

Abstract
Potential functions of pseudogenes on tumorigenesis and development of human malignancies have been gradually revealed recently. However, the specific regulation and intracellular events associated with pseudogenes have not been illustrated clearly in hepatocellular carcinoma (HCC). AKR1B10P1 is an isoform pseudogene of oncogenic AKR1B10, and is barely transcribed in normal hepatocytes. In this study, anomalous transcript of AKR1B10P1 was detected in both HCC tissues and cell lines, and is positively correlated with its parental genes. High level of AKR1B10P1 transcript is correlated with dismal clinicopathologic features, including large tumor dimension, high level of serum Alpha-fetoprotein (AFP), advanced TNM stages, tumor microsatellite formation and venous invasion. Loss-of and gain-of function assays demonstrated the exact impact of AKR1B10P1 on promoting HCC cell proliferation. Furthermore, transcription factor SOX4 was discovered facilitating the activation of AKR1B10P1 transcription, and was validated as a down-stream target degraded by tumor-suppressing miR-138. Meanwhile, we discovered the existence of a positive feedback from AKR1B10P1, by which miR-138 interacts with AKR1B10P1 via a competing endogenous RNA (ceRNA) way. Thus, we suggest a positive feedback loop of AKR1B10P1/miR-138/SOX4, promoting HCC cell proliferation. In summary, the AKR1B10P1/miR-138/SOX4 loop in HCC cells provides us potential and probable targets contributing to HCC prevention and therapeutic treatment.
Keywords: AKR1B10P1, miR-138, SOX4, feedback loop, cell proliferation
Introduction
Hepatocellular carcinoma (HCC) ranks the sixth incidence of human malignancies and leads to high mortality worldwide [1,2]. In spite of the progression in HCC treatment, the prognosis and over-all survival of HCC patients shows unappreciated outcomes [3,4]. Thus, innovative breakthrough for a better learning of HCC development is essential for further precise prediction of the prognosis and effectiveness on therapeutic treatment.
Pseudogenes are a group of DNA sequences generated from the duplication of their parental genes, accompanied with variety of mutations [5,6]. Initiately, pseudogenes were considered as non-functional ‘junk DNAs’ [7]. However, current evidence has gradually revealed the existence of pseudogenes transcripts, which are commonly composed with over 200 nucleotides and are regarded as a kind of long non-coding RNA (LncRNA) without protein-coding capacity [8].
As acknowledged, pseudogenes can exert competing endogenous RNA (ceRNA) effect as one of the most common functions. By binding with microRNAs (miRNAs), known as short non-coding RNAs with the length of about 22 nucleotides, pseudogenes competitively impacts the mRNA-protein translation or suppresses the degradation of mRNAs targeted by specific miRNAs, and induced stabilization of the targeted gene expression [9]. For instance, Glucosylceramidase Beta (GBA) is an oncogene in gastric cancer, targeted and suppressed by miR-212. GBAP1 is the pseudogene of GBA, and its transcript is increased in gastric cancer, particularly binding to miR-212 and protecting the expression of GBA [10]. Similarly, the activation of oncogenic MAPK pathway is stabilized by the ceRNA effects of both pseudogene KRASP1 and BRAFP1, by stabilizing the parental genes, KRAS and BRAF respectively, through binding miR-106b and miR-93 [11]. Whiltheless, disclosure of the exact functions of pseudogenes in HCC is limited yet.
Our previous study has revealed that Aldo-Keto Reductase Family 1 Member B10 (AKR1B10) was significantly highly expressed in HCC, exerting a remarkable enhancing effect on cell growth [12]. Considering the discovery of a potential indicator of HCC prevention and treatment, we further studied intensively, and discovered that the pseudogenes of AKR1B10, named AKR1B10P1, was tremendously transcribed in HCC tissues and cell lines, positively correlated with its parental gene. Whilst, abundant AKR1B10P1 transcript shows significant positive correlation with dismal clinicopathologic features in HCC patient, such as large tumor dimension, high serum Alpha-fetoprotein (AFP) expression, advanced TNM stages with tumor microsatellite and venous invasion. Functional experiments illustrated a remarkable enhancement of cell growth by AKR1B10P1. To figure out the regulation inducing AKR1B10P1 transcription activation, we explored the upstream transcription factors by using online prediction and followed molecular validation. SOX4 was screened out and confirmed as a transcription factor bind to the 5’-end sequence up AKR1B10P1 gene through Chromatin immunoprecipitation (ChIP) assay. We also discovered the down-regulation of miR-138, a tumor suppressor in HCC cells [13]. MiR-138 was found degrading SOX4 mRNA through direct interaction. Intriguingly, by analyzing and comparing the sequences of both AKR1B10P1 transcript and miR-138, we noticed the potential binding site between these two transcripts, on basis of which indicates a potential ceRNA effect. Thus, a feedback loop of AKR1B10P1/miR-138/SOX4 was proposed as a probable mechanism promoting HCC cell growth and tumor development. We supposed to suggest AKR1B10P1/miR-138/SOX4 as the innovative targets for HCC diagnosis and therapeutic treatment.
Materials and methods
Cell culture
HCC cell lines Hep3B, HepG2 and Huh7 were purchased from Shanghai Institutes for Biological Sciences, Chinese Academy of Science (Shanghai, China), The normal human hepatic cell line L02 was set as control. All cells above were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS), incubated at the environment temperature of 37°C, with 100 ug/ml streptomycin and 100 U/ml Penicillin in a humidified cell and an atmosphere of 5% CO2.
Clinical specimens
Ninety-three paired pathologically diagnosed HCC tumor specimens from patients conducted radical resection without preoperative therapy during 2015 to 2018 were collected along with the adjacent non-cancerous tissues, at the Department of General Surgery, Hepatobiliary Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. Informed consent was obtained and the study was approved by the Ethics Committee of Ruijin Hospital, Shanghai Jiaotong University School of Medicine, in accordance with the Declaration of Helsinki. Clinicopathologic features of the patients including gender, age, tumor size, number of lesions, grades et.al were collected.
