

Abstract
Recent studies have indicated that several circular RNAs (circRNAs) can affect the occurrence and development of hepatocellular carcinoma (HCC). Post-transcriptional methylation modifications, including 5-methylcytosine (m5C) modification, are closely related to the tumorigenesis of cancers. However, the map of m5C modification of circRNA in HCC remains to be investigated. In this study, we performed MeRIP-seq to identify m5C sites on circRNA of human HCC tissues and paired adjacent non-tumor tissues. Further, we analyzed the relationship between m5C and HCC. Moreover, we performed a bioinformatics analysis to predict the function of specific methylated transcripts. We found that there was a significant difference in m5C between HCC tissues and paired non-tumor tissues, suggesting potential critical roles of m5C of circRNA in HCC development. In addition, the Gene Ontology (GO) analysis results indicated that the unique distribution pattern of circRNA m5C in HCC was associated with specific metabolism-associated pathways. In conclusion, our findings suggest a possible association between HCC and m5C of circRNA. Additionally, our results provide new insights into a novel function of m5C RNA methylation of circRNA in HCC progression.
Keywords: circRNA, 5-methylcytosine, hepatocellular carcinoma, RNA methylation, MeRIP-seq
Introduction
Hepatocellular carcinoma (HCC) is one of the most widespread cancers with extremely poor prognosis. As the second common cause of tumor-related death worldwide, the disease contributes to nearly 662,000 deaths per year, most of which occur in sub-Saharan Africa and Asia [1-3]. Despite marked improvements in the treatment of HCC, incidence and mortality are still high, primarily due to the late diagnosis, early metastasis and high recurrence rates [4-6]. It is therefore imperative to acquire an in-depth understanding of the molecular mechanism and to identify new prognostic biomarkers and therapeutic targets that will allow for the development of improved HCC treatment strategies.
Circular RNAs (circRNAs), a class of non-coding RNAs (ncRNAs), was originally thought to be the result of the erroneously alternative splicing [7-9], but recent studies have been demonstrated that circRNAs play critical roles in cancers, including HCC [10-13]. CircRNAs can serve as circulating biomarkers for cancer diagnosis [10,14], and ongoing studies have revealed that circRNAs can regulate gene expression to promote or inhibit cancer at transcriptional, post-transcriptional, and translational levels [15,16]. Interestingly, a recent study shows that a part of circRNA that was originally thought to be untranslatable can be translated after RNA methylation [17,18]. However, the RNA methylation level, including circRNA methylation, has not been previously reported.
Increasing evidence has demonstrated that epigenetic dysregulation contributes to the initiation and progression of cancers, and posttranscriptional modification of RNA has received considerable critical attention in recent years [19]. N6-methyladenosine (m6A), the most prevalent internal mRNA modification, has been well researched in HCC and concerned studies revealed the molecular mechanism in tumors, including HCC [20,21]. Another post-transcriptional RNA modification, known as 5-methylcytosine (m5C), has been identified in stable and highly abundant tRNAs, rRNAs, and mRNAs [22-24]. Studies have shown that m5C modification is important for the stability of tRNA and plays a crucial role in the translation and structure of rRNA, which is dynamic, conservative, and tissue specific in mammals [25-29]. As a well-known modification with new epigenetic functions, m5C modification was proven to play essential roles in bladder cancer progression [30]. However, the distribution and the function of m5C modification in circRNA of HCC are largely unclear.
In the present study, we performed a global analysis of m5C of circRNA in human HCC and paired adjacent non-tumor tissues. The results showed some significant differences in the degree and location of methylation in samples of HCC tissues compared to adjacent tissues. Specifically, the degree of methylation was observed to be higher in HCC tissues than that in adjacent tissues, and these differentially methylated genes are widely derived from all chromosomes. Our findings suggest a possible association between HCC and m5C of circRNA.
Materials and methods
Sample material
Each pair of HCC and corresponding adjacent tissues was obtained from the same HCC patient. Further, these tissues were quickly frozen in liquid nitrogen for further preparation of RNA.
RNA preparation
Initially, we selected six biological replicates each in the HCC and adjacent tissues. Next, total RNA was extracted using TRIzol reagent (Invitrogen Corporation, CA, USA) as recommended by the manufacturer, followed by reducing ribosomal RNA content in total RNA with Ribo-Zero rRNA Removal Kit (Illumina, Inc., CA, USA). We then used NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA, USA) to measure the RNA concentration of each sample and used OD260/OD280 as the purity index. If the OD260/OD280 range was from 1.8 to 2.1, the RNA purity was qualified, and the QC Results were marked as a “Pass” (Table 1).
Table 1.
