

Abstract
Pathological remodeling of the right ventricular (RV) contributes to the mortality of pulmonary arterial hypertension (PAH) patients, and RV myocardial apoptosis and metabolism play decisive roles in RV remodeling. Qiliqiangxin (QLQX), a traditional Chinese medicine, has a cardio-protective effective on left ventricular remodeling. However, whether QLQX can decrease RV myocardial apoptosis, improve metabolism, and attenuate RV remodeling remain uncertain. This study investigated the effects of QLQX on RV remodeling, myocardial mitochondria, apoptosis, and metabolism reprogramming. RV remodeling was induced by intraperitoneal injection of Monocrotaline (MCT). We first discovered that QLQX improved hemodynamic parameters and inhibited MCT-induced RVH. Next, QLQX significantly attenuated RV remodeling which covered RV myocardial fibrosis, and RV capillary density. Furthermore, we uncovered that QLQX attenuated RV myocardial apoptosis. We also confirmed that QLQX reversed metabolic shift toward glycolysis which decreased the uptake of glucose showed by fluorodeoxyglucose F 18 positron emission tomography (18FDG-PET). Mechanistically, QLQX optimized mitochondrial function by ameliorating structural abnormality of mitochondria, reducing the release of cytochrome c from mitochondria, and upregulating the expression of SOD2. Mitochondria-dependent apoptosis and mitochondria-associated metabolism were involved in QLQX regulation of RV. Moreover, our study showed that PINK1/Parkin 2 pathway was involved in improving mitochondrial function. We concluded that QLQX could inhibit PAH-induced RV remodeling by decreasing mitochondria associated apoptotic pathway and reversing mitochondrial related metabolic shift. The PINK1/Parkin mitophagy pathway may play a key role in mitochondria protection.
Keywords: Right ventricular remodeling, pulmonary arterial hypertension, mitochondria function, apoptosis, metabolic shift, mitophagy
Introduction
Pulmonary arterial hypertension (PAH) is a fatal disorder of cardiovascular system with a prevalence of 15 per million people and the median survival is only 5 to 7 years once diagnosed [1-3]. PAH is a syndrome characterized by a progressive increase in pulmonary vascular resistance (PVR) and imposes a hemodynamic stress on the right ventricle (RV), which leads to RV overload and eventually give rise to RV pathological remodeling and RV failure (RVF). Available medical therapies are limited to reducing mean pulmonary arterial pressure (mPAP) and improving the quality of life in patients with PAH [4]. Nevertheless, these methods just scratches the surface against the progression of PAH [5]. More and more experiments have proved RVF as the decisive factor in the course of PAH independent of PVR. The animal experiment revealed that RV remodeling brought about the RVF [6]. Other experimental evidence has demonstrated that therapeutic intervention targeting the RV may also be worthwhile [7]. Therefore, reversing RV remodeling may be a new target for PAH treatment.
Recently, more studies have confirmed that apoptosis attributes to RV remodeling and eventually leads to RVF. Mitochondria have been shown to amplify apoptosis by releasing pro-apoptotic factors, such as cytochrome c into the cytoplasm [8]. Therefore, it is possible to regulate apoptosis by improving mitochondrial function. Moreover, mitochondrial function determines cardiomyocytes glucose metabolism [9,10]. Observational studies have confirmed that the capacity of glucose uptake in PAH patients increases [11,12] and in order to accommodate the increasing PVR, the RV metabolically shifts from glucose oxidation to anaerobic glycolysis [13,14]. It has been proven that enhancing glucose oxidation can restore RV function and improve RVH. This suggests that mitochondria therapy may offer a new approach to control RV myocardial apoptosis, improve glucose metabolism, and thus combat RV remodeling.
Previous studies have confirmed that selective mitochondria autophagy (mitophagy) can eliminate damaged mitochondria and prevent mitochondria pathological accumulation, which are associated with cardiac apoptosis and dysfunction [15,16]. Studies conducted by scholars from China, Japan, and the US conclude that autophagy and mitophagy could lower the chances of diabetic cardiomyopathy [17-19]. Additionally, some literature report that appropriate mitophagy level promotes cardiac metabolic reprogramming and metabolic shift toward aerobic oxidation [20,21].
Fuller understanding of the therapeutic power of Qiliqiangxin (QLQX) also requires thinking in traditional Chinese medicine doctrine. The drug can boost the “Qi”, the energy flow within a living entity, and warm the “Yang”, the positive side of dualistic monism. The latter helps remove meridian blockage during the development of heart failure caused by coronary heart disease and hypertension by promoting blood circulation [22]. Recently, it has been reported that QLQX combined with targeted drugs can not only reduce pulmonary artery pressure, improve clinical symptoms, but also protect vascular endothelial cell function. This combination, thereby, may improve cardiopulmonary function and increase treatment tolerance of chronic thromboembolic pulmonary hypertension. Researchers from our laboratory and others have reported that QLQX can attenuate left ventricular and pulmonary vascular remodeling in rats under post-MI congestive heart failure [23]. Previous studies have documented that QLQX could improve mitochondrial function, inhibit mitochondria-dependent apoptotic pathway in pressure overload and spontaneously hypertensive heart failure rats [22]. However, whether QLQX directly addresses RV remodeling remains unconfirmed. Here we hypothesize that QLQX can ameliorate the RV remodeling secondary to PAH by controlling mitochondria-dependent apoptosis and mitochondria-associated metabolism reprogramming. This study intends to bring our understanding of QLQX’s effectiveness against MCT-induced RVF to a deeper level. We aim to achieve it by (1) evaluating the effect of QLQX on RV remodeling including apoptosis, fibrosis and capillary density. (2) determining myocardial glucose uptake by 18F-FDG using small-animal PET imaging. (3) estimating the impact of QLQX on mitochondria structure and function. (4) calculating QLQX’s impact on the expression of mitophagy-related proteins, including PINK1, Parkin, and LC3B.