RT-qPCR assay, Western blot analysis and immunohistochemistry assay
RNA isolation was carried out from tissues and cells according to the instruction of TRIzol reagent (Invitrogen, USA), and the first-strand cDNA was synthesized by using High-Capacity cDNA Reverse Transcription Kit (ABI, USA). RT-primers of the mRNAs were synthesized by Sangon Biotech Company (Shanghai, China) (Table S1). Real-time quantitative polymerase chain reaction (RT-qPCR) was implemented according to the TaqMan Gene Expression Assays protocol (ABI, USA).
Antibody against SOX4 and the control GAPDH was purchased and manipulated following the manufactory instruction (Abcam, USA). The western blot analysis and immunohistochemistry assay were performed according to the similar methods we previously described [12]. The levels of protein expression were assigned to two experienced pathologists for blind examination following immunohistochemistry (IHC) assay, and were set into two groups as staining intensity graded subjectively: no to low staining (0~1+) and moderate to high staining (2+~3+).
Cell transfection
Hep3B cells in exponential phase were transfected with shRNA (GenePharma, Shanghai, China) for suppressing AKR1B10P1 transcript by transfection of lentiviral vector pLKO.1 (Addgene, Cambridge, USA), following the instruction, and the control ones were constructed. Puromycin was used for selecting the stable cells 48 hours after the infection. On the contrary, lentiviral vector pLV (Addgene, Cambridge, USA) was applied for ectopic expressing either AKR1B10P1 (pLV-AKR1B10P1) or SOX4 (pLV-SOX4), and the pLV-Null was set as the negative control. SOX4 depletion was conducted through shRNA in the same way as described above. The experiments for investigating SOX4 functions in Hep3B cells were conducted follow the similar protocol described in the text, and the results were presented in the form of Figures S1 and S2.
Hep3B cells over-expressing miR-138 (Hep3B/miR-138) were constructed through the mimic method according to the similar description in our previous study, followed by Dual-luciferase Reporter Assay respectively in pLV-SOX4 or pLV-Null treated cells, and the negative control ones were set (NigmiR).
Cell proliferation assay and cell cycle analysis
Hep3B cells (1×106) stably transfected were cultured in 96-well microtiter plates in triplicate and incubated at 37°C with an atmosphere of 5% CO2 for 5 days. Microplate computer software (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used for measuring the OD following the Cell Counting Kit-8 (CCK-8) assay kit protocol (Dojindo, Tokyo, Japan). The plot curves of cell proliferation were generated. Meanwhile, cells aforementioned were also treated in steps with ethanol fixation, RNase A treatment and propidium iodide staining. Flow cytometry detection by using FACSCalibur (Becton-Dickinson, Franklin Lakes, NJ, USA) were conducted. Cell populations at the G0/G1, S and G2/M phases were quantified through ModFit software (Becton-Dickinson). Cell debris and fixation artifacts were excluded.
Cell apoptosis rate measurement
Cell apoptosis rate was measured using PE-Annexin V Apoptosis Detection Kit I (BD Pharmingen, USA) according to the product instructions. Stable transfected Hep3B cells were resuspended in 1× Binding Buffer (1×106 cells/ml). 5 μl of FITC and 5 μl of PI were added into 100 μl of cell suspension, followed by 15 minutes incubation in darkness and 400 μl× Binding Buffer was added. The analysis of apoptosis was conducted by using flow cytometry (Becton Dickinson, USA). Both Annexin V-FITC-positive and PI-negative cells were considered as apoptosis ones.
Dual-luciferase reporter assay
Online tool of microcosm (http://mirecords.biolead.org) predicted miR-138 as a potential upstream regulator of SOX4 mRNA via directly interaction. A 202 bp sequence containing putative binding site of miR-138 was selected from the 3’-UTR of SOX mRNA, set along with the mutative sequence (Table S2). Simultaneously, dreamBase (http://www.sysu.edu.cn/403.html) was used and we recognized a short sequence (5’-ACACCAGC-3’) up the first ATG of AKR1B10P1 transcript probably binding with miR-138. Either the 3’-untranslated region (3’-UTR) of SOX4 mRNA or the AKR1B10P1 transcript containing the putative miR-138 binding site was intercepted, set along with the mutated ones (Table S2). Sequences above were cloned into pMIR-REPORT luciferase vectors (Promega, Madison, WI, USA), containing Firefly luciferase, and pRL-TK vectors containg Renilla luciferase used as control. The vectors were co-transfected into Hep3B cells overexpressing miR-138 and the control ones. The luciferase activation was measured by using Dual-Glo Luciferase assay system (Promega) 48 hours post-transfection.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was conducted for investigating the binding interaction between the AKR1B10P1 gene and the transcription factor SOX4. A total of 5×106 cells were cultured in each 10 cm dish and subjected to the protocol of ChIP by using ChIP-ITTM Kit (Active Motif). Chromatin was immune-precipitated with 2 μg of either SOX4 antibody (Abcam, USA) or IgG as the negative control. The extracted DNA followed was then analyzed through PCR and RT-qPCR by introducing the two sets of relative primers (Table S1) for amplification of the fragment including the promoter sequence of AKR1B10P1 gene.
Statistical analysis
Statistical analysis was carried out by using SPSS 20.0. P-values were calculated using an unpaired Student’s t-test and Fisher’s exact test. Differences were considered statistically significant at P-values < 0.05.
Results
Transcription activation of AKR1B10P1 in HCC cell lines and tissues
By exploring TCGA datasets, pseudogene AKR1B10P presents extremely low transcription in normal liver tissues and cells. However, in multiple human malignancies, including cholangiocarcinoma, lung squamous cell carcinoma and HCC, AKR1B10P1 presents detectable transcript products (Table S2 and Figure S1A, S1B). We further investigated starbase v3.0 project database (http://starbase.sysu.edu.cn/index.php) [14], and compared 374 HCC tissues and 50 normal liver tissues. As Figure S1 shown, a remarkable transcription activation in HCC tissues was illustrated (Fold change 13.64, P=1.0e-7).
