

Abstract
Systemic lupus erythematosus (SLE) is an autoimmune disease with multiple organ involvement. Lupus nephritis (LN) is a severe manifestation of the disease and the most common cause of mortality in SLE patients. The etiology of LN is multifactorial and accumulating evidence suggests that mitochondrial dysfunction contributes to LN initiation and progression. Mild mitochondrial uncoupler niclosamide ethanolamine salt (NEN) has recently been shown to be efficacious in the treatment of both diabetic kidney disease and non-diabetic adriamycin nephropathy. However, its role in autoimmune kidney disease has not been explored. Here, we report for the first time that NEN attenuated SLE and lupus nephritis in MRL/lpr mice. NEN treatment reduced urinary protein excretion and attenuated glomerular lesions in this model. NEN treatment also decreased urinary excretion of tubular injury biomarkers NGAL and Kim-1, restored renal tubule phenotypic alterations, inhibited tubular proliferation, and suppressed renal interstitial inflammation and fibrosis. In addition, NEN diet supplementation restored redox imbalance, promoted mitochondrial biogenesis, and improved energy dysregulation in the kidney. Importantly, NEN prevented the enlargement of lymph nodes and the spleen, and decreased serum anti-dsDNA antibody levels in the MRL/lpr mice. Therefore, our data suggest that this mild mitochondrial uncoupling agent has great potential for translational application as a novel therapy for autoimmune disease.
Keywords: Niclosamide ethanolamine salt, systemic lupus erythematosus, lupus nephritis, mitochondrial dysfunction
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disorder that is characterized by loss of tolerance to nuclear self-antigens and sustained overproduction of pathogenic autoantibodies. The clinical presentation of SLE is heterogeneous and multiple organs are involved [1]. As a severe manifestation of the disease, lupus nephritis (LN) is the most common cause of mortality in SLE patients. Despite advancement in diagnosis and treatment over the past decades, LN is still a challenge for clinicians [2]. Therefore, it is imperative to further study the pathogenesis of LN, with the intent of developing effective therapeutics for this pathology.
Although the etiology of LN is multifactorial and remains largely unknown, previous studies indicate that autoantibody production, immune complex deposition, complement activation, inflammation, fibrogenesis, aberrant cell proliferation, and apoptosis are all pathophysiological processes involved in LN [3]. In addition, metabolic pathways are increasingly considered to play a key role in autoimmune disease [4]. In patients with SLE and in lupus-prone mice, immune cells and target organs have metabolic abnormalities [5,6].
Mitochondria are the major site of cellular energy metabolism and participate in many cellular signal transduction pathways [7]. The kidney is abundant in mitochondria and accumulating evidence suggests that mitochondrial dysfunction contributes to LN initiation and progression [8]. Mitochondria are also the main source of reactive oxygen species (ROS). Studies indicate that a redox imbalance correlates with disease activity and mediates target organ damage in patients with SLE and lupus-prone mice [9]. Therefore, mitochondria are potential targets for the treatment of SLE and LN [10].
Niclosamide ethanolamine salt (NEN) is a mild mitochondrial uncoupler and is widely used as an anthelminthic drug [11]. Mitochondrial uncoupling is a key mechanism that regulates superoxide formation and mitochondrial biogenesis [12,13]. Previous studies demonstrate that NEN is efficacious in treating diabetic kidney disease and non-diabetic adriamycin nephropathy [14-16]. However, its role in autoimmune kidney disease has not been investigated to date. Therefore, this study seeks to determine the effects of NEN treatment in lupus-prone MRL/lpr mice, and explore the mechanisms involved.
Materials and methods
Animal experiments
Female MRL/lpr mice (3-4 weeks old) were purchased from Shanghai SLAC Laboratory Animal Company (Shanghai, China). Age matched female C57BL/6J mice were purchased from Guangdong Medical Laboratory Animal Center. All mice were housed in the Central Animal Facility at Shenzhen Graduate School of Peking University. At 12 weeks old, the MRL/lpr mice were allocated randomly into either the MRL/lpr group (SLE group, n = 6) or the MRL/lpr+NEN group (SLE+NEN, n = 6). Female C57BL/6J mice served as the control group (n = 6). The control and SLE groups received a standard diet, and the SLE+NEN group received a standard diet supplemented with 10 g/kg NEN (Hubei ShengTian HengChuang Biotech Co., Ltd, Wuhan, China) for 8 weeks. All animal procedures were approved by the Guangzhou University of Chinese Medicine Institutional Animal Care and Use Committee. All experiments were performed in accordance with relevant guidelines and regulations.
Tissue preparation
Mice were sacrificed following the 8 weeks of NEN supplementation. The spleen and lymph nodes (submaxillary, parotid, axillary, mesenteric, and inguinal) were isolated, weighed, and imaged. The kidneys were dissected and rinsed in phosphate buffered saline. A portion of the kidney was fixed in 10% formalin for pathological examination and immunohistochemical staining. The renal cortex (1 mm3) was fixed in 2.5% glutaraldehyde and then post-fixed in 1% osmic acid for transmission electron microscopy (TEM). The remaining renal tissues were immediately snap-frozen in liquid nitrogen and stored at -80°C for later analyses.