RNA quantification and quality assurance by NanoDrop ND-1000
| Patient ID | Sample type | OD260/280 Ratio | Conc. (ng/μl) | Volume (μl) | Quantity (μg) | QC result |
|---|---|---|---|---|---|---|
| 1 | paired non-tumor | 1.84 | 729.53 | 137.7 | 100.46 | Pass |
| 2 | paired non-tumor | 1.83 | 738.21 | 137.7 | 101.65 | Pass |
| 3 | paired non-tumor | 1.82 | 690.97 | 137.7 | 95.15 | Pass |
| 4 | paired non-tumor | 1.84 | 614.80 | 137.7 | 84.66 | Pass |
| 5 | paired non-tumor | 1.91 | 835.22 | 137.7 | 115.01 | Pass |
| 6 | paired non-tumor | 1.86 | 778.78 | 137.7 | 107.24 | Pass |
| 1 | HCC | 1.82 | 749.03 | 58.7 | 43.97 | Pass |
| 2 | HCC | 1.89 | 699.77 | 58.7 | 41.08 | Pass |
| 3 | HCC | 1.82 | 746.16 | 58.7 | 43.80 | Pass |
| 4 | HCC | 1.93 | 910.16 | 58.7 | 53.43 | Pass |
| 5 | HCC | 1.84 | 563.81 | 58.6 | 33.04 | Pass |
| 6 | HCC | 1.82 | 723.30 | 58.7 | 42.46 | Pass |
RNA MeRIP-seq library construction & sequencing
RNA MeRIP-seq was performed based on published experimental methods with some modifications [31]. Briefly, total RNA was lysed into 100 BP fragments and m5C RNA immunoprecipitation (IP) was performed with the GenSeqTM m5C RNA IP Kit (GenSeq Inc, China) according the manufacturer’s instructions.
Reading the mapping of sequencing and calling m5C
After sequencing, imaging analysis, base recognition, and quality control of the RNA by Illumina HiSeq 4000 Sequencer, the paired-end reads were harvested. The reads were performed with Q30. Next, we used cutadapt software (v1.9.3) for trimming of the 3’ adaptors, low-quality reads removal, and obtaining high-quality clean reads. Next, clean reads of input libraries were aligned to reference genome (GRCh38.gencode. v32) by STAR software [32]. CircRNAs were identified by DCC software using the STAR alignment results [33]. After that, clean reads of all libraries were aligned to the reference genome by Hisat2 software (v2.0.4) [34] followed by identifying methylated peaks on RNAs with MACS software [35]. Differentially methylated sites were identified by DiffReps software [36]. We have defined methylation sites with a foldchange > 2 (P ≤ 0.00001) as up-regulated in a particular sample. Peaks identified by both software and any overlap with exons of circRNA were identified and selected by in-house constructed scripts, which were further annotated accordingly (Figure 1).
Figure 1.
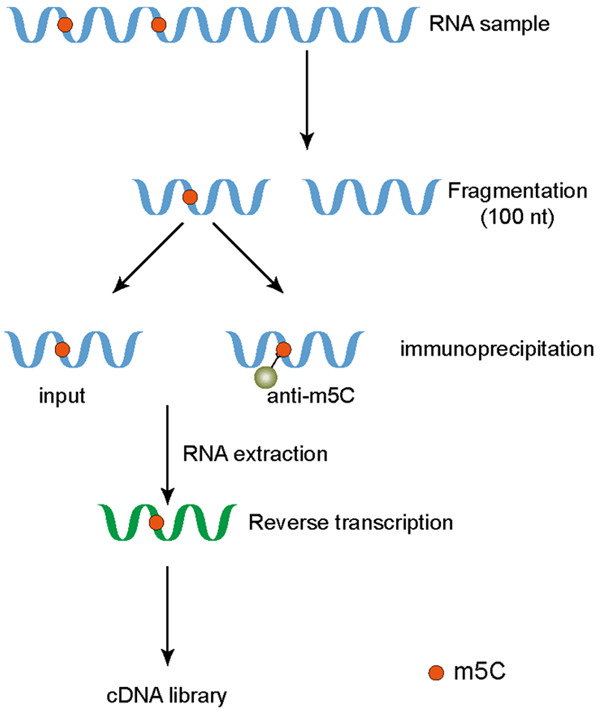
Graphical description of MeRIP-seq. RNA samples from cells were extracted and fragmented. One part was used as input for whole transcriptome sequencing, and the other part was co-immunoprecipitated with corresponding antibodies. Reverse transcription of the extracted RNA was performed to obtain cDNA libraries.
Statistics of the distribution characteristics of m5C peaks
We combined the m5C peaks identified in the six samples in the HCC group as the total m5C peaks of HCC, and the m5C peaks in adjacent tissue were treated in the same way, the common peaks of the two groups were identified using bed tools. The sequences of the vertices of the methylation peaks in the two groups (50 bp on each side of the vertices) were scanned using dreme software to find reliable motif sequence [37]. The E value is the enrichment p value times the number of candidate motifs tested. The smaller the E value of the motif, the higher the credibility of the motif. Then, according to the published method, we calculated the area where the m5C peak in the two groups was located and plotted it as a pie chart [38]. Heatmap.2 software package was used to perform clustering based on the log logfold enrichment (FE) value of each gene in twelve samples. The color represents the size of the logFE value. The closer the color in the figure is to red, the larger the logFE value. Subsequently, we used the histogram to show the enrichment degree of methylation on circRNA in the corresponding position of 24 chromosomes in the genome using circos software. The height of the histogram represents the size of the FE value.