Materials and methods
QLQX capsule ingredient
The ingredients of QLQX capsule include astragalus membranaceus, ginseng, aconite, salvia, zongzi, diarrhea, jade bamboo, cinnamon stick, safflower, fragrant rind, tangerine peel. To prepare for our study, we dissolved the capsule in sterile water at 0.1 g/concentration.
Monocrotaline rat model of RV remodeling
60 adult male Sprague-Dawley rats (body weight 220~250 g) were purchased from Beijing Vital River Laboratory Animal Technology Co. Ltd (Animal license number: SCXK (Beijing) 2017-0302). Animals were divided into three group: (1) Sham group (n=20): receiving intraperitoneal injection of saline for 6 weeks. (2) MCT group (n=20): receiving intraperitoneal monocrotaline (60 mg kg-1d-1; Sigma-Aldrich, St Louis, MO, USA) injection to induced RVF for week 1-4 and saline injection for week 5-6. (3) QLQX group (n=20): receiving MCT injection for week 1-4, then QLQX (0.1 g kg-1d-1) injection for week 5-6.
Hemodynamic evaluation
Intraperitoneal sodium pentobarbital injection was (40 mg kg-1) used for anesthetization in Sham, MCT and QLQX groups after 6 weeks of injection. Polygraph system (Power Lab 8/30; AD Instruments, Bella Vista, NSW, Australia) was used to measure the hemodynamic parameters including mPAP and RVSP. A polyethylene-50 catheter was inserted via the right external jugular vein into the pulmonary artery after tracheotomy, mPAP and RVSP were measured.
Assessment of RV hypertrophy index
Upon the completion of hemodynamic parameters measurement, we excised the hearts, separated them into RV walls, LV walls, and ventricular septum, and weighed all parts by wet weight. Lastly, we calculated the ratio of RV weight to LV plus septum weight (RV (LV+Sep)) for the RV hypertrophy index assessment.
Histopathology, immunohistochemistry, immunofluorescence, tunnel
During our experiment the heart tissues were fixed in 10% neutral formalin, embedded in paraffined, and sectioned by 4 μm for hematoxylin and eosin (HE) and Masson staining. Image pro plus 6.0 was used to quantify the ratio of fibrosis area to the entire surface. The capillary density and mitophagy-associated proteins were both determined by immunofluorescence staining. The paraffin-embedded RV sections (4 μm) were deparaffinized for citrate-EDTA antigen retrieval, and incubated with different primary antibodies: CD31 (1:500 dilution, ab24590, abcam), PGC-α (1:300 dilution, ab54481, abcam), LC3B (1:200 dilution, 3868T, cell signaling technology), PINK1 (1:200 dilution, ab23707, abcam), Parkin 2 (1:500 dilution, 2132S, cell signaling technology) and p62 (1:200 dilution, ab91526, abcam). Having been incubated overnight at 4°C, the RV sections were then washed with PBS and added with fluorescent secondary antibody. The images were generated by laser scanning confocal microscope (Olympus, FV1000) and analyzed by Image Pro Plus 6.0. For immunohistochemical staining, hydrogen peroxide (H2O2, 3%) was used for blockage. Then caspase 3 (1:500 dilution, BM3957, Bioss), Bax (1:500 dilution, BM3964, Bioss), Bcl-2 (1:500 dilution, BA0412, Bioss) antibodies were added to RV sections for a 60-minute incubation at 37°C. Finally, the RV sections were washed again with PBS for DAB colorimetry. Apoptosis was analyzed by the terminal-deoxynucleotidyl transferase-mediated 2’-deoxyuridine 5’-triphosphated nick-end-labeling (TUNEL) method (Roche, 11684795910).
Transmission electron microscopy
Fresh RV tissues were collected and cut into 1 mm3, prefixed in 2% glutaraldehyde for 2 h and then fixed in 1% osmium tetroxide, step by step dehydration with ethanol and then acetone. Later, samples were embedded in the epoxy resin at 37°C overnight, polymerized by propylene oxide at 60°C for 48 h. Lastly, samples were sliced into 60 nm sections, stained by lead citrate and uranium acetate, and observed with transmission electron microscope (JEOL, JEM-1400).
Western blot
RV samples were cut into pieces, underwent protein extraction by RIPA (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.1% SDS), quantified by BCA Protein Quantification Kit (Thermo Fisher, NCI3227CH). GAPDH was used as internal reference. SDS-PAGE separated the protein and electro transfer method was used. PVDF membrane (Millipore, 0.45 μm) were probed by mouse monoclonal SOD2 (1:2000 dilution, abcam), rabbit polyclonal Cyto-c (1:1000 dilution, Cell Signaling Technology) overnight. TBST buffer was washed for three times, 10 min each. Goat anti Mouse IgG as secondary antibodies (1:10000 dilution) was used to incubate the PVDF membrane at RT for 40 min and enhanced by chemiluminescence reagent (Millipore, WBKLS0500). Band intensities were determined by using Gel Image system ver.4.00 (Tanon, China). All measurements were repeated for at least three times. Measurements of cytosolic cytochrome c were achieved by subcellular fractionation and western blotting with cytochrome c antibody (1:1000 dilution, Cell Signaling Technology) as described previously.
18FDG-PET scanning and data analysis
Fasting 12 h rats were injected with 0.2 mL 18F-FDG (37 MBq; Beijing Atomic High-Tech Co., Ltd.) through the tail vein and then placed in a pre-numb box 30 minutes after 4%, 2 L/min isoflurane injection. An alkane flow was used to induce anesthesia. The anesthetized rats were fixed in a prone position on a scanning bed for MicroPET/CT (Siemens, Inveon) imaging, and anesthesia was maintained by 1.5 L/min 3% isoflurane flow. CT acquired 30 min firstly, PET acquired 10 min immediately thereafter. Siemens Inveon Research Workplace was used to register CT and PET images, and then areas of interest on the heart was marked to calculate standardized uptake values (SUV).
