

Abstract
Purpose: This study explored the effects of phosphofructokinase-1 (PFK1) on the radiosensitivity of colorectal cancer (CRC) in vivo and in vitro and the underlying mechanisms. Methods: Tissue samples from 48 patients with rectal cancer who had received neoadjuvant radiotherapy followed by surgery were analyzed. The expression of PFK1 in tissue samples was semi-quantitated by immunohistochemistry, and its relationship with clinicopathological features was analyzed. The effects of PFK1 knockdown on the survival, apoptosis, migration, and radiosensitivity of CRC cells were evaluated. Glycolysis-related indicators were used to examine glycolytic activity. The effects of PFK1 on the radiosensitivity of CRC in vivo were assessed by measuring tumor formation in nude mice. Results: PFK1 was overexpressed in rectal cancer and was higher in radiation-resistant tumors than in radiation-sensitive tumors. SiRNA-induced PFK1 silencing increased apoptosis and inhibited migration and proliferation of CRC cells. Knockdown of PFK1 made the CRC cells sensitive to ionizing radiation in vivo. Oligomycin partially restored the expression of PFK1, enhanced glycolysis, and reversed the enhanced radiosensitivity of CRC cells induced by siRNA-PFK1. Downregulation of PFK1 combined with irradiation inhibited growth of nude mice xenografts, which was related to an increase in apoptosis. Conclusions: Our study indicates that high expression of PFK1 is negatively correlated with radiosensitivity in CRC and likely accelerates the proliferation and migration of CRC cells. Downregulation of PFK1 may enhance the radiosensitivity of CRC cells in vivo and in vitro by inhibiting glycolysis.
Keywords: Colorectal cancer, phosphofructokinase-1, radiotherapy, HT29 cells, HCT116 cells, oligomycin A
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumors and the second leading cause of cancer-related death in the world [1]. According to the American Cancer Society, CRC ranked third in 2019 among all types of cancers in morbidity and mortality in both men and women [2]. In recent years, neoadjuvant and adjuvant radiotherapies have become the standard care for patients with locally advanced (T3-4) or lymph node-positive rectal cancer, even for patients with high-risk features who are considered poor or non-operative candidates.
However, some CRC patients who receive radiotherapy have poor prognosis due to radioresistance [3]. Improving the radiosensitivity of CRC and identifying mechanisms involved in radioresistance are current challenges in CRC treatment.
Reprogramming of energy metabolism is one of the important characteristics of malignant tumors [4]. Even in the presence of adequate oxygen levels, most malignant tumors obtain energy through glycolysis, which in turn produces lactic acid. This phenomenon is called the Warburg effect [5]. Positron emission tomography-computerized fluoroscopy with fluorodeoxyglucose as a tracer uses this high-glycolysis phenomenon for diagnosing diseases. In addition, some products of the Warburg effect can form an intracellular redox buffer network [6] that can increase the radiation resistance of tumor cells [7]. Reducing the glycolytic activity can reverse the radioresistance of many tumors [8]. However, studies on increasing the radiosensitivity of CRC by inhibiting glycolysis have not been reported.
PFK1 is a key enzyme in the aerobic glycolysis pathway [9] and a key driver of intracellular glucose flux [10]. There are three subtypes of PFK1 in human tissues: PFKM (muscle), PFKL (liver), and PFKP (platelets) [11], each of which is expressed to various degrees in tissues of the human body. In addition to the metabolic effects of PFK1 in tumors, high levels of PFK1 expression can promote the development, progression, and metastasis of cancer cells, which is a feature closely related to the prognosis of cancer patients [12-15]. Moreover, targeting 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatases, which are regulatory enzymes of allosteric actuators of PFK1, can enhance the radiosensitivity of tumor cells by inhibiting DNA repair [16]. However, the effects of PFK1 on radiosensitivity of CRC are not clear.
In this study, we investigated the effects of PFK1 on the proliferation, migration, and radiosensitivity of CRC cells to further elucidate the mechanisms by which PFK1 affects radiosensitivity in CRC. We first measured the PFK1 expression levels in 48 rectal cancer tissues and CRC cell lines by immunohistochemistry and analyzed the relationship between PFK1 expression and clinicopathological features. We explored the effects of PFK1 knockdown on glycolytic activity and radiosensitivity of CRC cells in vivo and in vitro. The results of this study provide a scientific basis for new strategies to increase the radiosensitivity of CRC.
Materials and methods
Patients and clinical tissue samples
This retrospective cohort study included 48 rectal cancer patients who underwent surgery, radiotherapy, and chemotherapy in the Third Xiangya Hospital between 2008 and 2016. The inclusion criteria were the following: (i) postoperative pathology confirmed as adenocarcinoma; (ii) no evidence of distant metastasis at diagnosis (TNM stage II-III); (iii) permission to use tissue specimens obtained by endoscopy before treatment in this research; (iv) negative surgical margins of resected specimens; and (v) neoadjuvant chemoradiotherapy before curative resection. The neoadjuvant radiotherapy consisted of 50 Gy in 25 fractions over 5 weeks. Based on their basic information and prognosis, the 48 patients were assigned to one of two groups: the radiation-sensitive group (n = 26) and the radiation-resistant group (n = 22). Pathologic responses to neoadjuvant radiotherapy were scored according to the tumor regression grade (TRG) developed by the American Joint Commission on Cancer and the College of American Pathology [17]. We defined patients with a TRG of 0 or 1 as the radiation-sensitive group and those with a TRG of 2 or 3 as the radiation-resistant group [18]. Tumor volume was assessed by using the gross tumor volume (GTV) for rectal cancer according to the Varian Treatment Planning System (Eclipse® External Beam Planning System, Version 11.03.1, Varian Medical Systems). The radiotherapy targets were delineated according to the Radiation Therapy Oncology Group consensus for rectal cancer. The GTV of stage III rectal cancer includes primary tumors and regional positive lymph nodes. All GTVs were examined by two oncologists and one radiologist.
The study was approved by the Medical Ethics Committee of the Third Xiangya Hospital of Central South University. Informed consent was obtained from all participating patients prior to initiation or enrollment in the present study.
Immunohistochemistry and scores
All rectal cancer samples were taken from primary tumors. The samples were fixed in formalin, embedded in paraffin, and cut into 4-μm sections. The sections were deparaffinized in xylene. followed by microwave treatment for 10 min in phosphate-buffered saline (PBS, pH 6.0). After cooling for 20 min and washing with PBS, the sections were incubated at room temperature for 10 min to inactivate endogenous peroxidase in the tissues. The sections were washed with PBS and incubated in non-immune animal serum for 60 min to reduce the non-specific binding of antibodies. The sections were incubated overnight at 4°C with anti-PFKM (1:300; sc-377346, Santa Cruz, CA, USA). Following the manufacturer’s instructions, detection of immunostaining was performed using the UltraSensitiveTM S-P kit (KALANG, Shanghai, China) with 3,3-diaminobenzidine (Santa Cruz, CA, USA) as the chromogen. Finally, slices were redyed with hematoxylin, dehydrated, cleared, and covered with glass. Two experienced pathologists, blinded to the clinicopathological status of the patients, read and scored the slides according to the immunohistochemical streptavidin-peroxidase method. Using this method, the expression of PFK1 was scored according to the proportion of positive cells and the staining intensity. The average percentage of positive tumor cells was examined at 400 × magnification in at least five random visual fields for each section. The samples were scored as follows: 0, the percentage of positive cells was less than 5%; 1, between 6% and 25%; 2, between 26% and 50%; 3, between 51% and 75%; and 4, more than 75%. The staining intensity was scored as follows: 0, no intracellular staining; 1, yellowish intracellular granules; 2, dark brown-orange intracellular granules; and 3, dark brown intracellular granules. The scores of positive tumor cells and staining intensity were multiplied to obtain the immunohistochemical staining score: negative (-), 0 points; mildly positive (+), 1 to 4 points; positive (++), 5 to 8 points; and strongly positive (+++), 9 to 12 points. Tumors graded (-) or (+) were regarded as having low expression, and tumors graded (++) or (+++) were regarded as having high expression. If the difference between the scores given by the two pathologists exceeded 3, the sample was reevaluated [19].
