

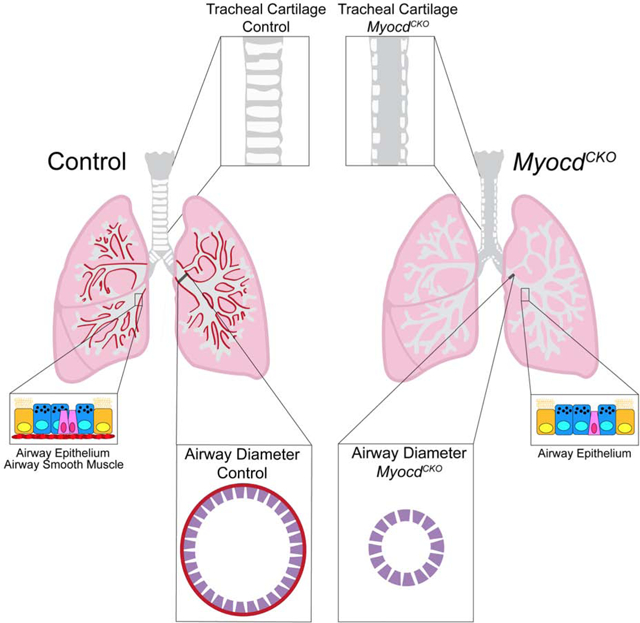
Summary
Airway smooth muscle is best known for its role as an airway constrictor in diseases such as asthma. However, its function in lung development is debated. A prevalent model, supported by in vitro data, posits that airway smooth muscle promotes lung branching through peristalsis and pushing intraluminal fluid to branching tips. Here, we test this model in vivo by inactivating Myocardin, which prevented airway smooth muscle differentiation. We found that Myocardin mutants show normal branching, despite absence of peristalsis. In contrast, tracheal cartilage, vasculature, and neural innervation patterns were all disrupted. Furthermore, airway diameter is reduced in the mutant, counter to the expectation that the absence of smooth muscle constriction would lead to a more relaxed and thereby wider airway. These findings together demonstrate that during development, while airway smooth muscle is dispensable for epithelial branching, it is integral for building the tracheal architecture and promoting airway growth.
eTOC blurb:
Young et al. find that the differentiation of airway smooth muscle is not required for lung branching morphogenesis in vivo. However, it is essential for the proper formation of tracheal architecture including cartilage patterning. In addition, it is essential for promoting airway size.
Introduction
Specification of the respiratory tract in mice begins at embryonic day 9 (E9) as marked by the expression of Nkx2.1, the earliest known genetic marker of the respiratory lineage (Lazzaro et al., 1991; Harris-Johnson et al., 2009;). By E9.5, the lung primordium initiates as two simple epithelial buds surrounded by mesenchyme. Following elongation of the primary buds, secondary branching ensues in a highly stereotypic pattern until approximately E16 (Metzger et al., 2008; Short et al., 2013). Strong evidence demonstrates that close interaction between the juxtaposed mesenchyme and epithelium is important for lung branching morphogenesis (Morrisey & Hogan, 2010).
Shortly after lung budding, diverse cell types emerge in the lung mesenchyme (Peng et al., 2013). For example, airway smooth muscle (ASM) can be detected starting at E10.5 as a small population of cells between the two lung buds (Hines et al., 2013). As development proceeds, ASM forms tightly packed bundles around the intrapulmonary airway epithelium. By E12.5, airway peristaltic contraction can be observed (Schittny et al., 2000). It has been hypothesized that ASM contraction drives peristalsis, which in turn is essential for maintaining airway fluid pressure and subsequently promoting lung growth (Jesudason et al., 2006).
This hypothesis was tested by manipulating ASM in ex vivo lung explant culture. For example, using pharmacological agents to activate or inhibit FGF signaling, SHH signaling, or L-type Ca2+ channels, both tip bifurcation and domain branching were disrupted (Kim et al., 2015; Goodwin et al., 2019). These results led the authors to conclude that ASM differentiation and/or contraction is crucial for lung branching morphogenesis, thereby supporting the hypothesis. However, this hypothesis has not been tested in vivo.
In contrast to the mouse intrapulmonary airway where ASM fully surrounds the epithelium, ASM is only present in the dorsal side of the trachea in juxtaposition to the cartilage on the ventral side. This complementary relationship is essential for proper tracheal function, where the contractility of the ASM maintains air pressure at inhalation, and the rigidity of the cartilage prevents tracheal collapse at exhalation. We have shown that this juxtaposition is established early during cell fate specification through crosstalk of the ASM and cartilage lineages (Hines et al., 2013). Recent studies showed that at a later stage, defects in ASM organization disrupted cartilage segmentation, suggesting continued crosstalk (Kishimoto et al., 2018; Yin et al., 2018).
In this study, we test the requirement of ASM in lung development by preventing ASM differentiation. This is achieved by inactivating Myocardin (Myocd), which encodes a transcription factor that is necessary for the specification of smooth muscle cell fate (Zheng, 2014). MYOCD binds co-activator serum response factor (SRF), and together they activate expression of key smooth muscle genes including actin alpha 2 (Acta2, also termed alpha smooth muscle actin), and transgelin (TagIn, also termed SM22α). We found that inactivating Myocd in the trachea and lung mesenchyme effectively prevented ASM differentiation. Using this mutant, we tested the in vivo role of differentiated ASM in branching morphogenesis and other aspects of lung development.
Results
Myocd inactivation led to loss of airway smooth muscle cells
To determine the specific role of differentiated smooth muscle cells for lung and trachea development, we inactivated Myocardin (Myocd) in the developing lung and tracheal mesenchyme by generating Tbx4rtTA;tetOcre;Myocdflox/flox mutant mice; hereafter referred to as MyocdCKO (Huang et al., 2008; Zhang et al., 2013). We activated rtTA by doxycycline administration starting at E5.5, which led to widespread cre activity in the lung and trachea mesenchyme at the onset of lung development (Zhang et al., 2013). At E12.5, shortly after the initiation of secondary branching morphogenesis, there was a clear loss of airway smooth muscle (ASM) cells in the MyocdCKO lung by immunostaining as compared to control (Figure 1A,B). Efficient gene inactivation and overall reduction of smooth muscle was confirmed by qRT analysis of Myocd, Acta2 and Tagln (Figure 1C). Interestingly, we observed selective loss of ASM while vascular smooth muscle (VSM) remained intact (Figure 1D–G). To determine if the differential requirement for Myocd in ASM and VSM is due to its differential expression in these tissues, we visualized Myocd transcripts using RNAscope in the E14.5 lung. Compared to robust Myocd expression in the ASM, only significantly reduced expression was observed in the VSM (Supplemental Figure 1). As Tbx4rtTA;tetOcre is active in both smooth muscle lineages when induced early as we did (Zhang et al., 2013), this suggests that Myocd is singularly required for ASM differentiation, but may work with other factors redundantly in the lung VSM.
Figure 1.
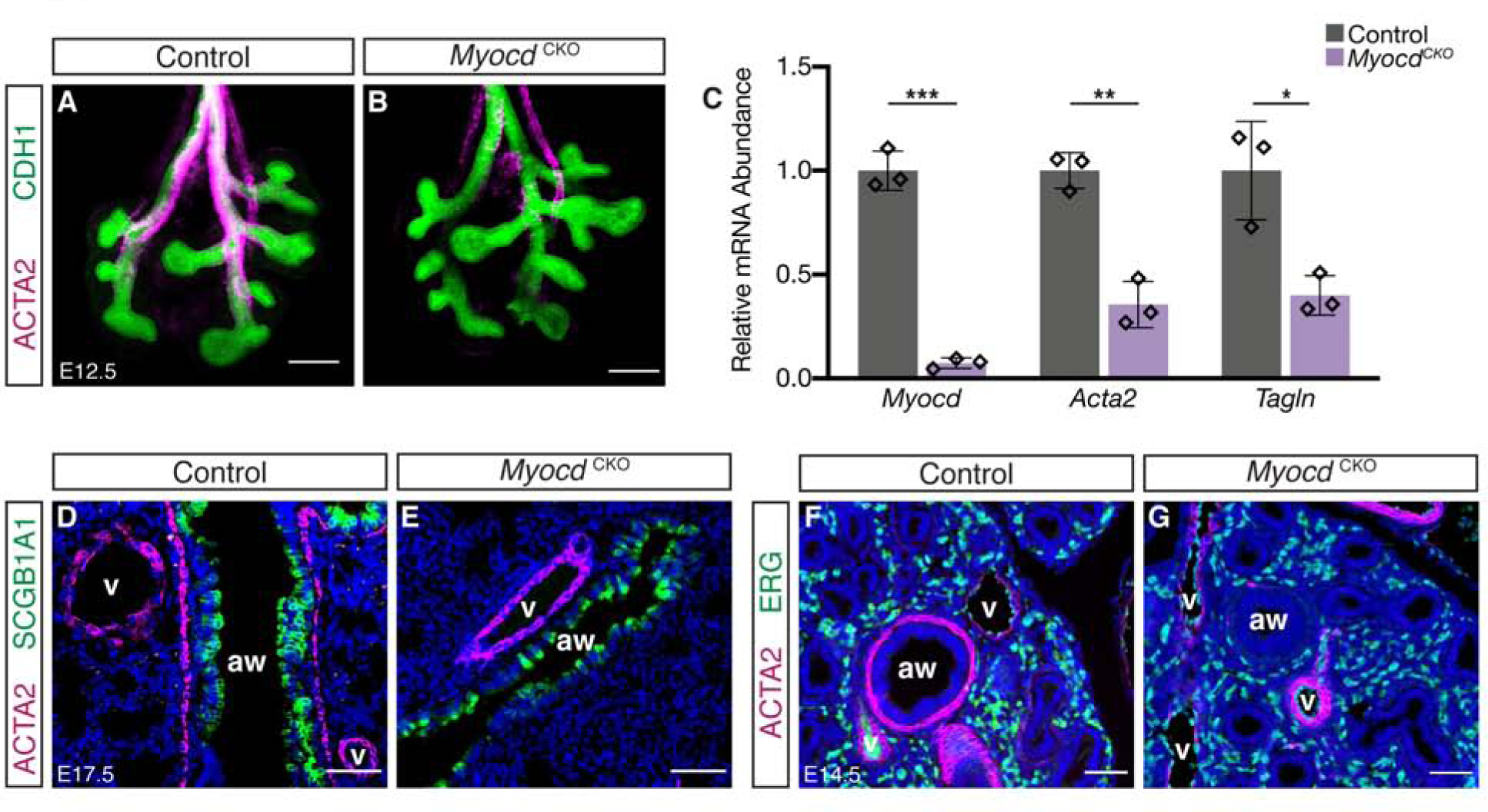
Airway smooth muscle differentiation is disrupted in MyocdCKO lungs. (A,B) Confocal images showing immunofluorescent detection of smooth muscle marker ACTA2 (magenta) and epithelium marker CDH1 (green) in whole lungs of control and MyocdCKO mice at E12.5, showing near complete absence of airway smooth muscle adjacent to airways in MyocdCKO. Scalebar: 200 μm. (C) qRT-PCR quantification of relative mRNA levels of Myocd and smooth muscle markers Acta2 and Tagln in control and MyocdCKO lungs at E12.5. Data are represented as individual points for each biological sample ± SD. *: p<0.05, **: p<0.005, ***: p<0.0005. (D-G) Immunofluorescent detection of ACTA2 (magenta), and club cell marker SCGB1A1 (green) (D,E) or endothelial marker ERG (green) (F,G) in lungs of control and MyocdCKO at E17.5 and E14.5, showing the near absence of the airway (aw) smooth muscle and the persistence of vascular (v) smooth muscle in MyocdCKO.