We detected the transcript level of AKR1B10P1 in three HCC cell lines (Hep3B, HepG2 and Huh7) and the control L02 cells. AKR1B10P1 showed about no transcription in L02 cells. However, in all the HCC cell lines, AKR1B10P1 presented a remarkable over-expression, and especially in Hep3B cells (Figure 1A).
Figure 1.
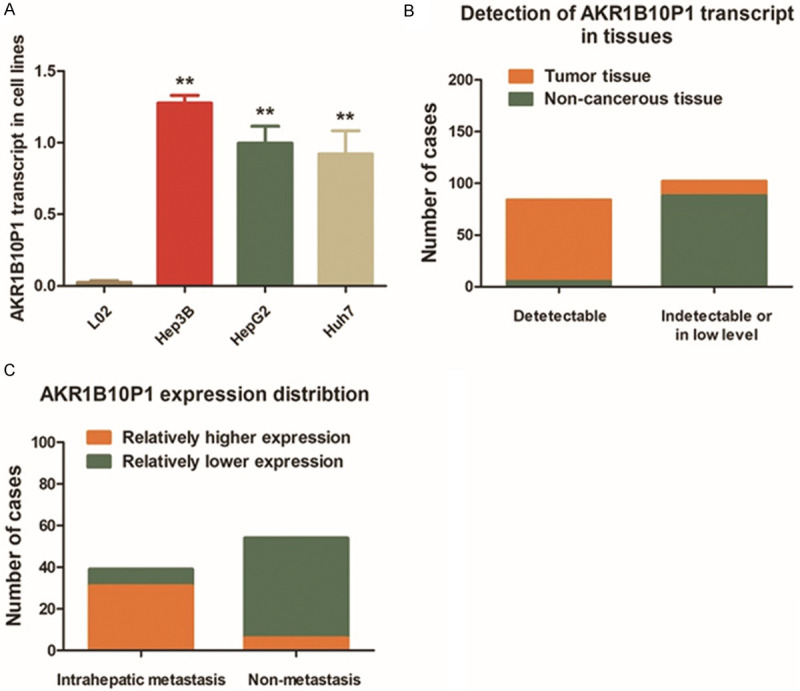
Pseudogene AKR1B10P1 is transcribed in HCC cell lines and tissues. A. RT-qPCR assay was applied to investigate the expression level of the transcript of pseudogene AKR1B10P1. As the results demonstrated, an detectable pseudogene AKR1B10P1 transcript at high level in HCC cell lines was observed. On the contrary, barely no obvious AKR1B10P1 transcript was detected in the control L02 cells (**P < 0.01). B. Paired specimens of 93 HCC cases were through RT-qPCR assay. AKR1B10P1 was remarkably transcribed in 84.84% of the HCC tumor tissues (79/93); while, only 5.38% (5/93) cases present a relatively low level AKR1B10P1 transcript in non-cancerous tissues. C. Intra-hepatic metastasis was validated in 37 patients out of the 97 cases through post-operative pathological examination. As presented, 83.78% (31/37) cases presented obviously higher AKR1B10P1 transcript.
Similar to HCC cells, AKR1B10P1 showed high expression in HCC tissues. Among the 93 HCC cases collected in our medical center, AKR1B10P1 was observed up-regulated in 84.84% (79/93) tumor specimens compared with the non-cancerous liver tissues. As for the non-cancerous tissues, only 5.38% (5/93) cases present low detectable AKR1B10P1 transcript (Figure 1B). Interestingly, in the 37 cases diagnosed with intra-hepatic metastasis, 83.78% cases (31/37) presented relatively higher AKR1B10P1 expression (Figure 1C). There findings prompts AKR1B10P1 is relate to HCC growth, development and metastasis.
High level of AKR1B10P1 transcript is correlated with dismal clinicopathologic features of the HCC patients
The clinicopathologic features of 93 HCC patients in our medical center were selected and analyzed. As presented in Table 1, there was no significant correlation between AKR1B10P1 transcription activation and the patient’s age, gender, virus control status or venous invasion. On the contrary, transcribed AKR1B10P1 was inclining to correlated with larger HCC tumor size (P < 0.05), more frequency of advanced TNM stages (P < 0.05), higher serum Alpha-fetoprotein (AFP) quantity (P < 0.01), incidence of tumor microsatellite formation (P < 0.01) and liver cirrhosis (P < 0.05).
Table 1.
Correlation between AKR1B10P1 transcript and clinicopathologic features in 93 HCC specimens
| Clinicopathologic parameters | AKR1B10P1 expression | P* | |
|---|---|---|---|
|
| |||
| Low (n=14) | High (n=79) | ||
| Age (years) | |||
| ≤50 | 9 | 40 | 0.396 |
| >50 | 5 | 39 | |
| Gender | |||
| Male | 9 | 42 | 0.564 |
| Female | 5 | 37 | |
| Diameter (cm) | |||
| ≤5 | 10 | 33 | 0.047 |
| >5 | 4 | 46 | |
| TNM stage | |||
| I~II | 8 | 28 | 0.045 |
| III~IV | 3 | 48 | |
| Tumor encapsulation | |||
| Absent | 7 | 33 | 0.574 |
| Present | 7 | 46 | |
| Tumor microsatellite formation | |||
| Absent | 11 | 30 | 0.007 |
| Present | 3 | 49 | |
| Venous invasion | |||
| No | 8 | 24 | 0.069 |
| Yes | 6 | 55 | |
| HBsAg | |||
| Negative | 5 | 13 | 0.132 |
| Positve | 9 | 68 | |
| AFP (ng/ml) | |||
| ≤400 | 12 | 12 | < 0.01 |
| >400 | 2 | 67 | |
| Cirrhosis | |||
| Absent | 5 | 7 | 0.016 |
| Present | 9 | 72 | |
AKR1B10P1 transcript level associated with clinicopathologic features in 93 HCC patients, including age, gender, tumor size, tumor stage (AJCC), tumor encapsulation, tumor microsatellite formation, vein invasion, HBsAg status, AFP level, and liver cirrhosis. Statistically significance was assessed by Fish’s exact text.