Statistics
All procedures were performed in triplicate. All data was presented as the mean ± standard deviation (SD). An independent-samples t-test, one-way analysis of variance (ANOVA) was conducted to evaluate the one-way layout data. P-values less than 0.05 were considered as significant. All analyses were performed using GraphPad Prism6.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
QLQX improved hemodynamic parameters and inhibited MCT-induced RVH
Pulmonary arterial hypertension (PAH) is defined as the mPAP greater than 25 mmHg at rest or greater than 30 mmHg during exercise. In this study, we used MCT to induce RV remodeling as previous studies have done [24-26]. We detected mPAP and RVSP variation in hemodynamic parameters in the MCT-induced model. In MCT injected rats, mPAP gradually increased from 14.36±3.58 mmHg at baseline to 31.95±3.27 mmHg, and RVSP increased from 21.78±1.97 mmHg to 51.75±2.52 mmHg after 4 weeks (Figure 1A-C). According to the PAH definition, we have successfully made the PAH model. In this study, QLQX treatment significantly lowered mPAP and RVSP by 43% and 26.5% (Figure 1B, 1C). High RV pressure secondary to PAH, which manifested by the elevating RV/[LV+S] ratios, can continuously induce RVH. Our study indicated that there was significantly less hypertrophy in QLQX group compared with MCT group (Figure 1D and 1E).
Figure 1.
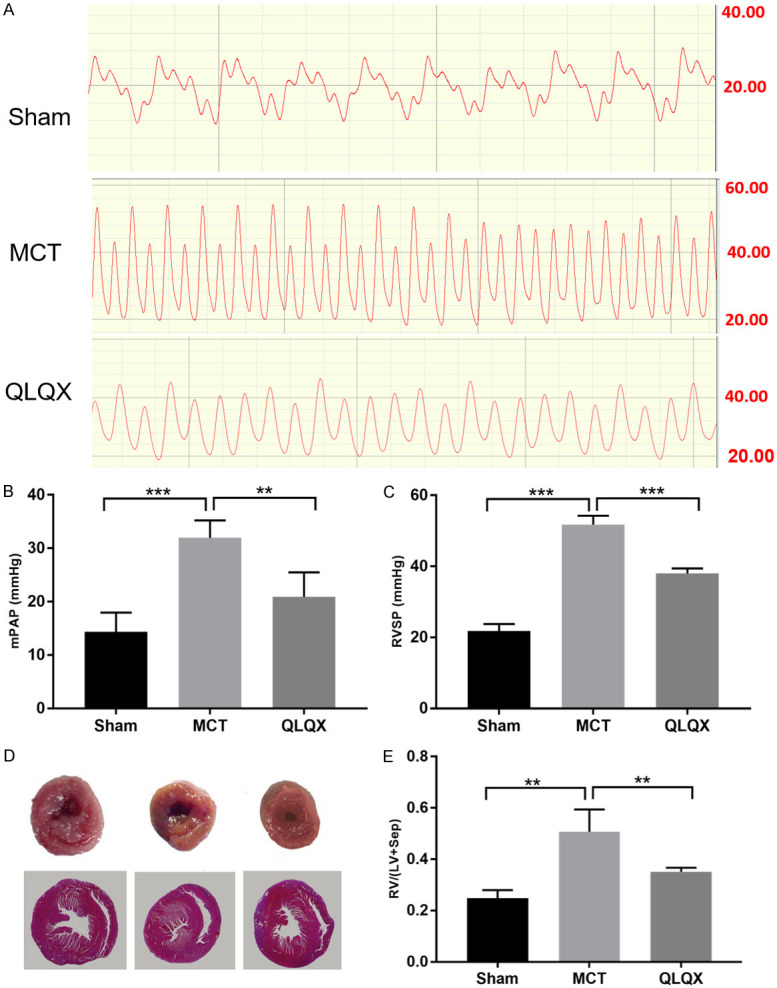
QLQX improved hemodynamic parameters and inhibited MCT-induced RVH. A. Representative images of the mean pulmonary arterial pressure (mPAP) in three groups. B. Qualification of the mPAP in three groups. The variation of mPAP was significantly increased in MCT group compared to Sham and QLQX groups. C. Right ventricular systolic pressure (RVSP). QLQX treatment remarkably decreased RVSP compared with MCT group. D. Representative images of the RV/[LV+Sep]. The ratio of RV weight to LV plus septum weight significantly increased in MCT group compared with QLQX. E. The ratio of free wall of RV weight to LV+Sep weight. The RV/[LV+Sep] ratio was significantly decreased in QLQX group compared to MCT. Sham, single intraperitoneal injection of saline group. MCT, monocrotaline induced SD rats right ventricular remodeling model group (60 mg kg-1); QLQX, Qiliqiangxin intraperitoneal injection (100 mg kg-1d-1) after the 4 weeks MCT induced RVF rats; RV, right ventricular; LV, left ventricular; Sep, septum. Data is presented as mean ± SD. ***P<0.0001; **P<0.001.
QLQX reversed RV remodeling in RVF
RV remodeling includes not only changes in cardiomyocyte morphology but also interstitial fibrosis and capillary density. In adult PAH cases, higher degree of fibrosis leads to worse prognosis [27]. As shown in the H&E staining image, the QLQX group demonstrated significantly improved RV myocardial shape, arrangement and nuclear morphology in RV wall compared to which from the MCT group rats (Figure 2A). Similarly, the pulmonary vessels under the effect of QLQX had larger vessel diameter and thinner vessel walls compared with the MCT group (Figure 2B). Meanwhile, the MCT group had significantly higher degree of fibrosis than which of the Sham, while QLQX treatment reversed such trend (Figure 2C and 2E). Finally, different from the decreasing micro vessel density in MCT group stained by CD31, QLQX treatment partly restored the density of RV capillaries (Figure 2D and 2F). All these results strongly suggested that QLQX could reverse RV remodeling in RVF.
Figure 2.