Light microscopy
Paraffin-embedded renal sections (4 μm) were stained with periodic acid-Schiff (PAS), hematoxylin and eosin (HE), and Masson trichrome. Using PAS staining, thirty glomeruli in each section were randomly selected under 40× magnification. Glomerular tuft area, number of nuclei, glomerular injury, and the degree of glomerulosclerosis were measured and evaluated. Glomerular injury was scored on a semiquantitative scale: 0 = normal [35 to 40 cells/glomerular cross section (GCS)]; 1 = mild [glomeruli with mild hypercellularity (41 to 50 cells/GCS)]; 2 = moderate [glomeruli with moderate hypercellularity (51 to 60 cells/GCS) including segmental and/or diffuse proliferative changes, hyalinosis], 3 = severe [glomeruli with severe hypercellularity (>60 cells/GCS) and/or segmental or global sclerosis and/or, necrosis and crescent formation]. The glomerulosclerosis index was scored on a scale of 0 to 4 based on the percentage of PAS staining positive area within the glomerulus (0, <5%; 1, 5%-25%; 2, 25%-50%; 3, 50%-75%; 4, >75%). The PAS staining was also used to evaluate tubular injury that was defined by the presence of tubular dilation, atrophy, intraluminal casts, loss of proximal tubular brush border, and tubular cell vacuolization and detachment. Twenty tubular areas in each section were randomly selected under 20× magnification and scored on a scale of 0 to 4 (0, <5%; 1, 5%-25%; 2, 25%-50%; 3, 50%-75%; 4, >75%). HE staining was used to evaluate the renal inflammatory infiltration index. Twenty inflammation regions under 40× magnification were randomly selected. The inflammatory infiltration index was determined according to the number of cell layers and graded on a scale of 0 to 3 (score: 0 = none; 1 = <5 cell layers; 2 = 5 to 10 cell layers; 3 = >10 cell layers). Masson trichrome staining was used to evaluate interstitial fibrosis. Fifteen interstitial fields in renal cortex were randomly selected under 20× magnification. Interstitial fibrosis was defined as the area occupied by positive interstitium and graded on a scale of 0 to 4 (0, <5%; 1, 5%-25%; 2, 25%-50%; 3, 50%-75%; 4, >75%).
Immunohistochemical staining
Immunohistochemical staining was performed on 4 μm thick renal sections. After antigen retrieval with citrate buffer, sections were incubated with primary antibodies against WT-1, p-E-cadherin (S838+S840), Ki-67, catalase, and superoxide dismutase 2 (SOD2) respectively. The sections were then washed and incubated with HRP-polymer conjugated anti-Mouse/Rabbit IgG complex. Staining was detected using a diaminobenzidine tetrahydrochloride solution and counterstain with hematoxylin. Twenty glomeruli and renal tubular areas were randomly selected to count WT-1 and Ki-67 positive cells.
Immunofluorescence staining
Frozen kidney sections (6 μm) were fixed with acetone for seconds. After rinsing, the sections were incubated with FITC conjugated primary antibodies against IgG and C3 respectively. Images were visualized and captured on a confocal microscope (LSM710, Carl Zeiss, Oberkochen, Germany). Ten to twenty images were randomly selected in each section to calculate the positive area ratio of IgG and C3 in glomerulus.
Transmission electron microscopy
TEM images were photographed by JEM-1400 (JEOL, Tokyo, Japan). Eight images per sample were analyzed using Image J software (National Institutes of Health, Bethesda, MD, USA) and the average podocyte foot process width (FPW) was calculated according to a previously described method [17]. Glomerular immune complex deposition in mesangial, subendothelial, and subepithelial regions was observed and photographed.
ELISA
Urinary neutrophil gelatinase-associated lipocalin (NGAL) and Kim-1 (R&D Systems, Minneapolis, MN, USA) levels were measured by ELISA according to the manufacturer’s instructions. The serum level of anti-dsDNA antibodies was also measured by ELISA, as previously reported [18]. Briefly, 96-well culture plates were pre-coated with 5 μg/ml calf thymus dsDNA (Sigma-Aldrich, USA) overnight. A mouse anti-dsDNA monoclonal antibody (EMD Millipore, Temecula, CA, USA) was used to prepare a reference standard curve. For analysis, absorbance was measured at 450 nm.
Urinary protein and H2O2 assay
Urine was collected using metabolic cages (Tecniplast, Buguggiate, Italy). Urinary protein was detected by Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA, USA). Urinary H2O2 level was determined using Amplex UltraRed reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Immunoblotting analysis
Renal tissues were lysed and prepared in sample loading buffer (Bio-Rad). The lysates were subjected to immunoblotting analysis according to previously described method [17]. Antibodies against p-E-cadherin (S838+S840) and E-cadherin were purchased from Abcam (Cambridge, UK). Antibodies against Vimentin, proliferating cell nuclear antigen (PCNA), p-4E-BP1 (Ser65), 4E-BP1, p-S6RP (Ser235/236), S6RP, p-Erk1/2 (Thr202/Tyr204), Erk1/2, p-p38 MAPK (Thr180/Tyr182), p38 MAPK, catalase, superoxide dismutase 2 (SOD2), VDAC, TOM20, COX IV, p-AMPK (Thr172), and AMPK were purchased from Cell Signaling Technology (Danvers, MA, USA). An antibody against Tfam was from Novus Biologicals (Littleton, CO, USA). β-actin antibody was from Sigma Aldrich (St. Louis, MO, USA). β-actin was used as the loading control.
mRNA analysis
Total RNA was extracted from renal tissues using the TRIzol Plus RNA purification kit (Invitrogen). First-strand cDNA was generated and quantitative real-time PCR (qPCR) was subsequently carried out in a QuantStudio 5 real-time PCR systems (Applied Biosystems, Foster City, CA, USA) as previously described [16]. Primers were synthesized by Sangon Biotechnology Company (Shanghai, China) and sequences are listed in Table 1. The amplification conditions were as follows: 95°C for 5 min followed by 45 cycles of 95°C for 15 s, 55°C for 15 s, and 72°C for 20 s. The relative mRNA expression was calculated by 2-ΔΔCT and normalized against the housekeeping gene β-actin.
Table 1.
Sequences of the primers for qPCR
| Gene | Primer Sequence (5’-3’) |
|---|---|
| Mouse FN | F: GCAGTGACCACCATTCCTG |
| R: CCTGTCTTCTCTTTCGGGTTCA | |
| Mouse COL I | F: CTGGAACAAATGGGCTCACTG |
| R: CAGGCTCACCAACAAGTCCTC | |
| Mouse COL III | F: ACAGCTGGTGAACCTGGAAG |
| R: ACCAGGAGATCCATCTCGAC | |
| Mouse TGF-β1 | F: CCGCAACAACGCCATCTATG |
| R: CTCTGCACGGGACAGCAAT | |
| Mouse IL-1β | F: TGACCTGGGCTGTCCTGATG |
| R: GGTGCTCATGTCCTCATCCTG | |
| Mouse IL-6 | F: AGATCTACTCGGCAAACC |
| R: CGTAGAGAACAACATAAGTCAG | |
| Mouse TNF-α | F: AGGCTGCCCCGACTACGT |
| R: GACTTTCTCCTGGTATGAGATAGCAAA | |
| Mouse MCP1 | F: AGGTGTCCCAAAGAAGCTGT |
| R: ACAGAAGTGCTTGAGGTGGT | |
| Mouse β-actin | F: GGACTCCTATGTGGGTGACG |
| R: AGGTGTGGTGCCAGATCTTC |
Results
NEN treatment reduced urinary protein excretion and attenuated glomerular lesions
Proteinuria is an important clinical manifestation of various renal diseases and glomerular lesions are the typical characteristics of LN. So we measured urinary protein excretion and evaluated glomerular pathological changes in our mouse model of SLE. Compared to control mice, urinary protein excretion significantly increased in the SLE group, as expected (Figure 1A). This increase was significantly reduced by 8 weeks of NEN treatment (Figure 1A). The SLE group presented with abnormally enlarged glomerular tuft area, and exhibited glomerular injury and glomerulosclerosis (Figure 1B-D and 1H). NEN treatment significantly attenuated these pathological changes (Figure 1B-D and 1H).
