

Abstract
Cyclic RGD peptides are well‐known ligands of integrins. The integrins αVβ3 and α5β1 are involved in angiogenesis, and integrin αVβ3 is abundantly present on cancer cells, thus representing a therapeutic target. Hence, synthetic and biophysical studies continuously are being directed towards the understanding of ligand‐integrin interaction. In this context, the development of versatile synthetic strategies to obtain fluorescent building blocks that can add molecular diversity and modular spectral characteristics while not compromising binding affinity or selectivity is a relevant task. An on‐resin intramolecular Suzuki–Miyaura cross‐coupling (SMC) between l‐ or d‐7‐bromotryptophan (7BrTrp) and a phenothiazine (Ptz) boronic acid affords fluorescent cyclic RGD pseudopeptides, c(RGD(W/w)Ptz). Ring closure by SMC establishes a phenothiazine–indole moiety with axial chirality. An array of eight novel compounds has been synthesized, among them one fluorescent compound with good affinity to integrin αVβ3. The fluorescence properties of the analogues can be efficiently tuned depending on the substituents in Ptz moiety even for fluorescence emission in the visible (red) spectral range.
Keywords: cross-coupling reactions, cyclopeptides, fluorescent peptide, integrin, phenothiazine
Shiny RGD peptides: Cyclic peptides containing the tripeptide sequence Arg‐Gly‐Asp (RGD) have been equipped with a phenothiazine‐indole biaryl moiety by Suzuki–Miyaura cross‐coupling. Modulation of the spectroscopic properties by substituents at the phenothiazine provides blue and red fluorescent RGD derivatives that selectively bind to integrin αVβ3 in the best case with nanomolar affinity.
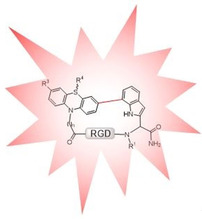
Introduction
Integrins are heterodimeric transmembrane glycoproteins, which mediate cell adhesion and migration phenomena.1 Several integrins recognize the sequence ‐Arg‐Gly‐Asp‐ in their ligands of the extracellular matrix. Peptides containing the RGD sequence have been shown to selectively bind to different integrins,2, 3 especially αVβ3 and α5β1, among others. In particular, αVβ3 is overexpressed in cancer cells, thus playing a pivotal role in controlling tumor angiogenesis and metastasis.4 Therefore, targeting αVβ3 has been showing promise to treat cancer.5, 6, 7, 8 The cyclization of RGD peptides was proven to be an efficient way of increasing their activity and selectivity towards different integrins by restricting their conformational space.9, 10 Cyclization also is an amenable strategy to improve metabolic stability.11, 12, 13, 14, 15, 16 Cilengitide c(RGDf(NMe)V), a small RGD containing cyclic peptide ligand has been the most successful example so far. Although Cilengitide failed in the first phase III trial for the treatment of glioblastoma, it is currently in phase II for treatment of other cancer types.17, 18 Noteworthy, Cilengitide shows promise when conjugated to radiopharmaceuticals.19 Proteins with RGD motif also show potential for selective integrin targeting.20, 21 Despite the relevance of the RGD sequence in cancer research, its mechanism of interaction with integrins is not fully understood. Therefore, synthetic as well as biophysical efforts are being directed towards the development of functional analogues. Fluorophor modification, while retaining bioactivity would allow their use in vitro and in vivo.22, 23, 24, 25, 26 In this regard, different fluorescent moieties have been incorporated in the cyclic RGD‐peptides as lateral chains such as derivatives of fluorescein (FITC),27 rhodamine (Cys5.5) and carbocyanine (cypate).28, 29 The use of fluorescent α‐amino acids like Trp is a general strategy for incorporation of a fluorescent probe in a peptide backbone, but its implementation is restricted owing to its low fluorescence emission intensity.30, 31, 32 We recently disclosed the synthesis of different Boc‐protected aryl tryptophan derivatives by Suzuki–Miyaura cross‐coupling in solution,33 and styryl tryptophan derivatives utilizing Mizoroki–Heck cross‐coupling.34 The use of an intramolecular Suzuki–Miyaura reaction performed on‐resin to form Trp‐Trp cyclic peptides showed the potential to improve biological properties by extending the π‐system.35 Moreover, Suzuki–Miyaura cross‐coupling resulted in a side chain‐to‐tail‐cyclized RGD peptides with biaryl moieties, providing a new dimension of structure–activity relationships.36 We additionally envisaged exploring the use of intramolecular Suzuki–Miyaura cross‐coupling on resin between 7‐bromotryptophan and phenothiazine (Ptz) boronic acids to build novel bioactive and fluorescent cyclic RGD derivatives, c(RGD(W/w)(7‐3‐Ptz)) (Scheme 1).