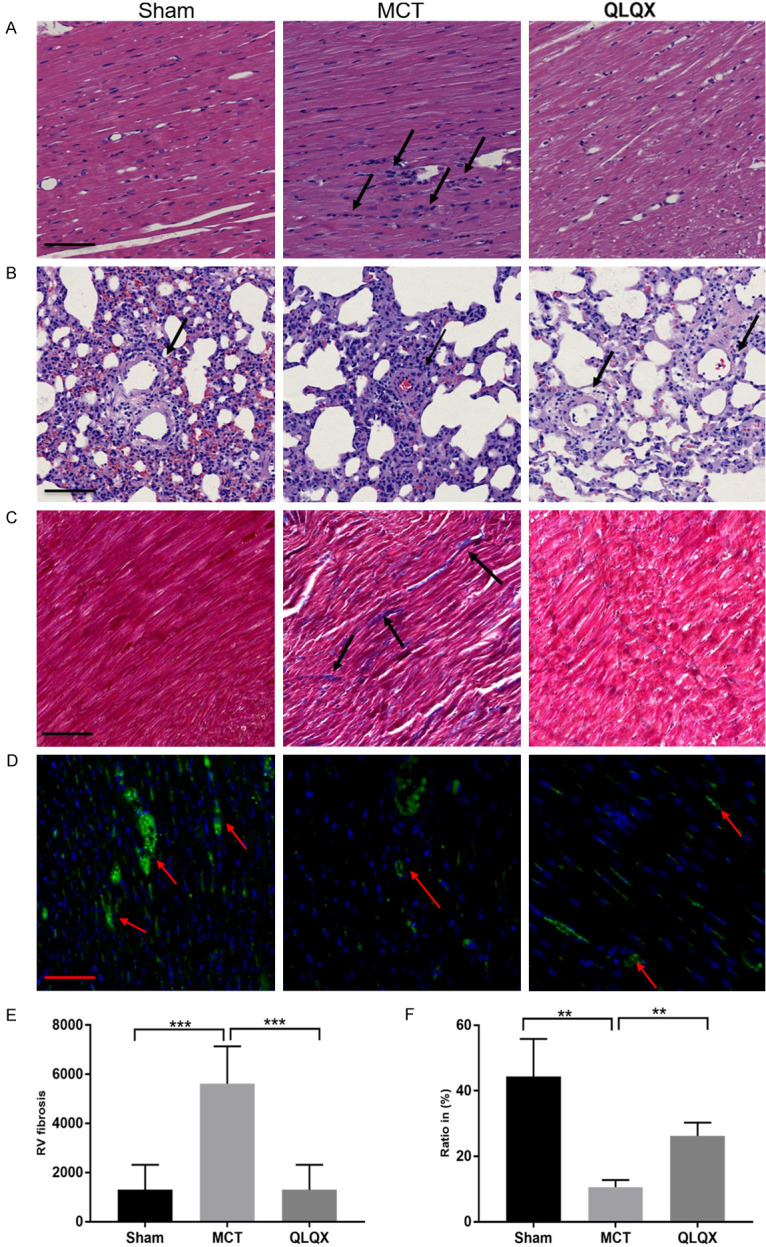
QLQX reversed RV remodeling in RVF. A. Hematoxylin and eosin staining of the fixed RV sections. QLQX treatment significantly improved RV myocardial shape, arrangement and nuclear morphometry in RV wall (black arrow). Scale bar, 50 μm. B. Hematoxylin and eosin staining of the fixed lung sections. The pulmonary vessels of QLQX group had larger blood diameter and thinner vessel wall compared with the MCT group (black arrow). Scale bar, 50 μm. C. Fibrotic areas were shown by Masson trichrome stain (black arrow). Scale bar, 50 μm. D. RV cryosections stained with CD31 (blood vessels stained by green, red arrow) and nuclei (4’, 6-diamidino-2-phenylindole; DAPI, blue). Scale bar, 50 μm. E. Fibrosis quantification (blue-stained areas) of RV cardiomyocytes. The MCT group had the significantly fibrosis degree than the Sham, while QLQX treatment diminished the extent of fibrosis. F. Capillaries to area (μm2) ratio. QLQX treatment partly restored the density of RV microvessel density compared with the MCT group. Original magnification, 100×; Scale bar, 50 μm. Data is presented as mean ± SD. ***P<0.0001; **P<0.001.
QLQX inhibited RV myocardial apoptosis
It has become clear that RV disease progression is associated with an early increase in RV apoptosis [28] which plays an important role in RVF. We then evaluated the apoptosis of RV cardiomyocytes by TUNEL staining. The MCT group had markedly stronger apoptotic signal than which of the Sham, while such signal was largely weakened by QLQX treatment (Figure 3A and 3C). Existing studies provided the view that mitochondria participated in the progress of cell apoptosis by releasing pro-apoptotic factors, such as the Bcl-2 family. We explored the effect of QLQX on Bcl-2 and Bax expression by immunohistochemical staining. Consistent with the TUNEL results, the MCT group had higher levels of cleaved caspase-3 and Bax, and lower expression of anti-apoptotic protein Bcl-2 than that of Sham and QLQX groups. Comparatively, QLQX treatment effectively reversed those results (Figure 3B, 3D-F).
Figure 3.

QLQX inhibited RV myocardial apoptosis and reversed metabolic shift. A. Representative images of TUNEL staining in three groups. TUNEL positive stains in bright green. Sham, red arrow; MCT, red oval; QLQX, red arrow. Nuclei stained blue with DAPI. Scale bar, 50 μm. B. Representative images of caspase 3, Bax and Bcl-2 staining in RV myocytes by immunohistochemical staining (red arrow). Scale bar, 50 μm. C. Quantitative changes of the incidence of myocardial apoptosis in RV among various treatment groups. RV myocytes after monocrotaline induced RVF increased apoptosis compared to Sham, while QLQX treatment group significantly decreased compared to MCT group. The percentages of TUNEL positive cells were calculated in 10 randomly chosen fields of each section at ×400 magnification. D-F. Quantification of caspase 3, Bax and Bcl-2 staining in RV myocytes. MCT group had a higher level of cleaved caspase-3, Bax and lower expression of anti-apoptotic protein Bcl-2 than that of control and QLQX groups, while QLQX reversed. G. The representative FDG-PET image was shown in Sham, MCT and QLQX group. FDG-PET detected and quantified the metabolic pattern reversing. Glucose uptake in RV (red arrows) was elevated in MCT group (red zone), while this accumulation was ameliorated after the QLQX treatment. Original magnification, 100×; Scale bar, 50 μm. Data is presented as mean ± SD. ***P<0.0001; **P<0.001; *P<0.05.