Joint analysis of transcriptomics and methylome
STAR software (v2.5.1b) was used to map high-quality reads to the genome (human gencode v32), then DCC software (v0.4.4) was used to detect and identify circular RNA. Subsequently, we annotated the identified circular RNA using the circBase database [39] and Circ2Traits [40], followed by data standardization and differential expression circRNA screening using edgeR software (v3.16.5) to obtain logCPM. We draw a scatter plot of the differential expression of methylated genes based on the average logCPM of the two groups.
GO and pathway enrichment analysis
Gene Ontology (GO) contains three parts: molecular functions, biological processes, and cellular components. We performed GO analysis (http://www.geneontology.org) to annotate and speculate on the likely role of these differentially methylated genes. GO terms with p value < 0.05 were considered statistically significant. Further, we have shown the most important items in the results.
Moreover, we performed KEGG pathway analysis, which is a process of mapping molecular data sets in genomics, transcriptomics, proteomics, and metabolomics onto the KEGG pathway to explain the biological functions of these molecules (https://david.ncifcrf.gov/). Differentially methylated circRNAs were used to perform KEGG to annotate and speculate pathways in which they may be involved. P value < 0.05 was considered as the threshold for significant enrichment. We have also listed pathways with methylated genes that were either up-regulated or down-regulated in HCC.
Results
General features of m5C methylation in human hepatocellular carcinoma and adjacent tissues
After 3’ adaptor-trimming and low-quality read removal by cutadapt software (v1.9.3), we found 6447 methylation clean reads in HCC tissues and 2840 methylation clean reads in adjacent tissues. Up to 2869 annotated genes (HCC tissues) and 1767 annotated genes (adjacent tissues) were mapped, respectively. We found that 4072 of the 6447 methylation sites only appeared in HCC tissues, while 465 of the 2840 methylation sites only appeared in adjacent tissues (Figure 2A and 2B). Moreover, the data suggested that the number of up-methylated transcript sites per gene (HCC tissues: 3.3 sites/gene, adjacent tissues: 3.72 sites/gene) was markedly higher than the methylated transcript sites that are present in the both samples (1.4 sites/gene).
Figure 2.
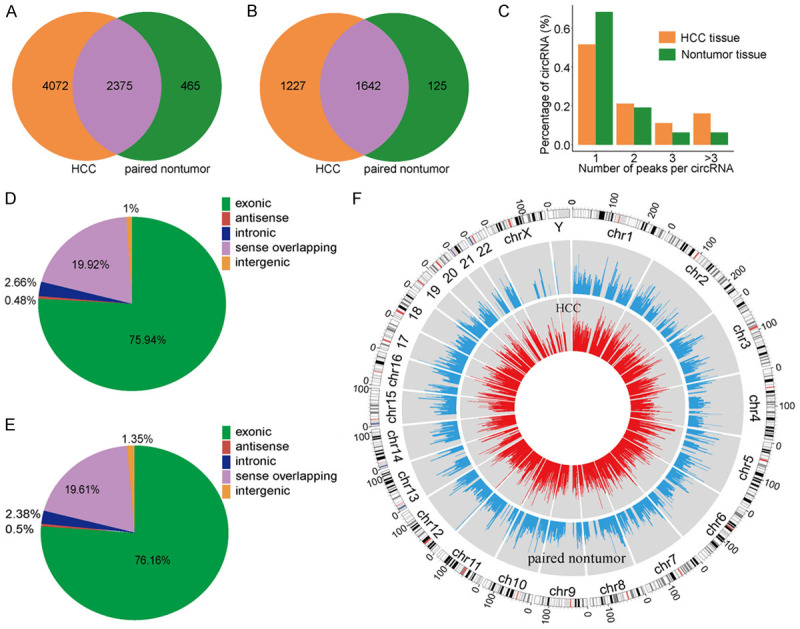
Overview of circRNA m5C in hepatocellular carcinoma (HCC) and adjacent tissues. A. Venn diagram of m5C peaks in HCC and adjacent tissues. B. Venn diagram of m5C genes in HCC and adjacent tissues. C. The number of methylation peaks in HCC and adjacent tissues on each circular RNA. It can be found that most circular RNAs have only one methylation peak. D, E. Pie chart of the source of methylated circRNA in HCC and adjacent non-tumor tissues. F. Visualization of m5C at the chromosome level in HCC and adjacent tissues.
Statistical analysis of the number of peaks on each circRNA
To determine the number of m5C peaks on each circRNA, we performed a statistical analysis of the methylation peaks and the corresponding circRNA (Figure 2C). The results showed that most of the circRNAs with methylation sites in HCC tissues had only one methylation peak (51.7%), and this ratio was higher in adjacent tissues (68.4%). The difference between the two groups was statistically significant (P < 0.0001). At the same time, the number of circRNAs with two or more methylation peaks on one circRNA was higher in HCC tissues than that in adjacent tissues (P < 0.0001).
Analysis of sources of circRNA methylation in HCC and adjacent tissues
Further, we summarized the source data of methylated circRNA and plotted the pie chart (Figure 2D and 2E). The results showed that most of the circRNAs were derived from exonic regions, followed by sense overlapping regions. Specifically, compared with adjacent tissues, HCC tissues had fewer methylated circRNAs derived from exons, and more methylated circRNAs from the sense overlapping regions. Additionally, there were fewer circRNAs derived from exons in HCC tissues, which may lead to decline of the number of circRNAs that can perform silencing and affect cell function.
