

Abstract
Waardenburg syndrome (WS) is a common syndromic hearing loss disease. A large group of patients affected by WS were found no mutations in the existed gene panel, indicating that there are still potential genes responsible for WS yet to be detected. In our previous study, we established an autosomal-dominant KIT (OMIM# 164920) mutation (c.2418T>A, p.Asp806Glu) pig pedigree which presented congenital bilateral severe sensorineural hearing loss and hypopigmentation, exact the same as human WS. Histological analysis showed nearly normal structures of the organ of Corti, stria vascularis (SV) and spiral neuron ganglions at E85. Scanning electron microscopy (SEM) exhibited that hair cells started to degenerate at E100, and totally gone at P1. Transmission electron microscope (TEM) showed disorganization of SV and disappearance of intermediate cells. The absence of endocochlear potentials also demonstrated the dysfunction of stria. Our study demonstrated that KIT mutation (c.2418T>A, p.Asp806Glu) interrupted the development of melanocytes in cochlea, which led to SV malformation and dysfunction, resulting in degeneration of hair cells and finally hearing loss. Therefore, KIT was highly supposed to be a newly found gene associated with WS and be added to the WS related gene screening panel clinically.
Keywords: KIT gene, Waardenburg syndrome, miniature pigs
Introduction
More than one hundred genes have been identified to cause hereditary hearing loss to date [1]. Waardenburg syndrome (WS) is one of the most common types of syndromic hearing loss, characterized by the combination of sensorineural hearing loss and hypopigmentation of various organs, like skin and hair, with or without heterochromia iridis [2]. WS accounts for 2-5% of congenital deafness with the prevalence of 1/42,000 [2] and is classified to 4 types (WS1, OMIM# 193500; WS2, OMIM# 193510; WS3, OMIM# 148820; WS4, OMIM# 277580) according to clinical presentation [3]. It is verified as an autosomal dominantly inherited disease caused by disorders of neural crest cells. PAX3 (OMIM# 606597), MITF (OMIM# 156845), SNAI2 (OMIM# 602150), SOX10 (OMIM# 602229), EDNRB (OMIM# 131244) and EDN3 (OMIM# 131242) are the several reported genes related to WS and have been added to the screening panel in recent years [3-5]. However, the etiology of the relative large group of WS patients remains unknown, indicating that there are still potential genes responsible for WS yet to be detected [4,5].
It is believed that all of the WS-associated genes acted in the common pathways that regulating the developmental process of neural crest-derived melanocytes by influencing MITF functions [5]. MITF is suggested to mutually interact with KITLG-KIT signaling in melanocyte development. It has been proved that activation of KIT by binding of its ligand KITLG (OMIM #184745) on the cell surface, leads to the activation of downstream MITF, and this signaling cascades are primarily involved in the development and migration of neural crest-derived melanocytes, including intermediate cells of SV [6-10]. In rodents, loss-of-function mutations of MITF led to the absence of melanocytes which gave rise to hypopigmentation and hearing loss, similar to the phenotypes in cases with KIT and KITLG mutations [6,10-12]. More importantly, Zazo Seco (2015) and Ogawa (2017) reported that mutations at KITLG could cause asymmetric and unilateral hearing loss and WS2 in humans, adding a new candidate to WS gene screening panel [13,14].
In addition to WS, there are other syndromes manifesting hearing loss and hypopigmentation due to melanocyte defects, like Piebaldism (OMIM# 172800). It is an autosomal dominant disorder characterized by a congenital white forelock, scattered normal pigmented and hypopigmented patches on the skin, which is caused by loss-of-function mutations in KIT, encoding the receptor of KITLG [7,13,14]. Sporadic cases of piebaldism with KIT mutations presented with congenital hearing loss, indicating that mutations at KIT and KITLG might be part of the same disorder [17,18].
KIT locates on 4q11-12 in humans, chromosome 5 in the mice, and 8p12 in pigs [19,20]. It encodes a 145-kD glycosylated transmembrane protein consisting of an extracellular domain, a transmembrane region, and a tyrosine kinase domain. The extracellular domain contains 5 Ig-like domain which functions in KITLG lignad binding, and receptor dimerization. Activation of KIT by binding of KITLG causes autophosphorylation at tyrosine residues and triggers the downstream signaling cascades [7,9,21,22]. Furthermore, several KIT mutations at tyrosine kinase domain including W(v), W(41), W(s), KIT(wads) have been identified in deaf mice and rats with decreased number of intermediate cells and malfunction of SV [23-28]. Thus, KIT is highly suspected as a potential gene underlies WS.
Animal model provides a useful tool to study the pathogenic mechanisms of deafness. Rodents are the most commonly used laboratory animals in otologic researches. However, huge differences in inner ear development, morphology and electrophysiology between human and mouse limit the application of mice as an ideal deafness model. According to our previous studies, Bama miniature pig, a natural Chinese miniature pig breed, is a more ideal mammalian model for otologic research [29-31]. Bama miniature pigs are characterized by their genetic stability and white coat color with large black patches around the head and buttocks. In addition to the genome, the size, composition, morphology and the function of auditory system highly assembles that of human. Pigs have a larger cochlea than rodents and are ideal for evaluating the fine anatomic changes of cochlear and audiological studies [29-31].
In our previous work, we performed random mutagenesis of the porcine genome by ENU treatment and successfully established a novel dominant mutant line with piebaldism phenotype. In this study, we identified c.2418T>A missense mutation in exon 17 of the KIT gene, resulting in amino acids substitution (p.Asp806Glu) in this model and named it as KITD806E/+ Porcine model. Auditory tests, morphological analysis from E85 to P1 and Endocochlear Potential (EP) recordings were performed aiming at exploring the role of KIT functioning in auditory system. Our study firstly described the pathological changes in cochlea in the KITD806E/+ porcine model and help us better understanding the mechanism of KIT associated hearing loss in human beings.
Materials and methods
Animals
Bama miniature pigs (Department of Laboratory Animal Science, College of Basic Medicine, Army Military Medical University, Chongqing, P. R. China) were bred under a specific pathogen-free environment. The experimental procedures and animal welfare were performed in accordance with the care and use of laboratory animals and the related ethical regulations of Army Military Medical University.