Scheme 1.

Proposed synthetic building block to obtained novel fluorescent (RGD(W/w)(7‐3‐Ptz)) derivatives through intramolecular C−C Suzuki–Miyaura cross‐coupling.
This approach would considerably increase the π‐system forming a Trp‐Ptz biaryl. Ptz has been previously applied in biochemistry as a marker for proteins, DNA, or other biomolecules.37 Since there are different reactive positions in phenothiazine (3, 7, S, ‐NH), they can be easily functionalized with different substituents, which are also compatible with Fmoc solid‐phase synthesis.38, 39, 40, 41, 42, 43, 44 Therefore, the tuning of the fluorescent and binding properties of the novel cyclic RGD pseudopeptides would be possible on demand. Ptz boronic acids were incorporated at the N‐terminus of the RGD sequence, while the l‐ or d‐7‐BrTrp residues were placed at the C‐terminus. Upon intramolecular Suzuki–Miyaura cross‐coupling on the resin a series of cyclic fluorescent (RGD(W/w)(7‐3‐Ptz)) peptides was obtained. Their binding to integrins αVβ3 and α5β1 was studied in vitro to elucidate the influence of the Trp‐Ptz fluorescent probe on the activity of the RGD peptides.
Results and Discussion
First, three new phenothiazine boronic acid derivatives (Ptz1, Ptz2, and Ptz3) were synthesized by borylation with B2Pin2 (Scheme 2).45 The spacer length between the phenothiazine nitrogen and amide can be varied to influence the overall conformation and, hence, was expected to modulate the affinity of the cyclic RGD‐peptides to the integrins. For that purpose, one methylene unit (Ptz1) or five methylene units (Ptz2) were introduced. In the case of compound 7, the order of the reaction steps was reversed because otherwise decarboxylation was observed during the borylation reaction. In all cases the reaction started from 3‐bromo‐10H‐phenothiazine 1. Compounds 2 and 5 were synthesized by nucleophilic substitution of bromide in 6‐bromohexanenitrile or ethyl bromoacetate, respectively, by 1. Hydrolysis of the nitrile or the ester group, resp., under basic conditions afforded the corresponding acids 3, 7, and 9.46
Scheme 2.
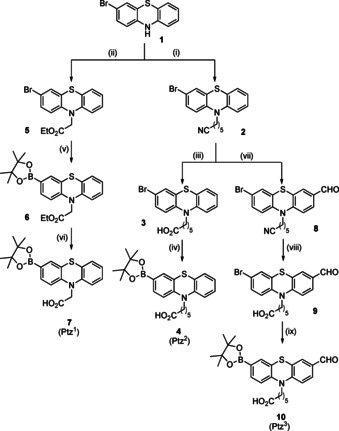
Reagents and conditions for the synthesis of Ptz1, Ptz2 and Ptz3: (i) 6‐bromohexanenitrile, KOH, NaI, DMF, 50 °C, 48 h, 71 %; (ii) Ethyl bromoacetate, KOH, NaI, DMF, 50 °C, 48 h, 46 %; (iii) MeOH/EtOH/KOH (4 m), reflux, 72 h, 72 %; (iv) & (v) B2Pin2 (2.2 equiv), PdCl2(PPh3)2 (0.02 equiv), KOAc (5 equiv), dry toluene (40 mL), 110 °C, Ar, 18 h, 70 %; (vi) MeOH/EtOH/KOH (eq) (4 m), 60 °C, 30 min, 33 %; (vii) DMF (1.1 equiv), 1,2‐DCE, POCl3 (1.2 equiv), 90 °C, 48 h, 46 %;42 (viii) MeOH/EtOH/KOH (4 m), reflux, 72 h, 29 %; (ix) B2Pin2 (2.2 equiv), PdCl2(PPh3)2 (0.02 equiv), KOAc (5 equiv), dry toluene (40 mL), 110 °C, Ar, 18 h, 93 %.
The formyl phenothiazine derivative (Ptz3, 10) was synthesized by Vilsmeier–Haack reaction using POCl3 and DMF.32 The incorporation of the aldehyde group in position 10 of the phenothiazine moiety would allow enlarging the conjugated π‐system, for example, by a Knoevenagel condensation causing a distinct bathochromic shift. Therefore, we expect that such derivatives could be used in vivo experiments.