Additionally, we used circos software to analyze the distribution of circRNA methylation sites on the chromosomes (Figure 2F). The results showed that the distribution of the methylation sites of circRNA on each chromosome was different between the two groups, with the X chromosome being the most evident. In addition, compared with the autosomes, the sex chromosomes in both groups were less methylated.
Motif analysis of methylation sites
Dreme software was used to scan the sequence of methylated peaks (50 bp on each side of the apex) of each group of samples to find meaningful motif sequences. The results are as shown in Figure 3A and 3B. Among the motifs measured in the HCC tissues, AHUACA (H = U/A/C) was observed to be the most common and reliable, that is, the most likely conserved methylation site motif (E-value = 5.7e-268). In the adjacent tissues, the most conserved motif was also the AHUACA (E value = 6.7e-080), same as the most reliable motif in HCC.
Figure 3.
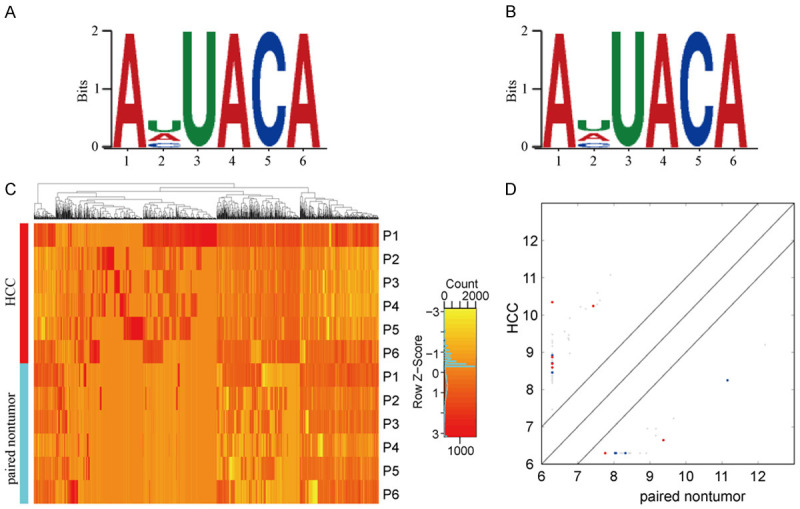
Characterization of methylation peaks and joint analysis of methylation and transcriptome. A. Motif sequence of m5C in hepatocellular carcinoma (HCC) group. B. Motif sequence of m5C in adjacent tissues group, which is consistent with the sequence in the HCC group. C. Cluster analysis of methylation in HCC and adjacent tissues. D. Scatter plot of relationship between gene expression level and methylation level, the Y-axis and X-axis represent the expression levels of genes in HCC and paired non-tumor, red dots represent genes with high methylation level, blue dots represent low methylation horizontal genes, while gray represents no significant difference.
Cluster analysis of differential methylation peaks
In addition, we performed a methylation heatmap analysis and cluster analysis of the total data (Figure 3C). The results of the cluster analysis showed that the difference in methylation degree can be used to distinguish the HCC group from the adjacent tissue group, that is, there were noticeable differences between the groups and relatively consistent within group. And these differences may be closely related to the occurrence and development of HCC. Overall, the frequency of methylation in the HCC group was much higher than that in the adjacent tissues. Specifically, a total of 2085 methylation sites were detected as up-regulated in HCC, while 813 methylation sites were up-regulated in adjacent tissues. We have listed the 20 circRNAs with the largest FC in Tables 2 and 3.
Table 2.
Top ten up-methylated peaks
| chrom | txStart | txEnd | GeneName | Foldchange |
|---|---|---|---|---|
| chr1 | 28463441 | 28463760 | PHACTR4 | 171.3 |
| chr1 | 179347921 | 179348500 | SOAT1 | 171.2 |
| chr6 | 32530601 | 32531080 | HLA-DRB1 | 152 |
| chr6 | 161039761 | 161040200 | MAP3K4 | 149.4 |
| chr15 | 43156641 | 43157100 | TMEM62 | 144 |
| chrX | 77642201 | 77642660 | ATRX | 138.3 |
| chr11 | 103303721 | 103304120 | DYNC2H1 | 115.3 |
| chr11 | 103281921 | 103282300 | DYNC2H1 | 86 |
| chr20 | 8744401 | 8744780 | PLCB1 | 81.7 |
| chr15 | 42294621 | 42294980 | AC012651.1 | 79.1 |
Table 3.
Top ten down-methylated peaks
| chrom | txStart | txEnd | GeneName | Foldchange |
|---|---|---|---|---|
| chr1 | 39354221 | 39354600 | MACF1 | 128.5 |
| chr18 | 24296101 | 24296700 | OSBPL1A | 118.8 |
| chr16 | 11730161 | 11730560 | TXNDC11 | 113.4 |
| chr11 | 20398841 | 20399300 | PRMT3 | 103.7 |
| chr16 | 53871821 | 53872140 | FTO | 99 |
| chr1 | 35347701 | 35348200 | SNORA62 | 95.7 |
| chr1 | 13719601 | 13720020 | PRDM2 | 95 |
| chr11 | 88309421 | 88309760 | CTSC | 93.9 |
| chr1 | 97517581 | 97517900 | DPYD | 89.9 |
| chrX | 63666941 | 63667280 | ARHGEF9 | 89.7 |
Effect of methylation on transcriptional expression
To investigate the effect of methylation on transcriptional expression, we combined analysis of transcriptome and methylation (Figure 3D). The difference in the transcription level of hypermethylated genes have been shown on Tables 4 and 5. The results show that whether in HCC or adjacent non-tumor tissues, hypermethylated genes tend to have lower transcriptional expression. Unfortunately, this trend is less noticeable due to the relatively small number of circRNAs and the relatively low frequency of methylation, so this hypothesis requires further validation.