Cell culture and treatments
Human CRC cell lines, SW480, SW620, HCT116, Colo205, HT29, caco-2, and LoVo, were purchased from the American Type Culture Collection, Manassas, VA, USA. The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum at 37°C in a 5% CO2 incubator. Oligomycin A was purchased from Selleck (Texas, USA). The cells were treated with oligomycin A at a concentration of 4 μM, which induced glycolysis but was not cytotoxic. Oligomycin A was added to the medium and cells were cultured for 12 h. Low-sugar DMEM and high-sugar DMEM were mixed at a ratio of 1:1. The final glucose concentration in the medium was 15 mM.
Cell transfection
HT29 and HCT116 cells were transfected with small interfering RNA (siRNA) targeting PFK1, siRNA-PFK1, using Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.), following the manufacturer’s recommendations. The cells were transfected for 24 h for further analysis. The sequences were as follows: PFKM-siRNA-1336 sense 5’-GGUUAAGCGUCUGGGAUAUTT-3’ and antisense 5’-AUAUCCCAGACGCUUAACCTT-3’; PFKM-siRNA-1611 sense 5’-GCCGGAGCUUCAUGAACAATT-3’ and antisense 5’-UUGUUCAUGAAGCUCCGGCTT-3’; PFKM-siRNA-2111 sense 5’-CUGACACAGCACUCAAUATT-3’ and antisense 5’-AUUGAGUGCUGUGUCAGCTT-3’; negative control sense 5’-UUCUCCGAACGUGUCACGUTT-3’ and antisense 5’-ACGUGACACGUUCGGAGAATT-3’.
Western blot
Total protein was extracted with immunoprecipitation buffer (Auragene, Changsha, China) and samples were centrifuged at 9000 rpm at 4°C for 10 min. The protein concentration was determined with a BCA protein assay kit (Auragene, Changsha, China). The proteins were separated by 10% SDS-PAGE and blotted onto nitrocellulose membranes, which were probed with anti-PFKM (1:500; sc-377346, Santa Cruz, CA, USA), followed by a horseradish peroxidase-conjugated secondary antibody (Auragene, Changsha, China). Protein bands were visualized with enhanced chemiluminescence and images were recorded (Auragene, Changsha, China). The signal intensity was analyzed with IPP6.0 software, and β-actin (1:500; sc-47778, Santa Cruz, CA, USA) was used as the internal control.
Cell migration assay
HT29 and HCT116 cells were inoculated in 6-well plates at a density of 1 × 105 cells/well and cultured until confluence reached more than 90%. A wound of about 1 mm width was made in the cell monolayer using the tip of a 20 μL sterile pipette tip, and the detached cells were removed by aspiration. The cells were incubated in DMEM containing 3% fetal bovine serum for 48 h. Three visual fields of the wound area were selected for each group and photographed under a microscope (Motic, Xiamen, China). The gel migration was calculated with the following formula: scratch healing rate = (1 - width between edges of scratches after incubation/width between edges of initial scratches) × 100%.
Cell proliferation assay
HT29 and HCT116 cells were incubated in 100 µL DMEM at a density of 3 × 103 cells/well in 96-well plates. Cell proliferation was evaluated with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay after incubation for 0, 24, 48, and 72 h, or for 24 h with different radiation doses (0, 2, 4, 6, and 8 Gy). MTT reagent (10 μL) was added to each well, and the cells were incubated at 37°C for another 4 h. The MTT solution was removed, and 150 µL dimethyl sulfoxide was added to each well to dissolve purple formazan crystals. The optical density (OD) was measured with a microplate reader (Bio-Rad, CA, USA) at a wavelength of 570 nm (OD570). Cell proliferation is expressed as a percentage of the control.
Colony formation assay
The cells were transfected with siRNA-PFK1 or pretreated with oligomycin A before X-ray irradiation with various doses (0, 2, 4, 6, and 8 Gy) and seeded in 10-cm tissue culture dishes. The dishes were incubated in fresh DMEM at 37°C in a humidified 5% CO2 incubator for 14 days. The colonies were fixed with 4% methanol in PBS and stained with Giemsa. Colonies of at least 10 cells were scored as survivors and counted with the naked eye or a stereoscopic microscope. Cell colony formation was calculated with the following formula: colony formation rate = (number of cell colonies formed/number of cells seeded) × 100%. The survival fraction was calculated as follows: survival fraction = (colony formation rate of experimental group/colony formation rate of control group) × 100%. The survival fraction of cells was fitted to the cell survival curve according to a linear-quadratic model (y = 1 - (1 - exp (-k × x))^N) using GraphPad software (La Jolla, CA, USA).
X-ray irradiation
The radiotherapy equipment was equipped with an American Varian TrueBeam linear accelerator (Varian, CA, USA) and radiation was emitted at a fixed dose rate of 4 Gy/min. Cells or mice were irradiated with various doses according to set parameters. The graded doses of X-rays used to irradiate cells were 0, 2, 4, 6, and 8 Gy.
Glycolysis analysis
Glycolytic activity was measured with a glucose assay kit, a lactic acid assay kit, an ATP assay kit, and a PFK1 activity kit (Nanjing, China). The kits were used to measure glucose consumption, lactic acid production, ATP production, and PFK1 activity, respectively, in HT29 and HCT116 cells following the manufacturer’s instructions.
Apoptosis assay
Apoptosis was detected in vitro by flow cytometry after staining with annexin V-fluorescein isothiocyanate (FITC)-propidium iodide with an apoptosis detection kit (Sigma, CA, USA) following the manufacturer’s instructions. Fluorescence was measured by flow cytometry (Becton Dickinson, FACSCanto II, NJ, USA). The flow cytometry data were analyzed with BD FACS Diva software v6.1.3 (BD Biosciences, NJ, USA).
Tissue apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) with an in situ Fluorescein TUNEL Cell Apoptosis Detection Kit, following the manufacturer’s instructions. Tissue was dehydrated, infiltrated and embedded, and then sliced and pasted. The slices were fully dewaxed and rehydrated. Protease K was added and samples were incubated at room temperature for 20 min. Then TUNEL reaction solution was added and samples were incubated at room temperature for 10-30 min. The marker was added and incubated for 1.5 h, and diaminobenzidine substrate solution was added. Images were captured in three fields of view of each slide with a Nikon microscope. Apoptotic nuclei were labeled by yellow or brown granules.
Animal assay
Animal experiments were approved by the animal center of Central South University (Changsha, China) and performed following international guidelines and procedures. To produce tumors in vivo, 1 × 106 HT29 cells or 1 × 106 HT29 cells transfected with siRNA-PFK1 or siRNA-NC were injected subcutaneously into the right flank of their abdomen of nude mice.
Tumor volume was measured at the time they became visible, approximately 1 week after injection. When the tumor volume reached 100 mm3, tumors in the experimental group were irradiated with 2 Gy. Tumor volumes were measured at 2-day intervals. After 25 days, the mice were sacrificed, the tumors were isolated, and tumor sizes were measured with Vernier calipers. Tumor volume was calculated with the following formula: tumor volume (mm3) = (the longest diameter) × (the shortest diameter)2/2.
Statistical analysis
All data were analyzed using Prism version 7.0 (GraphPad Software, Inc., La Jolla, CA, USA). Experimental values are expressed as mean ± SD. The quantitative data were compared using the Student t-test or univariate variance analysis, and the constituent ratio was compared using the chi-square test. The Kruskal-Wallis test was used to analyze primary GTV and its relationship with PFK1 protein levels, and the Mann-Whitney U test was used to analyze primary GTV and its relationship with the response of neoadjuvant radiotherapy. A P value less than 0.05 was considered to indicate statistical significance.