Ethics approval
All animal experiments and procedures were performed under the guidelines for the Care and Use of Laboratory Animals established by the Chongqing Association for Laboratory Animal Science and approved by the Animal Ethics Committee of Army Medical University, China.
Phenotype record, auditory brainstem response test and endocochlear potential recording
Phenotype of the F2 and F3 offspring, including the coat color, iris color and sex, were recorded. Auditory brainstem responses (ABRs) (Tucker-Davis-Technology RZ6) were evaluated as previously described [30]. Animals were anesthetized (3% pentobarbital sodium, 1 ml/kg, i.m.) and placed in a sound-proof chamber. The reference electrode was fixed in the lobulus auriculae of both ears, the recording electrode subcutaneously in the cranial vertex, and the grounding electrode in the nasal tip. Stimuli intensity descended from 90 to 30 dB SPL in a step of 10 dB with sweep number of 1,024 times. When approaching the threshold, a step of 5 dB was used to define the accurate threshold. Details of endocochlear potential (EP) measurement were consistant with our previous study [30].
Genome wide linkage analysis and family-based genome wide association analysis
The mutant founder was mated with 3 wild-type females to generate 29 G2 offspring. DNA samples from all 33 Bama miniature pigs (parents and offspring) were genotyped. Genotyping was performed using porcine SNP60 BeadChips (Illumina, Inc., USA), which contain 62,163 SNP markers. The raw data were filtered for those SNPs with a call rate >90% and a minor allele frequency ≥0.05. Genome wide linkage analysis was performed using the Merlin tool, and family-based genome wide association analysis was performed using PLINK software packages.
Mutation identification
Mutations in the candidate gene were detected by PCR and Sanger DNA sequencing of the exons and the exon-intron boundaries. DNA samples were prepared from 4 pigs, including 2 mutants and 2 normal individuals from the mutant lineage. Primers were designed to amplify 20 exons and the exon-intron flanking regions of gene KIT (Table S1). The primers were designed using Primer Premier 5.0 software. The 50 μl PCR reactions contained 1 μl genomic DNA, 11.5 μl Mix (Vazyme, China), 35.5 μl ddH2O and 1 μl each primer. The reactions were incubated at 94°C for 3 min; followed by 35 cycles of 94°C for 30 sec, 56°C for 35 sec and 72°C for 50 sec; and a final extension at 72°C for 8 min. The PCR products were visualized by agarose gel electrophoresis and purified by DNA gel extraction A according to the manufacturer’s instructions. The PCR products were sequenced on an ABI PRISM 3730 genetic analyzer, and the sequences were compared using the SeqMan software.
Celloidin embedding-hematoxylin-eosin Stain, scanning electron microscopy and transmission electron microscopy
The celloidin embedding-hematoxylin-eosin (CE-HE) stained cochlea sections, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) samples were made as our previous study [30,31]. The Bama miniature pigs were sacrificed in accordance with the Care and Use of laboratory animals, and the cochlea were dissected from the temporal bones and post-fixed in 4% paraformaldehyde or 2.5% glutaraldehyde at room temperature overnight. Cochlea were washed in 1% phosphate-buffered saline (PBS) and decalcified in 10% EDTA for approximately one month.
For the histological analysis, the cochlea were dehydrated using graded ethanol and dipped in graded celloidin. The cochlea were sectioned (15 μm) using a freezing microtome (Leica CM1900) and stained with hematoxylin and eosin. The sections were visualized under a Leica DMI3000 microscope. For the SEM analysis, decalcified cochlea was post-fixed for 2 h in 1% osmium, dehydrated in graded ethanol (50%-100%), and dried in a critical point dryer (HCP-2, Hitachi) using liquid CO2. Fixed sections were then coated using a sputter coater and examined under a scanning electron microscope. For the TEM analysis, after dehydrated in graded ethanol, cochlear tissues were then embedded in Epon resin and sectioned on an Reichart Ultracut E ultramicrotome (Boeckeler Instruments, Tucson, NM, USA). Ultrathin sections were mounted on formvar-coated slot grids, stained with lead citrate and uranyl acetate and examined via transmission electron microscope.
HGVS variant nomenclature
The Human Genome Variation Society recommendations were used to standardize the nomenclature of KIT mutant variants. RefSeq sequence NM_001044525.1 was used for the coding region transcript. And NP_001037990.1 was used for the amino acid sequence resulting from translation of this transcript.
Statistical analysis methods
The differences were analyzed using Student’s t-test for assessment of more than two groups on GraphPad Prism software. P<0.05 were considered as statistical significance.
Results
Hypopigmentation and deafness of KITD806E/+ pigs
The wild-type Bama miniature pigs exhbits white coat color with large black patches around the head and buttocks (Figure 1A). The mutant pigs showed pigmentation, including systemic white hair with small dark spots on the head and buttocks but normal irises (Figure 1B). For auditory tests, Click and 1-32 kHz tone-pip auditory brainstem response (ABR) were conducted both in the wild-type and mutant pigs. The wild-type pigs had an ABR waveforms similar to humans and its hearing thresholds were at 20 dB SPL in Click, and 20-60 dB SPL in 1-32 kHz tone-pips (Figure 1C, 1E). No identifiable waveforms could not be evoked in the mutant pigs at 90 dB SPL in all frequencies, demonstrating profound sensorineural hearing loss (Figure 1D, 1E). Moreover, the hypopigmentation feature was co-segregated with hearing loss.
Figure 1.

Hypopigmentation and deafness in KITD806E/+ pigs. A. The hair color of wild-type Bama pigs was white coat colored-belly with large black patches around the head and buttocks. B. The mutant pigs showed systemic white hair with small dark spots on the head and buttocks. C. ABR tests showed the wild-type pigs had a hearing threshold of 20 dB SPL in Click. D. ABR waveforms could not be induced in the mutant at 90 dB SPL, indicating profound hearing loss. E. Tone-pip ABR thresholds of wild-type and mutant pigs. The mutants remained irresponsive at 1-32 kHz frequencies.
Localization of the mutation to the KIT gene on chromosome 8
A gene mapping analysis was performed on 16 mutants and 17 controls using illumina 60K chips. In total, 61,565 single nucleotide polymorphisms (SNPs) had a call rate >95% and were annotated genomic positions in the Sscrofa10.2 assembly of the Sus scrofa genome. Parametric linkage analysis was performed, and the results indicated that the most significantly associated SNPs localized to a 31 Mb to 48 Mb region of chromosome 8 and the KIT gene (chr8: 43550231-43601377), which is associated with coat color in pigs, is also located on this segment (Figure 2A, 2B). Therefore, KIT was selected as a candidate gene responsible for the deafness and hypopigmentation observed in the mutant lineage.