Lung epithelial branching continued despite disruption of airway smooth muscle differentiation
MyocdCKO mutants died at birth. Lung size and gross morphology appeared normal at E18.5 (Figure S2A, B). To specifically address if ASM is essential for branching morphogenesis in vivo, we carried out either immunofluorescence staining with anti-ACTA2 and anti-CDH1 (also called E-Cadherin, an epithelium marker) antibodies (Figure 2A,B), or immunohistochemical staining with anti-CDH1 antibody at E14.5 (Figure S2C–F). The gross morphology of E14.5 MyocdCKO mutants from an independent source and distinct genetic background was also documented (Figure S2G,H). Quantification was carried out by counting the number of epithelial tips from the left lobes of either MyocdCKO and controls (N=11 for each). No statistically significant difference was found (Figure 2C).
Figure 2.
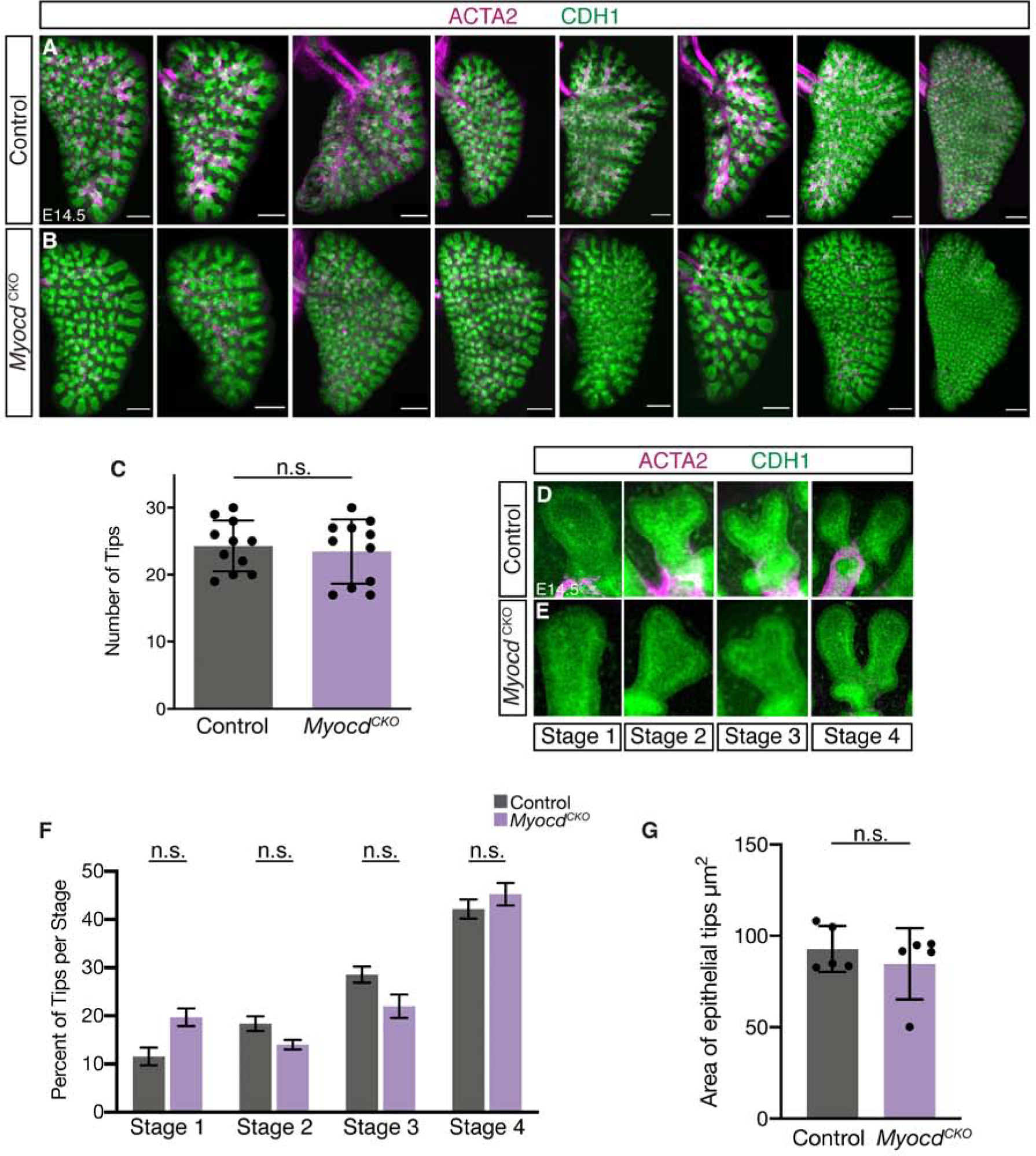
Loss of airway smooth muscle differentiation did not affect airway epithelial branching. (A,B) Maximum projection of z-stacks from confocal images showing immunofluorescent detection of ACTA2 (magenta) and CDH1 (green) in left lung lobes of control and MyocdCKO mice at E14.5, showing normal branching morphogenesis in MyocdCKO lungs. Columns represent control and MyocdCKO littermates. Scalebar: 250 μm. (C) Quantification of terminal epithelial tips in E14.5 control and MyocdCKO left lung lobes, N=11 each. Data are represented as individual points for each biological sample ± SD. (D-E) Representative images showing immunofluorescent detection of ACTA2 (magenta) and CDH1 (green) in the four stages of epithelial tip bifurcation in control and MyocdCKO lungs at E14.5. (F) Quantification of tips in each stage shown as a percentage of total tip number. Data are represented as individual points for each biological sample ± SD. (G) Quantification of epithelial tip areas shown as the average tip area per left lobe. Data are represented as mean ± SD.
We also addressed if there is any difference in the pattern of epithelial tips. A previous study defined four stages of bifurcation branching morphogenesis based on epithelial tip morphology (Kim et al., 2015). Epithelial buds begin as a single tip in stage one, flatten in stage two, form a cleft in stage three, and bifurcate into two daughter buds in stage four. We quantified the number of tips in each of the four stages of epithelial bifurcation and found no statistical difference between MyocdCKO lungs and controls, even though ASM is absent from the epithelial bifurcation clefts (Figure 2D–F). In addition, our quantification indicated that the epithelial bud tip area was not significantly altered in MyocdCKO lung compared to controls (Figure 2G). These data together led to the unexpected conclusion that despite severe disruption of ASM differentiation, the MyocdCKO lungs exhibited normal branching, suggesting that ASM differentiation is not required for branching morphogenesis.
In addition to investigating the role of smooth muscle differentiation, we also determined if smooth muscle contraction is essential for lung branching. Mylk (which encodes MLCK, myosin light chain kinase) is essential for ASM contraction in many tissues including when inactivated in the adult lung (Zhang et al., 2010). When inactivated during development, we found that Tbx4rtTA;tetOcre;Mylkfl/fl mutants (MylkCKO) exhibited normal branching pattern, despite a clear loss of MLCK (Figure S2I–L). This result suggests that similar to the finding from the ASM differentiation mutant, ASM contraction is not required for branching morphogenesis.
