAGAACTCTTAAAAC |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATTGGGTT | |
| miR-4517 | F: ACACTCCAGCTGGGAAATATGATGAAACTCACAG |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCTCAGCTG | |
| miR-942p | F: ACACTCCAGCTGGGCACATGGCCGAAACAGA |
| RT: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACTTCTC | |
| All | R: CTCAACTGGTGTCGTGGA |
| SDC1 | F: CCACCATGAGACCTCAACCC |
| R: GCCACTACAGCCGTATTCTCC | |
| U6 | F: CTCGCTTCGGCAGCACA |
| R: AACGCTTCACGAATTTGCGT | |
| GAPDH | F: TGTTCGTCATGGGTGTGAAC |
| R: ATGGCATGGACTGTGGTCAT |
F: forward primer; R: reverse primer; RT: reverse transcription.
Bisulfite sequencing PCR (BSP)
Genomic DNA was extracted from cells using TRIzol Reagent (Invitrogen), and then subjected to bisulfite conversion using an EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. The bisulfite-converted genomic DNA was used for a methylation analysis of the miR-665 promoter. The predicted methylation primers were designed according to “MethPrimer” (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi). The amplified fragments were cloned into a pGEMT Easy vector (Promega, Madison, WI, USA), and five clones were randomly selected for bisulfite sequencing. Percentage of methylation was calculated using QUMA (http://quma.cdb.riken.jp/top/index.html).
Cell transfection
MiR-665 mimics, small interfering RNA targeting SDC1 (siSDC1), and the negative control oligonucleotides (miR-NC and siNC) were synthesized by RiboBio Co., Ltd. (Guangzhou, China). The pcDNA3.1/SDC1 vector with the SDC1 coding region was successfully constructed by OriGene Technologies, Inc. (Beijing, China). A total of 2 × 105 VMSCs were plated into each well of six-well plates in triplicate, and then incubated overnight prior to transfection. All plasmid transfections were performed using Lipofectamine™ 2000 (Invitrogen, USA) according to instructions provided by the manufacturer.
Luciferase reporter assay
Among the predicted targets of miR-665, the 3’-UTR of SDC1 mRNA contained binding sites for miR-665 with reasonable scores. Therefore, SDC1 was selected as a potential target gene. Next, luciferase reporter assays were performed to determine whether miR-665 directly targets SDC1 mRNA. The SDC1 3’UTR segment containing the putative binding site for miR-665 was amplified and inserted into a pmirGLO vector (Promega), resulting in the SDC1 WT. Meanwhile, a mutation was introduced into the potential miR-665 binding sites (designated as MUT SDC1) by using the Quick Change Stratagene method. VMSCs (2 × 104 per well) were cultured in 24-well plates and then transfected with 50 nM miR-665 mimics or the NC, together with 0.5 µg of luciferase reporter vector containing the SDC1 WT or MUT SDC1, by using Lipofectamine 2000. Forty-eight hours later, luciferase activity was assessed with a Dual-Luciferase Reporter Assay System (Promega).
Western blot analysis
Briefly, total protein was extracted using a RIPA buffer (Beyotime Institute of Biotechnology, Shanghai, China) and the protein concentration in each sample was determined with a BCA assay kit (Beyotime Institute of Biotechnology). Next, an equal amount of protein (30 µg) from each sample was separated by 10% SDS-PAGE, and the protein bands were transferred onto PVDF membranes, which were subsequently blocked with 5% non-fat milk at room temperature for 1 h. The membranes were then incubated with primary antibodies against SDC1 and GAPDH (Abcam, Cambridge, UK) at 4°C overnight; after which, they were incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. The protein bands were visualized by using an enhanced Chemiluminescent Substrate kit (Pierce; Thermo Fisher Scientific, Inc.) and analyzed by Image-Pro Plus software version 6.0.
Statistical analysis
All in vitro experiments were performed at least three times and results are presented as a mean value ± standard deviation (SD). All statistical analyses were performed using SPSS Statistics for Windows, Version 19.0 software (IBM Corp., Armonk, NY, USA). Statistical differences between two groups were analyzed using the two-sided Student’s t-test or one-way analysis of variance (ANOVA), followed by Dunnett’s post hoc test. A p-value < 0.05 was considered to be statistically significant.