The l‐ and d‐7‐bromotryptophan derivatives were regioselectively obtained by enzymatic halogenation using immobilized cross‐linked enzyme aggregates (combiCLEAs) containing a FAD‐dependent tryptophan 7‐halogenase, as described previously.47 After Fmoc protection of 7‐bromotryptophan, it was loaded as the first amino acid on the Rink amide resin followed by assembly of the R(Pbf)GD(OtBu) sequence by SPPS using Fmoc chemistry and TBTU as the coupling reagent. Coupling of the corresponding phenothiazine derivatives was accomplished using in situ activation with Oxyma and DIC. The mixture was added to the resin in DMF and TMP and was shaken overnight, providing the linear peptides on the resin (11 a–g). In addition to the unmodified l‐ and d‐7‐bromotryptophan derivatives, the N‐methyl derivatives were incorporated, too, providing further structural diversity (Scheme 3).
Scheme 3.
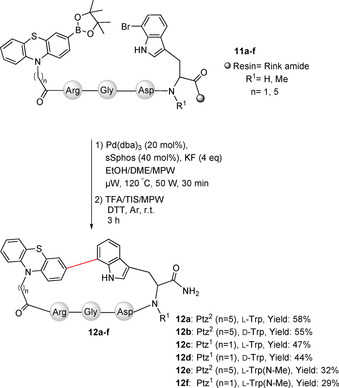
Peptide cyclization through Suzuki–Miyaura cross‐coupling.
The intramolecular Suzuki–Miyaura cross‐coupling was performed in a mixture of EtOH/DME/MPW (9:9:1), which was degassed in freeze–pump–thaw cycles. Pd(dba)3 (20 mol %) together with sSphos (sodium 2′‐dicyclohexylphosphino‐2,6‐dimethoxy‐1,1′‐biphenyl‐3‐sulfonate hydrate, 40 mol %) was used as catalyst, while KF was selected as the base. The reaction was activated by microwave at 120 °C by adaptation of a literature protocol (Scheme 3).48 Malononitrile and ammonium acetate were added to accomplish the on resin‐Knoevenagel condensation providing 12 g (Scheme 4).
Scheme 4.
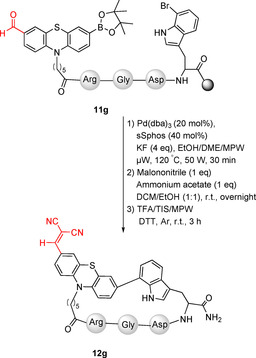
Synthesis of cyclopeptide 12 g with an additional electron‐withdrawing moiety by sequential on resin Suzuki/Knoevenagel reactions.
In the 2D‐NMR characterization, we detected that for some derivatives particular conformations in the (RGD(W/w)(7‐3‐Ptz) cycle were stabilized. Previously, Hoveyda et al. have shown that cyclopeptides with indole‐aryl moiety lead to atropisomerism, P and M, in the strained macrocycle, which reduces the rotation around the single bond of the biaryl moiety.49
Although it is a difficult task which depends on conformational stability, the atropisomers could be identified by NMR. In this work the P‐isomers could be successfully isolated as major atropisomers in the case of 12 c–f and characterized by NOESY experiments. The chemical shifts below 5 ppm of the α‐proton of tryptophan in the rigid cyclopeptides 12 c–f suggest a P‐biaryl isomer (Figure 1 and Supporting Information).49 NOE proximities were found between the NH1 and H2 of indole and both the α‐proton of aspartic acid and the amide proton of tryptophan, suggesting a more constrained cycle with the indole ring facing inside. It seems that the rigidity of the structure in the smaller cyclopeptides 12 c,d and those with N‐methylated tryptophan 12 e,f confers the observed conformational stability. On the other hand, for compounds 12 a,b containing a longer and more flexible linker, separate signals for the atropisomers were not observed.
Figure 1.
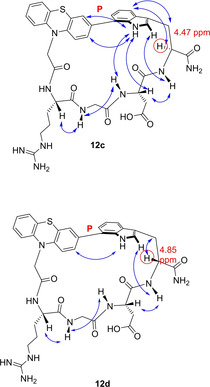
Major atropisomers of compounds 12 c, 12 d, according to 2D NMR (ROESY).
In summary, among eight new cyclopeptides were synthetized. The derivatives 12 a–c were obtained in moderate yields (44–58 %). Lower yields (29–32 %) were observed for the N‐methyl derivatives 12 e,f, where N‐methylation of tryptophan was performed on resin,47 presumably because of the lower reactivity of the secondary amine. In some cases (12 c–f) good diastereoselectivity was observed (dr: 85:15–92.5:7.5, Table 1) and the corresponding P‐atropisomers were isolated.
Table 1.