Figure 2.

Identification of the mutation within the c-KIT gene. A. Manhattan plot showing that the mutation-associated SNPs were all located on chromosome 8. Values of -log10 (observed value) >5.02 (blue horizontal line) are of chromosome-wide significance. B. Linkage analysis for autosomal dominant deafness and hypopigmentation. SNPs with significant linkage were located in the region of 31.0-48.0 Mb on chromosome 8 (LOD=7.8). C. Chromatograms of the genomic DNA sequence of the mutant versus the wild-type control. The open rectangle highlights the T to A transversion in exon 17 of c-KIT (c.2418T>A). D. The candidate mutation leads to a substitution of an aspartate by glutamate at amino acid 806 of the protein (p.D806E).
In order to screen the causative mutation in the KIT gene, all 20 exons and the exon-intron boundaries of KIT were PCR amplified and sequenced. PCR primers were listed in Table S1. A point mutation (T>A) was identified at CDS 2418 bp site on exon 17 which are co-segregated with the mutant phenotype, resulting in a conserved substitution of aspartate by glutamate at amino acid 806 of the protein (p.D806E) (Figure 2C, 2D). The mutation corresponds to position 2517 in exon 17 of the human KIT gene and is located within the tyrosine kinase domain where numbers of mutation were reported with piebaldism. These results strongly suggest that the KIT p.D806E mutation is linked to the deafness and hypopigmentation observed in the dominant inheritance lineage.
Cochlear morphology in KITD806E/+ pigs
Firstly, we compared the CE-HE images of cochlea of KITD806E/+ and KIT+/+ pigs at P1. Figure 3A-D showed the fine cochlear structure of KIT+/+ pigs, including the organ of Corti, Reissner’s membrane (RM), basilar membrane (BM), spiral ganglion neurons (SGNs) and SV. Similar to humans, the cochlea consists of three scala, including the scala vestibule (SVE), the scala media (SM) and the scala tympani (ST). The organ of Corti was lying on BM, and tectorial membrane (TM) was contacting with inner hair cells (IHCs) and outer hair cells (OHCs). However, the stereocilia of hair cells could not be observed clearly in CE-HE sections. No identifiable disformalties of cochlear fine structures were oberseved in KITD806E/+ pigs, no thinner SVs nor underdeveloped hair cells (Figure 3E-H).
Figure 3.
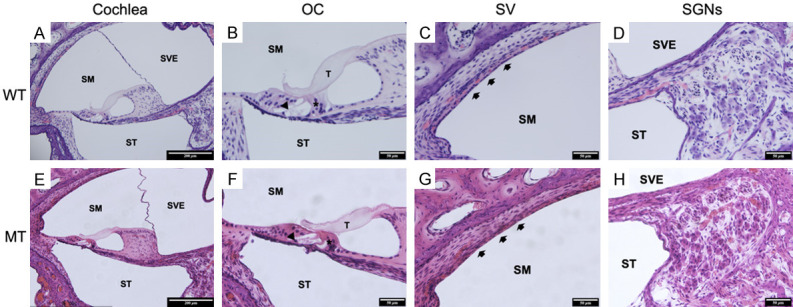
Generally normal structures of KITD806E/+ cochlea. A-D. Cochlea of wild type pigs showed structures similar to humans. A. Section from a wild-type pig (10×). The cochlea consists of three scala, including the scala vestibule (SVE), scala media (SM) and scala tympani (ST). The organ of Corti, Reissner’s membrane (RM), the basilar membrane (BM) and the stria vascularis (SV) are also shown. B. Histology of the organ of Corti from a wild-type pig (40×). The structure of the organ of Corti, including three rows of outer hair cells (OCHs), one row of inner hair cells (IHCs), a pillar cell, a phalangeal cell, a Deiters cell, a Hensen cell, a Claudius cell and a sulcus cell. C. Histology of the SV from a wild-type pig (40×). The SV contains three layers of cells. The thickness of the SV is normal. D. Histology of the spiral ganglion neurons (SGNs) from a wild-type pig (40×). E-H. Cochlea of mutants showed generally normal structures of tectorial membrane (T), inner hair cells (*), outer hair cells (arrow), SV (arrow heads) and SGNs.
Abnormal hair cell morphology in KITD806E/+ pigs
In order to inspect the pathological changes of hair cells through cochlear maturation, we seriesly observed the dynamic hair cells changes at E85, E100 and P1 via SEM. The structures of porcine cochlea were similar to human beings as one row of inner hair cells (IHCs) located in the inner most row toward the central region of the cochlea and three rows of outer hair cells (OHCs) located toward the peripheral region. IHCs and OHCs were separated by the head plate of inner pillar cells. The hair bundles of IHCs are arranged in a line. While the hair bundles of OHCs are positioned in a V-shaped pattern. The structures of hair cells at E85 were well developed (Figure 4A-C) and stayed the same through E100 (Figure 5A-C) and P1 (Figure 6A-C) in wild types. For the mutants, no significant abnormality was observed at E85 (Figure 4D-F). The stereocilia bundles began to degenerated in the basal and middle turns at E100 (Figure 5D-F). Then, pathological changes aggravated from E100 to P1, as stereocilia bundles could not be observed on the cuticular plates of all turns by P1 (Figure 6D-F).
Figure 4.