Loss of airway smooth muscle abrogated airway peristalsis
Airway peristalsis, presumably driven by ASM, has been postulated as a key component of ASM function in branching. To determine if peristalsis was affected in the MyocdCKO mutants, we dissected E12.5 lungs and cultured them at air-liquid interface under conditions that have been previously described to preserve peristalsis in the control (Schittny et al., 2000). At the time of harvest and start of culture, there was no significant difference in overall lung morphology between the MyocdCKO and control (Figure 3A,B). We confirmed that after 24 hours in culture, the MyocdCKO ex vivo lungs maintained a significant reduction in ASM compared to controls (Figure 3C–F). While control lungs underwent approximately 1±0.2 peristaltic contractions per minute, the MyocdCKO lungs never contracted (Figure 3G, Movie S1, Movie S2). This suggests that airway peristalsis is dependent on the presence of ASM, and the absence of peristalsis in MyocdCKO lungs does not affect branching morphogenesis.
Figure 3.

Loss of airway smooth muscle differentiation led to absence of airway peristalsis. (A-B) Representative control and MyocdCKO E12.5 lungs were dissected and imaged fresh, and (C,D) 24 hours after culture. Scalebar: 50 μm. (E,F) Representative maximum projection of z-stacks from confocal images showing immunofluorescent detection of ACTA2 (magenta) and CDH1 (green) in E12.5 cultured lungs, showing a lack of differentiated ASM in MyocdCKO lung. Scalebar: 50 μm. (G) Peristalsis was observed in control airways approximately once a minute, while MyocdCKO airways never contracted. Data are represented as individual points for each biological sample ± SD. ***: p<0.0001.
Cartilage segmentation occurs through cell aggregation
Previous findings from our lab showed that the juxtaposition of ASM and cartilage in the trachea is established early in development and crosstalk between cells of these two lineages impact their fate specification (Hines et al., 2013). However, the possible effect of ASM on cartilage condensation into segmented rings was not investigated. To begin addressing this question, we first characterized the timing of the wild-type process of cartilage segmentation. By wholemount RNA in situ hybridization of Col2a1, a marker for specified cartilage fate, tracheal cartilage cells emerge as a continuous band which start to be segmented at E13.5 (Figure S3A–D). We reasoned that the conversion from a sleeve to a segmented pattern may occur through either selected intersegmental cell death, or intersegmental trans-differentiation into non-cartilage fate, or convergence of cartilage cells into evenly spaced condensations. To distinguish among these possibilities, we first examined cell death. Immunostaining for apoptosis marker cleaved-Caspase 3 (CASP3) showed little cell death in the ventral mesenchyme, suggesting that mesenchymal cell death is unlikely a major contributor (Figure S3E). Next, we performed lineage tracing employing a Col2a1-cre to recombine a double reporter allele Rosa26R-loxp-tdTom-stop-loxp-eGFP (abbreviated RmTmG) (Ovchinnikov et al., 2000; Muzumdar et al., 2006). This system allowed us to trace the pattern of Col2a1-lineaged cells in the cartilage sleeve as they form segments. At E12.5, we observed that GFP expressing Col2a1-cre lineage labeled cells were positioned in an unsegmented band in the pre-condensation trachea before splitting into two bands in the main bronchi (Figure S3F). However, by E16.5, three days after cartilage segmentation, lineage labeled trachea exhibited an alternating pattern of GFP and tdTomato expressing cells (Figure S3G). This pattern persisted after birth (Figure S3H). The lineage tracing data argue against the possibility that some of the cartilage precursors in the continuous sleeve trans-differentiated into non-cartilage fate in the intersegmental region. Instead, these results together suggest that tracheal cartilage segmentation occurs via convergence of cartilage cells into evenly spaced C-shaped rings.
Airway smooth muscle differentiation is essential for proper tracheal architecture
Based on the above analysis, tracheal cartilage segmentation initiates at E13.5, after ASM differentiates and begins its phasic contractions at E11.5 (Schittny et al., 2000; Hines et al., 2013). To determine if loss of ASM differentiation in the MyocdCKO trachea would affect cartilage segmentation, we first stained E17.5 tracheas with alcian blue to outline the cartilage. The MyocdCKO mutant tracheas exhibited a near complete disruption of C-shaped rings, with stained nodules only on the lateral edge of the trachea in a segmented pattern (Figure 4A,B). This defect can be traced to E14.5, at the start of cartilage segmentation, as indicated by Col2a1 RNA in situ hybridization (Figure 4C,D). On sections, SOX9+ cartilage cells were still present in similar patterns in the control and MyocdCKO tracheas at E13.5, the start of cartilage segmentation (Figure 4E,F). After cartilage segmentation, there was a clear reduction of SOX9+ cells at E14.5 in the MyocdCKO (Figure 4G,H). By E17.5, the remaining SOX9+ cells were found in nodules at the lateral edge of the ventral portion of the trachea, similar to the pattern of wholemount alcian blue staining (Figure 4I,J). This severe reduction of cartilage may lead to collapsed trachea, a possible cause for the lethality of the MyocdCKO mutants at birth. Consistent with the findings from the MyocdCKO mutants, in the MylkCKO trachea where ASM remains present but its contraction was inhibited, tracheal cartilage pattern is disrupted (Figure S4A–D). These findings together indicate that ASM differentiation and contraction are required for tracheal cartilage segmentation.
Figure 4.

Loss of airway smooth muscle differentiation led to disrupted cartilage segmentation. (A,B) Whole mount alcian blue staining showed cartilage segmentation defects in MyocdCKO E18.5 tracheas. Scalebar: 50 μm. (C,D) Col2a1 wholemount RNA in situ hybridization revealed reduced chondrocytes in MyocdCKO E14.5 tracheas. Scalebar: 100 μm. (E-J) Immunofluorescent detection of ACTA2 (magenta) and cartilage marker SOX9 (green) expression in transverse sections of tracheas of control and MyocdCKO at E13.5 (E,F), E14.5 (G,H), and E17.5 (I,J). Scalebar: 50 μm for E-H and 100 μm for I,J. “t” indicates trachea and “e” indicates esophagus. (K) Heatmap of RNA-seq data showing groups of significantly downregulated genes, by an adjusted p-value less than 0.05, in the MyocdCKO E18.5 tracheas. (L) qRT-PCR quantification of relative mRNA levels of mechanosensitive genes Runx2, Spp1, Hapln1, and Itgbl1 in control and MyocdCKO tracheas at E18.5. *: p<0.05, **: p<0.005. (M) qRT-PCR quantification of relative mRNA levels of Sox9 and mechanosensitive genes Runx2, Spp1, and Hapln1 in control and MyocdCKO tracheas at E13.5. *: p<0.05. Data are represented as individual points for each biological sample ± SD.
To determine the molecular mechanism of tracheal cartilage malformation in the absence of ASM, we performed RNA-seq of control and MyocdCKO E18.5 tracheas. The significantly downregulated genes in the MycodCKO trachea consisted of genes in the categories of smooth muscle, cartilage, ion channels, neuronal regulation, and immune response (Figure 4K).
To determine possible molecular players mediating ASM effects on cartilage segmentation, we focused on mechanical pressure regulated genes. Previous studies have identified several genes that are regulated by mechanical pressure, and in turn play a role in chondrocytes and osteoblast development and maturation (Yoshida et al., 2004; Salingcarnboriboon et al., 2006). For example, Runx2 is a transcription factor that is activated during mechanical loading in osteoblasts and is also necessary for chondrocyte maturation. Runx2 activates Spp1 and Itgbl1, also mechanoresponsive genes expressed in chondrocytes (Felsenthal & Zelzer, 2017; Li et al., 2015). Hapln1 is an essential component of the cartilage extracellular matrix (ECM), and Hapln1 mutants die at birth due to impaired tracheal cartilage and subsequent respiratory failure (Watanabe & Yamada, 1999). Hapln1 also modulates hyaluronic acid signaling which has been shown to be important for mechanical responses in cartilage (Felsenthal & Zelzer, 2017). Itgbl1 has been shown to receive input from the ECM (Song et al., 2018). qRT-PCR analysis validated the RNA-seq data that the expression of mechanosensitive genes such as Runx2, Spp1, Hapln1, and Itgbl1 are downregulated in the MyocdCKO trachea compared to control at E18.5 (Figure 4L). However, the caveat is that their downregulation may be due to the significant reduction of SOX9+ cartilage cells at E18.5, as these genes are primarily expressed in the cartilage cells. To more rigorously determine if loss of ASM may affect these mechanosensitive genes, we examined their expression at E13.5, the stage before cartilage segmentation when there is still a comparable abundance of SOX9+ cells in the MyocdCKO and control tracheas. We found that while the expression of Sox9 in the MyocdCKO trachea at E13.5 is proportional to control, the expression of Runx2, Spp1, and Hapln1 is already downregulated (Figure 4M). Together, these findings suggest that in the absence of ASM, changes in mechanical pressure would alter the expression of mechanosensitive genes which in turn may affect cartilage segmentation.
To determine if the lack of ASM also affects the development of other cell types in the trachea, we assayed for the morphology of neurons and vessels. By anti-TUBB3 staining, a pan neuronal marker, several ganglia composed of intrinsic neurons are found to be distributed along the length of the dorsal trachea on the surface of smooth muscles (Figure S5A). While ganglia are still present in the MyocdCKO trachea, the cumulative cell body size are ~50% the total area of control ganglia (Figure S5B–E). These results suggest that ASM is important for proper tracheal innervation by intrinsic neurons during development.
To assess patterning of the tracheal vasculature, we used anti-ICAM2 staining, a pan endothelial marker. In the control, there is a net-like appearance of vasculature on the dorsal side while there is a striped organization of vasculature interspaced between the cartilage rings on the ventral side (Figure S5F–I). In the MyocdCKO trachea, vascular patterning on the dorsal side appeared normal, while vessels on the ventral side were no longer constrained in stripes in the absence of organized segmental cartilage rings (Figure S5J–M).
Airway smooth muscle is dispensable for the ability of lung epithelial cells to differentiate
The alveolar epithelium is comprised of alveolar epithelial type 1 cells (AT1) which line the alveolus and form the air-blood barrier, and alveolar epithelial type 2 (AT2) cells which secrete surfactants and reduce surface tension. A proper ratio of AT1 to AT2 cells is crucial for postnatal life. By immunostaining and qRT analysis, the number of AT1 and AT2 cells, and their ratio remained similar in MyocdCKO lungs at E18.5 compared to control (Figure S6A–D).
For a more comprehensive analysis of changes in MyocdCKO lungs, we performed RNA-seq in MyocdCKO and control E18.5 lungs. The significantly downregulated genes in the MycodCKO lung consists of genes related to smooth muscle cells, ciliated cells, club cells, and immune response (Figure 5A). By qRT analysis, we verified that the expression of several genes that are key markers of differentiated airway epithelial cell types were significantly downregulated in the MyocdCKO lung at E18.5 (Figure 5B). These include Scgb1a1 and Scgb3a2 for club cells; Foxj1, Mcidas and Myb for ciliated cells; Ascl1 and Calca for pulmonary neuroendocrine cells.
Figure 5.

Loss of airway smooth muscle differentiation did not affect the ability of airway epithelial cells to differentiate. (A) Heatmap of RNA-seq data showing selected groups of significantly downregulated genes, by an adjusted p-value less than 0.05, in the MyocdCKO E18.5 lung. (B) qRT-PCR quantification of relative mRNA levels of airway epithelial cell marker genes for general airway epithelium Sox2; club cells Scgb3a2 and Scgb1a1; ciliated cells Foxj1, Mcidas and Myb; pulmonary neuroendocrine cells Ascl1 and Calca; in control and MyocdCKO lungs at E18.5. Data are represented as individual points for each biological sample ± SD. *: p<0.05, **: p<0.005, ***: p<0.0005. (C,D) Immunofluorescent detection of ACTA2 (magenta) and SCGB1A1 (green). (E,F) Immunofluorescent detection of FOXJ1 (magenta) and smooth muscle marker TAGLN (green). (G-I) Immunofluorescent detection of CGRP (green) in control and MyocdCKO E18.5 lungs. Scalebars: 50μm.
Counter to the downregulation of airway epithelium marker expression by qRT-PCR, by immunostaining, airway epithelial cell markers such as SCGB1A1 for club cells, FOXJ1 for ciliated cells and CGRP for pulmonary neuroendocrine cells were detected at a similar intensity per cell in the MyocdCKO lung compared to control (Figure 5C–I). We also found that there was no difference in the balanced ratio of club and ciliated cells in either the MyocdCKO or MylkCKO compared to respective controls (Figure S6E–J). Additionally, while pulmonary neuroendocrine cells are present and remain innervated, the neuroepithelial cell body clusters were smaller in the MyocdCKO airway as compared to control (Figure 5G–I, Figure S6K–N).
Airway smooth muscle promotes airway circumferential growth
To resolve the discrepancy between qRT-PCR and immunostaining results of airway genes, we used immunostaining of thick longitudinal airway sections and 3D reconstruction of confocal images to visualize the entire thickness of the main intrapulmonary airway. Strikingly, the main airway was narrower in the E18.5 MyocdCKO lung compared to control, and quantification demonstrated statistical significance (Figure 6A–C). We can trace this phenotype to E14.5 by taking transverse sections of the primary bronchi at equivalent depths in the MyocdCKO and control (Figure 6D,E). Cell counting analysis revealed an approximately 40% decrease in the number of SOX2+ epithelial cells and a similar decrease in airway diameter in the MyocdCKO compared to control (Figure 6F,G). This is consistent with the decreased level of Sox2 expression by qRT-PCR analysis (Figure 5B). No change in cell death or apical-basal polarity was detected (Figure S7A–H). In comparison, there is a small, but statistically significant decrease in epithelial cell proliferation, shown by a reduction in the percentage of Ki67+SOX2+ proliferative airway cells in the total SOX2+ airway epithelial cells in the MyocdCKO airway compared to control (Figure 6H–J). Over time, this modest decrease in proliferation may accumulate and contribute to the clear difference in airway size at E18.5.
Figure 6.
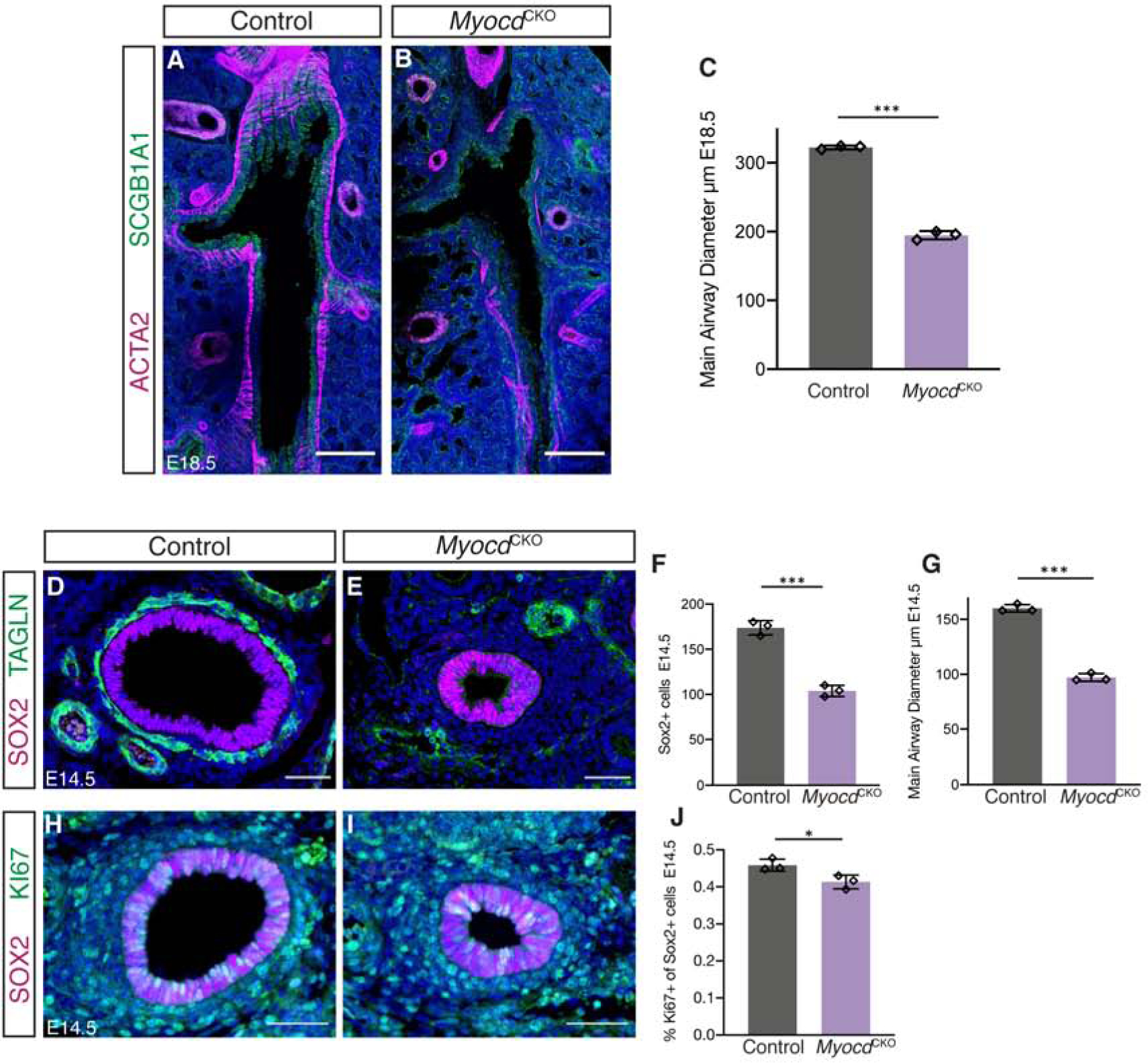
Loss of airway smooth muscle cell differentiation led to reduced airway size. (A,B) Confocal stacks covering the entire thickness of the main airways in 100μm thick cryosections of E18.5 left lobes showing immunofluorescent detection of ACTA2 (magenta) and SCGB1A1 (green). Scalebar: 200 μm. (C) Quantification showing reduced main airway diameter in the mutant at E18.5.***: p<0.0005. (D,E) Immunofluorescent detection of airway epithelial marker SOX2 (magenta) and TAGLN (green) in transverse sections taken at equivalent depths of the main airway in control and MyocdCKO lungs at E14.5. Scalebar: 50μm. (F) Quantification showing decreased number of SOX2+ cells in the MyocdCKO main airway at E14.5. ***: p<0.0005. (G) Quantification showing decreased main airway diameter in the mutant at E14.5. ***: p<0.0005. (H,I) Immunofluorescent detection of SOX2 (magenta) and proliferation marker KI67 (green) in transverse sections taken at similar depths of the main airway in control and MyocdCKO lungs at E14.5. Scalebar: 50μm. (J) Quantification showing decreased number of the percentage of Ki67+SOX2+ proliferating cells in total Sox2+ airway epithelial cells in the MyocdCKO main airway at E14.5. *: p<0.05. Data are represented as individual points for each biological sample ± SD.