Sequences, yields and IC50 values of the cyclic RGD‐peptides.
|
Cyclopeptide |
Yield (dr) |
IC50 (αVβ3)[a] [μm] |
IC50 (α5β1)[a] [μm] |
|---|---|---|---|
|
c(RGDW(7‐3Ptz2)) 12 a |
58 % (–) |
0.33±0.04 |
n.d. |
|
c(RGDw(7‐3Ptz2)) 12 b |
55 % (–) |
0.60±0.11 |
5.68±1.28 |
|
c(RGDW(7‐3Ptz1)) 12 c |
47 % (92.5:7.5) |
0.31±0.08 |
2.22±1.04 |
|
c(RGDw(7‐3Ptz1)) 12 d |
44 % (91.5:8.5) |
0.78±0.17 |
>10 |
|
c(RGD(NMe)W(7‐3Ptz2)) 12 e |
32 % (92.5:7.5) |
6.55±0.76 |
n.d. |
|
c(RGD(NMe)W(7‐3Ptz1)) 12 f |
29 % (85:15) |
4.52±0.53 |
>10 |
|
c(RGDw(7‐3Ptz2(CH(CN)2)) 12 g |
18 % (–) |
0.961±0.05 |
>10 |
|
c(RGDW(7‐3Ptz1(O))) 12 h |
16 % (–) |
0.052±0.009 |
5.93±0.89 |
|
c(RGDf(NMe)V) (Cilengitide) |
– |
0.0005±0.0002 |
0.0154±004 |
[a] Experimentally obtained by Elisa test (see Supporting Information). n.d: not determined
Finally, we explored the effect of the sulfoxide group in the binding properties to integrin.50 For that purpose, the P‐cyclopeptide 12 c was oxidized with 5 % hydrogen peroxide solution (1 mg mL−1) at room temperature obtaining 12 h a mixture 1:1 of diastereomeric sulfoxides (R and S) (Scheme 5).
Scheme 5.
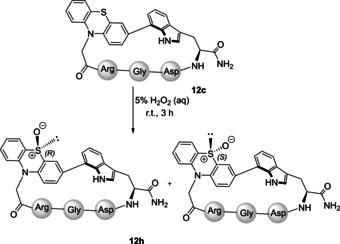
Oxidation of cyclopeptide c(RGDW(7‐3Ptz2)) 12 c to give the sulfoxide 12 h.
The binding affinities of the new cyclopeptides 12 a–h to the integrins αvβ3 and α5β1 were evaluated in a homogenous ELISA‐like solid‐phase binding assay as previously described, using Cilengitide as the positive control.51 In all cases the binding affinity to αvβ3 was higher than to α5β1 (Table 1). The highest affinity was obtained for cyclopeptide 12 h as a mixture of the R and S diastereomers (IC50=52 nm) with a good selectivity towards the integrin αVβ3 (≈110‐fold). The analogues with a shorter spacer Ptz1 12 c,d had similar affinities as the ones with the longer spacer Ptz2 12 a,b, respectively. According to the docking the spacer was placed toward the outside of the binding pocket and so it could not have an effect on affinity and it just influenced the rigidity of the compounds. The N‐methyl tryptophan derivatives 12 e,f displayed a 15–20‐fold decrease in the affinities compared to their unmethylated analogues 12 a–c (Table 1). Methyl substitution affects the peptide backbone conformation and hydrogen bonding ability, presumably leading to a less favorable orientation in the binding site. Interestingly, the l‐Trp derivatives 12 a–c have a 2‐fold higher binding affinity than the d‐Trp derivatives 12 b–d.
To rationalize the difference in binding affinity of the new derivatives in comparison to Cilengitide, and the effect of the oxidized derivative 12 h in comparison to its precursor 12 c, we performed docking and modeling studies.50, 51 Docking of Cilengitide to αVβ3 was performed and the resulting complex was similar to the Cilengitide‐αVβ3 X‐ray single‐crystal structure (Figure 2 A, PDB: 1l5g, Figure S51A).
Figure 2.
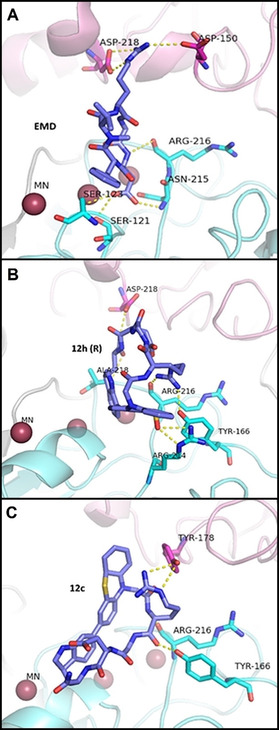
Model for the interaction between integrin αvβ3 and Cilengitide (A), R‐12 h (B), and 12 c (C) P‐atropisomers. The subunits are represented as follows: α‐ subunit (pink) and β‐subunit (light blue). All the inhibitors interact in the RGD‐binding pocket at the interface between α‐and β‐subunits. In the case of Cilengitide, the model of the crystal structure is presented. The corresponding Yasara homology model for Cilengitide interaction, as well as the superimposition between structures, are shown in the Figure S51.