Table 4.
Differences in transcription levels of hypermethylated genes in HCC
| Chrom | txStart | txEnd | Foldchange of methylation degree | Gene name | Catalog | logFC of transcription level |
|---|---|---|---|---|---|---|
| chr22 | 50392301 | 50392680 | 2.377727615 | PPP6R2 | exonic | 2.993041522 |
| chr12 | 18283241 | 18283620 | 4.178057554 | PIK3C2G | sense overlapping | -3.285974927 |
| chr13 | 99239741 | 99240140 | 3.469856459 | UBAC2 | exonic | 3.331288361 |
| chr12 | 18291141 | 18291720 | 3.284140969 | PIK3C2G | sense overlapping | -3.285974927 |
| chr19 | 46928421 | 46929020 | 2.087808603 | ARHGAP35 | sense overlapping | 5.560500054 |
| chr15 | 101366581 | 101367020 | 2.409874608 | PCSK6 | exonic | -3.667858726 |
| chr19 | 46924641 | 46925060 | 7.826530612 | ARHGAP35 | sense overlapping | 5.560500054 |
| chr1 | 59375381 | 59376280 | 3.686405338 | FGGY | exonic | -2.659245921 |
| chr10 | 95408001 | 95408580 | 2.082657517 | SORBS1 | exonic | -3.381399644 |
| chr10 | 95381881 | 95382260 | 4.489566613 | SORBS1 | exonic | -3.381399644 |
| chr1 | 59368641 | 59369520 | 2.024181757 | FGGY | exonic | -2.659245921 |
| chr17 | 2394181 | 2394760 | 3.462382445 | MNT | exonic | 3.472039848 |
| chr12 | 18287001 | 18287580 | 4.431796802 | PIK3C2G | sense overlapping | -3.285974927 |
| chr10 | 95401421 | 95401860 | 3.092629482 | SORBS1 | exonic | -3.381399644 |
| chr12 | 22656741 | 22657100 | 5.483965015 | ETNK1 | exonic | 3.553337593 |
| chr19 | 40857141 | 40857720 | 6.734767025 | CYP2A7 | sense overlapping | -3.274932432 |
| chr19 | 40851581 | 40852420 | 2.41031653 | CYP2A7 | sense overlapping | -3.274932432 |
Table 5.
Differences in transcription levels of hypermethylated genes in adjacent tissues
| Chrom | txStart | txEnd | Foldchange of methylation degree | Gene name | Catalog | logFC transcription level |
|---|---|---|---|---|---|---|
| chr19 | 40853241 | 40853820 | 7.616352201 | CYP2A7 | sense overlapping | -3.274932432 |
| chr1 | 59369381 | 59369880 | 2.810578662 | FGGY | exonic | -2.659245921 |
| chr14 | 88434281 | 88435160 | 2.993351064 | SPATA7 | exonic | -4.094387086 |
| chr6 | 116701881 | 116702440 | 2.510126582 | KPNA5 | exonic | 3.609326004 |
| chr10 | 95409361 | 95409700 | 3.820253165 | SORBS1 | exonic | -3.381399644 |
| chr19 | 40866861 | 40867480 | 2.089793642 | CYP2A7 | sense overlapping | -3.274932432 |
| chr18 | 13010681 | 13011320 | 2.085308057 | CEP192 | exonic | 3.962828068 |
| chr14 | 88435361 | 88436520 | 3.397698210 | SPATA7 | exonic | -4.094387086 |
| chr1 | 23053981 | 23054400 | 4.125000000 | KDM1A | exonic | 3.278760795 |
| chr1 | 219215281 | 219216640 | 2.846099789 | LYPLAL1 | sense overlapping | 3.666802455 |
GO analysis
To uncover the biological processes and molecular functions of differentially methylated circRNAs in HCC and adjacent tissues, we performed GO analysis on the data obtained by sequencing. For the biological processes (BP) category, we found that the genes with up-methylated m5C sites in HCC tissues were mainly related to cellular metabolic process, phosphorus metabolic process, and phosphate-containing compound metabolic process while the genes with down-methylated m5C were mainly related to cellular metabolic process, organelle organization, and cellular catabolic process. For the molecular functions (MF) category, the genes with up-methylated m5C were mainly related to catalytic activity, adenyl ribonucleotide binding, and ATP binding while the genes with down-methylated m5C are mainly related to catalytic activity, small conjugating protein-specific protease activity, and protein binding. For the cellular components (CC), methylation sites up-regulated in HCC tissues were mainly enriched in intracellular part, intracellular organelle, same as methylation sites upregulated in adjacent cancer tissue. The 10 most prominent categories are shown in the figures (Figure 4A-F).