Figure 1.

NEN reduced urinary protein excretion and attenuated glomerular lesions. (A) Determination of urinary protein excretion in each group. n = 6 per group. Measurement and evaluation of (B) glomerular tuft area, (C) glomerular injury index, and (D) glomerulosclerosis index in each group. n = 6 per group. (E, F) Total nuclei count and WT-1 positive cells per glomerulus in each group. n = 6 per group. (G) Measurement of FPW in different groups. n = 3 per group. (H, I) Representative images of PAS staining and WT-1 immunostaining in each group. Scale bar, 20 μm. (J) Representative TEM images showing pathological alterations of foot processes. Scale bar, 500 nm. **P<0.01 and ***P<0.001 vs. control group. #P<0.05, ##P<0.01 and ###P<0.001 vs. SLE group.
In the SLE group, the total number of nuclei per glomerulus increased compared to controls, while the number of podocytes (WT-1 positive cells) decreased significantly. These pathological changes were recovered by NEN treatment (Figure 1E, 1F, 1H and 1I). Foot process fusion was closely associated with proteinuria formation. Consistent with the urinary protein excretion results, increased FPW in the SLE group was attenuated by NEN treatment (Figure 1G and 1J).
As shown in Figure 2A-D, increased glomerular deposition of IgG and C3 was observed in the SLE group, which was significantly attenuated by NEN treatment. To explore the exact location of these immune complexes, we observed the ultrastructure of the glomerulus by TEM. These images revealed that electron dense deposits preferentially localized to the mesangial and subendothelial regions, and were occasionally found in the subepithelial region in SLE group (Figure 2E). After NEN treatment, the electron dense deposits were notably diminished and scarcely observed, particularly in the subepithelial region (Figure 2E).
Figure 2.
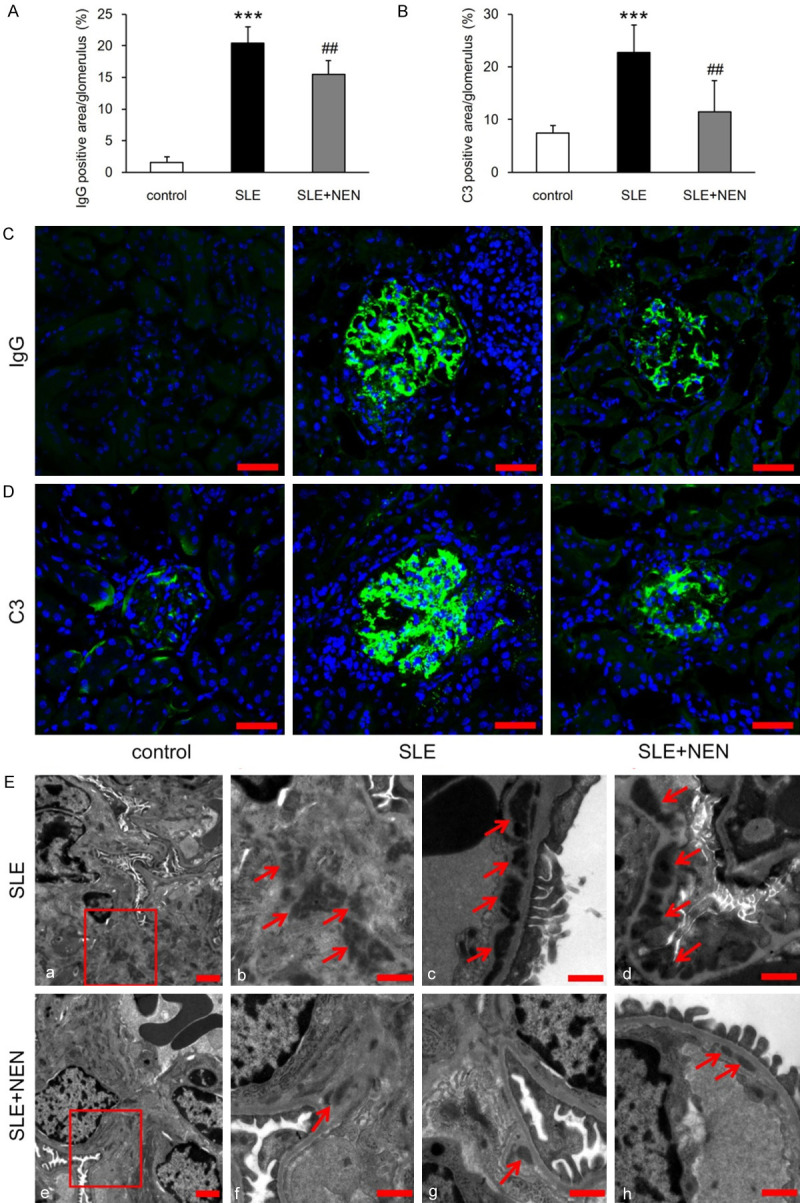
NEN ameliorated glomerular immune complex deposition. (A, B) Bar graphs indicating the analysis of IgG and C3 deposition in glomeruli from each group. (C, D) Representative immunofluorescence staining images of IgG and C3 in different groups. (E) Representative TEM images displaying the electron dense deposits in glomeruli of SLE and SLE+NEN groups. (a, b, e, f) Mesangial region; (c) Subendothelial region; (d) Subepithelial region; (g, h)Subendothelial region. Scale bar for (a and e), 2 μm; Scale bar for (b-d and f-h), 1 μm. n = 6 per group. ***P<0.001 vs. control group. ##P<0.01 vs. SLE group.