Results
PFK1 is overexpressed in rectal cancer, which is directly correlated with the response to radiation in rectal cancer patients
PFK1 was observed in the cytoplasm of the rectal cancer cells (Figure 1A). PFK1 was overexpressed in rectal cancer tissues, and its expression was closely correlated with TNM stage (P = 0.006) and primary GTV (P = 0.004), but it was not correlated with patient age, gender, or histopathological tumor grade (P > 0.05, Table 1). The immunohistochemical scores of PFK1 expression were significantly higher in the radiation-resistant group than in the radiation-sensitive group (P < 0.01; Figure 1B), and there was a statistically significant difference in histopathological grade (P = 0.038) and primary GTV (P = 0.017) between radiation-sensitive and radiation-resistant tumors (Table 2). There was no significant difference in age, sex, and TNM stage between the radiation-resistant and the radiation sensitive-group (Table 2). These results suggest that the expression of PFK1 might be closely related to the development of CRC and the resistance to radiation.
Figure 1.
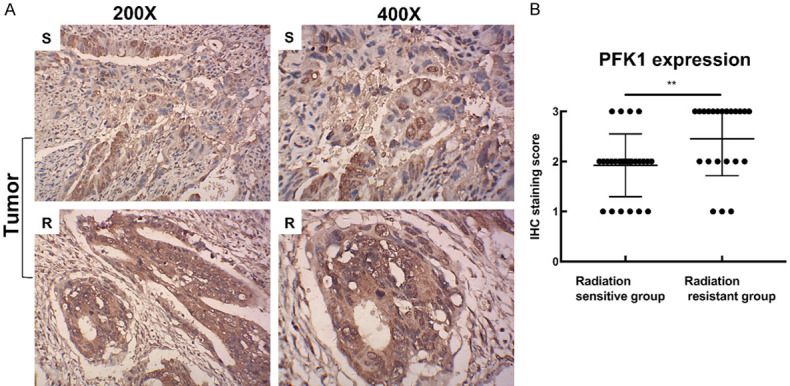
PFK1 expression is associated with the response to radiation in rectal cancer patients. A. Photomicrographs of PFK1 in rectal cancer tissues. B. Immunohistochemistry score of PFK1 expression in the radiation-sensitive and radiation-resistant groups. S, section of a radiation-sensitive tissue sample; R, section of a radiation-resistant tissue sample. CRC, colorectal cancer; PFK1, phosphofructokinase-1. **P < 0.01.
Table 1.
Correlations of PFK1 expression with the clinicopathological features of CRC
| Variable | Patients (n = 48) | PFK1 expression | P value | ||
|---|---|---|---|---|---|
|
| |||||
| + | ++ | +++ | |||
| Age, n (%) | 0.746 | ||||
| < 50 years | 18 | 4 (22.22) | 7 (38.89) | 7 (38.89) | |
| ≥50 years | 30 | 5 (16.67) | 15 (50.00) | 10 (33.33) | |
| Gender, n (%) | 0.526 | ||||
| Male | 33 | 7 (21.21) | 16 (48.48) | 10 (30.31) | |
| Female | 15 | 2 (13.33) | 6 (40.00) | 7 (46.67) | |
| TNM stage, n (%) | 0.006 | ||||
| II | 9 | 5 (55.56) | 3 (33.33) | 1 (11.11) | |
| III | 39 | 4 (10.26) | 19 (48.72) | 16 (41.02) | |
| Histopathological grade, n (%) | 0.767 | ||||
| High | 8 | 2 (25.00) | 4 (50.00) | 2 (25.00) | |
| Middle + low | 40 | 7 (17.50) | 18 (45.00) | 15 (37.50) | |
| Primary GTV, n, cm3 (median, [range]) | 48 | 9 | 22 | 17 | |
| 44.13 (30.30-56.08) | 68.34 (53.57-83.24) | 95.45 (72.56-122.87) | 0.004 | ||
CRC, colorectal cancer; PFK1, phosphofructokinase-1; GTV, gross tumor volume.
Table 2.
Correlations of clinicopathological features with the response to neoadjuvant radiotherapy of CRC
| Variables | Patients (n = 48) | Radiation sensitivity | P value | |
|---|---|---|---|---|
|
| ||||
| Radiation-sensitive group | Radiation-resistant group | |||
|
|
|
|||
| n = 26 | n = 22 | |||
| Age, n (%) | 0.881 | |||
| < 50 years | 18 | 10 (55.56) | 8 (44.44) | |
| ≥50 years | 30 | 16 (53.33) | 14 (46.67) | |
| Gender, n (%) | 0.938 | |||
| Male | 33 | 18 (54.55) | 15 (45.45) | |
| Female | 15 | 8 (53.33) | 7 (46.67) | |
| TNM stage, n (%) | 0.404 | |||
| II | 9 | 6 (66.67) | 3 (33.33) | |
| III | 39 | 20 (51.28) | 19 (48.72) | |
| Histopathological grade, n (%) | 0.038 | |||
| High | 8 | 7 (87.50) | 1 (12.50) | |
| Middle + low | 40 | 19 (47.50) | 21 (52.50) | |
| Primary GTV, n, cm3 (median [range]) | 48 | 54.825 (30.30-76.38) | 80.12 (58.67-122.87) | 0.017 |
CRC, colorectal cancer; GTV, gross tumor volume.
PFK1 expression in CRC cells was downregulated by transfection with siRNA
We measured PFK1 protein levels by western blot assay in the following seven human CRC lines: SW480, SW620, HCT116, Colo205, HT29, caco-2, and LoVo (Figure 2A). Among these cell lines, HT29 cells showed the highest expression of PFK1; HCT116 cells and Colo205 cells showed the second highest expression; and SW620 cells had the lowest expression. Since the proliferation rate of HCT116 cells was higher than that of Colo205 cells, we selected HT29 and HCT116 cells for transfection experiments. Three siRNA-PFK1s (siRNA-1336, siRNA-1611, and siRNA-2111) were transfected into HT29 and HCT116 cells. Compared with siRNA-NC-transfected and non-transfected control cells, intracellular PFK1 protein levels were decreased. The effects of siRNA-1336-PFK1 were the most significant (P < 0.001; Figure 2B). The knockdown of PFK1 by transfection with siRNA-1336-PFK1 in CRC cell lines was successful.
Figure 2.

PFK1 expression levels and the effects of siRNA on PFK1 expression in CRC cells. A. The expression levels of PFK1 in seven human CRC cell lines, as measured by western blot. HT29 cells exhibited the highest expression of PFK1 and SW620 cells exhibited the lowest expression. B. PFK1 expression levels in HT29 and HCT116 cells treated with three siRNA-PFK1s. SiRNA-1336-PFK1 exhibited significant inhibitory effects. Data are presented as mean ± SD (n = 3 independent experiments). Control, control group; siRNA-NC, negative control siRNA cells; PFK1, phosphofructokinase-1. **P < 0.01 and ***P < 0.001.
PFK1 knockdown can inhibit proliferation and migration and increase the apoptosis rate of human CRC cells
PFK1 was knocked down in HT29 and HCT116 cells by RNAi, and the effects on the survival, migration, and apoptosis were evaluated by MTT, cell migration, and apoptosis assays. In the MTT assay, we compared the effects after 24, 48, and 72 h. OD570 values of HT29 and HCT116 cells transfected with siRNA-PFK1 were significantly lower than in cells transfected with siRNA-NC (P < 0.05, Figure 3A), which indicated that PFK1 knockdown reduced cell survival. In the cell migration assay, the migration of HT29 and HCT116 cells was significantly suppressed after PFK1 knockdown compared with siRNA-NC-transfected and non-transfected control cells (P < 0.05, Figure 3B, 3C). Apoptosis of HT29 cells and HCT116 cells was measured by flow cytometry. After PFK1 knockdown, the apoptosis rate was significantly higher that in siRNA-NC-transfected and non-transfected control cells (Figure 4A, 4B). In summary, downregulation of PFK1 inhibits the proliferation and migration and promotes apoptosis of human CRC cells.