Figure 4.
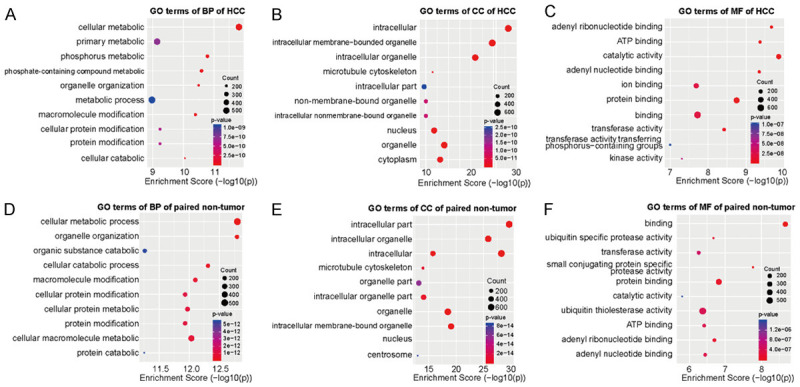
Gene ontology analyses of HCC and adjacent tissues. A-C. Biological processes (BP), cell component (CC), and molecular functions (MF) in HCC group, respectively. D-F. Biological processes, cell component, and molecular functions in adjacent tissues group, respectively. We have listed the 10 most significant terms in each figure.
Methylated circRNAs in different groups participate in different pathways
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed to identify pathways in which differentially methylated genes may be involved (Figure 5A and 5B). Analysis results showed that for genes with up-methylation in HCC tissues, they were mainly involved in ubiquitin-mediated proteolysis, endocytosis, and adherens junction. For genes with down-methylated m5C, they were mainly involved in endocytosis and protein processing in endoplasmic reticulum.
Figure 5.
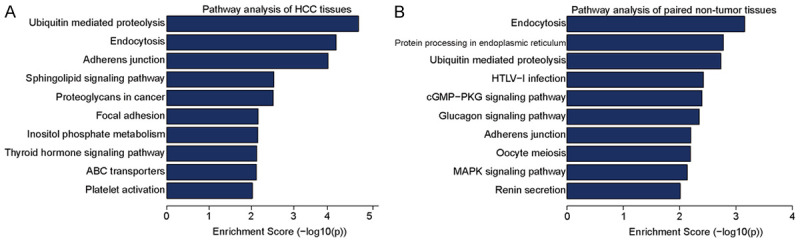
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of differentially methylated genes in hepatocellular carcinoma and adjacent tissues. A. Pathway analysis of differentially methylated genes in HCC group. B. Pathway analysis of differentially methylated genes in adjacent tissues group. We list the 10 most significant terms on each figure.
Discussion
As an important post-transcriptional modification, methylation has become a research hotspot in recent years. As a more comprehensive type of methylation, m6A, has been proven to play a critical role in many cancer-related pathways. Yongsheng Li et al, confirmed that m6A plays a vital role in 33 different types of cancer, including HCC [21,41]. As another type of methylation, m5C has also been shown to play key roles in cell functions, such as regulating RNA nucleation [29] and intergenerational transmission of acquired phenotypes [42], while also promoting the proliferation of bladder cancer by stabilizing mRNAs [30]. A recent article has proved that the mRNA of HCC tissue has significantly more m5c sites than that of adjacent tissues, and its hypermethylated mRNA is related to the abnormality of N-glycan biosynthesis [31]. However, m5C has a very extensive role in cells, and its distribution of circRNA in HCC tissues and its role in the pathogenesis of HCC have not been studied yet.
In this study, we sequenced the m5c methylation of circRNA in HCC and adjacent tissues using MeRIP-seq and compared them to identify differences between the two groups. We identified thousands of methylation peaks in circRNA and found significant differences in the number and distribution of methylation peaks between the two samples. Our results indicate that the frequency of methylation and the genes of methylation in HCC are far more than those in the adjacent tissues, which proves that there is a definite link between m5C and HCC. Analysis at the whole chromosome level showed that circRNAs derived from various chromosomes in HCC and adjacent tissues are significantly different, especially the X chromosome, indicating that m5C in HCC tissues is widely changed and may involve many pathways to influence the phenotype of HCC. Meanwhile, motif analysis showed that the sequences near the methylation sites in the two groups were consistent and relatively conserved, suggesting that there is a difference in the number of methylated writers rather than the types, which requires further rigorous experimentation to allow validation.
A previous study showed that circYAP can bind to its parent mRNA and inhibit YAP production, and YAP is an important protein in the process of tumorigenesis and development and is widely present in liver cancer and breast cancer [43]. Our results show that methylated circRNAs derived from exons are less abundant in HCC tissues. We therefore propose that the reduction of exon-derived circRNAs caused by methylation leads to a loss of inhibition of certain key proteins and promotes tumor development. In addition, we found that the degree of methylation in HCC tissues is positively correlated with the degree of corresponding gene expression, however, this trend is less noticeable and more samples are needed for further validation.