Effects of NEN on renal tubular injury
In addition to glomerular injury, renal tubular damage also occurs and contributes to LN progression [19]. In the SLE group, the tubular injury index increased significantly when compared to control mice (Figure 3A and 3D). Consistent with the pathological alterations, tubular injury biomarkers NGAL and Kim-1 were also significantly increased in the urine of these mice (Figure 3B and 3C). NEN treatment significantly reduced NGAL and Kim-1 excretion. However, tubular pathological injury was not significantly affected by NEN supplementation (Figure 3A-D).
Figure 3.

Effects of NEN treatment on renal tubular injury. A. Evaluation of renal tubular injury index in each group. B, C. Measurement of urinary NGAL and Kim-1 excretion in various groups. D. Representative images of tubule PAS staining. scale bar, 50 μm. n = 6 per group. ***P<0.001 vs. control group. ##P<0.01 and ###P<0.001 vs. SLE group.
NEN treatment restored renal tubule phenotypic alterations and inhibited tubular cell proliferation
Loss of E-cadherin expression is the hallmark of tubule phenotypic alterations [20]. E-cadherin plays a key role in the establishment and maintenance of epithelial cell polarity. Its phosphorylation promotes epithelial cell surface stability and adhesion, and participates in transmembrane signal transduction [21]. As shown in Figure 4E, p-E-cadherin was primarily expressed in tubular cells. Compared to the control group, lower expression of p-E-cadherin and E-cadherin, and increased vimentin/p-E-cadherin ratio were detected in the SLE group (Figure 4A-D). NEN treatment significantly upregulated p-E-cadherin expression and restored the ratio of vimentin to p-E-cadherin (Figure 4A, 4B and 4D).
Figure 4.
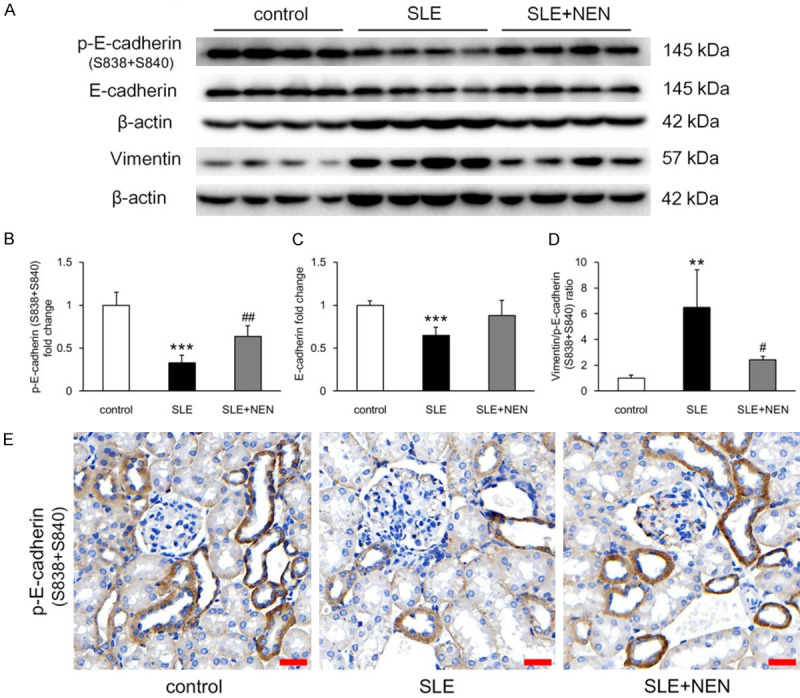
NEN treatment restored renal tubule phenotypic alterations. A. Immunoblotting bands shows p-E-cadherin (S838+S840), E-cadherin, and vimentin expression in each group. B-D. Quantitative analysis of p-E-cadherin (S838+S840), E-cadherin, and vimentin/p-E-cadherin (S838+S840) ratio in various groups. E. Representative images of p-E-cadherin (S838+S840) immunostaining in renal tubules of different groups. Scale bar, 20 μm. n = 4 per group. **P<0.01 and ***P<0.001 vs. control group. #P<0.05 and ##P<0.01 vs. SLE group.
TGF-β1 is a key mediator regulating cell phenotype alteration and proliferation. Compared to control mice, renal TGF-β1 mRNA level increased significantly in SLE mice, and this was significantly downregulated by NEN treatment (Figure 5A). Ki-67 and PCNA are markers to evaluate cell proliferative activity. In the SLE group, the number of Ki-67 positive tubular cells, evaluated by immunohistochemistry, and PCNA level in renal tissue, detected by immunoblotting, both increased remarkably compared to controls. These were both significantly reduced by NEN treatment (Figure 5B-E).
Figure 5.

NEN inhibited renal tubular proliferation. A. Determination of renal TGF-β1 mRNA expression level in each group. n = 6 per group. B. Counting of Ki-67 positive cells in renal tubules of different groups. n = 6 per group. C. Representative images of Ki-67 immunostaining in each group. Scale bar, 20 μm. D-M. Immunoblotting bands and quantitative analysis of p-4E-BP1 (Ser65), 4E-BP1, p-S6RP (Ser235/236), S6RP, p-Erk1/2 (Thr202/Tyr204), Erk1/2, p-p38 MAPK (Thr180/Tyr182), and p38 MAPK in each group. n = 4 per group. *P<0.05, **P<0.01 and ***P<0.001 vs. control group. #P<0.05, ##P<0.01 and ###P<0.001 vs. SLE group.
To determine the signaling pathways involved in renal tubular proliferation, we performed immunoblotting assays to detect p-4E-BP1 (Ser65), 4E-BP1, p-S6RP (Ser235/236), S6RP, p-Erk1/2 (Thr202/Tyr204), Erk1/2, p-p38 MAPK (Thr180/Tyr182), and p38 MAPK. Increased p-4E-BP1 (Ser65), 4E-BP1, and p-S6RP (Ser235/236) and decreased p-p38 MAPK (Thr180/Tyr182) expression was seen in SLE mice when compared to controls (Figure 5D, 5F-H and 5J). NEN supplementation significantly reduced p-4E-BP1 (Ser65) and 4E-BP1 but did not have a significant effect on the other signaling pathway proteins (Figure 5D and 5F-M).