P < 0.05.
Knock-down AKR1B10P1 suppresses cell proliferation of Hep3B cells and induces cell apoptosis
Knock-down AKR1B10P1 through shRNA was carried out in Hep3B cells, in which expression of AKR1B10P1 is the highest among the HCC cell lines studied (Figure 2A). The cell proliferation of Hep3B cells was remarkably impaired by knocking-down AKR1B10P1, and P value was < 0.05 for day 1~2 and was < 0.01 for day 2~4 (Figure 2B). According to the flow cytometric analysis, the definite arrest of cell cycles at G0/G1 phase was observed in Hep3B cells with AKR1B10P1 knock-down (Figure 2C, 2D). When AKR1B10P1 was knocked down, the percentage of the cells in G0/G1 phase was increased from 47.66% to 61.13% (P < 0.01). Whilst, the S phase and the G2/M phase were decreased respectively from 28.14% to 25.82% (P < 0.05) and 20.15% to 13.06% (P < 0.01).
Figure 2.

Knock-down AKR1B10P1 suppresses cell proliferation of Hep3B cells and induces cell apoptosis. A. AKR1B10P1 was knocked-down in Hep3B cells through shRNA transfection. RT-qPCR assay was used for validating the effect of the transfection. A significant defection of AKR1B10P1 expression was observed in the treated cells (**P < 0.01). B. CCK8 assay was applied for investigating the effect of AKR1B10P1 on cell proliferation. The Hep3B cell proliferation was significantly impaired by knocking-down AKR1B10P1. P value was < 0.05 for day 1~2 and was < 0.01 for day 2~4 (*P < 0.05, **P < 0.01). C. The representative histograms describes the cell cycle profiles of Hep3B cells by using flow cytometry. D. The cell cycle of Hep3B cells was arrested by knocking-down AKR1B10P1. Briefly, after knocking-down AKR1B10P1, the percentage of the cells in G0/G1 phase was increased from 47.66% to 61.13%; the S phase and the G2/M phase were decreased from 28.14% to 25.82% and 20.15% to 13.06% respectively. These results are means of three independent experiments ± SD. (*P < 0.05, **P < 0.01). E. The representative histograms describing cell apoptosis status in Hep3B cells through flow cytometry. F. The apoptosis rate of Hep3B cells was significantly increased from 8.96% to 28.04% (**P < 0.01) via knocking-down AKR1B10P1. The results are means of three independent experiments ± SD. (**P < 0.01).
The flow cytometric analysis was also utilized for cell apoptosis measurement. An increased apoptosis rate of Hep3B cells treated with AKR1B10P1 knock-down was observed, from 8.96% to 28.04% (P < 0.01) (Figure 2E, 2F).
SOX4 contributes to the activation of AKR1B10P1 transcription for the HCC cell proliferation
We collected a 3000 bp fragment upstream from the first ATG of AKR1B10P1 pseudogene, which could be regarded as the promoter region of AKR1B10P1 transcript. This sequence was used for predicting the potential binding site for transcription factor activating AKR1B10P1 transcription. According the results evaluated through the Database of Human Transcription Factor Targets (http://bioinfo.life.hust.edu.cn/hTFtarget#!/) and Gene-Cloud of Biotechnology Information (GCBI, https://www.gcbi.com.cn), we discovered that SOX4 is the probable transcription factor binding the promoter region (5’-GCAAACAAAGCC-3’, Chr. 10: 67749768-67749779) (Figure 3A). We detected the expression of SOX4 in HCC. Both the data from TCGA liver cancer database and the mRNA expression in HCC cell lines demonstrate a significant high expression of SOX4, which was positively related with AKR1B10P1 transcript level (Figure 3B-D). We knocked down SOX4 in Hep3B cells through shRNA method. A significant decrease of AKR1B10P1 transcript was noticed consequentially induced by knocking-down SOX4. However, no obvious change of SOX4 was detected by knocking-down AKR1B10P1 (Figure 3E).
Figure 3.

SOX4 activates the transcription of AKR1B10P1 in Hep3B cells. A. Evaluation and prediction was conducted through the Database of Human Transcription Factor Targets and Gene-Cloud of Biotechnology Information. As the result demonstrated, SOX4 is a transcription factor potentially binds to the promoter region of AKR1B10P1 gene (5’-GCAAACAAAGCC-3’, Chr. 10: 67749768-67749779). B. According to the result mining from TCGA liver cancer database, SOX4 shows a remarkable high expression in HCC tissues (P < 0.01). C. RT-qPCR assay was conducted to investigate the SOX4 expression in HCC cell lines. SOX4 was significantly highly expressed in HCC cell lines (**P < 0.01). D. Scatter plot was generated for presenting the relationship between SOX4 and AKR1B10P1 transcript. Expression of SOX4 mRNA was positively related with AKR1B10P1 transcript (Spearman R=0.4522, P < 0.0001). E. RT-qPCR assay was carried out for investigating the effect of knock-down on AKR1B10P1 transcription. Knock-down of SOX4 in Hep3B cells induced significant decrease of AKR1B10P1 transcript (**P < 0.01). F. ChIP assay was applied to investigate the interaction between SOX4 and the promoter region of AKR1B10P1 gene. IgG was used as the negative control. As the column chart, transcription factor SOX4 was confirmed binding to the specific sequence of the promoter region from SOX4 (**P < 0.01).
Findings above strongly supports the upstream activating effect of SOX4 on AKR1B10P1 transcription. On basis this, ChIP assay was carried out to investigate the interaction between these two genes. As Figure 3F demonstrates, transcription factor SOX4 exactly binds to the predicted site within the promoter region of AKR1B10P1 gene.