Results
OMST treatment counteracted BLM-induced cellular senescence in VSMCs
To investigate how OMST affects vascular senescence, we used BLM to establish a cellular senescence model using human VSMCs. As shown in Figure 1A, treatment with BLM resulted in shrunken cells that contained increased amounts of pigment when compared to control cells. Moreover, the adhesive ability of VSMCs was significantly decreased by BLM treatment (Figure 1B). Our data showed that the NAD+/NADH ratio played a key role in regulating cellular metabolism and energy production. Results from NAD+/NADH assays (Figure 1C) indicated that the NAD+/NADH ratio in the BLM group was significantly higher than that in the control group, but partially decreased after OMST treatment. We also assessed the effect of OMST in the cellular senescence model. Studies that employed immunostaining of γH2AX showed that OMST treatment obviously alleviated DNA damage (Figure 1D). SA-β-gal staining (Figure 1E, 1F) revealed that BLM induced an increase in the numbers of SA-β-Gal-positive cells when compared with those numbers in the control group, demonstrating that VSMC senescence had been successfully induced by BLM. Interestingly, the numbers of SA-β-Gal-positive cells were significantly reduced by OMST treatment. We next tested the effects of OMST on the proliferation and apoptosis of senescent VSMCs. Our results showed that the numbers of Edu-positive cells were significantly increased after OMST treatment (Figure 1G, 1H), and the increased level of apoptosis among VSMCs in the BLM group was significantly reduced by OMST treatment (Figure 1I, 1J). Taken together, these results showed that OMST inhibited VSMC senescence and promoted the proliferation of VSMCs.
Figure 1.

OMST treatment counteracted BLM-induced cellular senescence in human VSMCs. A. Cell morphology was observed in the BLM and control groups. We observed thattreatment with BLM resulted in shrunken cells that contained increased amounts of pigment when compared to control cells. B. Cell adhesion assays were performed using VSMCs in the BLM and control groups. C. The NAD+/NADH ratio was determined by an NAD+/NADH assay. D. Representative immunofluorescence images of nuclear γH2AX (cell nuclei: blue; γH2AX: red) in VSMCs from the control, BLM, and BLM+OMST groups. E, F. Cell senescence was evaluated by staining for SA-β-gal-positive cells. Representative photomicrographs showing SA-β-gal-positive cells (blue) among the VSMCs. G, H. Cell proliferation was assessed by the Edu flow cytometry assay. I, J. Flow cytometry with Annexin V/PI staining was used to analyze VSMC apoptosis. **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. BLM group. Abbreviations: VSMCs, vascular smooth muscle cells; BLM, bleomycin; OMST, olmesartan.
MiR-665 was expressed in response to BLM-induced VSMC senescence
To investigate the molecular mechanism by which OMST alleviates cellular senescence, VSMCs were treated with BLM alone or BLM+OMST; after which, the cells were harvested to examine alterations that occurred in their miRNA profiles. A total of 8 overlapping DEMs were identified in VSMCs via microarray analysis (Figure 2A, 2B). A GO analysis indicated that miR-665 and its targets were primarily grouped into the categories of ‘Organelle’ and ‘Molecular function’ (Figure 2C). Enriched pathways of miR-665 and its targets were primarily involved in ‘Proteoglycans in cancer’ and ‘Pathways in cancer’ (Figure 2D). We next performed PCR assays to validate the differential expression of the 8 miRNAs identified in the initial screening process. As indicated in Figure 2E, the level of miR-665 expression was significantly up-regulated, while the levels of miR-193-3p, miR-3133, miR-4517, and miR-942-3p expression were all notably downregulated in VSMCs with BLM-induced cellular senescence. However, only the altered levels of miR-665 and miR-3133 expression were reversed by OMST treatment. Because miR-665 showed a more obvious alteration than miR-3133, miR-665 was selected for further analysis.
Figure 2.
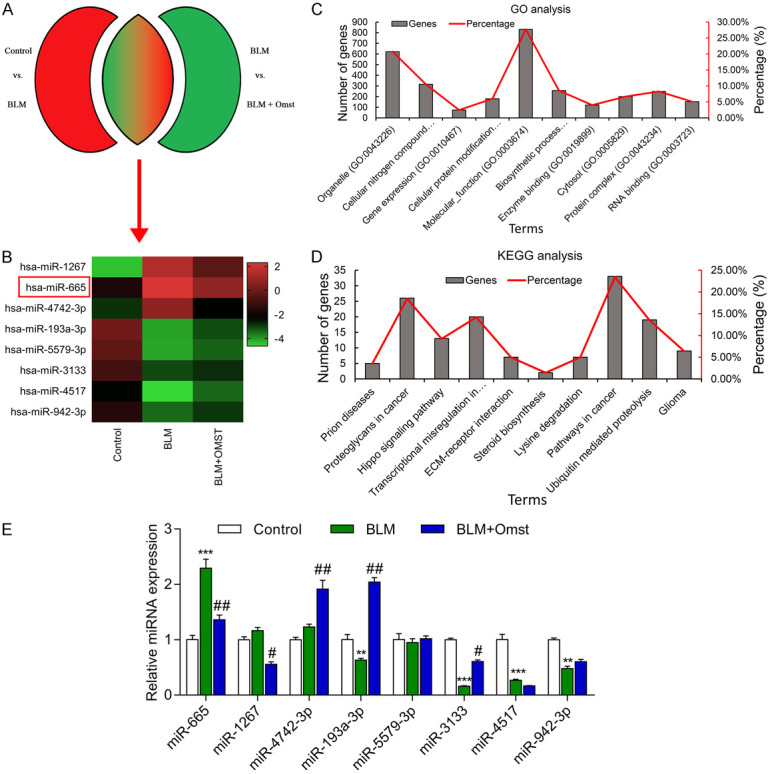
Identification of miR-665 expressed in response to BLM-induced senescence in VSMCs. (A, B) The expression levels of 8 miRNAs in VSMCs with BLM-induced senescence with and without OMST treatment as determined via microarray analysis. Histogram of (C) enriched GO terms and (D) KEGG signaling pathways of 8 miRNAs and their targets. (E) The 8 differentially expressed miRNAs were validated by quantitative real time PCR. **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. BLM group.