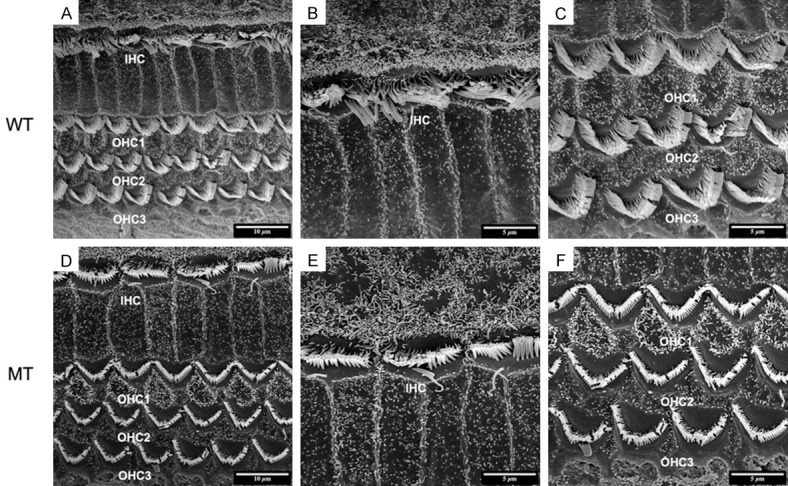
SEM image of basilar membrane at E85. A-C. Normal arranged of inner hair cells (IHC) and three rows of outer hair cells (OHC1, 2, 3) in wild-type pigs. A. Organ of corti are well developed at E85. The bundles of three rows of OHCs and one row of IHCs are regularly arranged on the basilar membrane (5000×). B. The hair bundles of IHCs are arranged in a line (10000×). C. While the hair bundles of OHCs are positioned in a V-shaped pattern. D-F. No significant abnormalities found in mutants.
Figure 5.
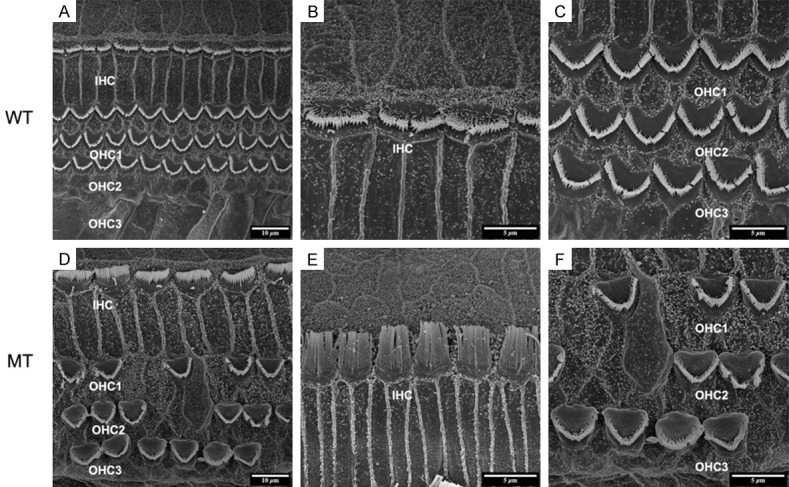
SEM image of basilar membrane at E100. A-C. Normal arranged of inner hair cells (IHC) and three rows of outer hair cells (OHC1, 2, 3) in wild-type pigs as E85. D-F. Stereocilia bundles began to degenerate in the basal and middle turns at E100. D. Missing or fused stereocilias of OHC1-3 in mutants (5000×). E. The hair bundles of IHCs are arranged in a line as E85 (10000×). F. The absence of OHCs and fusion of stereocilias were observed (10000×).
Figure 6.
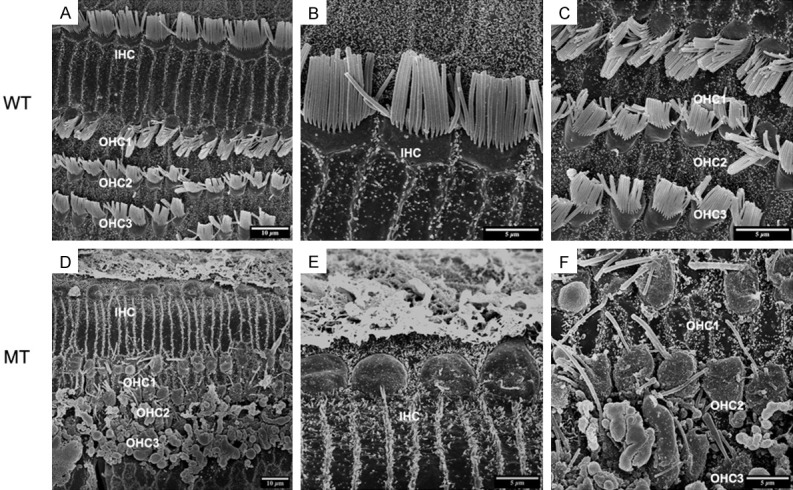
SEM image of basilar membrane at P1. A-C. Normal arranged of inner hair cells (IHC) and three rows of outer hair cells (OHC1, 2, 3) in wild-type pigs as E85 and E100. D-F. Progressively degenerated OHCs and IHCs. D. Deteriorative degeneration of the organ of Corti in the MT cochlea at P1 as absence of inner and outer hair cells was observed (5000×). E. Missing of stereocilias of IHCs (10000×). F. All of the three rows of OHCs were involved (10000×).
Disorganized structures and impaired function of stria vascularis in KITD806E/+ pigs
To investigate the pathological changes of SV in KITD806E/+ pigs, we examined the morphology of the SV by TEM and its function by recording EP. The TEM images revealed ultrastructural differences between the KIT+/+ and KITD806E/+ cochlea. For the wild-types, the SV consisted of three layers: the marginal cell (MC) layer, the intermediate cell (IC) layer and the basal cell (BC) layer. MCs were in the side of the scala media and BC of the spiral ligament. The MCs connected with each other via tight junctions and formed a barrier. Elongated processes of MCs interacted with the ICs and capillaries (Figure 7A-C). In KITD806E/+, ICs were visibly reduced in mutants and three layers of SV were disorganized (Figure 7D-F). EP of the middle turn of wild types was around 60 mV (Figure 8A) and was reduced to nearly 0-2 mV in KITD806E/+ pigs, which were statistically different (P<0.0001) (Figure 8B).
Figure 7.