Figure 3.

PFK1 knockdown inhibits the proliferation and migration of CRC cells. (A) OD570 values were detected by MTT assay in HT29 and HCT116 cells transfected with siRNA-PFK1 or siRNA-NC. Scratch wound healing assay measuring the migration of (B) HT29 and (C) HCT116 cells transfected with siRNA-NC or siRNA-PFK1. Data are presented as mean ± SD (n = 3 independent experiments). PFK1, phosphofructokinase-1; control, control group; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells. *P < 0.05, **P < 0.01, and ***P < 0.001.
Figure 4.

PFK1 knockdown promotes apoptosis of CRC cells. The apoptosis rate of (A) HT29 and (B) HCT116 cells treated with siRNA-NC or siRNA-PFK1 was measured by flow cytometry. Data are presented as mean ± SD (n = 3 independent experiments). PFK1, phosphofructokinase-1; control, control group; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells. *P < 0.05 and **P < 0.01.
PFK1 knockdown enhances radiosensitivity of human CRC cells
The effects of downregulation of PFK1 on the radiosensitivity of CRC cells were investigated. MTT assay was used to measure the survival rate of HT29 and HCT116 cells treated with various radiation doses (0, 2, 4, 6, and 8 Gy). The lethal dose for 25% of HT29 and HCT116 cells was about 2.7 Gy and 1.9 Gy, respectively; thus, 2 Gy was selected as the experimental radiation dose. The LD50 of HT29 and HCT116 cells was approximately 6.2 Gy and 4.2 Gy, respectively; therefore, 6 Gy and 4 Gy were selected as the positive control radiation doses for HT29 and HCT116 cells, respectively (Figure 5A). The survival of the cells after radiation exposure was also determined; the survival rate of PFK1 knockdown cells was significantly lower than that of siRNA-NC-transfected cells (Figure 5B).
Figure 5.
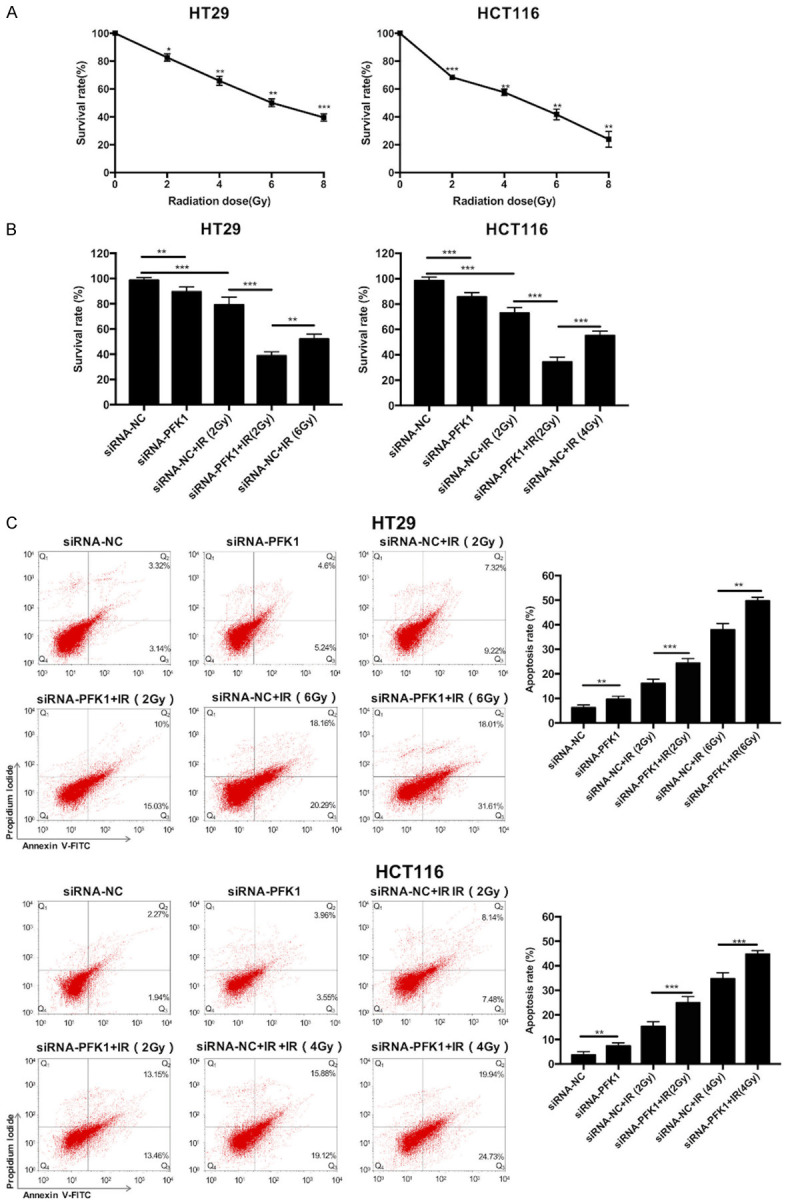
Effects of various treatments on the survival and apoptosis of CRC cells. A. The survival rate was detected by MTT assay in HT29 and HCT116 cells after various radiation doses (0, 2, 4, 6, and 8 Gy) for 24 h. The LD50 of HT29 and HCT116 cells was approximately 6.2 Gy and 4.2 Gy, respectively. B. The survival rate was detected using MTT assay in HT29 and HCT116 cells transfected with siRNA-NC or siRNA-PFK1 after treatment with or without radiation (2 Gy or 6 Gy) for 24 h. C. The apoptosis rate was detected by flow cytometry in siRNA-PFK1-transfected HT29 and HCT116 cells after exposure to irradiation. Data are presented as mean ± SD (n = 3 independent experiments). PFK1, phosphofructokinase-1; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells; IR, ionizing radiation. *P < 0.05, **P < 0.01, and ***P < 0.001.
In the apoptosis assay, the apoptosis rate of cells was increased after irradiation with 2 Gy, and the apoptosis rate increased in a radiation dose-dependent manner. Moreover, the apoptosis rate of PFK1 knockdown cells was higher than that of siRNA-NC-transfected control cells after exposure to radiation, and higher radiation doses caused higher apoptosis rates (Figure 5C).
To further determine the effects of PFK1 knockdown on the radiosensitivity of CRC cells, we performed a colony formation assay. In the absence or presence of irradiation, the colony survival fraction of PFK1 knockdown HT29 cells was significantly lower than that of siRNA-NC-transfected cells (Figure 6).
Figure 6.
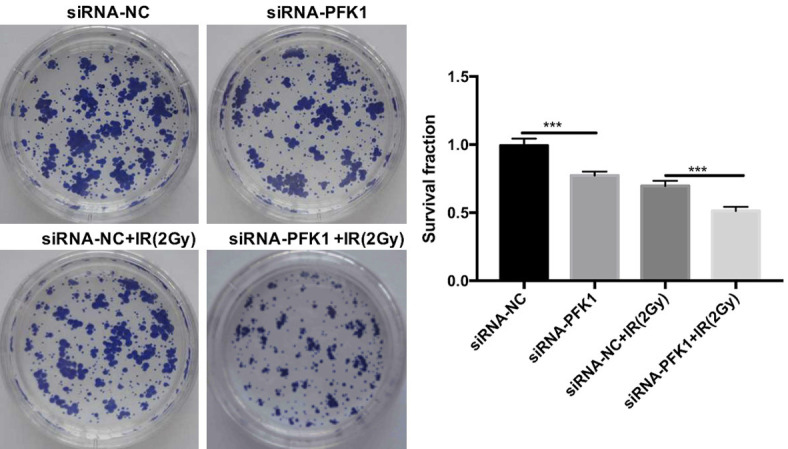
Effects of various treatments on the colony formation ability of CRC cells. The colony survival fraction was calculated in HT29 cells transfected with siRNA-NC or siRNA-PFK1 after exposure to 2 Gy irradiation. Data are presented as mean ± SD (n = 3 independent experiments). PFK1, phosphofructokinase-1; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells; IR, ionizing radiation. ***P < 0.001.