QLQX reversed metabolic shift toward glycolysis
Similar to LVF and being part of RV remodeling, metabolic remodeling also embodies the characteristics of RVH. This kind of remodeling can be identified and quantified in vivo by 18FDG-PET. Our study, likewise, used 18FDG-PET to evaluate the glucose metabolism. Glucose uptake was elevated in RV in MCT group, but this accumulation declined after the QLQX treatment (Figure 3G). The level of the PPAR-γ coactivator-1α (PGC-α), the master regulator of mitochondrial biogenesis and oxidative metabolism reduced in MCT group. The opposite was observed in QLQX group (Figure 4F, 4G).
Figure 4.
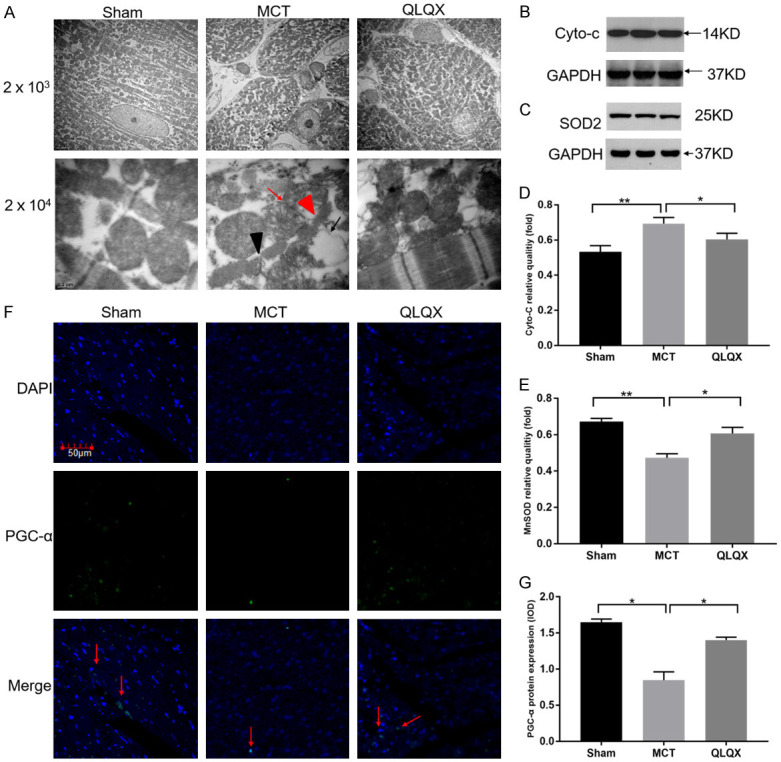
QLQX improved RV myocardial mitochondrial function in RVF. A. Representative TEM images of mitochondrial ultrastructure in RV myocytes. QLQX partly reversed the abnormal mitochondria ultrastructure in MCT-induced right ventricular failure group including mitochondrial swelling (red arrow), loss of mitochondrial cristae occurred with vacuoles formation in the matrix (black arrow), dissolution of the myofilaments (red triangle) and broken Z-lines (black triangle) in RV cardiomyocytes by transmission electron microscopy than in the Sham, which were significantly decreased in QLQX group. Scale bar, 2 μm (original magnification, 2000×); scale bar, 0.2 μm (original magnification, 20000×). B and C. The tissue lysates from the RV were separated on SDS-PAGE, and then immunoblotted with different specific antibodies. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in the immunoblot was shown as a loading control. B. Representative immunoblotting images of cytochrome C (Cyto-c) expression in three groups. C. Representative immunoblotting images of SOD2 expression in three groups. D. Quantification analysis of the Cyto-c expression level. Western blots showed that the increased Cyto-c expression in MCT can be significantly suppressed by QLQX. E. Western blots showed that the decreased MnSOD expression in MCT was significantly increased in QLQX group. F. Representative immunofluorescence staining images of PGC-α (PGC-α, green; DAPI, blue) in three groups (red arrow). Original magnification, 100×; scale bar, 50 μm. G. Quantification analysis of PGC-α expression in RV. Decreased PGC-α was observed in MCT compared to Sham and QLQX treatment by immunofluorescent staining. Integral optical density (IOD) are shown for five fields each rat for three rats. Data is presented as mean ± SD. **P<0.001; *P<0.05.
QLQX improved RV myocardial mitochondrial function in RVF
To explore the mechanism of ventricular hypertrophy and RV remodeling, we studied mitochondria, a function of cardiomyocytes substructure. The MCT group displayed more abnormalities than the Sham in terms of mitochondrial swelling, myofilaments dissolution, broken Z-lines in RV cardiomyocytes by transmission electron microscopy (TEM), and concurrent mitochondrial cristae loss and vacuoles formation in the matrix. Such abnormalities significantly lessened in QLQX group (Figure 4A). In order to investigate the effect of QLQX on mitochondrial function, we detected the cytochrome c (Cyto-c) from mitochondria and SOD2 expression by Western blot. Analysis showed that compared with the Sham levels, Cyto-c increased and SOD2 decreased in the MCT group, but after QLQX treatment, the expressions both reversed (Figures 4B-E, S2).
QLQX upregulated the mitophagy level in MCT-RVF rats
To explore the cause of mitochondrial dysfunction, we studied the mitophagy ability in the Sham, MCT and QLQX groups. Mitophagy can clear away the dysfunction and subsequent degradation of mitochondrial [30]. In our study, we observed mitochondrial autophagosome and mitophagy-associated protein via TME and immunofluorescence. In the context of RVF in QLQX treatment compared with MCT, some mitochondria or fragmented pieces were contained in lysosome, which suggests for mitophagy (Figure 5A). Additionally, our study observed attenuated LC3B expression in RV cardiomyocytes in MCT rats and enhanced LC3B expression in QLQX-treated rats (Figure 5B and 5F). To further clarified the molecular mechanism of mitophagy in RVF, we analyzed the mitophagy pathway protein, including tensin homolog (PTEN)-induced kinase 1 (PINK1), the mitochondrial phosphatase, cytosolic E3 ubiquitin ligase Parkin 2, Mitofusin 2 (MFN2), as well as p62, all of which have been implicated in PINK1/Parkin 2 mitophagy mitophagy. Immunohistochemistry and Immunofluorescence stain showed that the expressions of PINK1, Parkin 2, MFN2, p62 in RV myocardial from the MCT group were significantly lower than which from the Sham, while such condition recovered toward normal levels under QLQX treatment (Figures 5C-E, 5G-I, S1).
Figure 5.

QLQX upregulated the itophagy level in MCT-RVF rats. A. Representative TEM images of mitochondrial morphology in RV myocytes (red arrow). QLQX and Sham groups have more mitophagy represented by mitochondria or fragmented mitochondria containing in lysosome compared with MCT. Original magnification, 4000×; scale bar, 0.2 μm. B-E. The RV tissue were stained by immunofluorescence with different specific antibodies. B. Representative immunofluorescence staining images of RV tissue with LC3B (yellow arrow). C. Representative immunofluorescence staining images of RV tissue with PINK1 (white triangle). D. Representative immunofluorescent staining images of RV tissue with Parkin 2 (red triangle). E. Representative immunofluorescent staining images of RV tissue with p62 (white arrow). Original magnification, 100×; scale bar, 50 μm. F-I. Quantification analysis of LC3B, PINK1, Parkin 2 and p62 expression in RV. LC3B, PINK1, Parkin 2 and p62 expression levels were significantly decreased in MCT compared to Sham and QLQX treatment group by immunofluorescence staining. Integral optical density (IOD) are shown for five fields each rat for three rats. Data is presented as mean ± SD. ***P<0.0001; **P<0.001; *P<0.05.
Discussion
In our study, we discovered that QLQX played a constructive role in ameliorating RV remodeling by improving hemodynamics, cardiomyocyte morphology, capillary density, and RV interstitial fibrosis. Moreover, this study provides new mitochondria-oriented approach for treating MCT-induced RV remodeling via improving mitochondrial-dependent apoptosis and metabolic remodeling. Mitophagy causes mitochondrial dysfunction, which leads to apoptosis and metabolic remodeling. As for the mechanism behind, PINK1/Parkin 2 mitophagy may cause mitochondrial dysfunction which was associated with apoptosis and metabolic remodeling.
To a certain extent, metabolism derangement and disorders of mitochondrial function result in RVF. Several studies claim that right ventricular cardiomyocytes and vascular cells in PAH display a cancer-like metabolic shift, which can be identified by accelerated glycolysis and shrunken glucose oxidation. These appear to be good signs for cardiomyocytes, but in long-term dependence on anaerobic digestion for ATP generation could trigger RVF. Therapeutic targeting of metabolic abnormalities in PAH can restore oxidative phosphorylation and pulmonary vascular structure, and ease hypertrophy along with improving the contractility, hemodynamics, and cardiac output of the right ventricle, as many studies have demonstrated. Two clinical surveys report that rise in glucose uptake has to do with increased glycolysis in PAH patients [31-33]. Consistent with these findings, our study showed that the QLQX treatment of PAH effectively shifted metabolism towards aerobic respiration, which is indicated by glucose uptake, PGC-α decrease (Figures 3G, 4F, 4G). Moreover, it has been recognized that mitochondria metabolically adapt to the changing environment, and proper mitochondrial function is necessary supply stable energy flow to the heart. Studies suggest that metabolic deregulation is a sign of mitochondrial abnormalities, diminished ATP production and energy supply [20]. According to this relationship between metabolism and mitochondria, we further measured the effect of QLQX on mitochondrial structure by TEM, and its effect on mitochondrial function by assessing the mitochondrial biogenesis and the expression of Cyto-c and SOD2. PGC-α, the co-activators of the peroxisome proliferator activated receptors has a complex regulation of mitochondria biogenesis along with oxidative metabolism [29]. Pronounced PGC-α expressions represent greater mitochondrial mass and proliferation. Our study identifies that QLQX alleviates metabolism remodeling by raising PGC-α levels, thus optimizes mitochondrial structure and function (Figure 4F and 4G). Consistent with the effect of QLQX on metabolism, the abnormalities in mitochondrial ultrastructure and mitochondrial function were partly reversed by the drug (Figure 4A and 4B). In sum, on the one hand, mitochondrial dysfunction was the possible reason of the metabolism shift from aerobic oxidation to glycolysis. On the other hand, previous studies have revealed that functional abnormalities of cardiac mitochondria, triggered by intracellular stress such as oxidative stress, could accelerate apoptosis and mitochondria-dependent apoptosis pathway [8]. Under oxidative stress, the pro-apoptotic protein first translocates from the cytoplasm to the mitochondrial membrane, and forms a gap to facilitate the cytochrome c release from the mitochondrial membrane to the cytoplasm. Then, the pro-caspase 9 and caspase 3 were activated to catalyze downstream substrate. Finally, the preceding cascade amplification reaction leads to cell apoptosis. Bogaard et al. stated that RV failure was associated with myocardial apoptosis [34]. In human beings, apoptosis rates can increase from 1 in 104 in a normal heart to 1 in 400 in a heart failure [35,36]. Similarity, in the rat experiments, increasing myocardial apoptosis led to deteriorated cardiac function, therefore taking part in the pathological processes of heart failure [37]. In pulmonary artery banding and monocrotaline-induced right ventricular failure models, the myocardial apoptosis ratio upregulates [35]. Therefore, we closely monitored the RV myocardial apoptosis rates during this QLQX-targeted study. TUNEL staining indicates less cardiomyocyte apoptosis in QLQX group than in the MCT (Figure 3A). Consistent with existing studies, this result supports our hypothesis that the PAH involves apoptosis, and attenuated myocardial apoptosis reverses RV remodeling. Furthermore, considering the relationship between the mitochondria and apoptosis that we have mentioned above, we analyzed the mitochondrial apoptosis-related protein including Bax and Bcl-2. Both belong to the Bcl-2 gene family and are the regulator of cell apoptosis. The Bcl-2/Bax ratio determines the percentage of positive apoptotic cells [38,39]. The ratio in our study significantly dropped in RV cardiomyocytes in the MCT group compared with the Sham, but effectively recovered in QLQX group (Figure 3). That is to say, QLQX improved MCT-induced right ventricular remodeling via mitochondrial-dependent apoptosis pathway. Also considering the role of mitochondria in RV cardiomyocytes metabolism, QLQX may be a novel therapeutic pharmaceutical choice. It could alleviate RV remodeling in a mitochondrial approach, specifically, by inhibiting apoptosis and improving metabolic reprogramming with pulmonary arterial hypertension.
Furthermore, in order to understand the mechanism behind QLQX treatment, we focused on mitophagy in RV cardiomyocytes. On pathological conditions such as long-term excessive ischemia-hypoxia stimulation and pulmonary artery hypertension, damaged mitochondria accumulate in cardiomyocytes, which could lead to exacerbated apoptosis. Losing mitophagy control means that defective mitochondria will no longer be cleared away promptly. Therefore, mitophagy may be the possible protective mechanism underlying the effect of QLQX on RV apoptosis and cardiac metabolic shift. Under low cardiac stress, dysfunctional mitochondria can be efficiently removed by mitophagy. The mechanism is to be believed that oxidative stress products such as ROS can activate the PINK1/Parkin-development mitophagy pathway in degenerative and ischemic diseases [40,41]. Tensin homolog (PTEN)-induced kinase 1 (PINK1) phosphorylates Mitofusin 2 (MFN2), which in turn interacts with Parkin. Activated Parkin polyubiquitinates major autophagy adaptor proteins p62, mediates interaction with the microtubule-associated protein 1 light chain 3 (LC3B) and then swallows mitochondria and formats the autophagosome. From the ultrastructure of myocardial tissue, we observed that QLQX group had more autophagosomes around the mitochondria as same as the Sham group, while the MCT group had less autophagosomes (Figure 5A). Moreover, we observed amplified expressions of PINK1, Parkin 2, MFN2, p62 and LC3B in RV, indicating that QLQX efficiently accelerated the PINK1/Parkin pathway mitophagy (Figures 5B-I, S1). We may reason that under pulmonary arterial hypertension, dysfunctional mitophagy may no longer be able to clear the damaged and dead cardiomyocytes mitochondria, while QLQX can restore PINK1/Parkin-dependent mitophagy, so the clearing can resume its normal paces.
In conclusion, our study uncovered that QLQX could directly reverse RV remodeling secondary to PAH by lessening mitochondria-dependent apoptotic pathway and triggering metabolism shift from glycolysis to oxidation by restoring mitochondrial structure and function. The PINK1/Parkin-development mitophagy pathway might be the underlying mechanism to protect the mitochondria. QLQX might provide an alternative solution to pulmonary arterial hypertension. We welcome further studies to verify and expand our study.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Gra 81273 945).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804. doi: 10.1378/chest.08-0492. [DOI] [PubMed] [Google Scholar]
- 2.Haddad F, Ashley E, Michelakis ED. New insights for the diagnosis and management of right ventricular failure, from molecular imaging to targeted right ventricular therapy. Curr Opin Cardiol. 2010;25:131–140. doi: 10.1097/HCO.0b013e328335febd. [DOI] [PubMed] [Google Scholar]
- 3.Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right ventricular function in cardiovascular disease, part I: anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation. 2008;117:1436–1448. doi: 10.1161/CIRCULATIONAHA.107.653576. [DOI] [PubMed] [Google Scholar]
- 4.Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011;124:164–174. doi: 10.1161/CIRCULATIONAHA.110.983866. [DOI] [PubMed] [Google Scholar]
- 5.Lewis GD, Shah R, Shahzad K, Camuso JM, Pappagianopoulos PP, Hung J, Tawakol A, Gerszten RE, Systrom DM, Bloch KD, Semigran MJ. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116:1555–1562. doi: 10.1161/CIRCULATIONAHA.107.716373. [DOI] [PubMed] [Google Scholar]
- 6.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 7.Amsallem M, Sweatt AJ, Aymami MC, Kuznetsova T, Selej M, Lu H, Mercier O, Fadel E, Schnittger I, McConnell MV, Rabinovitch M, Zamanian RT, Haddad F. Right heart end-systolic remodeling index strongly predicts outcomes in pulmonary arterial hypertension: comparison with validated models. Circ Cardiovasc Imaging. 2017;10:e005771. doi: 10.1161/CIRCIMAGING.116.005771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adrain C, Martin SJ. The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Trends Biochem Sci. 2001;26:390–397. doi: 10.1016/s0968-0004(01)01844-8. [DOI] [PubMed] [Google Scholar]
- 9.Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation--a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta. 2011;1813:1333–1350. doi: 10.1016/j.bbamcr.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Fillmore N, Lopaschuk GD. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim Biophys Acta. 2013;1833:857–865. doi: 10.1016/j.bbamcr.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Can MM, Kaymaz C, Tanboga IH, Tokgoz HC, Canpolat N, Turkyilmaz E, Sonmez K, Ozdemir N. Increased right ventricular glucose metabolism in patients with pulmonary arterial hypertension. Clin Nucl Med. 2011;36:743–748. doi: 10.1097/RLU.0b013e3182177389. [DOI] [PubMed] [Google Scholar]
- 12.Bokhari S, Raina A, Rosenweig EB, Schulze PC, Bokhari J, Einstein AJ, Barst RJ, Johnson LL. PET imaging may provide a novel biomarker and understanding of right ventricular dysfunction in patients with idiopathic pulmonary arterial hypertension. Circ Cardiovasc Imaging. 2011;4:641–647. doi: 10.1161/CIRCIMAGING.110.963207. [DOI] [PubMed] [Google Scholar]
- 13.Zhabyeyev P, Gandhi M, Mori J, Basu R, Kassiri Z, Clanachan A, Lopaschuk GD, Oudit GY. Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res. 2013;97:676–685. doi: 10.1093/cvr/cvs424. [DOI] [PubMed] [Google Scholar]
- 14.Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–188. doi: 10.1161/CIRCRESAHA.113.301129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE, Abel ED. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res. 2015;56:546–561. doi: 10.1194/jlr.M055152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, Watanabe T, Morishita K, Okada H, Kawasaki M, Seishima M, Minatoguchi S. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. 2015;11:1146–1160. doi: 10.1080/15548627.2015.1051295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;11:1146–1160. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esteban-Martinez L, Sierra-Filardi E, Boya P. Mitophagy, metabolism, and cell fate. Mol Cell Oncol. 2017;4:e1353854. doi: 10.1080/23723556.2017.1353854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. 2019;124:1360–1371. doi: 10.1161/CIRCRESAHA.118.314607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun J, Zhang K, Xiong WJ, Yang GY, Zhang YJ, Wang CC, Lai L, Han M, Ren J, Lewith G, Liu JP. Clinical effects of a standardized Chinese herbal remedy, Qili Qiangxin, as an adjuvant treatment in heart failure: systematic review and meta-analysis. BMC Complement Altern Med. 2016;16:201. doi: 10.1186/s12906-016-1174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han A, Lu Y, Zheng Q, Zhang J, Zhao Y, Zhao M, Cui X. Qiliqiangxin attenuates cardiac remodeling via inhibition of TGF-beta1/Smad3 and NF-kappaB signaling pathways in a rat model of myocardial infarction. Cell Physiol Biochem. 2018;45:1797–1806. doi: 10.1159/000487871. [DOI] [PubMed] [Google Scholar]
- 24.Okada M, Harada T, Kikuzuki R, Yamawaki H, Hara Y. Effects of telmisartan on right ventricular remodeling induced by monocrotaline in rats. J Pharmacol Sci. 2009;111:193–200. doi: 10.1254/jphs.09112fp. [DOI] [PubMed] [Google Scholar]
- 25.Hessel MH, Steendijk P, den Adel B, Schutte CI, van der Laarse A. Characterization of right ventricular function after monocrotaline-induced pulmonary hypertension in the intact rat. Am J Physiol Heart Circ Physiol. 2006;291:H2424–2430. doi: 10.1152/ajpheart.00369.2006. [DOI] [PubMed] [Google Scholar]
- 26.Lourenço AP, Roncon-Albuquerque R, Brás-Silva C, Faria B, Wieland J, Henriques-Coelho T, Correia-Pinto J, Leite-Moreira AF. Myocardial dysfunction and neurohumoral activation without remodeling in left ventricle of monocrotaline-induced pulmonary hypertensive rats. Am J Physiol Heart Circ Physiol. 2006;291:H1587–1594. doi: 10.1152/ajpheart.01004.2005. [DOI] [PubMed] [Google Scholar]
- 27.Shehata ML, Lossnitzer D, Skrok J, Boyce D, Lechtzin N, Mathai SC, Girgis RE, Osman N, Lima JA, Bluemke DA, Hassoun PM, Vogel-Claussen J. Myocardial delayed enhancement in pulmonary hypertension: pulmonary hemodynamics, right ventricular function, and remodeling. AJR Am J Roentgenol. 2011;196:87–94. doi: 10.2214/AJR.09.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gores GJ, Flarsheim CE, Dawson TL, Nieminen AL, Herman B, Lemasters JJ. Swelling, reductive stress, and cell death during chemical hypoxia in hepatocytes. Am J Physiol. 1989;257:C347–354. doi: 10.1152/ajpcell.1989.257.2.C347. [DOI] [PubMed] [Google Scholar]
- 30.Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2016;311:L433–452. doi: 10.1152/ajplung.00128.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lundgrin EL, Park MM, Sharp J, Tang WH, Thomas JD, Asosingh K, Comhair SA, DiFilippo FP, Neumann DR, Davis L, Graham BB, Tuder RM, Dostanic I, Erzurum SC. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc. 2013;10:1–9. doi: 10.1513/AnnalsATS.201206-029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang W, Zhao L, Xiong CM, Ni XH, He ZX, He JG, Wilkins MR. Comparison of 18F-FDG uptake by right ventricular myocardium in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with congenital heart disease. Pulm Circ. 2012;2:365–372. doi: 10.4103/2045-8932.101651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med. 2013;91:1185–1197. doi: 10.1007/s00109-013-1064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campian ME, Verberne HJ, Hardziyenka M, de Bruin K, Selwaness M, van den Hoff MJ, Ruijter JM, van Eck-Smit BL, de Bakker JM, Tan HL. Serial noninvasive assessment of apoptosis during right ventricular disease progression in rats. J Nucl Med. 2009;50:1371–1377. doi: 10.2967/jnumed.108.061366. [DOI] [PubMed] [Google Scholar]
- 35.Hein S, Arnon E, Kostin S, Schönburg M, Elsässer A, Polyakova V, Bauer EP, Klövekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 36.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 37.Takemura G, Kanoh M, Minatoguchi S, Fujiwara H. Cardiomyocyte apoptosis in the failing heart--a critical review from definition and classification of cell death. Int J Cardiol. 2013;167:2373–2386. doi: 10.1016/j.ijcard.2013.01.163. [DOI] [PubMed] [Google Scholar]
- 38.Gross A. BCL-2 proteins: regulators of the mitochondrial apoptotic program. IUBMB Life. 2001;52:231–236. doi: 10.1080/15216540152846046. [DOI] [PubMed] [Google Scholar]
- 39.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 40.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.