NEN suppressed renal interstitial inflammation and fibrosis
Interstitial inflammation is a prominent pathological feature of LN and predicts deterioration of renal function [22]. Compared to control mice, the renal inflammatory infiltration index increased significantly in the SLE mice (Figure 6A and 6J). Consistent with inflammation, the mRNA expression levels of monocyte chemoattractant protein-1 (MCP-1), interleukin (IL)-1β, tumor necrosis factor α (TNF-α), and IL-6 were obviously upregulated (Figure 6C-F). NEN treatment inhibited the inflammatory infiltration and downregulated mRNA expression levels of MCP-1 and IL-1β (Figure 6A, 6J, 6C and 6D). However, the mRNA levels of TNF-α and IL-6 were not significantly reduced by NEN (Figure 6E and 6F).
Figure 6.
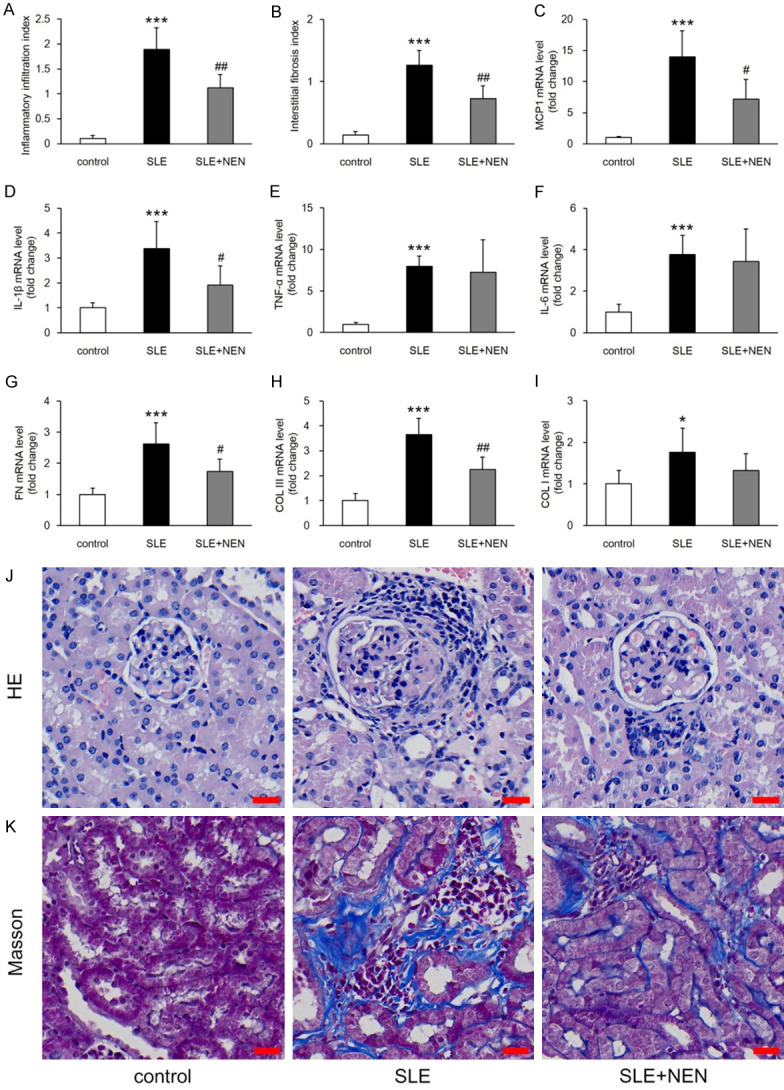
Suppressive effects of NEN on renal interstitial inflammation and fibrosis. A, B. Evaluation of inflammatory infiltration index and interstitial fibrosis index in each group. C-I. Bar graphs showing the renal mRNA expression levels of MCP-1, IL-1β, TNF-α, IL-6, FN, COL III, and COL I in each group. J, K. Representative images of HE staining for regions of inflammation and Masson trichrome staining for renal interstitium. Scale bar, 20 μm. n = 6 per group. *P<0.05 and ***P<0.001 vs. control group. #P<0.05 and ##P<0.01 vs. SLE group.
Renal interstitial fibrosis is associated with poor prognosis in various kidney diseases. Therefore, we evaluated the interstitial fibrosis index and measured fibrosis related mRNA expression, including fibronectin (FN), collagen (COL) III, and COL I. In agreement with an increased renal interstitial fibrosis index, the mRNA levels of FN, COL III, and COL I were also significantly increased in the SLE mice (Figure 6B, 6K and 6G-I). NEN treatment significantly reduced the interstitial fibrosis index and downregulated the mRNA levels of FN and COL III (Figure 6B, 6K, 6G and 6H). However, the mRNA level of COL I was not significantly affected by NEN (Figure 6I).
NEN treatment restored redox imbalance in the kidney
Catalase and SOD2 are important enzymes that regulate the intracellular redox state. Compared to control mice, the protein levels of renal catalase and SOD2 decreased and urinary H2O2 excretion increased remarkably in SLE mice (Figure 7A-D). NEN treatment restored the renal redox imbalance in lupus mice (Figure 7A-D). Immunohistochemical staining revealed that catalase and SOD2 were primarily expressed in renal tubules and scarcely in glomeruli (Figure 7E and 7F). This suggests that the renal tubules might be the site of oxidative stress in this disease.
Figure 7.

NEN recovered redox imbalance in kidney. A-C. Immunoblotting bands and quantitative analysis of catalase and SOD2 in each group. n = 4 per group. D. Bar graph showing the fold change of urinary H2O2 excretion in various groups. n = 6 per group. E, F. Representative immunohistochemistry images showing the expression of catalase and SOD2 in the kidney. Scale bar, 20 μm. **P<0.01 and ***P<0.001 vs. control group. ##P<0.01 and ###P<0.001 vs. SLE group.