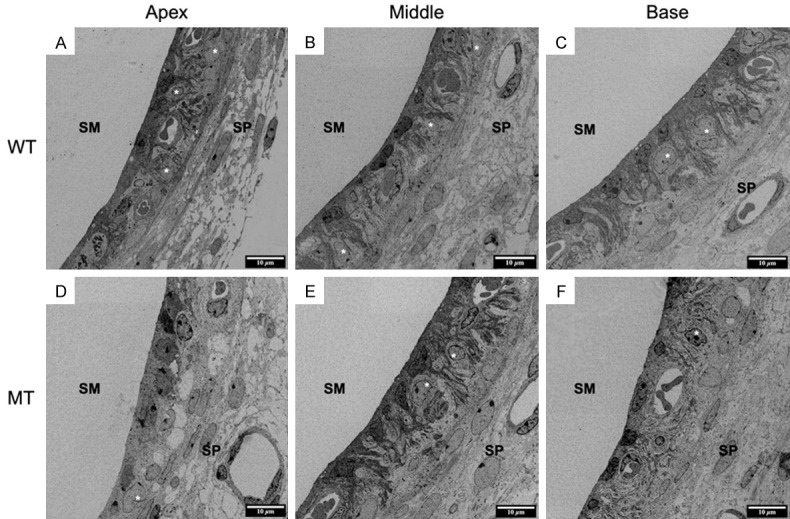
TEM image showed disorganization of SV in KITD806E/+ cochlea. (A-C) Normal structures of apical (A), middle (B) and basal (C) turn of wild type cochlea, consisting of marginal cells (MC), intermediate cells (IC) and basal cells (BC). MC contacted the scala media. And BC attached the spiral ligament. The MCs connected with each other via tight junctions and formed a barrier to block the endolymph. Elongated processes of MCs interacted with the ICs and capillaries. (D-F) Disorganized SVs and reduced ICs in mutants. MCs were aggregated and failed to interact with the ICs and capillaries. And ICs were reduced. (*) refers to intermediate cells.
Figure 8.
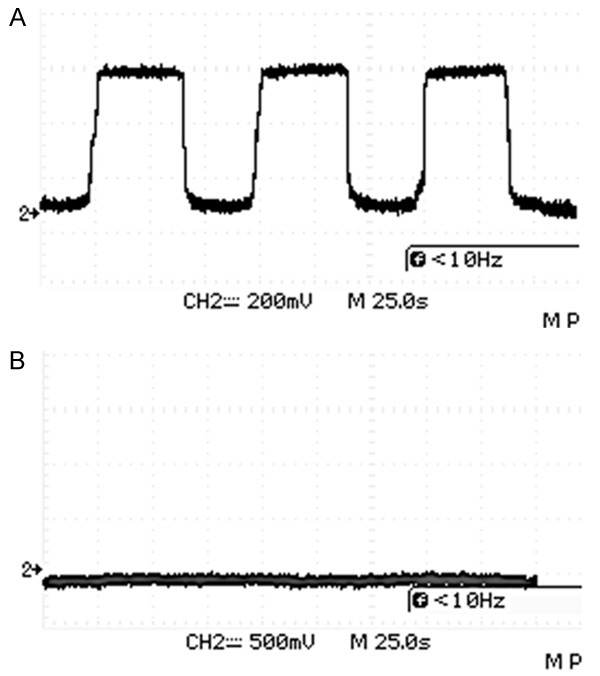
EPs in KITD806E/+ pigs were significantly lower than in wild type pigs. A. Approximately 60 mV of EPs in middle turn of wild type cochlea. B. EPs of mutants close to 0 in the same turn.
Discussion
In this study, we described the phenotype and the morphological changes from embryonic to postnatal stages found in the porcine KITD806E/+ model. The CE-HE images of KIT mutant cochlea was roughly normal, no disrupted hair cells nor disorganized SVs was found. SEM revealed that OHCs and IHCs began to degenerate since E100, and stereocilia bundles were totally disappeared at P1. Further TEM studies revealed reduced ICs and disorganized SV structure. EP at middle turn of KITD806E/+ cochlea was 0-2 mV compared to 60 mV in wild types. Our results demonstrated that the hearing loss in this KITD806E/+ porcine model was primarily caused by the malformation and dysfunction of the SVs and subsequent progressive degeneration of sensory epithelium through inner ear maturation. Our findings of this KITD806E/+ miniature pigs are similar to our previous reports about MITF-M mutant Rongchang porcine model [32,33].
It is known that hearing loss in WS is caused by disorders of melanocytes in the middle layer of the SV called intermediate cells, like abnormal proliferation, survival, migration, or differentiation of neural crest cells [4,5,34]. Various studies have proved that the histopathological changes of WS were primarily reduced number of intermediate cells in the SV both in patients and animal models, then subsequently degeneration of organ of Corti as its secondary alterations [4,34]. In the KITD806E/+ porcine cochlea, the reduction but not absence of intermediate cells in the SV was revealed and decreased EP was recorded. As the progressive apoptosis of hair bundles began at E100, the Reissner’s membrane and cochlear duct did not shown obvious changes after birth. Therefore, the pathological changes of KITD806E/+ pigs are not so severe as MITF-M mutant pigs [32,33].
Mice are mostly used as hereditary deafness model to date. However, the biological differences between mice and humans limit the use of these models in further therapy studies. Bama miniature pigs could be used as a useful model bridge the gap as the high similarity of the genetic and physiological characteristics and their inner ear structural with humans [29-31]. In mice and rats, mutations at KIT, also known as W locus, resulted in depigmentation of the skin and hair as well as deafness. Many mutant strains including W(s), W(v), KIT(wads) mice and W(s) rats had been demonstrated to interrupt development and migration of neural crest-derived melanocytes, resulting in decreased number of intermediate cells and malformation of SV which failed to generate normal EP and subsequently caused hearing loss [23-28]. These pathological changes are in accordance with KITD806E/+ pigs, indicating they might be part of the same disorder. However, unlike other KIT mutant rodents that cochlea-saccular retrogressions were mainly observed at postnatal period, our porcine models and humans shared a similar process that cochlear disorders began at embryonic stages [17,18,23-28]. Likewise, studies on KIT mutation cats and canines showed similar phenotypes as human, but the pathological changes had not been revealed [35-37].