PFK1 knockdown increased the apoptosis rate and decreased the survival rate and colony formation ability after exposure to radiation, indicating that PFK1 knockdown might enhance radiosensitivity of CRC cells. In our subsequent experiments, we investigated the underlying mechanisms.
PFK1 knockdown can inhibit glycolysis in human CRC cells
Glycolytic activity was measured in PFK1 knockdown and siRNA-NC-transfected control HT29 and HCT116 cells. As illustrated in Figure 7A, 7B, PFK1 knockdown suppressed glucose uptake and PFK1 activity and decreased the ability of the cells to produce lactic acid and ATP.
Figure 7.

PFK1 knockdown inhibits glycolysis in CRC cells. Changes in glucose consumption, lactate production, ATP levels, and PFK1 activity of (A) HT29 and (B) HCT116 cells transfected with siRNA-PFK1 or siRNA-NC. Data are presented as mean ± SD (n = 3 independent experiments). PFK1, phosphofructokinase-1; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells. ***P < 0.001.
Oligomycin A can reverse the PFK1 knockdown-induced increase in radiosensitivity in CRC cells by enhancing glycolysis
To study whether PFK1 knockdown enhanced the radiosensitivity of CRC cells by enhancing glycolysis, oligomycin A, an inhibitor of mitochondrial H+-ATP synthase, was used in HT29 cells. Oligomycin A increases glycolytic activity by increasing iPFK-2 activity and fructose 2,6-bisphosphate content, and it inhibits or delays apoptosis in cancer cells [20,21]. By western blot analysis, we found that with and without radiation exposure, PFK1 levels were partially restored by oligomycin A after knockdown (Figure 8A). Similarly, in the glycolysis assay, in siRNA-NC-transfected cells, oligomycin A significantly increased glycolytic activity, and in PFK1 knockdown cells, it reversed the reductions in glycolytic activity and PFK1 activity (Figure 8B). In the apoptosis assay, oligomycin A also reversed the increase in apoptosis in irradiated PFK1 knockdown cells (Figure 8C).
Figure 8.

Oligomycin A restores the glycolysis levels and reverses the increase in apoptosis induced by PFK1 knockdown combined with 4 Gy irradiation, thus delaying apoptosis. HT29 cells transfected with siRNA-NC or siRNA-PFK1 and co-treated with oligomycin after treatment with or without radiation (4 Gy) for 24 h. A. Expression of PFK1 was detected by Western blot in HT29 cells treated with siRNA-PFK1 and oligomycin A. B. Changes in glucose concentration, ATP levels, lactate production, and PFK1 activity in HT29 cells after various treatments. C. The apoptosis rate of HT29 cells after various treatments, as detected by flow cytometry. Data are presented as mean ± SD (n = 3 independent experiments). PFK1, phosphofructokinase-1; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells; Oligo, oligomycin A. *P < 0.05, **P < 0.01, and ***P < 0.001.
MTT assay results indicated that oligomycin A decreased the inhibitory effects of PFK1 knockdown on the proliferation of HT29 cells (Figure 9A). Furthermore, in the colony formation assay, after irradiation, the colony survival fraction in PFK1 knockdown cells was significantly lower than in siRNA-NC-transfected cells. Oligomycin A also restored the decreased colony survival fraction of PFK1 knockdown HT29 cells (Figure 9B, 9C). These data support the view that PFK1 knockdown in CRC cells inhibits cell survival and increases apoptosis by regulating glycolysis, resulting in enhanced radiosensitivity of CRC cells.
Figure 9.

Oligomycin A decreases the inhibitory effects of PFK1 knockdown on the survival and colony formation ability of HT29 cells with and without irradiation. A. In the absence or presence of irradiation, the cell survival rate was detected by MTT assay in siRNA-NC and PFK1 knockdown HT29 cells treated with or without oligomycin A. B and C. The colony survival fraction was detected using colony formation assay in HT29 cells treated with siRNA-NC, siRNA-PFK1, or siRNA-PFK1 plus oligomycin A after irradiation with 0, 2, 4, 6, or 8 Gy. Data are presented as mean ± SD (n = 3 independent experiments). PFK1, phosphofructokinase-1; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells; oligo, oligomycin A. *P < 0.05, **P < 0.01, and ***P < 0.001.
PFK1 knockdown combined with X-ray irradiation can inhibit the growth of transplanted tumors in nude mice by increasing apoptosis
To study the effects PFK1 knockdown on the radiosensitivity of CRC cells in vivo, HT29 cells, which exhibit high PFK1 expression, were selected for tumor transplantation experiments. Nude mice were subcutaneously inoculated with HT29 cells, PFK1 knockdown HT29 cells, or siRNA-NC-transfected HT29 cells. After 15 days, the tumor was treated with a single dose of 2 Gy. Tumor size was measured every 2 days. PFK1 knockdown significantly inhibited the growth of implanted tumors in mice with or without irradiation (P < 0.05, Figure 10A, 10B). The apoptosis rate of each group was measured by TUNEL staining on the 25th day. With or without irradiation, the apoptosis rate was higher in the PFK1 knockdown group than in the control group (P < 0.05, Figure 10C, 10D). These in vivo results confirm that PFK1 knockdown combined with X-ray irradiation inhibits the growth of xenografts in nude mice. These inhibitory effects may be related to an increased apoptosis rate.
Figure 10.
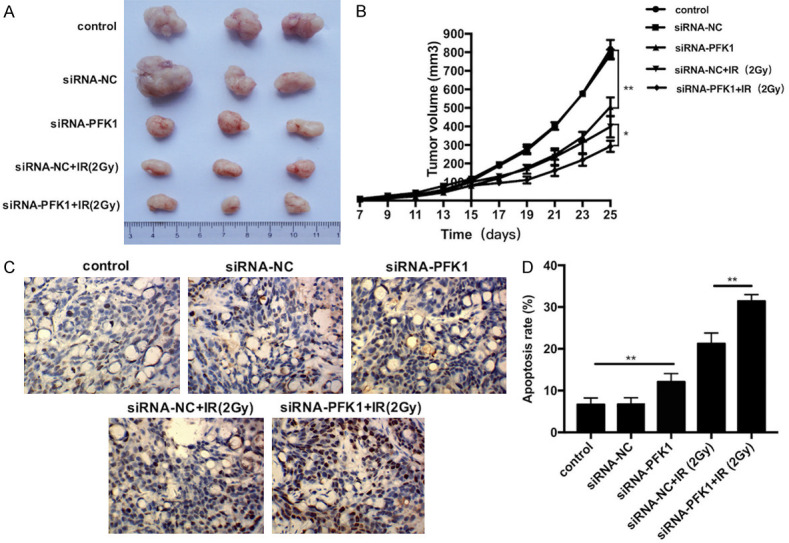
PFK1 knockdown enhances the radiosensitivity of CRC cells in vivo. A. Photographs of tumors derived from siRNA-PFK1-transfected or siRNA-NC-transfected HT29 cells at the end of the observation period. Irradiated and non-irradiated tissues at 15 days. B. Tumor growth curve in nude mice. After tumor cells were injected subcutaneously into the right flank of the abdomen of nude mice, the short and long diameters of the tumors were measured every 2 days, and tumor volumes (mm3) were calculated. C. Tumor tissues were stained with the indicated reagent after various treatments at the end of the observation period. Brown indicates apoptotic cells and blue indicates nuclei. D. The apoptosis rate was detected by TUNEL in implanted tumors under various treatments at the end of the observation period. Data are presented as mean ± SD (n = 5 per group). PFK1, phosphofructokinase-1; control, control group; siRNA-NC, negative control siRNA cells; siRNA-PFK1, PFK1 knockdown cells. *P < 0.05 and **P < 0.01.