When Cilengitide binds to αVβ3 (Figure 2 A), binding occurs at the interface between subunits αV and β3.51, 52 While the Arg and Asp side chains point into opposite directions making contacts with αV and β3, resp., the glycine residue lies at the interface between both subunits. In the Cilengitide‐αVβ3 complex, the Arg side chain is located in a narrow groove at the top of the propeller domain of the αV chain, where the guanidinium group is being held in place by a bidentate salt bridge to Asp218 and Asp150. On the other hand, the Asp group is completely buried in the complex.
Asp interacts with the β3 subunit through a hydrogen bond with the amide backbone of Tyr122 and Asn215 and contacts the aliphatic portion of Arg214. Additional contacts involve the hydrophobic portion of the Asp side chain and Cβ of Asn215. The glycine moiety makes several hydrophobic interactions with the αV subunit; the most critical one is the contact with the amide oxygen of Arg216.23, 53
Docking of P‐atropisomers 12 h (oxidized) and 12 c (non‐oxidized) revealed that both compounds bind at the same site as Cilengitide (Figure 2, and Figure S51), but the poses and the contacts with the receptor are different, which might explain the decreased affinity. The Arg moiety in R‐12 h is rotated towards the β‐subunit having an interaction with Tyr166 and Arg216 while the oxygen of the sulfoxide group interacts with Arg214. Trp is oriented towards the α‐subunit and interacts with Asp218 and Ala218.
In the case of the non‐oxidized 12 c, only two interactions were detected; one between the Arg moiety and Tyr178 of the α‐subunit, while the second between the Asp and Arg216 in the β‐subunit (Figure 2 C).
Docking of 12 f that displays low affinity to integrins, showed only one contact with Asn215 that is in agreement with the higher IC50 values (Table 1 and Figure SI52). As a conclusion, the sulfoxide group in R‐12 h favors the interaction of Ptz and Trp residues with the receptor. These stabilizing contacts increase the binding affinity towards αVβ3 integrin in the same pocket but through different interactions compared to Cilengitide.
Finally, the spectral characteristics of 12 h, its precursor 12 c, and the derivative 12 g were evaluated in DMSO because of the low solubility of synthesized compounds in water. (Figure 3). As expected, the absorption of Ptz‐RGD derivatives strongly depends on the substitution pattern. Compound 12 h with the oxidized Ptz (sulfoxide) gives rise to two intense absorptions at 286 and 308 nm while the UV spectrum of 12 c has a maximum at 262 nm and a second, weaker absorption band at 308 nm. Compound 12 g, which has an extended π‐system in position 10 of the Ptz shows two absorption bands at 268 and 318 nm, together with a new absorption band at 486 nm (Figure 3 A).
Figure 3.
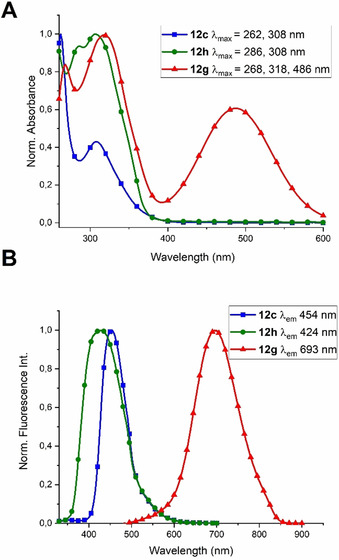
Spectral evaluation of cyclopeptides 12 c, 12 h, and 12 g in dimethyl sulfoxide. A) Normalized UV/Vis absorption spectra. B) Normalized fluorescence emission intensity spectra. All compounds were measured under the same experimental conditions with varying their excitation wavelength as follows: 12 c (λex=311 nm); 12 h (λex=312 nm), and 12 g (λex=489 nm).
The three compounds displayed interesting fluorescent properties (Figure 3 B). Importantly, 12 g emits in the red region (693 nm) upon excitation at 489 nm, while excitation of 12 c and 12 h at around 310 nm leads to fluorescence emission at 424 and 454 nm, respectively (Figure 3 B). These experiments demonstrate that RGD peptides containing the Ptz‐Trp moiety are suitable compounds to be used in spectroscopic experiments in the visible region and, more importantly, show promise for applications in in vivo imaging in the case of compound 12 g.