To test the possibility that ASM contractions may be necessary for establishing airway epithelial size, we examined airway size in the MylkCKO mutant lung compared to control. We found that there was no airway size defect in the MylkCKO lung, suggesting that differentiated ASM, but not contraction, is necessary for increasing the caliber of the airway tube (Figure S7I–K).
These findings led to the hypothesis that differentiated ASM may be a source of signaling factors that control the size of the airway epithelium. To uncover the molecular mechanism, we performed RNA-seq of MyocdCKO and control lungs at E13.5. We focused in on the signaling factors that are altered (Figure 7A). Among them, two key inhibitors of BMP signaling, Chrdl1 and Grem2, were downregulated. These changes stood out because active BMP signaling has been shown to constrain airway proliferation (Tadokoro et al.,2016). By qRT-PCR, we verified that the expression of Chrdl1 and Grem2 is significantly decreased in the MyocdCKO lung at E13.5 (Figure 7B). We analyzed BMP activity using anti-phosphorylated-SMAD1/5/8 (pSMAD1/5/8) immunostaining as a read-out of BMP activity. In concert with the decrease of BMP inhibitors, we observed a robust increase (p<0.0005) of pSMAD1/5/8 staining in the E13.5 main airway of the MyocdCKO mutant compared to control, demonstrating increased signaling (Figure 7C–G). These findings suggest that differentiated ASM is essential for promoting the circumferential growth of the airway, and differentiated ASM may act through regulating BMP signaling in this process.
Figure 7.

Loss of airway smooth muscle cell differentiation led to increased BMP activity in the main airway. (A) Heatmap of RNA-seq data showing groups of significantly downregulated genes, by an adjusted p-value less than 0.05, in the MyocdCKO E13.5 lung. (B) qRT-PCR quantification of relative mRNA levels of BMP4 antagonists Chrdl1 and Grem2 in control and MyocdCKO lungs at E13.5. *: p<0.05, ***: p<0.0005. (C-F) Confocal images of 20 μm thick cryosections of the main pulmonary airway in E13.5 left lung showing immunofluorescent detection of ACTA2 (magenta) and BMP activity indicator pSMAD1/5/8 (green). Dashed lines represent the outline of the main pulmonary airway. Scalebar: 200 μm. (E,F) Magnified images of the first proximal branches region of C, D, respectively. (G) Quantification showing increased pSMAD1/5/8 signal in the main airways in MyocdCKO compared to control. ***: p<0.0005. Data are represented as individual points for each biological sample ± SD.
Discussion
The ASM is a prominent site of pathogenesis in diseases such as asthma and COPD, and has been studied extensively in the context of pathogenesis. In comparison, its function in normal development is less understood. In this study, we used an in vivo genetic approach to test the role of ASM differentiation in lung development. Unexpectedly, our findings from the MyocdCKO challenged the notion that ASM, through its role in peristalsis, is essential for lung branching morphogenesis, a prevalent model supported by in vitro data (Schittny et al., 2000; Sparrow & Lamb, 2003; Kim et al., 2015; Goodwin et al., 2019). Instead, our data showed that disruption of ASM differentiation abolished peristalsis, but did not alter epithelial branching. Nevertheless, ASM differentiation is far from being dispensable. In the trachea, ASM is essential for the establishment of tracheal architecture, including cartilage segmentation, vascular patterning, and intrinsic neuron organization. The changes of these cell types in the mutant are either direct or indirect consequences of the loss of ASM. Given that in human, the juxtaposition of the ASM and cartilage is preserved deep into the lung, this role of ASM in establishing the cartilaginous airway architecture would prove significant in the human airway. An additional finding from the MyocdCKO mutant is that the differentiated ASM is required for circumferential growth of the airway. Postnatally in diseases such as asthma, increased number, size, and/or constriction of ASM cells is linked to airway narrowing. These findings suggest distinct roles of ASM in development versus pathogenesis.
The model that ASM is essential for branching morphogenesis follows the logic that differentiated ASM undergoes peristalsis. In the amniotic fluid filled fetal lung, peristalsis would push fluid into the distal tips to promote epithelial branching morphogenesis. This attractive model is supported by data from in vitro explant cultures. For example, experimentally increasing intraluminal pressure in culture led to increased branching (Nelson et al., 2017). Furthermore, disruption of ASM in culture via treatment with a series of pharmacological agents each led to disruption of branching ( Kim et al., 2015; Goodwin et al., 2019). We speculate that the discrepancy between these findings versus ours may be due to the difference between in vitro versus in vivo experimental settings. Furthermore, pharmacological agents used in the in vitro experiments have been shown to have effects not restricted to ASM. In our in vivo model, the genetic data demonstrate that even though ASM is lost and peristalsis is absent in the MyocdCKO lung, branching is not altered. This finding is consistent with two prior in vivo observations. First, in two separate studies, it was found that infiltration of amniotic fluid into the distal airway was observed starting at E16.5 (Buckley et al., 2005; Li et al., 2018). As branching morphogenesis has largely concluded by E16.5, it is unlikely that in vivo, amniotic fluid pressure on the distal tip would affect branching. Second, it was found that inhibition of retinoic acid signaling in vivo resulted in ectopic ASM differentiation into the distal airway. However, epithelial branching continued as normal (Chen et al., 2014). These findings, together with our data from the MyocdCKO mutant, suggest that presence and the amount of differentiated ASM and their function in peristalsis do not impact epithelial branching morphogenesis in vivo.
It should be noted that preventing ASM differentiation is a distinct operation from ablating ASM, and may have different impacts on the adjacent epithelium. In a recent study, genetic ablation of ACTA2+ cells was achieved via Acta2-cre mediated expression of diphtheria toxin receptor expression and supply of toxin in the ex vivo culture medium. This did not affect domain branch number or position, but led to reduced branch length and increased width (Goodwin et al., 2019). In the adult lung, after LGR6+ ASM cells were genetically ablated, the airway epithelial cells became defective in regeneration following naphthalene induced airway injury (Lee et al., 2017). These results are consistent with findings indicating that ASM is a niche and source of growth signals such as FGFs and WNTs for branching and activation of epithelial progenitors in adult injury repair (Mailleux et al., 2005; Volckaert et al., 2011; Zepp et al., 2017a).
For ASM function in tracheal cartilage segmentation, we hypothesize that this is achieved through mechanical load exerted by ASM constriction. The impact of mechanical force on cartilage formation has been demonstrated in axial and appendage skeletal development. For example, in limb development, mechanical pressure generated by skeletal muscles is essential for shaping the limb cartilage (Rolfe et al., 2014; Felsenthal & Zelzer, 2017). In the trachea, two independent recent studies showed that ASM-targeted inactivation of Kcnj13 which encodes a potassium channel, or Ror2 which encodes a WNT5a receptor, led to misalignment of the ASM, which in turn led to disrupted tracheal cartilage segmentation (Kishimoto et al., 2018; Yin et al., 2018). These findings suggest that disruption of tensile strengths of muscle could impair mechanical stability of the trachea and lead to cartilage condensation defects. For a possible molecular mechanism, work in chondrocytes outside of the trachea has demonstrated that mechanical load can regulate the expression of several genes that play key roles in cartilage formation, including Runx2, Spp1, and Hapln1 (Felsenthal & Zelzer, 2017a; X. Li et al., 2015; Salingcarnboriboon et al., 2006; Song et al., 2018; Watanabe & Yamada, 1999; Yoshida et al., 2004). Our data indicate that the expression of these genes is altered in the MyocdCKO trachea, consistent with the possibility that these molecular changes may contribute to the observed cartilage malformation.
While prevention of ASM differentiation did not affect branching morphogenesis, it led to an unexpected reduction of airway diameter. Airway diameter is directly proportional to airway conductance, and decreased airway conductance is a key feature of major lung diseases such as asthma. In Asthma, while decrease of airway conductance is largely due to airway constriction and mucus-based occlusion, one would expect that a narrower airway from development would add to the susceptibility to reduced conductance. However, little is known about how airway diameter is achieved through development, partly because many mutants that show abnormal airway diameter also show disrupted branching. The lack of a branching defect in the MyocdCKO lung allowed us to clearly demonstrate the airway diameter phenotype. Reduction of airway diameter only affected a small proportion of the whole lung, thereby did not alter overall lung size. Our finding that the reduced diameter is already present at E14.5 suggests that active circumferential growth occurs in parallel to branching morphogenesis.
In asthma, ASM hyperplasia or hypertrophy is linked to reduced airway size. Thus, the lack of differentiated ASM is expected to lead to relaxed, and thereby larger airway. Our finding that the MyocdCKO airway is smaller runs counter to expectation, and suggest that the role of ASM in development is distinct from that in the adult airway. We considered the possibility that mechanical force generated by differentiated ASM may be important for the stimulation of airway epithelial growth, similar to its role in cartilage condensation. However, our finding that the MylkCKO lung did not exhibit an airway size defect does not add credence to this possibility. Alternatively, as mentioned above, differentiated ASM is a source of signal and signaling regulators. We found that BMP4 antagonists Chrdl1 and Grem2 are significantly downregulated in the MyocdCKO lung. Single cell RNA-seq data from embryonic lungs shared on the LungGENS database revealed that Chrdl1 and Grem2 are selectively expressed in ASM and proliferative mesenchymal progenitors (Du et al., 2017). Furthermore, Bmpr1a and Bmpr1b, which encodes BMP receptors, are expressed in airway epithelial cells. The decrease in Chrdl1 and Grem2 are consistent with an increase in BMP activity in the proximal airway, as demonstrated by pSMAD staining. BMP signaling has been shown to influence airway and alveolar epithelial repair (Chung et al., 2018; Tadokoro et al., 2016; Zepp et al., 2017b). Specifically, BMP constrains the proliferation of airway cells and this can be released by BMP antagonists in the context of airway repair (Tadokoro et al., 2016). Our results suggest the possibility that ASM differentiation promotes airway diameter growth through regulating signaling. Taken together, the in vivo findings in this study revealed unexpected roles of ASM during the building of a functional lung.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xin Sun (xinsun@ucsd.edu). All RNA-seq fastq and processed files are publicly available at the NIH NCBI GEO database accession number GSE143394.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Myocdflox, Tbx4rtTA, and tetOcre alleles and transgenic lines have been described previously (Huang et al., 2009; Zhang et al., 2013). Myocdflox/+ mice were bred to Tbx4rtTA;tetOcre;Myocdflox/flox to generate trachea and lung mesenchyme specific knockouts. The Mylkflox/flox allele has been described previously (He et al., 2008). Mylkflox/flox mice were bred to Tbx4rtTA;tetOcre;Mylkflox/+ to generate trachea and lung mesenchyme specific knockouts. Mice were housed and all experimental procedures were performed in an American Association for Accreditation of Laboratory Animal Care-accredited laboratory animal facility at the University of California San Diego (UCSD). Noon of the day on which a virginal plug was detected was considered to be E0.5. Prenatal rtTA activity was induced by doxycycline (dox) administration starting at gestational day 5.5 by feeding pregnant females with dox food (625 mg/kg; Test-Diet, Richmond, IN, USA).
METHOD DETAILS
Tissue preparation and immunostaining
Whole E12.5, E13.5, E14.5, E17.5, and E18.5 embryos, tracheas, and lungs were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) diluted in PBS. Samples were either stained wholemount or embedded in paraffin or frozen in OCT (Electron Microscopy Sciences) for sectioning. Whole lungs and sections were immunostained using standard protocols. For pSMAD1/5/8 staining, the Tyramide SuperBoost™ Kit (ThermoFisher) was used followed by TrueBlack® Lipofuscin Autofluorescence Quencher (Biotium) staining following these product’s published protocols. Slides were mounted with either Vectashield (Vector Labs) and visualized/photographed using a Zeiss AxioImager.A2 microscope and AxioCam MRc camera. Whole mount images and thick cryo sections were mounted using Citifluor (Ted Pella, Inc.) and visualized/photographed using Zeiss 880 Airyscan and/or Leica Sp8 confocal microscopes and cameras. Images were processed using ImageJ.
Quantitative RT-PCR (qRT-PCR)
RNA was isolated from embryonic whole lungs or tracheas using an RNEasy micro kit (Qiagen), respectively, as per the manufacturer’s protocol. Either 250μg, 500μg, or 1000μg of RNA was reverse transcribed into cDNA using the iScript Reverse Transcriptase (Bio-Rad Laboratories). For qRT-PCR, 10ng of cDNA was amplified using gene-specific primers and iTaq SYBR green Supermix (Bio-Rad Laboratories) on a Bio-Rad CFX Connect real-time PCR machine. For each gene, at least three technical replicates and three biological replicates were assayed. Data were analyzed with the change in cycle threshold (Ct) value method. Statistical significance was determined using Student’s t-test.
Analysis of peristalsis in ex vivo embryonic lungs
Lungs from E12.5 mouse embryos were dissected in sterile PBS and cultured on porous membranes (nucleopore polycarbonate track-etch membrane, 8 mm pore size, 25 mm diameter; Whatman) at air-liquid interphase of DMEM/F12 medium supplemented with antibiotics (50 units/ml of penicillin and streptomycin). Ex vivo lungs were cultured in an incubator at 37°C in 5% CO2 for 24 hours. Cultured lungs were imaged and videoed for 10 mins on a 37°C warm plate. Peristaltic contractions were calculated by observing the most proximal bronchial branch junction in the right lung. After imaging, ex vivo cultured lungs were then fixed in 4% paraformaldehyde (Electron Microscopy Sciences) diluted in PBS and immunostained using standard protocols.
RNAscope in situ hybridization
RNAscope Multiplex Fluorescent v2 Assay (ACD) was used to perform in situ hybridization of Myocd in E14.5 wild-type lungs. Tissue was prepared as a fresh-frozen sample following the published RNAscope protocol, then sectioned 10 μm thick. The Myocd probe was stained with the Opal 520 fluorophore. These sections were then counter stained with DAPI and the ACTA2 antibody as described above.
Whole-Mount in situ hybridization
Embryonic lungs were dissected in PBS, fixed in 4% paraformaldehyde overnight at 4°C, and then dehydrated to 100% MeOH. Whole-mount in situ hybridization was carried out by using an established protocol (Abler et al., 2011).
RNA-seq analysis
Control and MyocdCKO E13.5 lungs, E18.5 tracheas, and E18.5 lungs were dissected in PBS and immediately stored in TRIzol Reagent (ThermoFisher Scientific). Tissues were then lysed using a TissueLyser (Qiagen), and total mRNA was extracted using standard TRIzol RNA extraction protocol. mRNA was cleaned using the RNeasy Micro Kit (Qiagen). cDNA libraries were made for each individual lung or trachea using Illumina TruSeq RNA Library Prep Kit V2 (Illumina) and sequenced on a SR75 run on the HiSeq4000 platform (Illumina). Sequences in FASTQ files were aligned to the mouse reference genome using STAR (Dobin et al., 2013). Reads were counted using featureCounts and differential expression analysis was performed using DeSeq2 (Liao et al., 2014; Love et al., 2014). Heatmaps were generated using HeatMapper software (Babicki et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Cell counts were quantified manually using the ImageJ Cell Counter feature. For each sample, six different sections from the same lung were quantified and the average was taken to represent the count for each sample. Area was also measured in ImageJ. For E14.5 epithelial tips, area was calculated for each tip and the average was taken as the tip area for each left lobe. For tracheal ganglia, the ganglia were measured individually and then added together to represent the total ganglia area per trachea. Airway diameter measurements were measured in ImageJ. For thick sections, diameter was measured by drawing a straight line through the diameter of the main pulmonary airways and calculating the length. For transverse sections of the circular main airway, diameter was measured by drawing a straight line through the circular airway. For each sample, three different sections from the same lung at similar depths were quantified and the average was taken to represent the diameter for each sample. pSMAD1/5/8 fluorescence activity was measured for the first proximal branches as pixel intensity for the pSMAD1/5/8 channel (green) in the confocal stacks. Statistical significance was determined using Student’s t-test, and presented as mean ± standard deviation.
DATA AND CODE AVAILABILITY
All RNA-seq fastq and processed files are publicly available at the NIH NCBI GEO database accession number GSE143394. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE143394
Supplementary Material
Movie S1. Supplemental to Figure 3. Peristalsis video for control lungs cultured for 24 hours. Video is a total of 6 seconds condensed from 10 minutes of filming.
Movie S2. Supplemental to Figure 3. Peristalsis video for MyocdCKO lungs cultured for 24 hours. Video is a total of 6 seconds long, condensed from 10 minutes of filming.
Key Resources Table
| REAGENT | SOURCE | IDENTIFIER |
|---|---|---|
| ANTIBODIES | ||
| mouse anti-ACTA2; 1:200 | Sigma-Aldrich | C6198 |
| rabbit anti-CDH1; 1:200 | Cell Signaling | 3195S |
| rabbit anti-cleaved Caspase 3; 1:100 | Cell Signaling Technology | 9661 |
| rabbit anti-CGRP; 1:200 | Sigma-Aldrich | C8198 |
| rabbit anti-ERG; 1:100 | Abcam | ab92513 |
| mouse anti-FOXJ1; 1:200 | ThermoFisher Scientific | 14-9965-80 |
| mouse anti-HOPX; 1:50 | Santa Cruz | sc-398703 |
| mouse anti-ICAM2; 1:200 | BD Pharmingen | 553326 |
| rabbit anti-Ki67; 1:200 | Abcam | ab15580 |
| mouse anti-MLCK; 1:200 | Sigma | M7905 |
| rabbit anti-pMLC (Myosin light chain phospho S20); 1:200 | Abcam | ab2480 |
| Rabbit anti-pSMAD1/5/8; 1:200 | Cell Signaling Technology | 9516 |
| rabbit anti-pro-SPC; 1:200 | Sigma-Aldrich | AB3786 |
| rabbit anti-SCGB1A1; 1:200 | Seven Hills Bioreagents | WRAB-3950 |
| mouse anti-SOX2; 1:50 | Santa Cruz | sc-365823 |
| rabbit anti-SOX9; 1:500 | Sigma-Aldrich | AMAB90795 |
| rabbit anti-TAGIN; 1:200 | Abcam | ab227566 |
| biotin anti-TUBB3; 1:100 | R&D Systems | BAM1195 |
| Biotin anti-rabbit; 1:250 | ThermoFisher | 31820 |
| Cy3 anti-mouse; 1:250 | Jackson Immunoresearch | 115-165-003 |
| Cy3 anti-rabbit; 1:250 | Jackson Immunoresearch | 111-165-003 |
| FITC anti-rabbit; 1:250 | ThermoFisher Scientific | A-11008 |
| FITC anti-streptavidin; 1:250 | ThermoFisher Scientific | S11223 |
| RNAscope® probes | ||
| Mm-Myocd | Advanced Cell Diagnostics | 581051 |
| STAINING REAGENTS | ||
| TrueBlack® Lipofuscin Autofluorescence Quencher | Biotium | 23007 |
| Alexa Fluor™ 488 Tyramide SuperBoost™ Kit, streptavidin | ThermoFisher | B40932 |
| EXPERIMENTAL MODELS: ORGANISMS/STRAINS | ||
| Myocdflox | Gift from M. Parmacek | N/A |
| Mylkflox | Gift from M. Zhu | N/A |
| Tbx4rtTA | Gift from Wei Shi | N/A |
| tetO-cre | The Jackson Laboratory | JAX:006234, RRID:IMSR_JAX:006234 |
| SOFTWARE | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| STAR | Dobin et al., 2013 | N/A |
| featureCounts | Liao et al., 2014 | N/A |
| DeSeq2 | Love et al., 2014 | N/A |
| HeatMapper | Babicki et al., 2016 | N/A |
| Prism 8 | Graph Pad Software | https://www.graphpad.com/scientific-software/prism/ |
| DEPOSITED DATA | ||
| RNA-seq data | NIH NCBI GEO | GSE143394 |
| OLIGONUCLEOTIDES | ||
| qPCR Primer: Actb Forward: CGGCCAGGTCATCACTATTGGCAAC | This Paper | N/A |
| qPCR Primer: Actb Reverse: GCCACAGGATTCCATACCCAAGAAG | This Paper | N/A |
| qPCR Primer: Myocd Forward: CTACCCTGGGATGCACCAAAC | This Paper | N/A |
| qPCR Primer: Myocd Reverse: AGGGGATAGAGGGGTATTGCT | This Paper | N/A |
| qPCR Primer: Acta2 Forward: CATGTACCCAGGCATTGCTGAC | This Paper | N/A |
| qPCR Primer: Acta2 Reverse: TGCTGGAAGGTAGACAGCGAAG | This Paper | N/A |
| qPCR Primer: Tagln Forward: GCAGTGCAGAGGACTCTAATGG | This Paper | N/A |
| qPCR Primer: Tagln Reverse: AAGGCCAATGACGTGCTTCC | This Paper | N/A |
| qPCR Primer: Runx2 Forward: AACGATCTGAGATTTGTGGGC | This Paper | N/A |
| qPCR Primer: Runx2 Reverse: CCTGCCTGGGATTTCTTGGTT | This Paper | N/A |
| qPCR Primer: Spp1 Forward: AGCAAGAAACTCTTCCAAGCAA | This Paper | N/A |
| qPCR Primer: Spp1 Reverse: GTGAGATTCGTCAGATTCATCCG | This Paper | N/A |
| qPCR Primer: Itgbl1 Forward: TGGGAAGTGTTACTGTGGAAAC | This Paper | N/A |
| qPCR Primer: Itgbl1 Reverse: GTCCCCAGTTGGATCAACATC | This Paper | N/A |
| qPCR Primer: Hapln1 Forward: AAATGTGAGGTGATTGAAGGGC | This Paper | N/A |
| qPCR Primer: Hapln1 Reverse: GCGTCCCAGTCGTGGAAAG | This Paper | N/A |
| qPCR Primer: Sox9 Forward: GCCACGGAACAGACTCACAT | This Paper | N/A |
| qPCR Primer: Sox9 Reverse: GGACCCTGAGATTGCCCAGA | This Paper | N/A |
| qPCR Primer: Sox2 Forward: TAGAGCTAGACTCCGGGCGATGA | This Paper | N/A |
| qPCR Primer: Sox2 Reverse: TTGCCTTAAACAAGACCACGAAA | This Paper | N/A |
| qPCR Primer: Scgb1a1 Forward: ATGAAGATCGCCATCACAATCAC | This Paper | N/A |
| qPCR Primer: Scgb1a1 Reverse: GGATGCCACATAACCAGACTCT | This Paper | N/A |
| qPCR Primer: Scgb3a2 Forward: GACAGGACTGAAGAAGTGTGTGG | This Paper | N/A |
| qPCR Primer: Scgb3a2 Reverse: GGAGGTTGTTCACGTAGCAAAGG | This Paper | N/A |
| qPCR Primer: Foxj1 Forward: CTCCTATGCCACTCTCATCTGC | This Paper | N/A |
| qPCR Primer: Foxj1 Reverse: GACAGGTTGTGGCGGATGGAAT | This Paper | N/A |
| qPCR Primer: Mcidas Forward: GTGGAAGTCCTTTTCGGGATGC | This Paper | N/A |
| qPCR Primer: Mcidas Reverse: TAGGAGACGCTTCGGTTCCGAG | This Paper | N/A |
| qPCR Primer: Myb Forward: AGACCCCGACACAGCATCTA | This Paper | N/A |
| qPCR Primer: Myb Reverse: CAGCAGCCCATCGTAGTCAT | This Paper | N/A |
| qPCR Primer: Ascl1 Forward: TCTGGCAAGATGGAGAGTGGAGC | This Paper | N/A |
| qPCR Primer: Ascl1 Reverse: AAAGAAGCAGGCTGCGGGAG | This Paper | N/A |
| qPCR Primer: Calca Forward: CCTTTCCTGGTTGTCAGCATCTTG | This Paper | N/A |
| qPCR Primer: Calca Reverse: CTGGGCTGCTTTCCAAGATTGAC | This Paper | N/A |
| qPCR Primer: Chrdl1 Forward: AACCTCCAAGCCAAAACTTTGA | This Paper | N/A |
| qPCR Primer: Chrdl1 Reverse: CCAGTGCTACTTTTCTGGTTGTC | This Paper | N/A |
| qPCR Primer: Grem2 Forward: GGTAGCTGAAACACGGAAGAA | This Paper | N/A |
| qPCR Primer: Grem2 Reverse: TCTTGCACCAGTCACTCTTGA | This Paper | N/A |
| qPCR Primer: Hopx Forward: CCACGCTGTGCCTCATCGCA | This Paper | N/A |
| qPCR Primer: Hopx Reverse: GGCCTGGCTCCCTAGTCCGT | This Paper | N/A |
| qPCR Primer: Sftpc Forward: ATGGACATGAGTAGCAAAGAGGT | This Paper | N/A |
| qPCR Primer: Sftpc Reverse: CACGATGAGAAGGCGTTTGAG | This Paper | N/A |
Highlights:
Inactivating Myocd prevented airway smooth muscle differentiation and peristalsis.
Preventing airway smooth muscle differentiation did not alter epithelial branching.
Preventing airway smooth muscle differentiation disrupted tracheal architecture.
Preventing airway smooth muscle differentiation led to reduced airway diameter.
Acknowledgements
We thank the members of the Sun lab for insightful discussions. We thank Dr. Michael Parmacek and Dr. Minsheng Zhu for sharing mouse strains. Confocal imaging was performed at the UCSD School of Medicine Microscopy Core, supported by NINDS NS047101. RNA-seq was conducted at the UCSD IGM Core. This work was supported by National Heart, Lung, and Blood Institute grants R01 HL142215, HL143256, HL122406 and HL119946 (to X.S.). R.Y. has received support from the NSF GRFP #DGE-1747503 and the Laboratory of Genetics at U.W. Madison NIH Training Grant #5T32GM007133.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Buckley SMK, Waddington SN, Jezzard S, Lawrence L, Schneider H, Holder MV, … Coutelle C (2005). Factors influencing adenovirus-mediated airway transduction in fetal mice. Molecular Therapy, 12(3), 484–492. 10.1016/j.ymthe.2005.02.020 [DOI] [PubMed] [Google Scholar]
- Chen F, Marquez H, Kim YK, Qian J, Shao F, Fine A, … Cardoso WV (2014). Prenatal retinoid deficiency leads to airway hyperresponsiveness in adult mice. Journal of Clinical Investigation, 124(2), 801–811. 10.1172/JCI70291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung MI, Bujnis M, Barkauskas CE, Kobayashi Y, & Hogan BLM (2018). Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development (Cambridge), 145(9), 1–10. 10.1242/dev.163014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Kitzmiller JA, Sridharan A, Perl AK, Bridges JP, Misra RS, … Xu Y (2017). Lung Gene Expression Analysis (LGEA): An integrative web portal for comprehensive gene expression data analysis in lung development. Thorax, 72(5), 481–484. 10.1136/thoraxjnl-2016-209598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenthal N, & Zelzer E (2017a). Mechanical regulation of musculoskeletal system development, 9, 4271–4283. 10.1242/dev.151266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenthal N, & Zelzer E (2017b). Mechanical regulation of musculoskeletal system development. Development, 144(23), 4271–4283. 10.1242/dev.151266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin K, Mao S, Guyomar T, Miller E, Radisky DC, Košmrlj A, & Nelson CM (2019). Smooth muscle differentiation shapes domain branches during mouse lung development. Development (Cambridge, England), (October). 10.1242/dev.181172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris-Johnson KS, Domyan ET, Vezina CM, & Sun X (2009). β-Catenin promotes respiratory progenitor identity in mouse foregut. Proceedings of the National Academy of Sciences of the United States of America, 106(38), 16287–16292. 10.1073/pnas.0902274106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines E. a, Jones MN, Verheyden JM, Harvey JF, & Sun X (2013). Establishment of smooth muscle and cartilage juxtaposition in the developing mouse upper airways. Proceedings of the National Academy of Sciences of the United States of America, 110(48), 19444–19449. 10.1073/pnas.1313223110/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1313223110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines E. a, Jones MN, Verheyden JM, Harvey JF, Sun XX, Wei C, … Morrisey EE (2013). Establishment of smooth muscle and cartilage juxtaposition in the developing mouse upper airways. Proceedings of the National Academy of Sciences of the United States of America, 110(48), 19444–19449. 10.1073/pnas.1313223110/-/DCSupplemental.www.pnas.org/cgi/doi/10.1073/pnas.1313223110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Cheng L, Li J, Chen M, Zhou D, Lu MM, … Parmacek MS (2008). Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. Journal of Clinical Investigation, 118(2). 10.1172/JCI33304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesudason EC, Smith NP, Connell MG, Spiller DG, White MRH, Fernig DG, & Losty PD (2006). Peristalsis of airway smooth muscle is developmentally regulated and uncoupled from hypoplastic lung growth. American Journal of Physiology. Lung Cellular and Molecular Physiology, 291(4), L559–65. 10.1152/ajplung.00498.2005 [DOI] [PubMed] [Google Scholar]
- Kim HY, Pang MF, Varner VD, Kojima L, Miller E, Radisky DC, & Nelson CM (2015). Localized Smooth Muscle Differentiation Is Essential for Epithelial Bifurcation during Branching Morphogenesis of the Mammalian Lung. Developmental Cell, 34(6), 719–726. 10.1016/j.devcel.2015.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto K, Tamura M, Nishita M, Minami Y, Yamaoka A, Abe T, … Morimoto M (2018). Synchronized mesenchymal cell polarization and differentiation shape the formation of the murine trachea and esophagus. Nature Communications, 9(2816). 10.1038/s41467-018-05189-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro D, Price M, Felice D,M, Lauro D,R, … Di Lauro R (1991). The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development (Cambridge, England), 113(4), 1093–1104. [DOI] [PubMed] [Google Scholar]
- Lee JH, Tammela T, Hofree M, Choi J, Marjanovic ND, Han S, … Kim CF (2017). Anatomically and Functionally Distinct Lung Mesenchymal Populations Marked by Lgr5 and Lgr6. Cell, 170(6), 1149–1163.e12. 10.1016/j.cell.2017.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang Z, Chu Q, Jiang K, Li J, & Tang N (2018). The Strength of Mechanical Forces Determines the Differentiation of Alveolar Epithelial Cells. Developmental Cell, 44(3), 297–312.e5. 10.1016/j.devcel.2018.01.008 [DOI] [PubMed] [Google Scholar]
- Li X, Du X, Li D, Kong P, Sun Y, Liu P, … Feng Y (2015). ITGBL1 Is a Runx2 Transcriptional Target and Promotes Breast Cancer Bone Metastasis by Activating the TGF b Signaling Pathway, 75(16), 3302–3314. 10.1158/0008-5472.CAN-15-0240 [DOI] [PubMed] [Google Scholar]
- Mailleux AA, Kelly R, Veltmaat JM, De Langhe SP, Zaffran S, Thiery JP, & Bellusci S (2005). Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development, 132(9), 2157–2166. 10.1242/dev.01795 [DOI] [PubMed] [Google Scholar]
- Mandar Deepak Muzumdar Bosiljka Tasic, Miyamichi Kazunari, Li Ling, and L. L (2006). A Gobal Double-Flourescent Cre Reporter Mouse. Genesis, 224(September), 219–224. 10.1002/dvg [DOI] [PubMed] [Google Scholar]
- Metzger RJ, Klein OD, Martin GR, & Krasnow MA (2008). The branching programme of mouse lung development. Nature, 453(7196), 745–750. 10.1038/nature07005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisey EE, & Hogan BLM (2010). Preparing for the First Breath: Genetic and Cellular Mechanisms in Lung Development. Developmental Cell, 18(1), 8–23. 10.1016/j.devcel.2009.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Gleghorn JP, Pang M, Jaslove JM, Goodwin K, Varner VD, … Stone HA (2017). Microfluidic chest cavities reveal that transmural pressure controls the rate of lung development, 4328–4335. 10.1242/dev.154823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovchinnikov DA, Deng JM, Ogunrinu G, & Behringer RR (2000). Col2a1- directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis, 26(2), 145–146. [DOI] [PubMed] [Google Scholar]
- Peng T, Tian Y, Boogerd CJ, Lu MM, Kadzik RS, Stewart KM, … Morrisey EE (2013). Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature, 500(7464), 589–592. 10.1038/nature12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe RA, Nowlan NC, Kenny EM, Cormican P, Morris DW, Prendergast PJ, … Murphy P (2014). Identification of mechanosensitive genes during skeletal development: Alteration of genes associated with cytoskeletal rearrangement and cell signalling pathways. BMC Genomics, 15(1). 10.1186/1471-2164-15-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salingcarnboriboon R, Tsuji K, Komori T, Nakashima K, Ezura Y, & Noda M (2006). Runx2 Is a Target of Mechanical Unloading to Alter Osteoblastic Activity and Bone Formation in Vivo, 147(5), 2296–2305. 10.1210/en.2005-1020 [DOI] [PubMed] [Google Scholar]
- Schittny JC, Miserocchi G, & Sparrow MP (2000). Spontaneous peristaltic airway contractions propel lung liquid through the bronchial tree of intact and fetal lung explants. American Journal of Respiratory Cell and Molecular Biology, 23(1), 11–18. 10.1165/ajrcmb.23.1.3926 [DOI] [PubMed] [Google Scholar]
- Short K, Hodson M, & Smyth I (2013). Spatial mapping and quantification of developmental branching morphogenesis. Development (Cambridge), 140(2), 471–478. 10.1242/dev.088500 [DOI] [PubMed] [Google Scholar]
- Song EK, Jeon J, Jang DG, Kim HE, Sim HJ, Kwon KY, … Park TJ (2018). ITGBL1 modulates integrin activity to promote cartilage formation and protect against arthritis, 7486(October), 1–16. [DOI] [PubMed] [Google Scholar]
- Sparrow MP, & Lamb JP (2003). Ontogeny of airway smooth muscle: Structure, innervation, myogenesis and function in the fetal lung. Respiratory Physiology and Neurobiology, 137(2–3), 361–372. 10.1016/S1569-9048(03)00159-9 [DOI] [PubMed] [Google Scholar]
- Tadokoro T, Gao X, Hong CC, Hotten D, & Hogan BLM (2016). BMP signaling and cellular dynamics during regeneration of airway epithelium from basal progenitors. Development, 143(5), 764–773. 10.1242/dev.126656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volckaert T, Dill E, Campbell A, Tiozzo C, Majka S, Bellusci S, & Langhe S. P. De. (2011). Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. The Journal of Clinical Investigation, 121(11), 4409–4419. 10.1172/JCI58097.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, & Yamada Y (1999). Mice lacking link protein develop dwarfism and craniofacial abnormalities. Nature Genetics, 21(2), 225–229. 10.1038/6016 [DOI] [PubMed] [Google Scholar]
- Yin W, Kim H, Wang S, Gunawan F, Wang L, Kishimoto K, … Stainier DYR (2018). The potassium channel KCNJ13 is essential for smooth muscle cytoskeletal organization during mouse tracheal tubulogenesis. Nature Communications, 1–13. 10.1038/s41467-018-05043-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, … Komori T (2004). Runx2 and Runx3 are essential for chondrocyte maturation , and Runx2 regulates limb growth through induction of Indian hedgehog, 952–963. 10.1101/gad.1174704.and [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, & Morrisey EE (2017a). Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell, 170(6), 1134–1148.e10. 10.1016/j.cell.2017.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, & Morrisey EE (2017b). Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell, 170(6), 1134–1148.e10. 10.1016/j.cell.2017.07.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, Peng YJ, Zhang GS, He WQ, Qiao YN, Dong YY, … Zhu MS (2010). Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. Journal of Biological Chemistry, 285(8), 5522–5531. 10.1074/jbc.M109.062836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Menke DB, Jiang M, Chen H, Warburton D, Turcatel G, … Shi W (2013). Spatial-temporal targeting of lung-specific mesenchyme by a Tbx4 enhancer. BMC Biology, 11, 111 10.1186/1741-7007-11-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X-L (2014). Myocardin and smooth muscle differentiation. Archives of Biochemistry and Biophysics, 543, 48–56. 10.1016/j.abb.2013.12.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Supplemental to Figure 3. Peristalsis video for control lungs cultured for 24 hours. Video is a total of 6 seconds condensed from 10 minutes of filming.
Movie S2. Supplemental to Figure 3. Peristalsis video for MyocdCKO lungs cultured for 24 hours. Video is a total of 6 seconds long, condensed from 10 minutes of filming.
Data Availability Statement
All RNA-seq fastq and processed files are publicly available at the NIH NCBI GEO database accession number GSE143394. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE143394