Discussion
Radiotherapy is an important treatment for CRC, but tumor radioresistance often leads to a poor prognosis. Therefore, the identification and characterization of the molecular mechanisms that cause tumor radioresistance, as well as the identification of therapeutic targets to improve radiosensitivity, are important research objectives. In recent years, the glycolytic phenotype of malignant tumors has been found to be closely related to their chemical and radiation resistance [8,22]. Hexokinase, pyruvate kinase, and PFK1-three key enzymes of glycolysis-promote glycolysis [9] and are overexpressed in many tumors [23-26]. Inhibition of hexokinase or pyruvate kinase can diminish tumor radioresistance [24,27-30]. However, studies of the expression of PFK1 in CRC cells and its effects on the response to radiation are rare. Here, we present one of the first studies to investigate the relationship between PFK1 and radiotherapy resistance in CRC and to elucidate the underlying mechanisms.
PFK1 is highly expressed in certain malignant tumor tissues, e.g., glioma, bladder cancer, and cervical cancer [10,11,31,32], and is associated with the risk of malignancy [33]. Ahsan et al. [12] found that the PFKM gene region 12q13.11 is closely associated with the risk of breast cancer. In the present study, we examined the tissue samples of 48 patients with rectal cancer by immunohistochemistry. We found that PFK1 was overexpressed in rectal cancer tissues, and that PFK1 expression was closely correlated with TNM stage and primary GTV. In addition, we found that PFK1 was expressed to various degrees in seven human CRC cell lines. These results indicate that PFK1 may play a key role in the development and progression of CRC. PFKP expression has been found to be increased in oral cancer and is associated with tumor tissue differentiation and lymph node metastasis, and downregulation of PFKP expression inhibited autophagy, proliferation, migration, and invasion of oral cancer cells [25]. In the present study, we downregulated the expression of PFK1 in HCT116 and HT29 cells (which highly express PFK1) by RNAi. PFK1 knockdown significantly inhibited cell proliferation and migration and promoted apoptosis. Li et al. [10] similarly reported that upregulation of TAp73, a key regulator of PFK1, promoted the Warburg effect and enhanced cell proliferation in CRC, liver cancer, and other tumors. Wang et al. [33] found that miR-128, which directly targets the 3’ untranslated region of PFK1, negatively regulated the expression of PFKL in lung cancer cells and inhibited colony formation and tumor growth. These findings suggested that PFK1 can enhance the proliferation and invasion of cancer tissues and could thus play an important role in the occurrence and development of CRC.
To adapt to adverse microenvironments, most tumors obtain energy through glycolysis, which is rapid and inefficient, rather than by oxidative phosphorylation [5,34]. PFK1, the second key enzyme of glycolysis, is the most important regulatory factor in the glycolysis pathway and promotes the Warburg effect [11,35]. In the present study, we decreased glucose uptake, lactic acid and ATP production, and PFK1 activity in CRC cells by downregulating PFK1 expression, thus inhibiting glycolysis. These results suggest that PFK1 knockdown could inhibit glucose metabolism in CRC cells. The high glycolytic activity of malignant tumors not only provides a growth advantage but also contributes to tumor radioresistance [36-38]. Tumor tissues produce lactic acid due to active glycolysis [39], and the accumulation of lactic acid can promote tumor recurrence, metastasis, and radiotherapy resistance [40,41]. In the present study, we found that PFK1 knockdown enhanced the radiosensitivity of CRC cells, which may have been the result of an increase in apoptosis and inhibition of colony formation and cell proliferation. Through tumor forming experiments in nude mice, we found that PFK1 knockdown combined with X-ray irradiation inhibited the growth of implanted tumors in mice by promoting apoptosis. Our data are consistent with previous findings that epigallocatechin-3-gallate enhanced the inhibition of sorafenib on the growth of hepatocellular carcinoma cells in xenotransplantation mice by directly inhibiting the activity of PFK [35]. However, the specific mechanism leading to the enhanced radiosensitivity of CRC caused by PFK1 remains unclear.
To verify whether PFK1 knockdown enhanced the radiosensitivity of CRC cells by reducing the levels of glycolysis, we treated human CRC HT29 cells with oligomycin A, an inhibitor of mitochondrial F0F1ATP synthase that can induce and enhance glycolysis in a variety of cancer cells [42-44]. Oligomycin A also inhibited TNF-induced apoptosis [21] and delayed apoptosis by decreasing the intracellular release of cytochrome c [20]. Moreover, oligomycin A activated AMP-dependent protein kinase in hatched monocytes, increased iPFK-2 activity and fructose 2,6-bisphosphate content, and increased glycolytic activity. Fructose 2,6-bisphosphate is one of the strongest activators of PFK1 [45]. In our experiments, oligomycin A partially reversed the downregulation of PFK1, the decrease in glycolytic and PFK1 activity, and the enhancement of radiosensitivity triggered by PFK1 knockdown combined with radiation treatment in HT29 cells. These results are similar to those reported by Bhatt et al. [36], who found that a mitochondrial respiratory regulator (dinitrophenol) enhanced the radiosensitivity of tumors by inducing glycolysis. We also found that the expression of PFK1 in the radiation-resistant group was upregulated in patients with rectal cancer and that there was a statistical difference in the histopathological grade and primary GTV between the radiation-resistant group and the radiation-sensitive group, a result that differs from those of Cui et al. [46]. This discrepancy might be explained by the small sample size of high-grade rectal cancer specimens in our study. These results suggest that PFK1 activity is a determinant of the radiosensitivity of CRC that may enhance tumor radiosensitivity and improve the prognosis of patients by inhibiting glycolysis and lactic acid production. Therefore, PFK1 may be used as a target for the radiosensitization of CRC.
We wanted to further explore the downstream molecular mechanisms of PFK1 knockdown affecting the radiosensitivity of CRC cells. Bhatt et al. [36] found that elevation of glycolysis results in increased expression of repair proteins Rad51 and Ku70, which appears to facilitate the DNA repair in tumor cells and to be partly responsible for radioresistance. Moreover, PFK1 promoted the connection between Yes-associated protein (YAP)/tafazzin (TAZ) with its transcriptional cofactors TEADs, and maintained the cell proliferation induced by YAP/TAZ. The expression of YAP/TAZ, important transcriptional coactivators regulating proliferation, survival, and self-renewal, was associated with glycolysis, and the activity of YAP/TAZ was regulated by glycolysis to promote cancer cell proliferation and tumorigenesis [47]. Recent research demonstrated that the radiosensitivity of pancreatic cancer was increased upon inhibition of YAP/TAZ activity [48]. Therefore, we hypothesize that PFK1 knockdown might enhance radiosensitivity by reducing YAP/TAZ expression and facilitating the DNA repair; however, further experiments are needed to confirm this hypothesis.
In addition, the effects of PFK1 knockdown on CRC radiosensitivity are not well understood, which may be related to the siRNA transfection efficiency. In future studies, we aim to use shRNA instead of siRNA to silence PFK1 for better and more stable sensitization.
In conclusion, high PFK1 expression is negatively correlated with radiosensitivity in CRC and likely accelerates the proliferation and migration of CRC cells. PFK1 knockdown may enhance the radiosensitivity of CRC cells in vivo and in vitro by inhibiting glycolysis. Therefore, downregulation of PFK1 may be a new therapeutic strategy for patients with radioresistant CRC.
Acknowledgements
We would like to thank LetPub (www.letpub.com) for providing linguistic assistance during the preparation of this manuscript.
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 3.Lorimer PD, Motz BM, Kirks RC, Boselli DM, Walsh KK, Prabhu RS, Hill JS, Salo JC. Pathologic complete response rates after neoadjuvant treatment in rectal cancer: an analysis of the national cancer database. Ann Surg Oncol. 2017;24:2095–2103. doi: 10.1245/s10434-017-5873-8. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:111. doi: 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson KA, Schell JC, Rutter J. Pyruvate and metabolic flexibility: illuminating a path toward selective cancer therapies. Trends Biochem Sci. 2016;41:219–230. doi: 10.1016/j.tibs.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen H, Hau E, Joshi S, Dilda PJ, McDonald KL. Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism. Mol Cancer Ther. 2015;14:1794–1804. doi: 10.1158/1535-7163.MCT-15-0247. [DOI] [PubMed] [Google Scholar]
- 8.Shimura T, Noma N, Sano Y, Ochiai Y, Oikawa T, Fukumoto M, Kunugita N. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother Oncol. 2014;112:302–307. doi: 10.1016/j.radonc.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Al Hasawi N, Alkandari MF, Luqmani YA. Phosphofructokinase: a mediator of glycolytic flux in cancer progression. Crit Rev Oncol Hematol. 2014;92:312–321. doi: 10.1016/j.critrevonc.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Li L, Li W, Chen T, Bin Z, Zhao L, Wang H, Wang X, Xu L, Liu X, Wang D, Li B, Mak TW, Du W, Yang X, Jiang P. TAp73-induced phosphofructokinase-1 transcription promotes the Warburg effect and enhances cell proliferation. Nat Commun. 2018;9:4683. doi: 10.1038/s41467-018-07127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoneberg T, Kloos M, Bruser A, Kirchberger J, Strater N. Structure and allosteric regulation of eukaryotic 6-phosphofructokinases. Biol Chem. 2013;394:977–993. doi: 10.1515/hsz-2013-0130. [DOI] [PubMed] [Google Scholar]
- 12.Ahsan H, Halpern J, Kibriya MG, Pierce BL, Tong L, Gamazon E, McGuire V, Felberg A, Shi J, Jasmine F, Roy S, Brutus R, Argos M, Melkonian S, Chang-Claude J, Andrulis I, Hopper JL, John EM, Malone K, Ursin G, Gammon MD, Thomas DC, Seminara D, Casey G, Knight JA, Southey MC, Giles GG, Santella RM, Lee E, Conti D, Duggan D, Gallinger S, Haile R, Jenkins M, Lindor NM, Newcomb P, Michailidou K, Apicella C, Park DJ, Peto J, Fletcher O, dos Santos Silva I, Lathrop M, Hunter DJ, Chanock SJ, Meindl A, Schmutzler RK, Müller-Myhsok B, Lochmann M, Beckmann L, Hein R, Makalic E, Schmidt DF, Bui QM, Stone J, Flesch-Janys D, Dahmen N, Nevanlinna H, Aittomäki K, Blomqvist C, Hall P, Czene K, Irwanto A, Liu J, Rahman N, Turnbull C Familial Breast Cancer Study. Dunning AM, Pharoah P, Waisfisz Q, Meijers-Heijboer H, Uitterlinden AG, Rivadeneira F, Nicolae D, Easton DF, Cox NJ, Whittemore AS. A genome-wide association study of early-onset breast cancer identifies PFKM as a novel breast cancer gene and supports a common genetic spectrum for breast cancer at any age. Cancer Epidem Biomar. 2014;23:658–669. doi: 10.1158/1055-9965.EPI-13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L, Wang Y, Bai R, Yang K, Tian Z. MiR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1alpha regulation. Oncogenesis. 2016;5:e224. doi: 10.1038/oncsis.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo X, Zhang X, Wang T, Xian S, Lu Y. 3-Bromopyruvate and sodium citrate induce apoptosis in human gastric cancer cell line MGC-803 by inhibiting glycolysis and promoting mitochondria-regulated apoptosis pathway. Biochem Biophys Res Commun. 2016;475:37–43. doi: 10.1016/j.bbrc.2016.04.151. [DOI] [PubMed] [Google Scholar]
- 15.Prasad CP, Sodergren K, Andersson T. Reduced production and uptake of lactate are essential for the ability of WNT5A signaling to inhibit breast cancer cell migration and invasion. Oncotarget. 2017;8:71471–71488. doi: 10.18632/oncotarget.17277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafsson NMS, Farnegardh K, Bonagas N, Ninou AH, Groth P, Wiita E, Jonsson M, Hallberg K, Lehto J, Pennisi R, Martinsson J, Norstrom C, Hollers J, Schultz J, Andersson M, Markova N, Marttila P, Kim B, Norin M, Olin T, Helleday T. Targeting PFKFB3 radiosensitizes cancer cells and suppresses homologous recombination. Nat Commun. 2018;9:3872. doi: 10.1038/s41467-018-06287-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mace AG, Pai RK, Stocchi L, Kalady MF. American joint committee on cancer and college of American pathologists regression grade: a new prognostic factor in rectal cancer. Dis Colon Rectum. 2015;58:32–44. doi: 10.1097/DCR.0000000000000266. [DOI] [PubMed] [Google Scholar]
- 18.Huang MY, Lee HH, Tsai HL, Huang CW, Yeh YS, Ma CJ, Huang CM, Chen CY, Huang JJ, Wang JY. Comparison of efficacy and safety of preoperative chemoradiotherapy in locally advanced upper and middle/lower rectal cancer. Radiat Oncol. 2018;13:53. doi: 10.1186/s13014-018-0987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaemmerer D, Peter L, Lupp A, Schulz S, Sanger J, Baum RP, Prasad V, Hommann M. Comparing of IRS and Her2 as immunohistochemical scoring schemes in gastroenteropancreatic neuroendocrine tumors. Int J Clin Exp Pathol. 2012;5:187–194. [PMC free article] [PubMed] [Google Scholar]
- 20.Santamaria G, Martinez-Diez M, Fabregat I, Cuezva JM. Efficient execution of cell death in non-glycolytic cells requires the generation of ROS controlled by the activity of mitochondrial H+-ATP synthase. Carcinogenesis. 2006;27:925–935. doi: 10.1093/carcin/bgi315. [DOI] [PubMed] [Google Scholar]
- 21.Shchepina LA, Pletjushkina OY, Avetisyan AV, Bakeeva LE, Fetisova EK, Izyumov DS, Saprunova VB, Vyssokikh MY, Chernyak BV, Skulachev VP. Oligomycin, inhibitor of the F-0 part of H+-ATP-synthase, suppresses the TNF-induced apoptosis. Oncogene. 2002;21:8149–8157. doi: 10.1038/sj.onc.1206053. [DOI] [PubMed] [Google Scholar]
- 22.Chen FL, Zhuang MK, Zhong CM, Peng J, Wang XZ, Li JY, Chen ZX, Huang YH. Baicalein reverses hypoxia-induced 5-FU resistance in gastric cancer AGS cells through suppression of glycolysis and the PTEN/Akt/HIF-1 alpha signaling pathway. Oncol Rep. 2015;33:457–463. doi: 10.3892/or.2014.3550. [DOI] [PubMed] [Google Scholar]
- 23.Zhong JT, Zhou SH. Warburg effect, hexokinase-II, and radioresistance of laryngeal carcinoma. Oncotarget. 2017;8:14133–14146. doi: 10.18632/oncotarget.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng MB, Wang HH, Guo WH, Wu ZQ, Zeng XL, Zaorsky NG, Shi HS, Qian D, Niu ZM, Jiang B, Zhao LJ, Yuan ZY, Wang P. Targeting pyruvate kinase M2 contributes to radiosensitivity of non-small cell lung cancer cells in vitro and in vivo. Cancer Lett. 2015;356:985–993. doi: 10.1016/j.canlet.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 25.Chen G, Liu H, Zhang Y, Liang J, Zhu Y, Zhang M, Yu D, Wang C, Hou J. Silencing PFKP inhibits starvation-induced autophagy, glycolysis, and epithelial mesenchymal transition in oral squamous cell carcinoma. Exp Cell Res. 2018;370:46–57. doi: 10.1016/j.yexcr.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F, Gong Z, Guo C, Li X, Deng H, Cao K, Zhou M, Xiang B, Li X, Li Y, Li G, Xiong W, Zeng Z. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37:87. doi: 10.1186/s13046-018-0758-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X, Liu M, Sun H, Wang F, Xie X, Chen X, Su J, He Y, Dai Y, Wu H, Shen L. HK2 is a radiation resistant and independent negative prognostic factor for patients with locally advanced cervical squamous cell carcinoma. Int J Clin Exp Pathol. 2015;8:4054–4063. [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Shen L, Chen X, Qian Y, Zhou Q, Wang Y, Li K, Liu M, Zhang S, Huang X. High expression of PKM2 as a poor prognosis indicator is associated with radiation resistance in cervical cancer. Histol Histopathol. 2015;30:1313–1320. doi: 10.14670/HH-11-627. [DOI] [PubMed] [Google Scholar]
- 29.Katagiri M, Karasawa H, Takagi K, Nakayama S, Yabuuchi S, Fujishima F, Naitoh T, Watanabe M, Suzuki T, Unno M, Sasano H. Hexokinase 2 in colorectal cancer: a potent prognostic factor associated with glycolysis, proliferation and migration. Histol Histopathol. 2017;32:351–360. doi: 10.14670/HH-11-799. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Ma Y, Wang P, Song Z, Liu B, Sun X, Zhang H, Yu J. Knockdown of PKM2 enhances radiosensitivity of non-small cell lung cancer. Cell Biochem Biophys. 2015;73:21–26. doi: 10.1007/s12013-015-0567-y. [DOI] [PubMed] [Google Scholar]
- 31.Sun CM, Xiong DB, Yan Y, Geng J, Liu M, Yao XD. Genetic alteration in phosphofructokinase family promotes growth of muscle-invasive bladder cancer. Int J Biol Markers. 2016;31:e286–293. doi: 10.5301/jbm.5000189. [DOI] [PubMed] [Google Scholar]
- 32.Lee JH, Liu R, Li J, Zhang C, Wang Y, Cai Q, Qian X, Xia Y, Zheng Y, Piao Y, Chen Q, de Groot JF, Jiang T, Lu Z. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat Commun. 2017;8:949. doi: 10.1038/s41467-017-00906-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Mei Q, Ai YQ, Li RQ, Chang L, Li YF, Xia YX, Li WH, Chen Y. Identification of lung cancer oncogenes based on the mRNA expression and single nucleotide polymorphism profile data. Neoplasma. 2015;62:966–973. doi: 10.4149/neo_2015_117. [DOI] [PubMed] [Google Scholar]
- 34.Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y, Zhang S, Gong Z, Wei F, Yang L, He Y, Zhou M, Li X, Li G, Xiong W, Zeng Z. Role of long non-coding RNAs in glucose metabolism in cancer. Mol Cancer. 2017;16:130. doi: 10.1186/s12943-017-0699-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S, Wu L, Feng J, Li J, Liu T, Zhang R, Xu S, Cheng K, Zhou Y, Zhou S, Kong R, Chen K, Wang F, Xia Y, Lu J, Zhou Y, Dai W, Guo C. In vitro and in vivo study of epigallocatechin-3-gallate-induced apoptosis in aerobic glycolytic hepatocellular carcinoma cells involving inhibition of phosphofructokinase activity. Sci Rep. 2016;6:28479. doi: 10.1038/srep28479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhatt AN, Chauhan A, Khanna S, Rai Y, Singh S, Soni R, Kalra N, Dwarakanath BS. Transient elevation of glycolysis confers radio-resistance by facilitating DNA repair in cells. BMC Cancer. 2015;15:335. doi: 10.1186/s12885-015-1368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu L, Sun Y, Li J, Wang Y, Zhu Y, Shi Y, Fan X, Zhou J, Bao Y, Xiao J, Cao K, Cao P. Silencing the Girdin gene enhances radio-sensitivity of hepatocellular carcinoma via suppression of glycolytic metabolism. J Exp Clin Cancer Res. 2017;36:110. doi: 10.1186/s13046-017-0580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu G, Li YI, Gao X. Overexpression of microRNA-133b sensitizes non-small cell lung cancer cells to irradiation through the inhibition of glycolysis. Oncol Lett. 2016;11:2903–2908. doi: 10.3892/ol.2016.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voelxen NF, Blatt S, Knopf P, Henkel M, Appelhans C, Righesso LAR, Pabst A, Goldschmitt J, Walenta S, Neff A, Mueller-Klieser W, Ziebart T. Comparative metabolic analysis in head and neck cancer and the normal gingiva. Clin Oral Investig. 2018;22:1033–1043. doi: 10.1007/s00784-017-2185-0. [DOI] [PubMed] [Google Scholar]
- 40.Fujiwara S, Wada N, Kawano Y, Okuno Y, Kikukawa Y, Endo S, Nishimura N, Ueno N, Mitsuya H, Hata H. Lactate, a putative survival factor for myeloma cells, is incorporated by myeloma cells through monocarboxylate transporters 1. Exp Hematol Oncol. 2015;4:12. doi: 10.1186/s40164-015-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hao J, Graham P, Chang L, Ni J, Wasinger V, Beretov J, Deng J, Duan W, Bucci J, Malouf D, Gillatt D, Li Y. Proteomic identification of the lactate dehydrogenase A in a radioresistant prostate cancer xenograft mouse model for improving radiotherapy. Oncotarget. 2016;7:74269–74285. doi: 10.18632/oncotarget.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Untereiner AA, Pavlidou A, Druzhyna N, Papapetropoulos A, Hellmich MR, Szabo C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem Pharmacol. 2018;149:174–185. doi: 10.1016/j.bcp.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 44.Lopez-Rios F, Sanchez-Arago M, Garcia-Garcia E, Ortega AD, Berrendero JR, Pozo-Rodriguez F, Lopez-Encuentra A, Ballestin C, Cuezva JM. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007;67:9013–9017. doi: 10.1158/0008-5472.CAN-07-1678. [DOI] [PubMed] [Google Scholar]
- 45.Marsin AS, Bouzin C, Bertrand L, Hue L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J Biol Chem. 2002;277:30778–30783. doi: 10.1074/jbc.M205213200. [DOI] [PubMed] [Google Scholar]
- 46.Cui Y, Yang X, Shi Z, Yang Z, Du X, Zhao Z, Cheng X. Radiomics analysis of multiparametric MRI for prediction of pathological complete response to neoadjuvant chemoradiotherapy in locally advanced rectal cancer. Eur Radiol. 2019;29:1211–1220. doi: 10.1007/s00330-018-5683-9. [DOI] [PubMed] [Google Scholar]
- 47.Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M, Grifoni D, Pession A, Zanconato F, Guzzo G, Bicciato S, Dupont S. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J. 2015;34:1349–1370. doi: 10.15252/embj.201490379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gopal U, Mowery Y, Young K, Pizzo SV. Targeting cell surface GRP78 enhances pancreatic cancer radiosensitivity through YAP/TAZ protein signaling. J Biol Chem. 2019;294:13939–13952. doi: 10.1074/jbc.RA119.009091. [DOI] [PMC free article] [PubMed] [Google Scholar]