As for the condition in human beings, KIT is a causitive gene of piebaldism, an autosomal dominant disorder that interupts neural crest-derived melanocyte development and migration resulting in congenital white forelock and hypopigmented patches of skin where melanocytes are absent [15,16,38,39]. In sporadic cases, piebaldism has been found to be associated with congenital hearing loss, the same manifestations as WS [17,18,40]. Hulten et al. (1987) described a Pakistani case of presumed homozygous piebaldism. The proband manifested the congenital absence of pigmentation in the eyes, skin and hair, deafness, hypotonia and delayed developmental milestones with his parents both being affected by piebaldism. However the disease locus had not been studied [40]. Kilsby et al. (2013) described another Pakistani case of presumed homozygous piebaldism manifesting albinism and deafness caused by homozygous deletion of exons 20 and 21 in KIT, resulting in loss of the final 77 amino acids of the full 976 amino acid protein. Both of his parents are heterozygotes for this mutation [18]. Spritz et al. (1998) reported a heterozygous mutation of the KIT gene (p.Arg796Gly) in a South African girl of Xhosa stock, presenting piebaldism and congenital profound hearing loss [17]. Hamadah et al. (2019) reported a family with expanded syndrome of piebaldism with p.Ala608Asp induced unilateral deafness in only one patient. The above reported cases with KIT mutations manifested hypopigmentation associated with congenital deafness, overlapping the features of WS, and were analogous to our KITD806E/+ porcine model (align to p.Asp810Glu in human) and all above mutations located in tyrosine kinase domain coding region of KIT (Figure 9).
Figure 9.
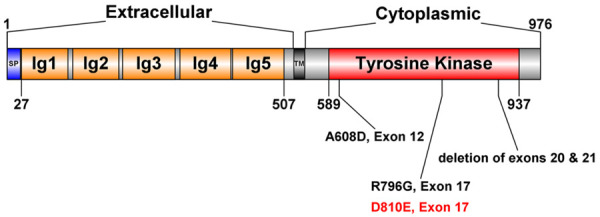
Representation of the structure of KIT, illustrating the reported mutations in patients with hearing loss (A608D, R796G, deletion of exons 20 and 21). SP, signal peptide; TM, transmembrane domain. D810E in human aligns to D806E in pigs (red).
KIT plays a role, partly overlapping MITF, in cell survival, proliferation, differentiation and migration of neural crest cells, which are the precursors of melanocytes, hematopoietic stem cells, germ cells, mast cells and the interstitial cells of Cajal [9,21,41-44]. In humans, gain-of-function mutations in the KIT gene have been associated with many neoplastic diseases. While loss-of-function KIT mutations are associated with piebaldism, due to abnormal development of neural crest-derived melanocytes [11,23,24]. It is not unexpected piebaldism occasionally associated with deafness, since intermediate cells of SV are neural crest-derived. The similar mechanism was confirmed in MITF associated WS of both human and animal models [6,8-11,33,46].
Previous reports indicate that ion channels in the SV facilitate K+ recycling in the cochlea, and help maintain a high level of K+ in the endolymph, which is the fundation of sensory hair cell mechano-transduction. K+ circulation in cochlea consists of following processes: K+ flowing from the endolymph into the perilymph via the sensory hair cells, K+ being uptaken from the perilymph by fibrocytes of the spiral ligament, K+ funneling into the basal and intermediate cells of the SV via gap junctions, K+ outflowing from the intermediate cells to the intrastrial fluid, and K+ being secreted into the endolymph by the marginal cells of the SV. We speculated that the KITD806E/+ mutation disrupted the balance of K+ homeostasis because of the lack of intermediate cells in the SV. As a result, EP was nearly completely absent because of the lack of K+ in the scala media. Moreover, hair cell transduction was inhibited which further impacted the depolarization of hair cells and release of neurotransmitters, ultimately resulting in deafness [47,48].
Furthermore, the absence of intermediate cells of SV would lead to accumulated K+ in intrastrial fluid, resulting in disorders of strial capillaries. This would cause increase of endothelial permeability, and subsequently result in series of changes in ischemia and hypoxia of inner ear [27] which led to the degeneration of well-developed hair cells as we found in E100 KITD806E/+ cohclea.
However, the molecular mechanisms by which KIT mutations causing hereditary deafness remain unknown. KITLG was a newly identified gene associated with WS [13,14]. Reports have shown that the binding of KIT with its ligand KITLG would activate downstream MITF via the Ras-Erk signaling pathway, the PI3 kinase survival pathway and miRNAs [8,24,48-50]. Other studies also showed that MITF was necessary for KIT expression in melanoblasts [7,23]. It was reported that nearly all WS-associated genes would influence MITF expression and function by viarous pathways [5,6,10,12]. As a key role of KITLG/KIT-MITF signaling in neural crest-derived melanocyte development, KIT could be a candidate for WS [6,9,21,45]. Therefore, we speculated that the KITD806E/+ mutation in Bama miniature pigs would affect MITF expression, which led to pigmentation defects and deafness. Additional analysis of the MITF gene and KITLG/KIT-MITF signaling pahtway is underway to better understand its role in the KITD806E/+ deafness model.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (NSFC#81570933; 81670940; 81970897), Key International (Regional) Joint Research Program of National Natural Science Foundation of China (NSFC#81820108009).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Shearer AE, Hildebrand MS, Smith RJ. Hereditary hearing loss and deafness overview. GeneReviews®. 1993 [Google Scholar]
- 2.Nayak CS, Isaacson G. Worldwide distribution of Waardenburg syndrome. Ann Otol Rhinol Laryngol. 2003;112:817–820. doi: 10.1177/000348940311200913. [DOI] [PubMed] [Google Scholar]
- 3.Song J, Feng Y, Acke FR, Coucke P, Vleminckx K, Dhooge IJ. Hearing loss in Waardenburg syndrome: a systematic review. Clin Genet. 2016;89:416–425. doi: 10.1111/cge.12631. [DOI] [PubMed] [Google Scholar]
- 4.Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 5.Tachibana M. A cascade of genes related to Waardenburg syndrome. J Investig Dermatol Symp Proc. 1999;4:126–129. doi: 10.1038/sj.jidsp.5640195. [DOI] [PubMed] [Google Scholar]
- 6.Vandamme N, Berx G. From neural crest cells to melanocytes: cellular plasticity during development and beyond. Cell Mol Life Sci. 2019;76:1919–1934. doi: 10.1007/s00018-019-03049-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, Fisher DZ, Fisher DE. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 2000;14:301–312. [PMC free article] [PubMed] [Google Scholar]
- 8.Alexeev V, Yoon K. Distinctive role of the cKit receptor tyrosine kinase signaling in mammalian melanocytes. J Invest Dermatol. 2006;126:1102–10. doi: 10.1038/sj.jid.5700125. [DOI] [PubMed] [Google Scholar]
- 9.Rönnstrand L. Signal transduction via the stem cell factor receptor/c-Kit. Cell Mol Life Sci. 2004;61:2535–2548. doi: 10.1007/s00018-004-4189-6. [DOI] [PubMed] [Google Scholar]
- 10.Ritter KE, Martin DM. Neural crest contributions to the ear: implications for congenital hearing disorders. Hear Res. 2019;376:22–32. doi: 10.1016/j.heares.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saleem MD. Biology of human melanocyte development, Piebaldism, and Waardenburg syndrome. Pediatr Dermatol. 2019;36:72–84. doi: 10.1111/pde.13713. [DOI] [PubMed] [Google Scholar]
- 12.Hou L, Pavan WJ. Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: do all roads lead to Mitf? Cell Res. 2008;18:1163–1176. doi: 10.1038/cr.2008.303. [DOI] [PubMed] [Google Scholar]
- 13.Zazo Seco C, Serrão De Castro L, Van Nierop JW, Morín M, Jhangiani S, Verver EJ, Schraders M, Maiwald N, Wesdorp M, Venselaar H, Spruijt L, Oostrik J, Schoots J Baylor-Hopkins Center for Mendelian Genomics. Van Reeuwijk J, Lelieveld SH, Huygen PL, Insenser M, Admiraal RJ, Pennings RJ, Hoefsloot LH, Arias-Vásquez A, De Ligt J, Yntema HG, Jansen JH, Muzny DM, Huls G, Van Rossum MM, Lupski JR, Moreno-Pelayo MA, Kunst HP, Kremer H. Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2. Am J Hum Genet. 2015;97:647–660. doi: 10.1016/j.ajhg.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogawa Y, Kono M, Akiyama M. Pigmented macules in Waardenburg syndrome type 2 due to KITLG mutation. Pigment Cell Melanoma Res. 2017;30:501–504. doi: 10.1111/pcmr.12597. [DOI] [PubMed] [Google Scholar]
- 15.Spritz RA, Giebel LB, Holmes SA. Dominant negative and loss of function mutations of the c-kit (mast/stem cell growth factor receptor) proto-oncogene in human piebaldism. Am J Hum Genet. 1992;50:261–269. [PMC free article] [PubMed] [Google Scholar]
- 16.Murakami T, Fukai K, Oiso N, Hosomi N, Kato A, Garganta C, Barnicoat A, Poppelaars F, Aquaron R, Paller AS, Ishii M. New KIT mutations in patients with piebaldism. J Dermatol Sci. 2004;35:29–33. doi: 10.1016/j.jdermsci.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Spritz RA, Beighton P. Piebaldism with deafness: molecular evidence for an expanded syndrome. Am J Med Genet. 1998;75:101–103. doi: 10.1002/(sici)1096-8628(19980106)75:1<101::aid-ajmg20>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 18.Kilsby AJ, Cruwys M, Kukendrajah C, Russell-Eggitt I, Raglan E, Rajput K, Loshe P, Brady AF. Homozygosity for piebaldism with a proven KIT mutation resulting in depigmentation of the skin and hair, deafness, developmental delay and autism spectrum disorder. Clin Dysmorphol. 2013;22:64–67. doi: 10.1097/MCD.0b013e32835e8ce5. [DOI] [PubMed] [Google Scholar]
- 19.Johansson Moller M, Chaudhary R, Hellmén E, Höyheim B, Chowdhary B, Andersson L. Pigs with the dominant white coat color phenotype carry a duplication of the KIT gene encoding the mast/stem cell growth factor receptor. Mamm Genome. 1996;7:822–830. doi: 10.1007/s003359900244. [DOI] [PubMed] [Google Scholar]
- 20.Yarden Y, Kuang WJ, Yang-Feng T, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hemesath TJ, Price ER, Takemoto C, Badalian T, Fisher DE. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature. 1998;391:298–301. doi: 10.1038/34681. [DOI] [PubMed] [Google Scholar]
- 22.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature. 1988;335:88–89. doi: 10.1038/335088a0. [DOI] [PubMed] [Google Scholar]
- 23.Cable J, Barkway C, Steel KP. Characteristics of stria vascularis melanocytes of viable dominant spotting (Wv Wv) mouse mutants. Hear Res. 1992;64:6–20. doi: 10.1016/0378-5955(92)90164-i. [DOI] [PubMed] [Google Scholar]
- 24.Kitamura K, Sakagami M, Umemoto M, Takeda N, Doi K, Kasugai T, Kitamura Y. Strial dysfunction in a melanocyte deficient mutant rat (ws/ws rat) Acta Otolaryngol. 1994;114:177–181. doi: 10.3109/00016489409126038. [DOI] [PubMed] [Google Scholar]
- 25.Cable J, Huszar D, Jaenisch R, Steel KP. Effects of mutations at the W locus (c-kit) on inner ear pigmentation and function in the mouse. Pigment Cell Res. 1994;7:17–32. doi: 10.1111/j.1600-0749.1994.tb00015.x. [DOI] [PubMed] [Google Scholar]
- 26.Hoshino T, Mizuta K, Gao J, Araki S, Araki K, Takeshita T, Wu R, Morita H. Cochlear findings in the white spotting (Ws) rat. Hear Res. 2000;140:145–156. doi: 10.1016/s0378-5955(99)00192-6. [DOI] [PubMed] [Google Scholar]