Conclusions
We have synthesized and characterized novel bioactive fluorescent RGD cyclopeptides by incorporating phenothiazine and 7‐bromotryptophan. They display good to moderate affinity to integrin αvβ3, and intriguing fluorescence properties. The affinity towards the αvβ3 integrin is higher than to α5β1 with moderate to good selectivity. The two diastereoisomers of the sulfoxide‐containing inhibitor 12 h gave a 52 nm IC50 value for binding to integrin αvβ3. Based on the in silico docking results, R‐12 h involves the sulfoxide oxygen in binding to the integrin. Extending the π‐system of the phenothiazine moiety provides a bioactive RGD peptide with a fluorescence emission at 693 nm (λex=486 nm) that may be suitable for in vivo imaging.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully acknowledge the award of a Georg Forster Fellowship of the Alexander von Humboldt Foundation, Germany to E.G. Open access funding enabled and organized by Projekt DEAL.
E. Ghabraie, I. Kemker, N. Tonali, M. Ismail, V. I. Dodero, N. Sewald, Chem. Eur. J. 2020, 26, 12036.
References
- 1. Nieberler M., Reuning U., Reichart F., Notni J., Wester H. J., Schwaiger M., Weinmüller M., Räder A., Steiger K., Kessler H., Cancers 2017, 9, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Samanen J., Ali F., Romoff T., Calvo R., Serrenson E., Vasko J., Storer B., Berry D., Bennett D., Strohsacker M., Powers D., Stadel J., Nichols A., J. Med. Chem. 1991, 34, 3114–4125. [DOI] [PubMed] [Google Scholar]
- 3. Gurrath M., Müller G., Kessler H., Aumailley M., Timpl R., Eur. J. Biochem. 1992, 210, 911–921. [DOI] [PubMed] [Google Scholar]
- 4. Zitzmann S., Ehemann V., Schwab M., Cancer Res. 2002, 62, 5139–5143. [PubMed] [Google Scholar]
- 5. Hwang R., Varner J. V., Hematol. Oncol. Clin. North Am. 2004, 18, 991–1006. [DOI] [PubMed] [Google Scholar]
- 6. Bello L., Francolini M., Marthyn P., Zhang J. P., Carroll R. S., Nikas D. C., Strasser J. F., Villani R., Cheresh D. A., Black P. M., Neurosurgery 2001, 49, 380–389. [DOI] [PubMed] [Google Scholar]
- 7. Jin H., Varner J., Brit. J. Cancer 2004, 90, 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dayam R., Aiello F., Deng J., Wu Y., Garofalo A., Chen X., Neamati N., J. Med. Chem. 2006, 49, 4526–4534. [DOI] [PubMed] [Google Scholar]
- 9. Aumailley M., Gurrath M., Müller G., Calvete J., Timpl R., Kessler H., FEBS Lett. 1991, 291, 50–54. [DOI] [PubMed] [Google Scholar]
- 10. Haubner R., Finsinger D., Kessler H., Angew. Chem. Int. Ed. Engl. 1997, 36, 1374–1389; [Google Scholar]; Angew. Chem. 1997, 109, 1440–1456. [Google Scholar]
- 11.