There is already a lot of evidence that m5C is involved in many cellular functions, such as regulating RNA nucleus [29], affecting cell differentiation [44], regulating stem cell function, and stress [45] etc. Through further bioinformatics analysis, GO terms of differentially methylated genes and KEGG pathways in the two groups were determined. The up-regulated methylated genes in HCC tissues were identified to be involved in various aspects of cell function, such as energy and protein metabolism and endocytosis, which indicates that the cellular functions affected by methylation are extensive, and the mechanism involved needs to be extensively studied. These results suggest that there is a need for more research to facilitate a detailed understanding of the m5C.
Conclusion
In summary, our study reveals for the first time the differences in m5C between HCC tissues and adjacent tissues, both in quantity and distribution. We conducted an in-depth bioinformatics analysis to show the distribution and possible functions of m5C in HCC. However, the functions of m5C in cells are extensive, and presently we do not know enough about, so more experiments are needed to determine the role and mechanism of m5C, which will eventually provide new treatment targets for HCC.
Acknowledgements
This study was supported by funds from the National Natural Science Foundation of China (81671958 and 81902832); Science and Technology Innovation Talents in Henan Universities (19HASTIT003); Henan Medical Science and Technology Research Project of 2018 (SBGJ2018002); the Science and Technology Research Project of Henan Province (192102310117); Key Scientific Research Project of Henan Higher Education Institutions of China (21A320026). And the study has been approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University, and the patients’ written informed consent was obtained before the study began. The ethics committee approval number is 2019-KY-21. In addition, we thank the patients who participated in this study and Cloud-Seq Biotech Ltd. Co. (Shanghai, China) for the MeRIP-Seq service.
Disclosure of conflict of interest
None.
References
- 1.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–1314. doi: 10.1016/S0140-6736(18)30010-2. [DOI] [PubMed] [Google Scholar]
- 2.Cai K, Li T, Guo L, Guo H, Zhu W, Yan L, Li F. Long non-coding RNA LINC00467 regulates hepatocellular carcinoma progression by modulating miR-9-5p/PPARA expression. Open Biol. 2019;9:190074. doi: 10.1098/rsob.190074. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 4.Portolani N, Coniglio A, Ghidoni S, Giovanelli M, Benetti A, Tiberio GA, Giulini SM. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Ann Surg. 2006;243:229–235. doi: 10.1097/01.sla.0000197706.21803.a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–1763. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y, Huan L, Wu Y, Bao C, Chen B, Wang L, Huang S, Liang L, He X. LncRNA ID2-AS1 suppresses tumor metastasis by activating the HDAC8/ID2 pathway in hepatocellular carcinoma. Cancer Lett. 2020;469:399–409. doi: 10.1016/j.canlet.2019.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73:3852–3856. doi: 10.1073/pnas.73.11.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cocquerelle C, Mascrez B, Hetuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB J. 1993;7:155–160. doi: 10.1096/fasebj.7.1.7678559. [DOI] [PubMed] [Google Scholar]
- 9.Danan M, Schwartz S, Edelheit S, Sorek R. Transcriptome-wide discovery of circular RNAs in archaea. Nucleic Acids Res. 2012;40:3131–3142. doi: 10.1093/nar/gkr1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Yu F, Li P. Circular RNAs: characteristics, function and clinical significance in hepatocellular carcinoma. Cancers (Basel) 2018;10:258. doi: 10.3390/cancers10080258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu L, Chen Q, Yao T, Li T, Ying S, Hu Y, Guo J. Hsa_circ_0005986 inhibits carcinogenesis by acting as a miR-129-5p sponge and is used as a novel biomarker for hepatocellular carcinoma. Oncotarget. 2017;8:43878–43888. doi: 10.18632/oncotarget.16709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun HD, Xu ZP, Sun ZQ, Zhu B, Wang Q, Zhou J, Jin H, Zhao A, Tang WW, Cao XF. Down-regulation of circPVRL3 promotes the proliferation and migration of gastric cancer cells. Sci Rep. 2018;8:10111. doi: 10.1038/s41598-018-27837-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bachmayr-Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation--exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep. 2015;5:8057. doi: 10.1038/srep08057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z, Yang J, Fan J, Liu L, Qin W. Hsa_circ_0001649: a circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark. 2016;16:161–169. doi: 10.3233/CBM-150552. [DOI] [PubMed] [Google Scholar]
- 15.Peng L, Chen G, Zhu Z, Shen Z, Du C, Zang R, Su Y, Xie H, Li H, Xu X, Xia Y, Tang W. Circular RNA ZNF609 functions as a competitive endogenous RNA to regulate AKT3 expression by sponging miR-150-5p in Hirschsprung’s disease. Oncotarget. 2017;8:808–818. doi: 10.18632/oncotarget.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng L, Yuan XQ, Li GC. The emerging landscape of circular RNA ciRS-7 in cancer (review) Oncol Rep. 2015;33:2669–2674. doi: 10.3892/or.2015.3904. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y, Wong CC, Xiao X, Wang Z. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017;27:626–641. doi: 10.1038/cr.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.AbouHaidar MG, Venkataraman S, Golshani A, Liu B, Ahmad T. Novel coding, translation, and gene expression of a replicating covalently closed circular RNA of 220 nt. Proc Natl Acad Sci U S A. 2014;111:14542–14547. doi: 10.1073/pnas.1402814111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delaunay S, Frye M. RNA modifications regulating cell fate in cancer. Nat Cell Biol. 2019;21:552–559. doi: 10.1038/s41556-019-0319-0. [DOI] [PubMed] [Google Scholar]