NEN promoted mitochondrial biogenesis and improved energy dysregulation in the kidney
The structural integrity of mitochondria is essential for maintaining their physiological function. Tfam is a transcription factor and plays a critical role in mitochondrial biogenesis [23]. In SLE mice, Tfam levels and mitochondrial components, including voltage-dependent anion channel (VDAC), transporter of outer mitochondrial membrane (TOM20), and cytochrome oxidase (COX) IV, were decreased (Figure 8A-E). As the guardian of metabolism and mitochondrial homeostasis, AMP-activated protein kinase (AMPK) was activated in response to mitochondrial deficiency in the kidneys of SLE mice (Figure 8A, 8F and 8G). NEN treatment significantly increased the expression of Tfam and mitochondrial components, improving energy dysregulation in the kidneys of these mice (Figure 8A-G).
Figure 8.
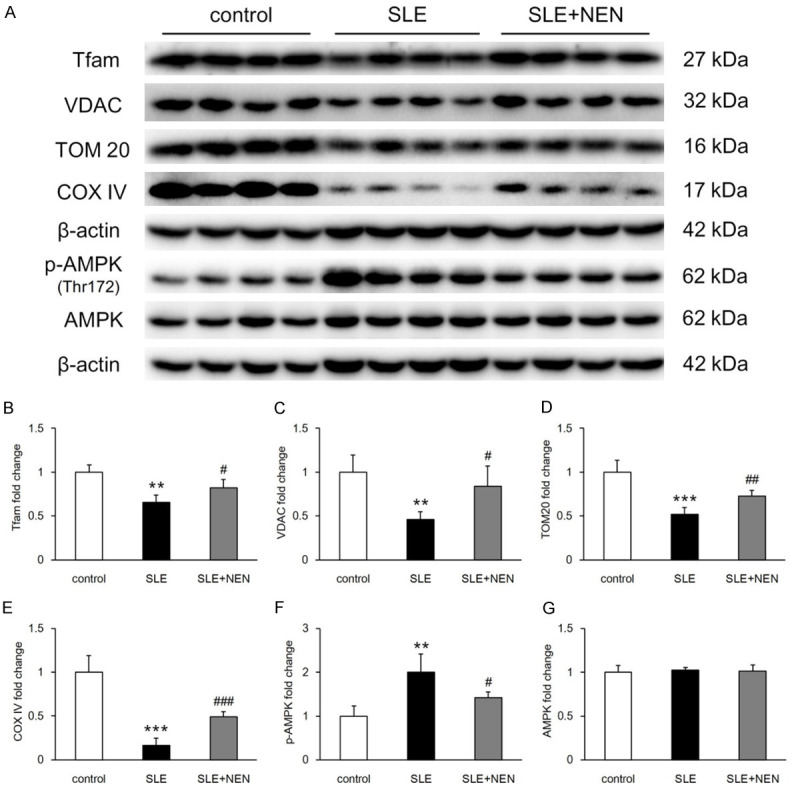
NEN promoted mitochondrial biogenesis and improved energy dysregulation in the kidney. A. Immunoblotting images of Tfam, VDAC, TOM20, COX IV, p-AMPK (Thr172), and AMPK in renal tissues of various groups. B-G. Bar graphs showing the quantitative analysis of these proteins. n = 4 per group. **P<0.01 and ***P<0.001 vs. control group. #P<0.05, ##P<0.01 and ###P<0.001 vs. SLE group.
NEN treatment prevented the enlargement of lymph nodes and the spleen, and decreased serum anti-dsDNA antibody levels
The lymph nodes and spleen are important organs in the regulation and initiation of immune responses. In the SLE group, abnormally enlarged spleens and lymph nodes were observed, which was notably prevented by NEN treatment (Figure 9A-D). Consistent with these changes in the spleen and lymph nodes, increased serum anti-dsDNA antibody levels in SLE group were significantly decreased by NEN (Figure 9E).
Figure 9.
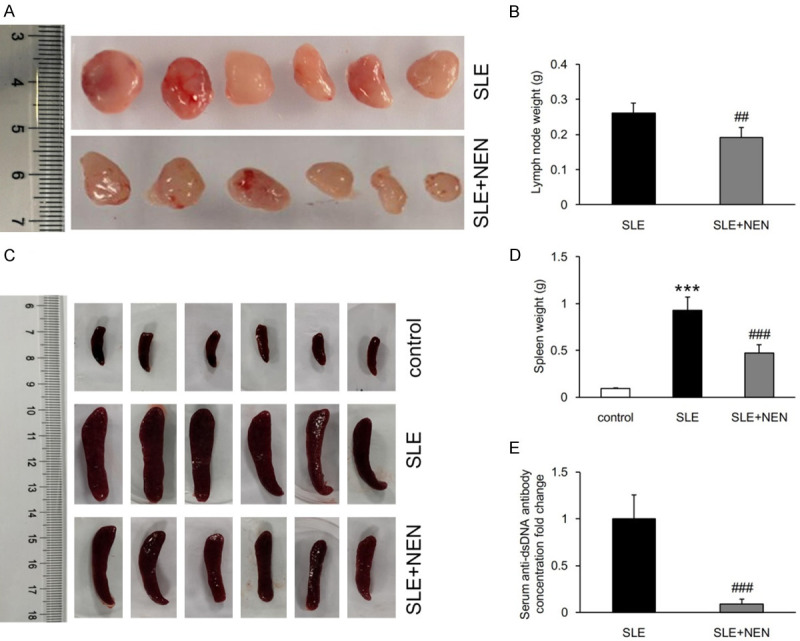
NEN prevented the enlargement of lymph nodes and spleen, and decreased serum anti-dsDNA antibody level. A, B. Representative images of lymph nodes from SLE and SLE+NEN group mice, and bar graphs showing the results of weight statistics. C, D. Images of the spleen and the weight statistics in various groups mice. E. Measurement of serum anti-dsDNA antibody levels in SLE and SLE+NEN group mice. n = 6 per group. ***P<0.001 vs. control group. ##P<0.01 and ###P<0.001 vs. SLE group.
Discussion
In this study, we report for the first time that NEN treatment is efficacious in attenuating SLE and LN. Potential mechanisms involved in its protective action include regulation of redox state, promotion of mitochondrial biogenesis, and modulation of the immune response.