- 11a. Chatterjee J., Rechenmacher F., Kessler H., Angew. Chem. Int. Ed. 2013, 52, 254–269; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 268–283; [Google Scholar]
- 11b. Becker A., von Richter O., Kovar A., Scheible H., van Lier J. J., Johne A., J. Clin. Pharmcol. 2015, 55, 815–824. [DOI] [PubMed] [Google Scholar]
- 12. Koivunen E., Wang B., Ruoslahti E., Nat. Biotechnol. 1995, 13, 265–270. [DOI] [PubMed] [Google Scholar]
- 13. Assa-Munt N., Jia X., Laakkonen P., Ruoslahti E., Biochemistry 2001, 40, 2373–2378. [DOI] [PubMed] [Google Scholar]
- 14. Burkhart D. J., Kalet B. T., Coleman M. P., Post G. C., Koch T. H., Mol. Cancer. Ther. 2004, 3, 1593–1604. [PubMed] [Google Scholar]
- 15. Kolhar P., Kotamraju V. R., Hikita S. T., Clegg D. O., Ruoslahti E., J. Biotechnol. 2010, 146, 143–146. [DOI] [PubMed] [Google Scholar]
- 16. Hwang D. S., Sim S. B., Cha H., Biomaterials 2007, 28, 4039–4046. [DOI] [PubMed] [Google Scholar]
- 17. Marelli U. K., Rechenmacher F., Sobahi T. R. A., Mas-Moruno C., Kessler H., Front. Oncol. 2013, 3, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Bretschi M., Cheng C., Witt H., Dimitrakopoulou-Strauss A., Strauss L. G., Semmler W., Bauerle T., J. Cancer Res. Clin. Oncol. 2013, 139, 573–583; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Bretschi M., Merz M., Komljenovic D., Berger M. R., Semmler W., Bauerle T., Oncol. Rep. 2011, 26, 843–851. [DOI] [PubMed] [Google Scholar]
- 19. Nabors L. B., Mikkelsen T., Hegi M. E., Ye X., Batchelor T., Lesser G., Peereboom D., Rosenfeld M. R., Olsen J., Brem S., Fisher J. D., Grossman S. A., Cancer 2012, 118, 5601–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sonntag M. H., Schill J., Brunsveld L., ChemBioChem 2017, 18, 441–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sankaran S., Cavatorta E., Huskens J., Jonkheijm P., Langmuir 2017, 33, 8813–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiong J. P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D. L., Joachimiak A., Goodman S. L., Arnaout M. A., Science 2001, 294, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiong J. P., Stehle T., Zhang R., Joachimiak A., Frech M., Goodman S. L., Arnaout M. A., Science 2002, 296, 151–155. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Dechantsreiter M. A., Planker E., Mathä B., Lohof E., Hölzemann G., Jonczyk A., Goodman S. L., Kessler H., J. Med. Chem. 1999, 42, 3033–3040; [DOI] [PubMed] [Google Scholar]
- 24b. Mas-Moruno C., Rechenmacher F., Kessler H., Anti-Cancer Agents Med. Chem. 2010, 10, 753–768; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24c. Sheldrake H. M., Patterson L. H., J. Med. Chem. 2014, 57, 6301–6315. [DOI] [PubMed] [Google Scholar]
- 25. Reardon D. A., Neyns B., Weller M., Tonn J. C., Nabors L. B., Stupp R., Future Oncol. 2011, 7, 339–354. [DOI] [PubMed] [Google Scholar]
- 26. Nabors L. B., Fink K. L., Mikkelsen T., Grujicic D., Tarnawski D. H. N., Mazurkiewicz M., Salacz M., Ashby L., Zagonel V., Depenni R., Neuro-Oncology 2015, 17, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Zheng Y., Ji S., Czerwinski A., Valenzuela F., Pennington M., Liu S., Bioconjugate Chem. 2014, 25, 1925–1941; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Cheng Z., Wu Y., Xiong Z. M., Gambhir S. S., Chen X. Y., Bioconjugate Chem. 2005, 16, 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ye Y. P., Bloch S., Xu B. G., Achilefu S., J. Med. Chem. 2006, 49, 2268–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schottelius M., Laufer B., Kessler H., Wester H. J., Acc. Chem. Res. 2009, 42, 969–980. [DOI] [PubMed] [Google Scholar]
- 30. Harkiss A. H., Sutherland A., Org. Biomol. Chem. 2016, 14, 8911–8921. [DOI] [PubMed] [Google Scholar]
- 31. Xiong L., Yu M., Cheng M., Zhang M., Zhang X., Xu C., Li F., Mol. BioSyst. 2009, 5, 241–243. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Kevan L., Steen L. B., Chem. Phys. Lett. 1975, 34, 184–188; [Google Scholar]
- 32b. Klein R., Tatischeff I., Chem. Phys. Lett. 1977, 51, 333–338; [Google Scholar]
- 32c. Wilkinson F., Garner A., Photochem. Photobiol. 1978, 27, 659–670; [Google Scholar]
- 32d. Bryant F. D., Santus R., Grossweiner L. I., J. Phys. Chem. 1975, 79, 2711–2716; [Google Scholar]
- 32e. Bent D. V., Hayon E., J. Am. Chem. Soc. 1975, 97, 2612–2619. [DOI] [PubMed] [Google Scholar]
- 33. Frese M., Schnepel C., Minges H., Voss H., Feiner R., Sewald N., ChemCatChem 2016, 8, 1799–1803. [Google Scholar]
- 34. Gruß H., Belu C., Bernhard L. M., Merschel A., Sewald N., Chem. Eur. J. 2019, 25, 5880–5883. [DOI] [PubMed] [Google Scholar]
- 35. García-Pindado J., Willemse T., Goss R., Maes B. U. W., Giralt E., Ballet S., Teixidó M., Biopolymers 2018, 109, e23112. [DOI] [PubMed] [Google Scholar]
- 36. Kemker I., Schnepel C., Schröder D. C., Marion A., Sewald N., J. Med. Chem. 2019, 62, 7417–7430. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Tierney M. T., Sykora M., Khan S. I., Grinstaff M. W., J. Phys. Chem. B 2000, 104, 7574–7576; [Google Scholar]
- 37b. Tierney M., Grinstaff M. W., J. Org. Chem. 2000, 65, 5355–5359; [DOI] [PubMed] [Google Scholar]
- 37c. Nakadan N., Imabayashi S. I., Watanabe M., Langmuir 2004, 20, 8786–8791; [DOI] [PubMed] [Google Scholar]
- 37d. Wagner C., Wagenknecht H. A., Chem. Eur. J. 2005, 11, 1871–1876. [DOI] [PubMed] [Google Scholar]
- 38. Alagille D., Baldwin R. M., Tamagnan G. D., Tetrahedron Lett. 2005, 46, 1349–1351. [Google Scholar]
- 39.