- 20.Amort T, Rieder D, Wille A, Khokhlova-Cubberley D, Riml C, Trixl L, Jia XY, Micura R, Lusser A. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 2017;18:1. doi: 10.1186/s13059-016-1139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, Wang Q, Li X, Zhang Y, Xu J. Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types. Mol Cancer. 2019;18:137. doi: 10.1186/s12943-019-1066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006;34:721–733. doi: 10.1093/nar/gkj471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agris PF. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008;9:629–635. doi: 10.1038/embor.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaefer M, Pollex T, Hanna K, Lyko F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009;37:e12. doi: 10.1093/nar/gkn954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gigova A, Duggimpudi S, Pollex T, Schaefer M, Kos M. A cluster of methylations in the domain IV of 25S rRNA is required for ribosome stability. RNA. 2014;20:1632–1644. doi: 10.1261/rna.043398.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaefer M, Pollex T, Hanna K, Tuorto F, Meusburger M, Helm M, Lyko F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010;24:1590–1595. doi: 10.1101/gad.586710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma S, Yang J, Watzinger P, Kotter P, Entian KD. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013;41:9062–9076. doi: 10.1093/nar/gkt679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol. 2012;19:900–905. doi: 10.1038/nsmb.2357. [DOI] [PubMed] [Google Scholar]
- 29.Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, Li A, Wang X, Bhattarai DP, Xiao W, Sun HY, Zhu Q, Ma HL, Adhikari S, Sun M, Hao YJ, Zhang B, Huang CM, Huang N, Jiang GB, Zhao YL, Wang HL, Sun YP, Yang YG. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017;27:606–625. doi: 10.1038/cr.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Li A, Sun BF, Yang Y, Han YN, Yuan X, Chen RX, Wei WS, Liu Y, Gao CC, Chen YS, Zhang M, Ma XD, Liu ZW, Luo JH, Lyu C, Wang HL, Ma J, Zhao YL, Zhou FJ, Huang Y, Xie D, Yang YG. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol. 2019;21:978–990. doi: 10.1038/s41556-019-0361-y. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Q, Zheng Q, Yu X, He Y, Guo W. Overview of distinct 5-methylcytosine profiles of messenger RNA in human hepatocellular carcinoma and paired adjacent non-tumor tissues. J Transl Med. 2020;18:245. doi: 10.1186/s12967-020-02417-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics. 2016;32:1094–1096. doi: 10.1093/bioinformatics/btv656. [DOI] [PubMed] [Google Scholar]
- 34.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen L, Shao NY, Liu X, Maze I, Feng J, Nestler EJ. diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One. 2013;8:e65598. doi: 10.1371/journal.pone.0065598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo GZ, MacQueen A, Zheng G, Duan H, Dore LC, Lu Z, Liu J, Chen K, Jia G, Bergelson J, He C. Unique features of the m6A methylome in arabidopsis thaliana. Nat Commun. 2014;5:5630. doi: 10.1038/ncomms6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–1670. doi: 10.1261/rna.043687.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosal S, Das S, Sen R, Basak P, Chakrabarti J. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet. 2013;4:283. doi: 10.3389/fgene.2013.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Peng C, Chen J, Chen D, Yang B, He B, Hu W, Zhang Y, Liu H, Dai L, Xie H, Zhou L, Wu J, Zheng S. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18:127. doi: 10.1186/s12943-019-1053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y, Zhang X, Shi J, Tuorto F, Li X, Liu Y, Liebers R, Zhang L, Qu Y, Qian J, Pahima M, Liu Y, Yan M, Cao Z, Lei X, Cao Y, Peng H, Liu S, Wang Y, Zheng H, Woolsey R, Quilici D, Zhai Q, Li L, Zhou T, Yan W, Lyko F, Zhang Y, Zhou Q, Duan E, Chen Q. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat Cell Biol. 2018;20:535–540. doi: 10.1038/s41556-018-0087-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu N, Yuan Z, Du KY, Fang L, Lyu J, Zhang C, He A, Eshaghi E, Zeng K, Ma J, Du WW, Yang BB. Translation of yes-associated protein (YAP) was antagonized by its circular RNA via suppressing the assembly of the translation initiation machinery. Cell Death Differ. 2019;26:2758–2773. doi: 10.1038/s41418-019-0337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flores JV, Cordero-Espinoza L, Oeztuerk-Winder F, Andersson-Rolf A, Selmi T, Blanco S, Tailor J, Dietmann S, Frye M. Cytosine-5 RNA methylation regulates neural stem cell differentiation and motility. Stem Cell Reports. 2017;8:112–124. doi: 10.1016/j.stemcr.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortes-Garrido R, Gkatza N, Dietmann S, Frye M. Stem cell function and stress response are controlled by protein synthesis. Nature. 2016;534:335–340. doi: 10.1038/nature18282. [DOI] [PMC free article] [PubMed] [Google Scholar]