Additionally, as the supplementary materials for further supporting the upstream regulation of AKR1B10P1 by SOX4, we investigated the expression and functions of SOX4 in HCC cells (Figure S2). Briefly, we conducted IHC assay for measuring SOX4 expression in those 93 paired HCC specimens. Obviously, SOX4 is highly expressed in HCC tissues compared with the non-cancerous liver tissues (Figure S2A, S2B). Meanwhile, Kaplan-Meier plot generated for indicated the overall survival (OS) according to GC patients’ follow-up information indicated that high SOX4 level in HCC tissue is associated with poor outcome of the patients with low OS rate (Figure S2C). As for the impact on cell proliferation, the CCK8 assay showed a significant inhibition of cell proliferation by knocking-down SOX4, along with the increase of cell apoptosis (from 10.28% to 26.54%) (Figure S2D, S2E). Re-introducing AKR1B10P1 through lentiviral vector pLV rescued the suppression of HCC cell proliferation and suppressed the cell apoptosis (from 26.39% to 11.12%) induced by SOX4 knock-down (Figure S2F, S2G).
MiR-138 negatively regulates SOX4 expression in HCC cells post-transcriptionally
Since the increase of SOX4 in HCC cells along with the AKR1B10P1 transcription, we wonder the upstream regulation of SOX4, especially in the post-transcriptional ways. MiR-138 was predicted as one of potential regulators upstream SOX4, by using microcosm online software (https://www.microcosm.com/). According to the prediction, miR-138 was supposed to directly bind to the 3’-UTR of SOX4 mRNA (Figure 4A).
Figure 4.
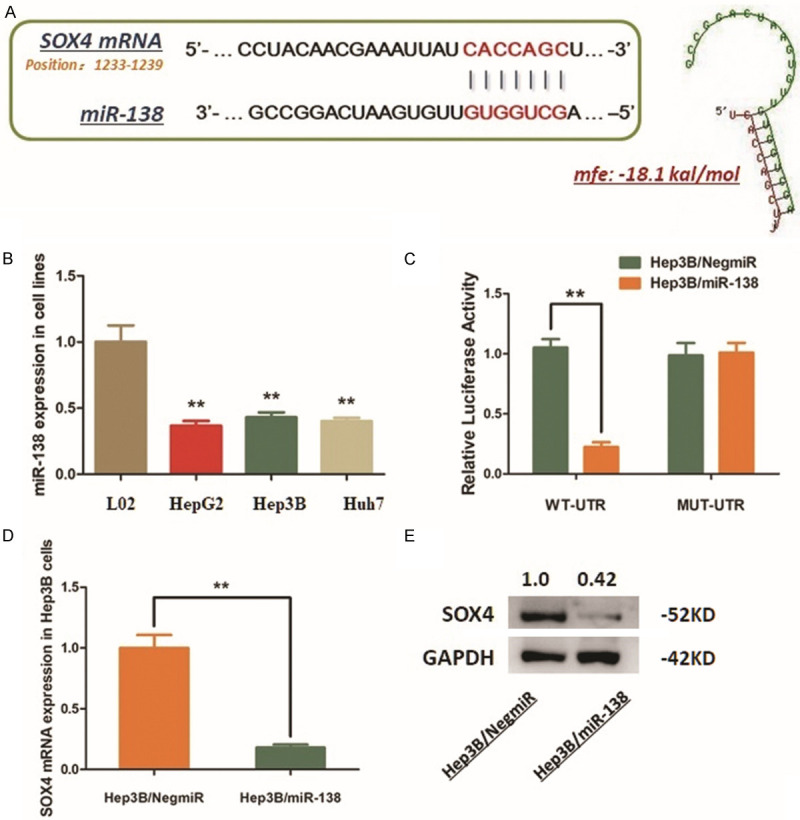
MiR-138 negatively regulates SOX4 expression in Hep3B cells via the directly interaction with SOX4 mRNA. A. MiR-138 was predicted as the up-stream regulator of SOX4 in Hep3B cells by interacting with the 3’-UTR of SOX4 mRNA. The minimum free energy (Mfe) hybridization is calculated as: -18.1 kal/mol. B. MiR-138 was down-regulated in Hep3B cells (**P < 0.01). C. The direct interaction between SOX4 and miR-138 was detected by Dual-luciferase reporter assay. Ectopic expression of miR-138 in Hep3B cells (Hep3B/miR-138) significantly declined the luciferase signal of wildtype binding sequence, compared with the negative control (Hep3B/NigmiR). The signal suppression induced by miR-138 was defected in Hep3B cells transfected with mutated binding sequence (**P < 0.01). D. RT-qPCR assay demonstrated that the mRNA expression of SOX4 was significantly decreased by introducing miR138 into Hep3B cells (**P < 0.01). E. Western blot analysis indicated that the SOX4 protein was significantly decreased by introducing miR-138 into Hep3B cells.
We measured the expression status of miR-138 in different HCC cell lines, and find an extreme decrease of miR-138 expression compared with the control L02 cells (Figure 4B). MiR-138 mimics was transfected into Hep3B cells. And, the vectors containing the fragment of 3’-UTR from SOX4 mRNA (WT-UTR) and the control luciferase vectors containing the mutated sequence (MUT-UTR) were constructed. The following dual-luciferase reporter assay was conducted. As the results shown, ectopic expression of miR-138 in Hep3B cells (Hep3B/miR-138) significantly declined the luciferase signal of SOX4/pMIR/WT, compared with the negative control (Hep3B/NigmiR). The signal suppression induced by miR-138 was defected in Hep3B cells transfected with mutated binding sequence (Figure 4C). The results above demonstrate that miR-138 directly binds to and degenerates SOX4 mRNA.
AKR1B10P1/miR-138/SOX4 consist a positive feedback loop promoting HCC growth
As we confirmed, miR-138 negatively regulates SOX4 expression in HCC, and SOX4 transcriptionally activates AKR1B10P1 and then promotes HCC growth. On basis of this potential pathway, we further investigate the structure of AKR1B10P1 transcript. We noticed a sequence located at the site from 780 bp to 802 bp to the 3’ end of the AKR1B10P1 transcript provides a potential binding site for miR-138, which prompts AKR1B10P1 sponging miR-138 in a classic ceRNA way (Figure 5A). As the RT-qPCR assay indicated, the expression of miR-138 is negatively correlated with AKR1B10P1 transcript (Figure 5B).
Figure 5.
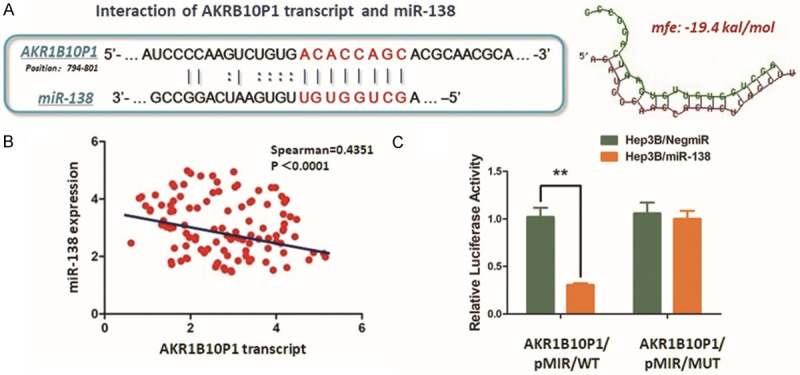
AKR1B10P1 transcript sponges miR-138 to exert stabilization effect on SOX4 expression. A. Predicted binding site of AKR1B10P1 transcript potentially interacts with the seed sequence of miR-138. The minimum free energy (Mfe) hybridization is calculated as: -19.4 kal/mol. B. RT-qPCR was conducted for measuring the expression of miR-138. As the scatter plot demonstrated, expression of miR-138 was negatively related with AKR1B10P1 transcript (Spearman R=0.4351, P < 0.0001). C. The direct interaction between AKR1B10P1 and miR-138 was validated by applying Dual-luciferase reporter assay. Hep3B cells were transfected with the predicted binding site either wildtype (AKR1B10P1/pMIR/WT) or mutated (AKR1B10P1/pMIR/MUT). Up-regulation of miR-138 significantly reduced the luciferase signal of the Hep3B cells of AKR1B10P1/pMIR/WT, compared with the negative control (Hep3B/NigmiR); And also, Hep3B cells of AKR1B10P1/pMIR/MUT site abolished this suppressive effect (**P < 0.01).
According to the results of dual luciferase reporter assay, we observed a significant decrease of the luciferase signal in Hep3B/miR-138 cells post-transfection AKR1B10P1/pMIR/WT vectors into these cells. While, the AKR1B10P1/pMIR/MUT vector transfection did not induce significant signal changes (Figure 5C). Hence, findings above strongly support the ceRNA effect of ARK1B10P1 on miR-138 within the HCC cells.
Thus, we suggest the existence of the regulating pattern of AKR1B10P1/miR-138/SOX4. In brief, AKR1B10P1 transcription is activated by SOX4 in the promotion of HCC growth; Simultaneously, AKR1B10P1 exerts the stabilization effect on SOX4 expression as a positive feedback loop through sponging miR-138 (Figure 6).
Figure 6.
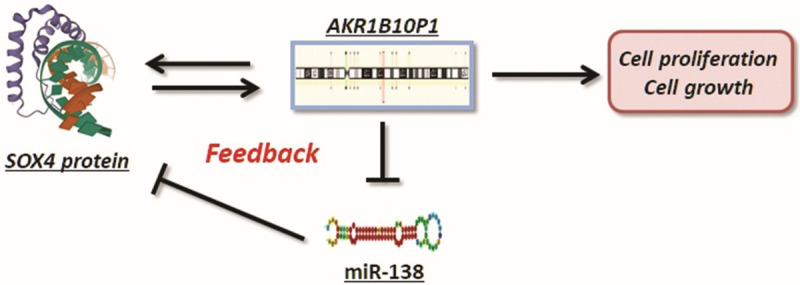
AKR1B10P1/miR-138/SOX4 consist a positive feedback loop promoting HCC growth. The summary of the positive feedback regulation of AKR1B10P1/miR-138/SOX4 loop, which promotes HCC growth. Briefly, AKR1B10P1 transcription is activated by SOX4 in the promotion of HCC growth; Meanwhile, AKR1B10P1 stabilizes SOX4 expression as a positive feedback loop through interacting with miR-138 as a molecular sponge.
Discussion
HCC is an extremely aggressive human malignancy with active ability in tumor growth, invasiveness and metastasis, which leads to poor outcome of the patients and high mortality [15,16]. Cell proliferation is one of the pivotal indicators reflecting the tumor cell ability of growth, adaption and tumor development [17,18]. However, the mechanisms promoting HCC cell growth and tumor development in HCC research remains limitation for intensive understanding.
According to the reviewed literatures, there exist quite a lot of pseudogenes, which has been uncovered exerting particular functions either promoting or suppressing tumorigenesis and tumor growth. For example, pseudogene PTENP1, whose parental gene is PTEN, exerts tumor inhibiting effort through binding oncogene miR-17, miR-19 and miR-21, consequentially suppresses human malignancies, such as clear cell renal carcinoma, oral squamous carcinoma and gastric cancer [19,20]. On the contrary, the pseudogene of protein products of the regenerating islet-derived (REG), named REGP, plays the enhancing function in colorectal cancer cell proliferation and tumorigenicity, associated with poor outcome of patients, through forming an RNA-DNA triplex and recruiting BRIP1to inhibit the anti-cancer related transcription intracellular [21].
As for human liver cancer, especially HCC, the exact functions of pseudogenes remain to be illustrated. One specific example is PCNA pseudogene transcript 1 (PCNAP1). PCAN was discovered exerting ceRNA effect on miR-153, by which consequentially stabilizes the cccRNA/HBV axis (cccRNA, the covalently closed circular DNA), which induces the HBV viral persistence in hepatocytes and tumor initiation [22]. On the contrary, pseudogene of INTS6 (INTS6P1, Integrator complex subunit 6 pseudogene 1) inhibits the cell growth suppression in HCC through interaction with onco-miR-17-5p, which is an upstream negative regulator of its parental gene INTS6 [23].
AKR1B10P1 (chr10:67750284-67751225) is a processed pseudogene of AKR1B10, which is originated from mRNA retro-transposition. According to our investigation, this gene lacks introns and presents barely detectable transcript in normal hepatocytes. However, the RT-qPCR assay definitely gave out a significant expression of AKR1B10P1 transcript in both HCC tissues and cell lines, prompting an anomalous activation of AKR1B10P1 transcription in the process of HCC.
As we discovered through the functional experiments, AKR1B10P1 transcript plays an important role in HCC cell proliferation. Knock-down AKR1B10P1 significant impairs the cell growth by arresting the cell cycle in G0/G1 phase and induces cell apoptosis. Along with this discovery, we confirmed the absolute incline of active tumor growth, frequent invasion and aggressive metastasis with high expression of AKR1B10P1, according to the analysis of clinicopathologic features.
The activation of AKR1B10P1 transcription is a particular intracellular event of HCC cells. Exploration on the AKR1B10P1 gene revealed the potential binding site from the promoter region with SOX4, a member of the group C subclass in the SOX family, functioning as an essential transcription factor involves in the regulation of cell stemness, differentiation and progenitor development [24]. Accumulating evidence has revealed that SOX4 participates in multiple pathways concerning with tumorigenesis and progress, including Wnt, TGF-β and PI3K signaling, and has been identified as one of the oncogenes amplified in different cancers like breast cancer, ovarian cancer and lung cancer [25,26]. In our study, the investigation confirmed the high expression of SOX4, positively correlated with AKR1B10P1 transcript. By carrying out ChIP assay, we verified the direct binding between SOX4 protein and the promoter of AK1B10P1 gene in HCC cells. Moreover, knock-down SOX4 significantly decreased the transcript of AKR1B10P1. Findings above definitely indicates that pseudogene AKR1B10P1 is activated by SOX4 in the HCC cells.
MiR-138 is a tumor suppressing non-coding RNA post-transcriptionally regulating its targets in different human malignancies, including bladder cancer, gastric cancer and colorectal cancer [27-29]. Combining with the prediction by the online tools and the Dual-luciferase reporter assay, we confirmed miR-138 as an up-streaming regulator inducing degradation of SOX4 mRNA. Considering the low expression of miR-138 in HCC cells, we suppose that miR-138/SOX4 is the pivotal factors directing AKR1B10P1 transcription.
Notably, as we discovered, the regulation between miR-138/SOX4 and AKR1B10P1 is not in a one way mode. Through the structure analysis of AKR1B10P1 transcript, we found a specific sequence, which potentially binds with miR-138 as a competing region against the degradation of SOX4 mRNA. The Dual-luciferase reporter assay was then used and revealed the direct interaction between AKR1B10P and miR-138. Hence, our findings demonstrate an innovative loop of AKR1B10P1/miR-138/SOX4 positively feedbacks the transcription activation and promote HCC growth.
By understanding the positive feedback loop, we give out a thorough view of the anomalous transcription activation of pseudogene AKR1B10P1, and the related enhancement of HCC cell proliferation and tumor growth. The ceRNA effect of AKR1B10P1 competitively binds miR-138 and exerts the stabilization of SOX4 expression, and SOX4 functions as the active transcription factor facilitates the transcription of AKR1B10P1.
In summary, our study demonstrates a reliable positive feedback loop promoting HCC, and these three genes might be targets for innovative prevention and therapeutic treatment of HCC.
Acknowledgements
The authors thank Xinping Ren, Lei Dong, Zhiyuan Wu for providing valuable technical supports and assistance. This study was kindly supported by grants from the following: National Natural Science Foundation of China (No. 81602544); Shanghai Pujiang Talent Project (No. 18PJD029); Research physician project from Shanghai Jiao Tong University School of Medicine (20191901).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Vilgrain V, Pereira H, Assenat E, Guiu B, Ilonca AD, Pageaux GP, Sibert A, Bouattour M, Lebtahi R, Allaham W, Barraud H, Laurent V, Mathias E, Bronowicki JP, Tasu JP, Perdrisot R, Silvain C, Gerolami R, Mundler O, Seitz JF, Vidal V, Aube C, Oberti F, Couturier O, Brenot-Rossi I, Raoul JL, Sarran A, Costentin C, Itti E, Luciani A, Adam R, Lewin M, Samuel D, Ronot M, Dinut A, Castera L, Chatellier G SARAH Trial Group. Efficacy and safety of selective internal radiotherapy with yttrium-90 resin microspheres compared with sorafenib in locally advanced and inoperable hepatocellular carcinoma (SARAH): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2017;18:1624–1636. doi: 10.1016/S1470-2045(17)30683-6. [DOI] [PubMed] [Google Scholar]
- 2.Sartorius K, Sartorius B, Aldous C, Govender PS, Madiba TE. Global and country underestimation of hepatocellular carcinoma (HCC) in 2012 and its implications. Cancer Epidemiol. 2015;39:284–290. doi: 10.1016/j.canep.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Pinyol R, Montal R, Bassaganyas L, Sia D, Takayama T, Chau GY, Mazzaferro V, Roayaie S, Lee HC, Kokudo N, Zhang Z, Torrecilla S, Moeini A, Rodriguez-Carunchio L, Gane E, Verslype C, Croitoru AE, Cillo U, de la Mata M, Lupo L, Strasser S, Park JW, Camps J, Sole M, Thung SN, Villanueva A, Pena C, Meinhardt G, Bruix J, Llovet JM. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut. 2019;68:1065–1075. doi: 10.1136/gutjnl-2018-316408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu; Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341. e1323. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sasidharan R, Gerstein M. Genomics: protein fossils live on as RNA. Nature. 2008;453:729–731. doi: 10.1038/453729a. [DOI] [PubMed] [Google Scholar]