MiR-665 was up-regulated during BLM-induced VSMC senescence due to aberrant DNA methylation
To explore whether the aberrant overexpression of miR-665 was due to demethylation, BSP was used to examine the promoter CpG Island of miR-665. As shown in Figure 3A, 3B, the CpG Island of the miR-665 promoter was predicted. A subsequent BSP analysis (Figure 3C) showed that the methylation status of the miR-665 promoter was reduced from 69.23% in the BLM group to 35.39% in the control group, which might explain the increased miR-665 expression after BLM treatment.
Figure 3.
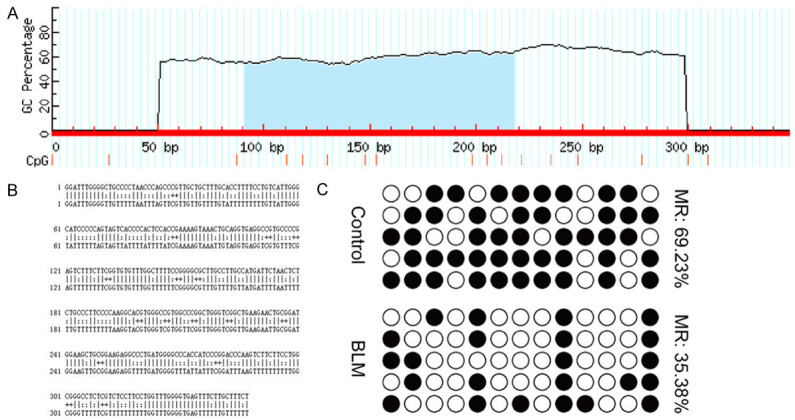
Analysis of the miR-665 promoter in VSMCs with BLM-induced senescence. A, B. The CpG Island of the miR-665 promoter was predicted, and primers were designed using MethPrimer. C. BSP results and the relative methylation ratio of each CpG site in the miR-665 promoter region in BLM-induced VSMCs are shown. The solid circles represent methylated CpG sites, while hollow circles represent non-methylated CpG sites.
Up-regulation of miR-665 reversed the effects of OMST on BLM-induced VSMC senescence
To investigate how miR-665 participates in the process by which OMST alleviates BLM-induced VSMC senescence, a miR-665 mimic was used to examine the effects of molecular manipulation of miR-665 on VSMC senescence. As shown in Figure 4A, the level of miR-665 in the BLM+OMST group was ~2-fold higher than that in the mimic control. Subsequently, we also found that the decreases in NAD+/NADH ratio (Figure 4B), numbers of SA-β-Gal-positive cells (Figure 4C, 4D), and γH2AX immunostaining (Figure 4E) in the BLM+OMST group could all be significantly reversed by miR-665 overexpression. In addition, transfection with the miR-665 mimic remarkably abolished the effect of OMST on the proliferation (Figure 4F, 4G) and apoptosis (Figure 4F-H) of VSMCs with BLM-induced senescence. There results indicated that OMST counteracted BLM-induced cellular senescence by down-regulating miR-665.
Figure 4.

Up-regulation of miR-665 reversed the effects of OMST on BLM-induced VSMC senescence. BLM-induced VSMCs were treated with OMST with or without miR-665 mimic transfection. A. MiR-665 expression was determined by quantitative real time PCR. B. The NAD+/NADH ratio was determined by a NAD+/NADH assay. C, D. Cell senescence was evaluated by staining for SA-β-gal-positive cells. Representative photomicrographs showing SA-β-gal-positive cells (blue) among VSMCs. E. Representative immunofluorescence images of nuclear γH2AX (cell nuclei: blue; γH2AX: red) in VSMCs from the control, BLM, BLM+OMST, and BLM+OMST+miR-665 mimic groups. F-H. Cell proliferation and cell apoptopsis was assessed by the Edu flow cytometry assay and Annexin V/PI staining flow cytometry assay. ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. BLM group; &P < 0.05 vs. BLM+OMST group.