Glomerular injury is the basis for pathologic classification in LN [24]. In the current study, SLE mice had increased total nuclei in their glomeruli. Considering the loss of podocytes observed, we hypothesized that this increase may be due to proliferation of mesangial or endothelial cells or infiltrating inflammatory cells. NEN treatment not only prevented an increase in cell nuclei counts in the glomeruli but also attenuated podocyte loss. Therefore, NEN may exert different effects on different cell types.
Although glomerular lesions are the focus of LN research, prominent tubular injury also impacts renal outcome [25]. In the normal kidney, renal tubular cells exhibit a characteristic phenotype and proliferate at a relatively slow rate [26]. In the stressed kidney of lupus mice, renal tubules undergo phenotypic alterations and cells are hyperproliferative. TGF-β1 is known to mediate tubular cell phenotypic alteration and proliferation [27]. Therefore, an upregulation in TGF-β1 mRNA level may explain the pathological changes in our model. We also explored cell signaling pathways that may participate in tubular cell growth and proliferation. The data demonstrated that multiple pathways are involved and the translation repressor protein, 4E-BP1, may be a potential therapeutic target of NEN.
In addition to glomerular and tubular injury, interstitial inflammation also plays an important role in LN progression. In SLE mice, inflammatory cell infiltration was observed in the renal interstitium. Their distribution was predominantly surrounding the blood vessels and the glomeruli, particularly at the vascular pole. Consistent with this increase in inflammatory cells, the mRNA levels of inflammation related cytokines including MCP-1, IL-1β, TNF-α, and IL-6, were upregulated. NEN treatment significantly downregulated the expression of MCP-1 and IL-1β, suggesting that NEN has inhibitory effects on inflammation. However, the mechanisms behind this require further studies.
Interstitial fibrosis is a common feature of various chronic kidney diseases. It is a sign of chronicity in LN progression and a predictor of poor prognosis. A previous study indicated that renal fibrosis is associated with defective fatty acid oxidation in renal tubular cells and that targeting the metabolic defect may prevent renal fibrosis [28]. It is well known that mitochondrial uncoupling increases the oxidation of fatty acids. So the preventing effects of NEN on interstitial fibrosis could be at least partly attributed to its uncoupling action on mitochondria in renal tubular cells.
Oxidative stress is implicated in LN progression. Consistent with previous studies [29], the kidneys in SLE mice had a redox imbalance, demonstrated by decreased catalase and SOD2 expression, and increased urinary H2O2 excretion. Mitochondrial uncouplers are able to dissipate protonmotive forces and greatly reduce ROS production [12]. Therefore, we hypothesized that the protective effect of NEN on redox imbalance might be associated with its uncoupling function and ability to upregulate catalase. In addition, Tfam loss, mitochondrial components deficiency, and energy homeostasis dysregulation also occurred in the kidneys of SLE mice. Mice with tubule-specific deletion of Tfam developed severe mitochondrial loss and energy deficit, progressing to renal fibrosis and failure [30]. NEN’s upregulation of Tfam may explain its positive role in mitochondrial biogenesis and regulation of energy metabolism. This also strongly suggests that mitochondrial integrity is crucial in maintaining function. However, the underlying molecular mechanisms remain unclear.
The spleen and lymph nodes are involved in B and T cells mediated immune responses. Splenomegaly and lymphadenopathy are the manifestations of immune disorders in SLE mice [31]. In this study, we found that NEN treatment notably reduced the enlarged spleen and lymph nodes. So we postulated that NEN may exert immunomodulatory effects on B cell activation. As expected, the increased serum anti-dsDNA antibody level in SLE mice was significantly reduced by NEN treatment. In line with the level of serum anti-dsDNA antibodies, glomerular immune complex deposits in SLE mice were also attenuated by NEN. Taking these results into account, we conclude that the renal improvement induced by NEN treatment is associated with its immunomodulatory effects.
In summary, we demonstrate here the efficacy of NEN treatment in SLE and LN. These findings suggest that this mild mitochondrial uncoupling agent has great potential for translational application as a novel therapy for autoimmune disease.
Acknowledgements
This study was supported by grants from National Natural Science Foundation of China (81673794), Shenzhen Science and Technology Project (JCYJ20190812183603627), and Sanming Project of Medicine in Shenzhen (SZSM201512040).
Disclosure of conflict of interest
None.
References
- 1.Durcan L, O’Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet. 2019;393:2332–2343. doi: 10.1016/S0140-6736(19)30237-5. [DOI] [PubMed] [Google Scholar]
- 2.Yu F, Haas M, Glassock R, Zhao MH. Redefining lupus nephritis: clinical implications of pathophysiologic subtypes. Nat Rev Nephrol. 2017;13:483–495. doi: 10.1038/nrneph.2017.85. [DOI] [PubMed] [Google Scholar]
- 3.Yung S, Chan TM. Mechanisms of kidney injury in lupus nephritis - the role of anti-dsDNA antibodies. Front Immunol. 2015;6:475. doi: 10.3389/fimmu.2015.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeshima Y, Iwasaki Y, Fujio K, Yamamoto K. Metabolism as a key regulator in the pathogenesis of systemic lupus erythematosus. Semin Arthritis Rheum. 2019;48:1142–1145. doi: 10.1016/j.semarthrit.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Morel L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol. 2017;13:280–290. doi: 10.1038/nrrheum.2017.43. [DOI] [PubMed] [Google Scholar]
- 6.Yang SK, Zhang HR, Shi SP, Zhu YQ, Song N, Dai Q, Zhang W, Gui M, Zhang H. The role of mitochondria in systemic lupus erythematosus: a glimpse of various pathogenetic mechanisms. Curr Med Chem. 2020;27:3346–3361. doi: 10.2174/0929867326666181126165139. [DOI] [PubMed] [Google Scholar]