- 27.Fujimura T, Suzuki H, Shimizu T, Tokui N, Kitamura T, Udaka T, Doi Y. Pathological alterations of strial capillaries in dominant white spotting W/Wv mice. Hear Res. 2005;209:53–59. doi: 10.1016/j.heares.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 28.Ruan HB, Zhang N, Gao X. Identification of a novel point mutation of mouse proto-oncogene c-kit through N-ethyl-N-nitrosourea mutagenesis. Genetics. 2005;169:819–831. doi: 10.1534/genetics.104.027177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi HJ, Guo W, Wu N, Li JN, Liu HZ, Ren LL, Liu PN, Yang SM. The temporal bone microdissection of miniature pigs as a useful large animal model for otologic research. Acta Oto-Laryngologica. 2014;134:26–33. doi: 10.3109/00016489.2013.835866. [DOI] [PubMed] [Google Scholar]
- 30.Guo W, Yi H, Ren L, Chen L, Zhao L, Sun W, Yang SM. The morphology and electrophysiology of the cochlea of the miniature pig. Anat Rec (Hoboken) 2015;298:494–500. doi: 10.1002/ar.23095. [DOI] [PubMed] [Google Scholar]
- 31.Guo W, Yi H, Yan Z, Ren L, Chen L, Zhao LD, Ning Y, He DZZ, Yang SM. The morphological and functional development of the stria vascularis in miniature pigs. Reprod Fertil Dev. 2017;29:585–593. doi: 10.1071/RD15183. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Guo W, Ren L, Yang M, Zhao Y, Guo Z, Yi H, Li M, Hu Y, Long X, Sun B, Li J, Zhai S, Zhang T, Tian S, Meng Q, Yu N, Zhu D, Tang G, Tang Q, Ren L, Liu K, Zhang S, Che T, Yu Z, Wu N, Jing L, Zhang R, Cong T, Chen S, Zhao Y, Zhang Y, Bai X, Guo Y, Zhao L, Zhang F, Zhao H, Zhang L, Hou Z, Zhao J, Li J, Zhang L, Sun W, Zou X, Wang T, Ge L, Liu Z, Hu X, Wang J, Yang S, Li N. A de novo silencer causes elimination of MITF-M expression and profound hearing loss in pigs. BMC Biol. 2016;14:1–15. doi: 10.1186/s12915-016-0273-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen W, Hao QQ, Ren LL, Ren W, Lin HS, Guo WW, Yang SM. Cochlear morphology in the developing inner ear of the porcine model of spontaneous deafness. BMC Neurosci. 2018;19:1–8. doi: 10.1186/s12868-018-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tachibana M, Kobayashi Y, Matsushima Y. Mouse models for four types of Waardenburg syndrome. Pigment Cell Res. 2003;16:448–454. doi: 10.1034/j.1600-0749.2003.00066.x. [DOI] [PubMed] [Google Scholar]
- 35.Greibrokk T. Hereditary deafness in the Dalmatian: relationship to eye and coat color. J Am Anim Hosp Assoc. 1994;30:170–176. [Google Scholar]
- 36.Strain GM. Deafness prevalence and pigmentation and gender associations in dog breeds at risk. Vet J. 2004;167:23–32. doi: 10.1016/s1090-0233(03)00104-7. [DOI] [PubMed] [Google Scholar]
- 37.David VA, Menotti-Raymond M, Wallace AC, Roelke M, Kehler J, Leighty R, Eizirik E, Hannah SS, Nelson G, Schäffer AA, Connelly CJ, O’Brien SJ, Ryugo DK. Endogenous retrovirus insertion in the KIT oncogene determines White and White spotting in domestic cats. G3 (Bethesda) 2014;4:1881–1891. doi: 10.1534/g3.114.013425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ezoe K, Holmes SA, Ho L, Bennett CP, Bolognia JL, Brueton L, Burn J, Falabella R, Gatto EM, Ishii N, Moss C, Pittelkow MR, Thompson E, Ward KA, Spritz RA. Novel mutations and deletions of the KIT (steel factor receptor) gene in human piebaldism. Am J Hum Genet. 1995;56:58–66. [PMC free article] [PubMed] [Google Scholar]
- 39.Hamadah I, Chisti M, Haider M, Al Dosssari H, Alhumaidan R, Meyer BF, Wakil SM. A novel KIT mutation in a family with expanded syndrome of piebaldism. JAAD Case Reports. 2019;5:627–631. doi: 10.1016/j.jdcr.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hulten MA, Honeyman MM, Mayne AJ, Tarlow MJ. Homozygosity in piebald trait. J Med Genet. 1987;24:568–571. doi: 10.1136/jmg.24.9.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 42.Besmer P, Manova K, Duttlinger R, Huang EJ, Packer A, Gyssler C, Bachvarova RF. The kit-ligand (steel factor) and its receptor c-kit/W: Pleiotropic roles in gametogenesis and melanogenesis. Development. 1993;119:125–137. [PubMed] [Google Scholar]
- 43.Cable J, Jackson IJ, Steel KP. Mutations at the W locus affect survival of neural crest-derived melanocytes in the mouse. Mech Dev. 1995;50:139–150. doi: 10.1016/0925-4773(94)00331-g. [DOI] [PubMed] [Google Scholar]
- 44.MacKenzie MA, Jordan SA, Budd PS, Jackson IJ. Activation of the receptor tyrosine kinase kit is required for the proliferation of melanoblasts in the mouse embryo. Devel Biol. 1997;192:99–107. doi: 10.1006/dbio.1997.8738. [DOI] [PubMed] [Google Scholar]
- 45.Phung B, Sun J, Schepsky A, Steingrimsson E, Rönnstrand L. C-KIT signaling depends on microphthalmia-associated transcription factor for effects on cell proliferation. PLoS One. 2011;6:e24064. doi: 10.1371/journal.pone.0024064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aoki H, Tomita H, Hara A, Kunisada T. Conditional deletion of kit in melanocytes: white spotting phenotype is cell autonomous. J Invest Dermatol. 2015;135:1829–1838. doi: 10.1038/jid.2015.83. [DOI] [PubMed] [Google Scholar]
- 47.Chen J, Zhao HB. The role of an inwardly rectifying K+ channel (Kir4.1) in the inner ear and hearing loss. Neuroscience. 2014;265:137–146. doi: 10.1016/j.neuroscience.2014.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Locher H, de Groot JC, van Iperen L, Huisman MA, Frijns JH, Chuva de Sousa Lopes SM. Development of the stria vascularis and potassium regulation in the human fetal cochlea: insights into hereditary sensorineural hearing loss. Dev Neurobiol. 2015;75:1219–40. doi: 10.1002/dneu.22279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roskoski R. Structure and regulation of Kit protein-tyrosine kinase - the stem cell factor receptor. Biochem Biophys Res Commun. 2005;338:1307–1315. doi: 10.1016/j.bbrc.2005.09.150. [DOI] [PubMed] [Google Scholar]
- 50.Rachmin I, Fisher DE. Microphthalmia-associated transcription factor phosphorylation: cross talk between GSK3 and MAPK signaling. Pigment Cell Melanoma Res. 2019;32:345–347. doi: 10.1111/pcmr.12765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.