SDC1 was a direct target of miR-665
We analyzed the expression of SDC1 protein in VSMCs after transfection with the miR-665 mimics or NC. As expected, miR-665 mimics transfection downregulated the expression of SDC1 protein (Figure 5A). We then used TargetScan software to perform a bioinformatics analysis that sought to identify the potential target genes of miR-665. Among the predicted target genes, SDC1 was reported to be associated with aging and senescence, and was therefore selected for further study. As shown in Figure 5B, a potential miR-665 binding site was identified on the 3’UTR of SDC1 mRNA. Therefore, a luciferase reporter assay was conducted to further examine whether miR-665 might regulate SDC1 expression. Our results showed that the relative luciferase activity in the SDC1 WT was significantly downregulated by transfection with the miR-665 mimic, while no obvious change occurred after transfection with the SDC1 MUT, confirming that miR-665 directly binds to the 3’UTR of SDC1 mRNA (Figure 5C).
Figure 5.
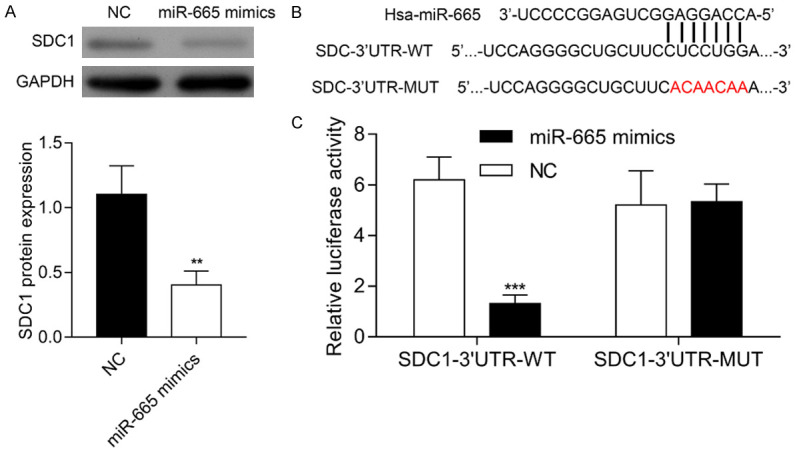
SDC1 was a direct target of miR-665. A. A western blot analysis was performed to detect the expression of SDC1 protein in VMSCs after transfection with miR-665 mimics or the NC. B. The predicted binding site of miR-665 on the 3’-UTR of human SDC1 mRNA. A mutant binding site was constructed and the red letters indicate mutated nucleotides. C. Relative luciferase activity was evaluated. **P < 0.01, ***P < 0.001 vs. NC. Abbreviations: UTR, untranslated region; NC, negativecontrol; WT, wild type; MUT, mutant.
Downregulation of SDC1 reversed the effects of OMST on BLM-induced VSMC senescence
To investigate whether SDC1 regulates the ability of OMST to alleviate BLM-induced VSMC senescence, VSMCs in the BLM+OMST group were transfected with siSDC1 to validate its biological function. A western blot analysis (Figure 6A) and quantitative real time PCR analysis (Figure 6B) showed that SDC1 expression was significantly downregulated in VSMCs with BLM-induced VSMC senescence, but was partially restored after OMST treatment. Notably, siSDC1 transfection remarkably suppressed SDC1 expression in the BLM+OMST group. In agreement with miR-665 overexpression, we also found that siSDC1 transfection abolished the effects that OMST exerted on the NAD+/NADH ratio (Figure 6C), numbers of SA-β-Gal-positive cells (Figure 6D, 6E), immunostaining of γH2AX (Figure 6F), numbers of Edu-positive cells (Figure 6G, 6H), and apoptosis (Figure 6G-I). There results indicated that OMST counteracted BLM-induced cellular senescence by up-regulating SDC1.
Figure 6.

Downregulation of SDC1 reversed the effects of OMST on BLM-induced VSMC senescence. BLM-induced VSMCs were treated with OMST with or without siSDC1 transfection. A. SDC1 protein expression was detected by western blot analysis. B. SDC1 mRNA expression was determined by quantitative real time PCR. C. The NAD+/NADH ratio was determined by an NAD+/NADH assay. D, E. Cell senescence was evaluated by staining for SA-β-gal-positivecells. Representative photomicrographs showing SA-β-gal-positive cells (blue) among VSMCs. F. Representative immunofluorescence images of nuclear γH2AX (cell nuclei: blue; γH2AX: red) in VSMCs from the control, BLM, BLM+OMST, and BLM+OMST+siSDC1 groups. G-I. Cell proliferation and cell apoptopsis was assessed by the Edu flow cytometry assay and Annexin V/PI staining flow cytometry assay. **P < 0.01, ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. BLM group; &P < 0.05 vs. BLM+OMST group.
OMST counteracted BLM-induced cellular senescence via miR-665 targeting of SDC1
A rescue experiment was performed to analyze whether the direct targeting of SDC1 by miR-665 accounted for the ability of OMST to counteract BLM-induced cellular senescence. As shown in Figure 7A, 7B, the miR-665 mimic significantly suppressed SDC1 expression in VMSCs, while transfection with the pcDNA3.1/SDC1 vector restored SDC1 expression in the cells. As expected, SDC1 overexpression remarkably reversed the effects of miR-665 overexpression on the NAD+/NADH ratio (Figure 7C), numbers of SA-β-Gal-positive cells (Figure 7D, 7E), immunostaining of γH2AX (Figure 7F), numbers of Edu-positive cells (Figure 7G, 7H), and cellular apoptosis (Figure 7G-I). Collectively, our findings suggest that the targeting of SDC1 by miR-665 is the molecular mechanism by which OMST alleviates BLM-induced cellular senescence.
Figure 7.

OMST counteracted BLM-induced cellular senescence via miR-665 targeting of SDC1. BLM-induced VSMCs were treated with OMST plusmiR-665 mimics + SDC1 transfection. A. SDC1 protein expression was detected by western blotting. B. SDC1 mRNA expression was determined by quantitative real time PCR. C. The NAD+/NADH ratio was determined by a NAD+/NADH assay. D, E. Cell senescence was evaluated by staining for SA-β-gal-positive cells. Representative photomicrographs showing SA-β-gal-positive cells (blue) among VSMCs. F. Representative immunofluorescence images of nuclear γH2AX (cell nuclei: blue; γH2AX: red) in VSMCs from the control, BLM, BLM+OMST, BLM+OMST+miR-665 mimic, and BLM+OMST+miR-665 mimic+SDC1 groups. G-I. Cell proliferation and cell apoptopsis was assessed by the Edu flow cytometry assay and Annexin V/PI staining flow cytometry assay. ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01 vs. BLM group; &P < 0.05 vs. BLM+OMST group; @P < 0.05 vs. BLM+OMST+miR-665 mimic group.
Discussion
While the anti-atherosclerotic effects of OMST are well acknowledged, the molecular mechanism by which OMST regulates vascular senescence has remained unclear. In this study, we showed that OMST inhibits VMSC senescence caused by BLM. We detected the changes in miRNA expression that occurred in response to effective OMST treatments, and hypothesized that DEMs might be involved in the mechanisms of induced cellular senescence. We also found that the levels of miR-665 expression in VMSCs with BLM-induced senescence were significantly higher than those in control VMCSs, but were obviously reduced after OMST treatment, as validated by quantitative real time PCR. Moreover, we found that CpG sites in the promoter region of the miR-665 gene were extensively demethylated in cells with BLM-induced senescence, and there was a concomitant up-regulation of miR-665 expression. He et al. [23] reported that DNA methylation plays an important and complex role in regulating miRNA expression. Our results indicated that the up-regulation of miR-665 in cells with BLM-induced senescence was due, at least in part, to the demethylation of CpG sequences in the miR-66 gene promoter region. Moreover, our data also suggest that OMST alleviates BLM-mediated VMSC senescence by downregulating miR-665 expression via reversing demethylation of the miR-665 promoter (Figure 8).
Figure 8.
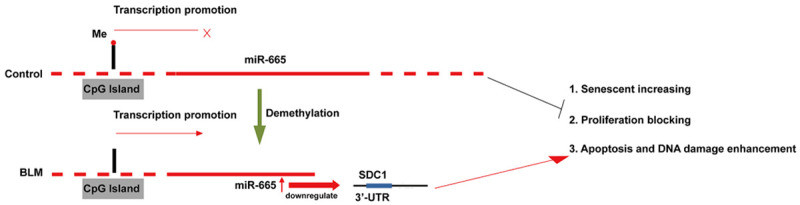
A diagram showing the mechanism for up-regulation of miR-665 in BLM-induced cellular senescence.
Functional assays demonstrated that overexpression of miR-665 reversed the inhibitory effects of OMST on BLM-induced VSMC senescence, as reflected by an elevated NAD+/NADH ratio, stronger immunostaining of γH2AX, increased numbers of SA-β-Gal-positive cells, and a higher apoptosis rate. Previous studies revealed that miR-665 was associated with cell proliferation, differentiation, and migration [24-26]. Li et al. [27] showed that ectopic expression of miR-665 promoted apoptosis ininflammatory bowel disease tissue that was exposed to different inflammatory stimuli. Recent studies have investigated whether inhibition of miR-665 might be a potential therapeutic approach for reducing inflammation and apoptosis in the clinical treatment of intestinal ischemia/reperfusion [28], intervertebral disc degeneration [29], coronary microvessel dysfunction, and heart failure [30]. Interestingly, miR-665 was also reported to inhibit VSMC proliferation, invasion, and migration that occurs in atherosclerosis [31]. From these viewpoints, miR-665 might be an important regulator of BLM-mediated VMSC senescence, and could serve as a biomarker for monitoring the pharmacological effects of OMST in treating atherosclerosis.
Targetscan software was used to predict the target genes of miR-665. A further analysis revealed that the 3’UTR of miRNA-665 has a binding site that enables it to directly regulate SDC1 expression. Syndecan-1 (SDC1) is a transmembrane heparan sulfate proteoglycan, and an extracellular matrix receptor involved in intercellular communication and proliferation, as well as angiogenesis and cell metastasis [32,33]. Similar to the effect of miR-665 overexpression, we observed that downregulation of SDC1 also reversed the effects of OMST on BLM-induced VSMC senescence. In accordance with these data, the significance of SDC1 expression has been studied in processes such as arteriogenesis [34], homeostasis [35], and tumorigenesis [36,37]. Although a direct association between OMST-alleviated BLM-mediated VMSC senescence and SDC1 expression has not been reported, alterations in SDC1 expression have been suggested as a mechanism for senescence induced by genotoxic stress [38] and ionizing radiation [39].
Moreover, our rescue experiments further confirmed that OMST counteracted BLM-induced cellular senescence via miR-665 targeting of SDC1. As an inhibitor of angiotensin II-induced VSMC migration, OMST has been demonstrated to suppress atherosclerosis progression by mediating the Src and mitogen-activated protein kinase pathways [40], and has recently been approved by the FDA for use in treating cardiovascular diseases [16]. Based on our study, we hypothesize that the inhibitory effect of OMST on BLM-mediated VMSC senescence is due to an upregulation of SDC1 resulting from inhibition of miR-665.
In summary, our study, for the first time, identified miR-665 as a critical molecule that regulates the ability of OMST to decrease BLM-mediated VMSC senescence by targeting SDC1 expression. Our in vitro experiments revealed that miR-665 overexpression significantly reversed the effects of OMST on BLM-mediated VMSC senescence. The inhibitory effects of miR-665 on BLM-mediated VMSC senescence might result from miR-665 targeting and reducing SDC1 gene expression. Taken together, the miR-665/SDC1 axis might be a novel therapeutic target for treating atherosclerosis.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81774241), the Science and Technology Planning Project of Guangdong Province of China (No. 2017ZC0207), and the Medical Scientific and Technologic Research Foundation of Guangdong Province of China (No. 2017029).
Disclosure of conflict of interest
None.
References
- 1.Chistiakov DA, Grechko AV, Myasoedova VA, Melnichenko AA, Orekhov AN. The role of monocytosis and neutrophilia in atherosclerosis. J Cell Mol Med. 2018;22:1366–1382. doi: 10.1111/jcmm.13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu X, Ma BD, Liu S, Liu J, Ma BX. Long noncoding RNA LINC00341 promotes the vascular smooth muscle cells proliferation and migration via miR-214/FOXO4 feedback loop. Am J Transl Res. 2019;11:1835–1842. [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, Gray K, Kumar S, Clarke M, Bennett M. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132:1909–1919. doi: 10.1161/CIRCULATIONAHA.115.016457. [DOI] [PubMed] [Google Scholar]
- 4.Liu T, Xu J, Guo JL, Lin CY, Luo WM, Yuan Y, Liu H, Zhang J. YAP1 up-regulation inhibits apoptosis of aortic dissection vascular smooth muscle cells. Eur Rev Med Pharmacol Sci. 2017;21:4632–4639. [PubMed] [Google Scholar]
- 5.Luo Z, Xu W, Ma S, Qiao H, Gao L, Zhang R, Yang B, Qiu Y, Chen J, Zhang M, Tao B, Cao F, Wang Y. Moderate autophagy inhibits vascular smooth muscle cell senescence to stabilize progressed atherosclerotic plaque via the mTORC1/ULK1/ATG13 signal pathway. Oxid Med Cell Longev. 2017;2017:3018190. doi: 10.1155/2017/3018190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1alpha-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. 2015;35:1963–1974. doi: 10.1161/ATVBAHA.115.305896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, Miyauchi H, Orimo M, Okada S, Takamura M, Nagai T, Kaneko S, Komuro I. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation. 2006;114:953–960. doi: 10.1161/CIRCULATIONAHA.106.626606. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto T, Katayama I. Vascular changes in bleomycin-induced scleroderma. Int J Rheumatol. 2011;2011:270938. doi: 10.1155/2011/270938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang X, Wang X, Xie X, Zeng S, Li Z, Xu X, Yang H, Qiu F, Lin J, Diao Y. Kallistatin protects against bleomycin-induced idiopathic pulmonary fibrosis by inhibiting angiogenesis and inflammation. Am J Transl Res. 2017;9:999–1011. [PMC free article] [PubMed] [Google Scholar]
- 10.Koca SS, Ozgen M, Dagli AF, Gozel N, Ozercan IH, Isik A. The protective effects of bevacizumab in bleomycin-induced experimental scleroderma. Adv Clin Exp Med. 2016;25:249–253. doi: 10.17219/acem/32484. [DOI] [PubMed] [Google Scholar]
- 11.Liu T, Gonzalez De Los Santos F, Zhao Y, Wu Z, Rinke AE, Kim KK, Phan SH. Telomerase reverse transcriptase ameliorates lung fibrosis by protecting alveolar epithelial cells against senescence. J Biol Chem. 2019;294:8861–8871. doi: 10.1074/jbc.RA118.006615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu XM, Jin YN, Ma L. Olmesartan medoxomil reverses glomerulosclerosis in renal tissue induced by myocardial infarction without changes in renal function. Exp Ther Med. 2014;8:105–109. doi: 10.3892/etm.2014.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masuda T, Muto S, Fujisawa G, Iwazu Y, Kimura M, Kobayashi T, Nonaka-Sarukawa M, Sasaki N, Watanabe Y, Shinohara M, Murakami T, Shimada K, Kobayashi E, Kusano E. Heart angiotensin II-induced cardiomyocyte hypertrophy suppresses coronary angiogenesis and progresses diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2012;302:H1871–1883. doi: 10.1152/ajpheart.00663.2011. [DOI] [PubMed] [Google Scholar]
- 14.Chrysant SG. Effectiveness of the fixed-dose combination of olmesartan/amlodipine/hydrochlorothiazide for the treatment of hypertension in patients stratified by age, race and diabetes, CKD and chronic CVD. Expert Rev Cardiovasc Ther. 2013;11:1115–1124. doi: 10.1586/14779072.2013.827449. [DOI] [PubMed] [Google Scholar]
- 15.Brousil JA, Burke JM. Olmesartan medoxomil: an angiotensin II-receptor blocker. Clin Ther. 2003;25:1041–1055. doi: 10.1016/s0149-2918(03)80066-8. [DOI] [PubMed] [Google Scholar]
- 16.Zhuang RJ, Jin WD, Wang XY, Wu XM. Identification and characterization of the druggable kinase targets of olmesartan and its analogues from a systematic kinase-chemical interaction profile in atherosclerosis. J Mol Graph Model. 2018;80:211–216. doi: 10.1016/j.jmgm.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 17.Zhou Y, Tian F, Wang J, Yang JJ, Zhang T, Jing J, Chen YD. Efficacy study of olmesartan medoxomil on coronary atherosclerosis progression and epicardial adipose tissue volume reduction in patients with coronary atherosclerosis detected by coronary computed tomography angiography: study protocol for a randomized controlled trial. Trials. 2016;17:10. doi: 10.1186/s13063-015-1097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gong X, Shao L, Fu YM, Zou Y. Effects of olmesartan on endothelial progenitor cell mobilization and function in carotid atherosclerosis. Med Sci Monit. 2015;21:1189–1193. doi: 10.12659/MSM.892996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sievers P, Uhlmann L, Korkmaz-Icoz S, Fastner C, Bea F, Blessing E, Katus HA, Preusch MR. Combined treatment with olmesartan medoxomil and amlodipine besylate attenuates atherosclerotic lesion progression in a model of advanced atherosclerosis. Drug Des Devel Ther. 2015;9:3935–3942. doi: 10.2147/DDDT.S85203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng Y, Zhou M, Zhou W. MicroRNA-30e regulates TGF-beta-mediated NADPH oxidase 4-dependent oxidative stress by Snai1 in atherosclerosis. Int J Mol Med. 2019;43:1806–1816. doi: 10.3892/ijmm.2019.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan P, Wang YJ, Li S, Wang Y, He JY, Chen YY, Deng HQ, Huang W, Zhan JK, Liu YS. The PI3K/Akt/mTOR pathway regulates the replicative senescence of human VSMCs. Mol Cell Biochem. 2016;422:1–10. doi: 10.1007/s11010-016-2796-9. [DOI] [PubMed] [Google Scholar]
- 23.He XX, Kuang SZ, Liao JZ, Xu CR, Chang Y, Wu YL, Gong J, Tian DA, Guo AY, Lin JS. The regulation of microRNA expression by DNA methylation in hepatocellular carcinoma. Mol Biosyst. 2015;11:532–539. doi: 10.1039/c4mb00563e. [DOI] [PubMed] [Google Scholar]
- 24.Hu Y, Yang C, Yang S, Cheng F, Rao J, Wang X. miR-665 promotes hepatocellular carcinoma cell migration, invasion, and proliferation by decreasing Hippo signaling through targeting PTPRB. Cell Death Dis. 2018;9:954. doi: 10.1038/s41419-018-0978-y. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Prashad N. miR-665 targets c-MYC and HDAC8 to inhibit murine neuroblastoma cell growth. Oncotarget. 2018;9:33186–33201. doi: 10.18632/oncotarget.26046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao L, Jin H, Zheng Y, Mao Y, Fu Z, Li X, Dong L. DANCR-mediated microRNA-665 regulates proliferation and metastasis of cervical cancer through the ERK/SMAD pathway. Cancer Sci. 2019;110:913–925. doi: 10.1111/cas.13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M, Zhang S, Qiu Y, He Y, Chen B, Mao R, Cui Y, Zeng Z, Chen M. Upregulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell Death Dis. 2017;8:e2699. doi: 10.1038/cddis.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Wang G, Feng D, Zu G, Li Y, Shi X, Zhao Y, Jing H, Ning S, Le W, Yao J, Tian X. Targeting the miR-665-3p-ATG4B-autophagy axis relieves inflammation and apoptosis in intestinal ischemia/reperfusion. Cell Death Dis. 2018;9:483. doi: 10.1038/s41419-018-0518-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan H, Zhao L, Song R, Liu Y, Wang L. microRNA-665 promotes the proliferation and matrix degradation of nucleus pulposus through targeting GDF5 in intervertebral disc degeneration. J Cell Biochem. 2018;119:7218–7225. doi: 10.1002/jcb.26888. [DOI] [PubMed] [Google Scholar]
- 30.Chen C, Fan J, Wang D. MiR-665 aggravates heart failure via suppressing CD34-mediated coronary microvessel angiogenesis. China Circ J. 2017 doi: 10.18632/aging.101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li K, Pan J, Wang J, Liu F, Wang L. MiR-665 regulates VSMCs proliferation via targeting FGF9 and MEF2D and modulating activities of Wnt/beta-catenin signaling. Am J Transl Res. 2017;9:4402–4414. [PMC free article] [PubMed] [Google Scholar]
- 32.Couchman JR, Woods A. Syndecans, signaling, and cell adhesion. J Cell Biochem. 1996;61:578–584. doi: 10.1002/(sici)1097-4644(19960616)61:4<578::aid-jcb11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 33.Mise I, Vucic M. Comparison of syndecan-1 immunohistochemical expression in lobular and ductal breast carcinoma with nodal metastases. Anal Cell Pathol (Amst) 2018;2018:9432375. doi: 10.1155/2018/9432375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang GL, Weitz K. Impaired arteriogenesis in syndecan-1(-/-) mice. J Surg Res. 2015;193:22–27. doi: 10.1016/j.jss.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Pal-Ghosh S, Tadvalkar G, Stepp MA. Alterations in corneal sensory nerves during homeostasis, aging, and after injury in mice lacking the heparan sulfate proteoglycan syndecan-1. Invest Ophthalmol Vis Sci. 2017;58:4959–4975. doi: 10.1167/iovs.17-21531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stepp MA, Pal-Ghosh S, Tadvalkar G, Rajjoub L, Jurjus RA, Gerdes M, Ryscavage A, Cataisson C, Shukla A, Yuspa SH. Loss of syndecan-1 is associated with malignant conversion in skin carcinogenesis. Mol Carcinog. 2010;49:363–373. doi: 10.1002/mc.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujii T, Shimada K, Tatsumi Y, Fujimoto K, Konishi N. Syndecan-1 responsive microRNA-126 and 149 regulate cell proliferation in prostate cancer. Biochem Biophys Res Commun. 2015;456:183–189. doi: 10.1016/j.bbrc.2014.11.056. [DOI] [PubMed] [Google Scholar]
- 38.Zingoni A, Cecere F, Vulpis E, Fionda C, Molfetta R, Soriani A, Petrucci MT, Ricciardi MR, Fuerst D, Amendola MG, Mytilineos J, Cerboni C, Paolini R, Cippitelli M, Santoni A. Genotoxic stress induces senescence-associated ADAM10-dependent release of NKG2D MIC ligands in multiple myeloma cells. J Immunol. 2015;195:736–748. doi: 10.4049/jimmunol.1402643. [DOI] [PubMed] [Google Scholar]
- 39.Liakou E, Mavrogonatou E, Pratsinis H, Rizou S, Evangelou K, Panagiotou PN, Karamanos NK, Gorgoulis VG, Kletsas D. Ionizing radiation-mediated premature senescence and paracrine interactions with cancer cells enhance the expression of syndecan 1 in human breast stromal fibroblasts: the role of TGF-beta. Aging (Albany NY) 2016;8:1650–1669. doi: 10.18632/aging.100989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kyotani Y, Zhao J, Tomita S, Nakayama H, Isosaki M, Uno M, Yoshizumi M. Olmesartan inhibits angiotensin II-Induced migration of vascular smooth muscle cells through Src and mitogen-activated protein kinase pathways. J Pharmacol Sci. 2010;113:161–168. doi: 10.1254/jphs.09332fp. [DOI] [PubMed] [Google Scholar]