- 7.Quiros PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17:213–226. doi: 10.1038/nrm.2016.23. [DOI] [PubMed] [Google Scholar]
- 8.Davidson A. What is damaging the kidney in lupus nephritis? Nat Rev Rheumatol. 2016;12:143–153. doi: 10.1038/nrrheum.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pravda J. Systemic lupus erythematosus: pathogenesis at the functional limit of redox homeostasis. Oxid Med Cell Longev. 2019;2019:1651724. doi: 10.1155/2019/1651724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee HT, Wu TH, Lin CS, Lee CS, Wei YH, Tsai CY, Chang DM. The pathogenesis of systemic lupus erythematosus - from the viewpoint of oxidative stress and mitochondrial dysfunction. Mitochondrion. 2016;30:1–7. doi: 10.1016/j.mito.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Chen W, Mook RA Jr, Premont RT, Wang J. Niclosamide: beyond an antihelminthic drug. Cell Signal. 2018;41:89–96. doi: 10.1016/j.cellsig.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berry BJ, Trewin AJ, Amitrano AM, Kim M, Wojtovich AP. Use the protonmotive force: mitochondrial uncoupling and reactive oxygen species. J Mol Biol. 2018;430:3873–3891. doi: 10.1016/j.jmb.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerqueira FM, Laurindo FR, Kowaltowski AJ. Mild mitochondrial uncoupling and calorie restriction increase fasting eNOS, akt and mitochondrial biogenesis. PLoS One. 2011;6:e18433. doi: 10.1371/journal.pone.0018433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han P, Shao M, Guo L, Wang W, Song G, Yu X, Zhang C, Ge N, Yi T, Li S, Du H, Sun H. Niclosamide ethanolamine improves diabetes and diabetic kidney disease in mice. Am J Transl Res. 2018;10:1071–1084. [PMC free article] [PubMed] [Google Scholar]
- 15.Han P, Zhan H, Shao M, Wang W, Song G, Yu X, Zhang C, Ge N, Yi T, Li S, Sun H. Niclosamide ethanolamine improves kidney injury in db/db mice. Diabetes Res Clin Pract. 2018;144:25–33. doi: 10.1016/j.diabres.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Han P, Yuan C, Wang Y, Wang M, Weng W, Zhan H, Yu X, Wang T, Li Y, Yi W, Shao M, Li S, Yi T, Sun H. Niclosamide ethanolamine protects kidney in adriamycin nephropathy by regulating mitochondrial redox balance. Am J Transl Res. 2019;11:855–864. [PMC free article] [PubMed] [Google Scholar]
- 17.Sun H, Wang W, Han P, Shao M, Song G, Du H, Yi T, Li S. Astragaloside IV ameliorates renal injury in db/db mice. Sci Rep. 2016;6:32545. doi: 10.1038/srep32545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, Liang Y, Yang N. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-kappaB activation. Int Immunopharmacol. 2013;17:116–122. doi: 10.1016/j.intimp.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 19.Yu F, Wu LH, Tan Y, Li LH, Wang CL, Wang WK, Qu Z, Chen MH, Gao JJ, Li ZY, Zheng X, Ao J, Zhu SN, Wang SX, Zhao MH, Zou WZ, Liu G. Tubulointerstitial lesions of patients with lupus nephritis classified by the 2003 international society of nephrology and renal pathology society system. Kidney Int. 2010;77:820–829. doi: 10.1038/ki.2010.13. [DOI] [PubMed] [Google Scholar]
- 20.Venkov CD, Link AJ, Jennings JL, Plieth D, Inoue T, Nagai K, Xu C, Dimitrova YN, Rauscher FJ, Neilson EG. A proximal activator of transcription in epithelial-mesenchymal transition. J Clin Invest. 2007;117:482–491. doi: 10.1172/JCI29544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McEwen AE, Maher MT, Mo R, Gottardi CJ. E-cadherin phosphorylation occurs during its biosynthesis to promote its cell surface stability and adhesion. Mol Biol Cell. 2014;25:2365–2374. doi: 10.1091/mbc.E14-01-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alsuwaida AO. Interstitial inflammation and long-term renal outcomes in lupus nephritis. Lupus. 2013;22:1446–1454. doi: 10.1177/0961203313507986. [DOI] [PubMed] [Google Scholar]
- 23.Picca A, Lezza AM. Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion. 2015;25:67–75. doi: 10.1016/j.mito.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Bajema IM, Wilhelmus S, Alpers CE, Bruijn JA, Colvin RB, Cook HT, D’Agati VD, Ferrario F, Haas M, Jennette JC, Joh K, Nast CC, Noel LH, Rijnink EC, Roberts ISD, Seshan SV, Sethi S, Fogo AB. Revision of the international society of nephrology/renal pathology society classification for lupus nephritis: clarification of definitions, and modified national institutes of health activity and chronicity indices. Kidney Int. 2018;93:789–796. doi: 10.1016/j.kint.2017.11.023. [DOI] [PubMed] [Google Scholar]
- 25.Leatherwood C, Speyer CB, Feldman CH, D’Silva K, Gomez-Puerta JA, Hoover PJ, Waikar SS, McMahon GM, Rennke HG, Costenbader KH. Clinical characteristics and renal prognosis associated with interstitial fibrosis and tubular atrophy (IFTA) and vascular injury in lupus nephritis biopsies. Semin Arthritis Rheum. 2019;49:396–404. doi: 10.1016/j.semarthrit.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomasova D, Anders HJ. Cell cycle control in the kidney. Nephrol Dial Transplant. 2015;30:1622–1630. doi: 10.1093/ndt/gfu395. [DOI] [PubMed] [Google Scholar]
- 27.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 28.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bona N, Pezzarini E, Balbi B, Daniele SM, Rossi MF, Monje AL, Basiglio CL, Pelusa HF, Arriaga SMM. Oxidative stress, inflammation and disease activity biomarkers in lupus nephropathy. Lupus. 2020;29:311–323. doi: 10.1177/0961203320904784. [DOI] [PubMed] [Google Scholar]
- 30.Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, Palmer M, Susztak K. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 2019;30:784–799. e785. doi: 10.1016/j.cmet.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akizuki S, Ishigaki K, Kochi Y, Law SM, Matsuo K, Ohmura K, Suzuki A, Nakayama M, Iizuka Y, Koseki H, Ohara O, Hirata J, Kamatani Y, Matsuda F, Sumida T, Yamamoto K, Okada Y, Mimori T, Terao C. PLD4 is a genetic determinant to systemic lupus erythematosus and involved in murine autoimmune phenotypes. Ann Rheum Dis. 2019;78:509–518. doi: 10.1136/annrheumdis-2018-214116. [DOI] [PubMed] [Google Scholar]