- 6.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sur S, Saha S, Tisa LS, Bothra AK, Sen A. Characterization of pseudogenes in members of the order Frankineae. J Biosci. 2013;38:727–732. doi: 10.1007/s12038-013-9356-1. [DOI] [PubMed] [Google Scholar]
- 8.Han L, Yuan Y, Zheng S, Yang Y, Li J, Edgerton ME, Diao L, Xu Y, Verhaak RGW, Liang H. The Pan-Cancer analysis of pseudogene expression reveals biologically and clinically relevant tumour subtypes. Nat Commun. 2014;5:3963. doi: 10.1038/ncomms4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lou W, Ding B, Fu P. Pseudogene-derived lncRNAs and their miRNA sponging mechanism in human cancer. Front Cell Dev Biol. 2020;8:85. doi: 10.3389/fcell.2020.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma G, Liu H, Du M, Zhang G, Lin Y, Ge Y, Wang M, Jin G, Zhao Q, Chu H, Gong W, Zhang Z. A genetic variation in the CpG island of pseudogene GBAP1 promoter is associated with gastric cancer susceptibility. Cancer. 2019;125:2465–2473. doi: 10.1002/cncr.32081. [DOI] [PubMed] [Google Scholar]
- 11.Karreth FA, Reschke M, Ruocco A, Ng C, Chapuy B, Leopold V, Sjoberg M, Keane TM, Verma A, Ala U, Tay Y, Wu D, Seitzer N, Velasco-Herrera Mdel C, Bothmer A, Fung J, Langellotto F, Rodig SJ, Elemento O, Shipp MA, Adams DJ, Chiarle R, Pandolfi PP. The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell. 2015;161:319–332. doi: 10.1016/j.cell.2015.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Zhou Y, Fei X, Chen X, Chen Y. Biostatistics mining associated method identifies AKR1B10 enhancing hepatocellular carcinoma cell growth and degenerated by miR-383-5p. Sci Rep. 2018;8:11094. doi: 10.1038/s41598-018-29271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang W, Zhao LJ, Tan YX, Ren H, Qi ZT. MiR-138 induces cell cycle arrest by targeting cyclin D3 in hepatocellular carcinoma. Carcinogenesis. 2012;33:1113–1120. doi: 10.1093/carcin/bgs113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 16.Wong MC, Jiang JY, Goggins WB, Liang M, Fang Y, Fung FD, Leung C, Wang HH, Wong GL, Wong VW, Chan HL. International incidence and mortality trends of liver cancer: a global profile. Sci Rep. 2017;7:45846. doi: 10.1038/srep45846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood CE, Hukkanen RR, Sura R, Jacobson-Kram D, Nolte T, Odin M, Cohen SM. Scientific and regulatory policy committee (SRPC) review: interpretation and use of cell proliferation data in cancer risk assessment. Toxicol Pathol. 2015;43:760–775. doi: 10.1177/0192623315576005. [DOI] [PubMed] [Google Scholar]
- 18.Goodlad RA. Quantification of epithelial cell proliferation, cell dynamics, and cell kinetics in vivo. Wiley Interdiscip Rev Dev Biol. 2017;6 doi: 10.1002/wdev.274. [DOI] [PubMed] [Google Scholar]
- 19.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang R, Guo Y, Ma Z, Ma G, Xue Q, Li F, Liu L. Long non-coding RNA PTENP1 functions as a ceRNA to modulate PTEN level by decoying miR-106b and miR-93 in gastric cancer. Oncotarget. 2017;8:26079–26089. doi: 10.18632/oncotarget.15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yari H, Jin L, Teng L, Wang Y, Wu Y, Liu GZ, Gao W, Liang J, Xi Y, Feng YC, Zhang C, Zhang YY, Tabatabaee H, La T, Yang RH, Wang FH, Yan XG, Farrelly M, Scott R, Liu T, Thorne RF, Guo ST, Zhang XD. LncRNA REG1CP promotes tumorigenesis through an enhancer complex to recruit FANCJ helicase for REG3A transcription. Nat Commun. 2019;10:5334. doi: 10.1038/s41467-019-13313-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng J, Yang G, Liu Y, Gao Y, Zhao M, Bu Y, Yuan H, Yuan Y, Yun H, Sun M, Gao H, Zhang S, Liu Z, Yin M, Song X, Miao Z, Lin Z, Zhang X. LncRNA PCNAP1 modulates hepatitis B virus replication and enhances tumor growth of liver cancer. Theranostics. 2019;9:5227–5245. doi: 10.7150/thno.34273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng H, Ishida M, Li L, Saito A, Kamiya A, Hamilton JP, Fu R, Olaru AV, An F, Popescu I, Iacob R, Dima S, Alexandrescu ST, Grigorie R, Nastase A, Berindan-Neagoe I, Tomuleasa C, Graur F, Zaharia F, Torbenson MS, Mezey E, Lu M, Selaru FM. Pseudogene INTS6P1 regulates its cognate gene INTS6 through competitive binding of miR-17-5p in hepatocellular carcinoma. Oncotarget. 2015;6:5666–5677. doi: 10.18632/oncotarget.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.She ZY, Yang WX. SOX family transcription factors involved in diverse cellular events during development. Eur J Cell Biol. 2015;94:547–563. doi: 10.1016/j.ejcb.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 25.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno CS. SOX4: the unappreciated oncogene. Semin Cancer Biol. 2019 doi: 10.1016/j.semcancer.2019.08.027. S1044-579X(18)30145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y, Pan ZG, Shu L, Li QJ. Podocalyxin-like, targeted by miR-138, promotes colorectal cancer cell proliferation, migration, invasion and EMT. Eur Rev Med Pharmacol Sci. 2018;22:8664–8674. doi: 10.26355/eurrev_201812_16631. [DOI] [PubMed] [Google Scholar]
- 28.Pang L, Li B, Zheng B, Niu L, Ge L. miR-138 inhibits gastric cancer growth by suppressing SOX4. Oncol Rep. 2017;38:1295–1302. doi: 10.3892/or.2017.5745. [DOI] [PubMed] [Google Scholar]
- 29.Blanca A, Sanchez-Gonzalez A, Requena MJ, Carrasco-Valiente J, Gomez-Gomez E, Cheng L, Cimadamore A, Montironi R, Lopez-Beltran A. Expression of miR-100 and miR-138 as prognostic biomarkers in non-muscle-invasive bladder cancer. APMIS. 2019;127:545–553. doi: 10.1111/apm.12973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.