- 39a. Mietzsch F., Angew. Chem. 1954, 66, 363–371; [Google Scholar]
- 39b. Ionescu M., Mantsch H., Adv. Heterocycl. Chem. 1967, 8, 83–113; [Google Scholar]
- 39c. Bodea C., Silberg I., Adv. Heterocycl. Chem. 1968, 9, 321–460; [DOI] [PubMed] [Google Scholar]
- 39d. Okafor C. O., Heterocycles 1977, 7, 391–427; [Google Scholar]
- 39e. Eckstein Z., Urbanski T., Adv. Heterocycl. Chem. 1979, 23, 1–53. [DOI] [PubMed] [Google Scholar]
- 40. Dumitriu G. M., Bîcu E., Belei D., Rigo B., Dubois J., Farce A., Ghinet A., Bioorg. Med. Chem. Lett. 2015, 25, 4447–4452. [DOI] [PubMed] [Google Scholar]
- 41. Belei D., Dumea C., Samson A., Farce A., Dubois J., Bîcu E., Ghinet A., Bioorg. Med. Chem. Lett. 2012, 22, 4517–4522. [DOI] [PubMed] [Google Scholar]
- 42. Dumitriu G. M., Ghinet A., Bîcu E., Rigo B., Dubois J., Farce A., Belei D., Bioorg. Med. Chem. Lett. 2014, 24, 3180–3185. [DOI] [PubMed] [Google Scholar]
- 43. Levi L., Müller T. J. J., Chem. Soc. Rev. 2016, 45, 2825–2846. [DOI] [PubMed] [Google Scholar]
- 44. Ou C., Zhang J., Zhang X., Yang Z., Chen M., Chem. Commun. 2013, 49, 1853–1855. [DOI] [PubMed] [Google Scholar]
- 45. Zhu X. Q., Dai Z., Yu A., Wu S., Cheng J. P., J. Phys. Chem. B 2008, 112, 11694–11707. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y., Ballard C. E., Zheng S. L., Gao X., Ko K. C., Yang H., Brandt G., Lou X., Tai P. C., Luc C. D., Wanga B., Bioorg. Med. Chem. Lett. 2007, 17, 707–711. [DOI] [PubMed] [Google Scholar]
- 47. Frese M., Sewald N., Angew. Chem. Int. Ed. 2015, 54, 298–301; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 302–305. [Google Scholar]
- 48. Afonso A., Feliu L., Planas M., Tetrahedron 2011, 67, 2238–2245. [Google Scholar]
- 49. Shinohara T., Deng H., Snapper M. L., Hoveyda A. H., J. Am. Chem. Soc. 2005, 127, 7334–7336. [DOI] [PubMed] [Google Scholar]
- 50. Barker P. L., Bullens S., Bunting S., Burdick D. J., Chan K. S., Deisher T., Eigenbrot C., Gadek T. R., Gantzos R., Lipari M. T., Muir C. D., Napier M. A., Pitti R. M., Padua A., Quan C., Stanley M., Struble M., Tom J. Y. K., Burnier J. P., J. Med. Chem. 1992, 35, 2040–2048. [DOI] [PubMed] [Google Scholar]
- 51. Kapp T. G., Rechenmacher F., Neubauer S., Maltsev O. V., Cavalcanti-Adam E. A., Zarka R., Sci. Rep. 2017, 7, 39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morris G. M., Goodsell D. S., Haliday R. S., Huey R., Hart W. E., Belew R. K., Olson A. J., J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- 53. Bella J., Humphries M. J., BMC Struct. Biol. 2005, 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary