

Abstract
Glucocorticoids (GCs), key mediators of stress signals, are also potent wound healing inhibitors. To understand how stress signals inhibit wound healing, we investigated the role of membranous glucocorticoid receptor (mbGR) by using cell-impermeable BSA-conjugated dexamethasone. We found that mbGR inhibits keratinocyte migration and wound closure by activating a Wnt-like phospholipase (PLC)/ protein kinase C (PKC) signaling cascade. Rapid activation of mbGR/PLC/PKC further leads to activation of known biomarkers of nonhealing found in patients, β-catenin and c-myc. Conversely, a selective inhibitor of PKC, calphostin C, blocks mbGR/PKC pathway, and rescues GC-mediated inhibition of keratinocyte migration in vitro and accelerates wound epithelialization of human wounds ex vivo. This novel signaling mechanism may have a major impact on understanding how stress response via GC signaling regulates homeostasis and its role in development and treatments of skin diseases, including wound healing. To test tissue specificity of this nongenomic signaling mechanism, we tested retinal and bronchial human epithelial cells and fibroblasts. We found that mbGR/PLC/PKC signaling cascade exists in all cell types tested, suggesting a more general role. The discovery of this nongenomic signaling pathway, in which glucocorticoids activate Wnt pathway via mbGR, provides new insights into how stress-mediated signals may activate growth signals in various epithelial and mesenchymal tissues.
INTRODUCTION
In addition to being major stress signals, glucocorticoids (GCs) are widely used therapeutic agents in treatment of both local and systemic inflammatory disorders. Prolonged therapeutic use of GCs has numerous side effects including potent inhibition of wound healing (Lee et al., 2005; Stojadinovic et al., 2007; Vukelic et al., 2011). Wound healing involves a complex, multistep process that requires an intricate balance of various signaling pathways to restore epidermal barrier. Aberrant signaling in response to injury leads to impairment of epithelialization, common for major types of chronic wounds (Brem et al., 2007; Pastar et al., 2014; Stojadinovic et al., 2005). In spite of epidemic proportions of nonhealing wounds, development of new therapies is impeded by lack of understanding regarding mechanisms that inhibit healing (Eming et al., 2014).
We found that GCs are synthesized in epidermis and regulate inflammatory response during acute injury (Slominski et al., 2007; Slominski et al., 2005a; Slominski et al., 2005b; Slominski et al., 2004; Stojadinovic et al., 2007; Vukelic et al., 2011), whereas stress was found to impair wound healing (Stojadinovic et al., 2012). Emerging evidence that local production in skin may contribute to systemic GCs and vice versa suggests substantial cross-talk between local and systemic hypothalamic-pituitary-adrenal axis that may influence healing outcomes (Jozic et al., 2014, Jozic et al., 2015; Skobowiat and Slominski, 2015; Slominski et al., 2015).
The membranous fraction of glucocorticoid receptor (mbGR) was found to regulate the activity of many signaling molecules (Almawi and Melemedjian, 2002; Chen and Farese, 1999; Samarasinghe et al., 2012; Strehl and Buttgereit, 2014; Strehl et al., 2011; Vernocchi et al., 2013). These nongenomic changes do not require direct interaction of glucocorticoid receptor (GR) with a promoter. Instead, they activate secondary messenger systems to generate biological responses within minutes, serving as a priming event to prepare cells for subsequent genomic activity (Almawi and Melemedjian, 2002; Chen and Farese, 1999; Strehl and Buttgereit, 2014; Strehl et al., 2011; Vernocchi et al., 2013). Our recent discovery that GR localizes to the plasma membrane of keratinocytes adds to the complexity and suggests presence of additional pathways in skin (Stojadinovic et al., 2013).
Wnt pathway is essential for many aspects of skin development, physiology, and pathology (He et al., 1998). Previous microarray analyses of GC-treated keratinocytes, demonstrated upregulation of protein kinase C (PKC) (Stojadinovic et al., 2007), which regulates glycogen synthase kinase 3-β (GSK-3β) activity (Christian et al., 2002; Goode et al., 1992; Hart et al., 1998; Hinoi et al., 2000; Nakamura et al., 1998). We found that β-catenin acts as a coregulator of GR-mediated transcriptional regulation of wound-inducible keratin genes and EGF-mediated migration (Lee et al., 2005; Radoja et al., 2000; Stojadinovic et al., 2005; Stojadinovic et al., 2007). Furthermore, we found that in epidermis of chronic wounds, activation of β-catenin, and consequentially c-myc, is associated with a nonhealing phenotype (Stojadinovic et al., 2005). Thus, we postulate that activation of mbGR may activate a Wnt-like pathway, contributing to inhibition of wound closure. Indeed, we found that targeting mbGR by dexamethasone (Dex)-BSA leads to rapid activation of phospholipase-γ (PLCγ) and protein kinase C (PKC) that, in turn, leads to phosphorylation of GSK-3β, activation of nuclear β-catenin and subsequent overexpression of c-myc. This nongenomic signaling functionally impairs keratinocytes resulting in inhibition of migration and wound closure that can be reversed by selective inhibitors of PKC. Furthermore, we found that this signaling pathway functions in various cells and tissues, suggesting its more general role in integrating stress signals (mediated by GCs) into growth signals (mediated by Wnt pathway).
RESULTS
GCs promote nuclear localization of β-catenin through protein kinase C pathway
To test if GCs activate β-catenin, we stimulated primary human keratinocytes (HEKs) with Dex in the presence/absence of GR antagonist, Ru486, and evaluated localization of the phosphorylated β-catenin through indirect immunofluorescence (Ogiwara et al., 1998; Stojadinovic et al., 2005). As expected, treatment with lithium chloride resulted in activation and nuclearization of β-catenin (Figure 1a and b). Similarly, treatment with Dex led to a robust nuclearization of β-catenin, which could be blocked by pretreatment with GR antagonist, Ru486, suggesting that GCs may activate β-catenin pathway (Figure 1c and d). To determine if observed activation of β-catenin is mediated by PKC, we pretreated HEKs in the presence or absence of calphostin C (CC), a known inhibitor of PKC (Ogiwara et al., 1998). Pretreatment with CC blocked the Dex-mediated activation of β-catenin, as evidenced by its absence from the nucleus of HEKs (Figure 1e). These findings suggest that GCs can mediate rapid effects, resulting in nuclear localization of β-catenin through activation of the PKC pathway.
Figure 1. GCs promote nuclearization of β-catenin through PKC.
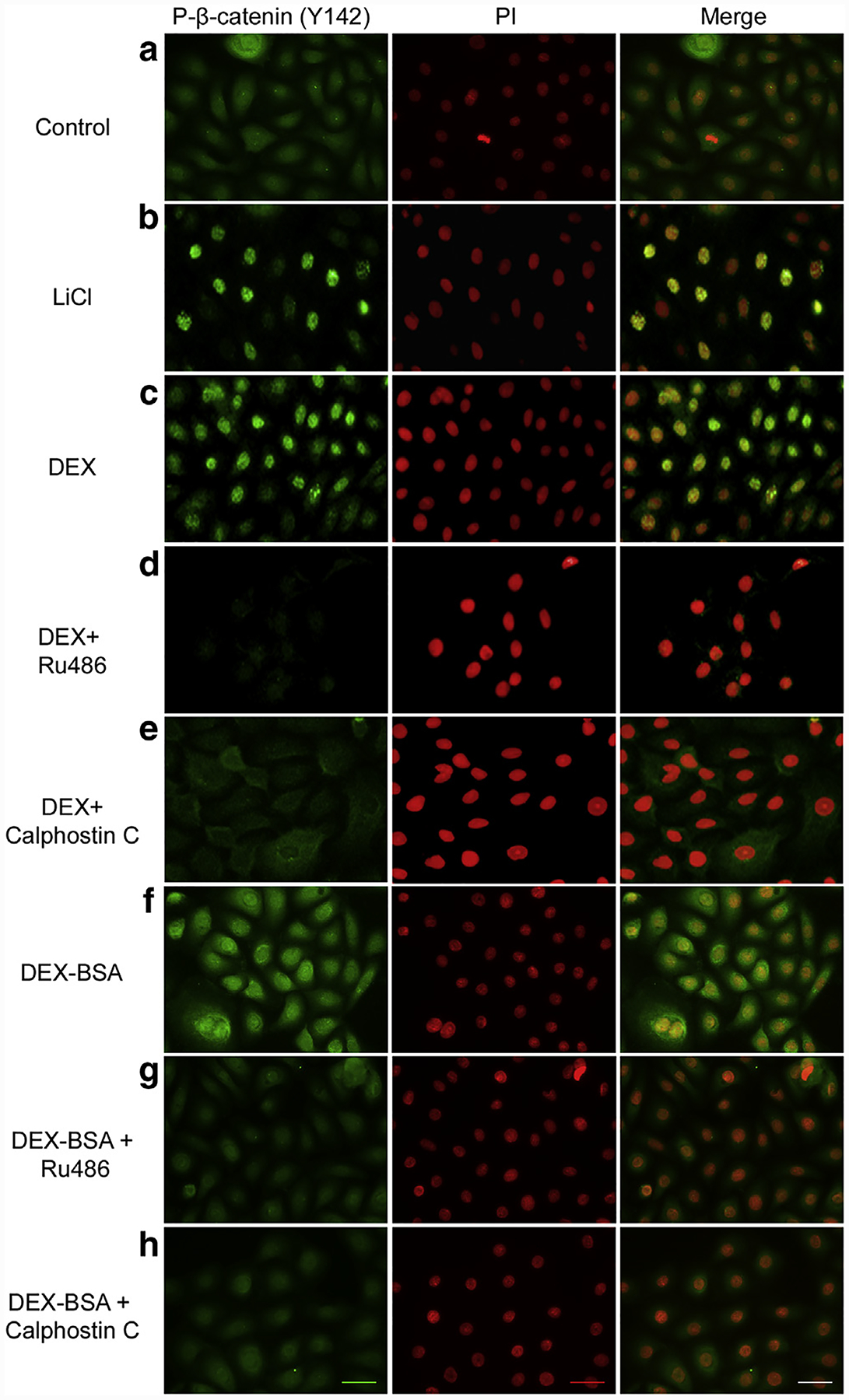
Primary human keratinocytes control (a) or stimulated with either 30 mM LiCl (b), 1 μM Dex (c) or 100 nM Dex-BSA (f) for 24 hours in presence of 1 μM Ru486 (d and g) or 0.2 μM CC (e and h) are shown. Presence of phospho-β-catenin (Y142) was visualized by immunofluorescence (green; left panels). Nuclei were visualized by staining with PI (red; middle panels) and relative nuclearization was assessed by merging the two images using ImageJ (with co-localization appearing yellow; right panels). CC, calphostin C; Dex, dexamethasone; Dex-BSA, BSA-conjugated dexamethasone; GC, glucocorticoid; LiCl, lithium chloride; PI, propidium iodide; PKC, protein kinase C. Scale bar = 50 μm.
Selective activation of membranous glucocorticoid receptor by BSA-conjugated dexamethasone does not contribute to transcriptional regulation of genomic glucocorticoid receptor
We and others found previously that a subpopulation of the GR is localized at the plasma membrane (Bartholome et al., 2004; Stojadinovic et al., 2013; Strehl et al., 2011). We used Dex-BSA to functionally characterize the mbGR because the large BSA moiety prevents entry into cells and biding to their intracellular receptors (Hu et al., 2010; Nahar et al., 2015; Samarasinghe et al., 2012). To test if mbGR is involved in activating β-catenin, we stimulated HEKs with Dex-BSA in the presence/absence of Ru486 and CC (GR and PKC antagonists, respectively) and evaluated localization of the phosphorylated β-catenin through indirect immunofluorescence. Dex-BSA treatment led to nuclearization of β-catenin, which was reversed by GR and PKC antagonists (Figure 1, f–h). We then treated HEKs with 1 μM Dex (D) or 100 nM Dex-BSA (DB) for 10, 15, 30, 45, 60, or 120 minutes and assessed activation of GR by immunoblotting of GR phosphorylation at S211 (Wang et al., 2002). We observed rapid activation of GR, within first 10 minutes, that reached its peak between 30 and 45 minutes after stimulation with either ligand (Supplementary Figure S1a online). To discern genomic from nongenomic effects, we treated HEKs with either Dex or Dex-BSA in the presence or absence of a transcriptional inhibitor, actinomycin D, and tested the expression of GILZ (GC response element-induced leucine zipper) via real-time PCR, which we found to be among the first transcriptionally regulated genes by GCs in HEKs (Stojadinovic et al., 2007). As expected, Dex stimulated induction of GILZ, whereas Dex-BSA showed no change in GILZ expression (Supplementary Figure S1b online), suggesting that Dex-BSA-mbGR does not contribute to genomic effects by directly regulating gene expression. Furthermore, we treated keratinocytes with Dex or Dex-BSA and analyzed phosphorylated GR (S211) localization after subcellular fractionation and observed that treatment of keratinocytes with Dex stimulated nuclear translocation of phosphorylated GR, whereas treatment with Dex-BSA did not. There were no detectable changes in nuclearization of total GR as shown by immunofluorescence staining of HEKs after treatment with Dex or Dex-BSA (Supplementary Figure S1c online). Collectively, these results suggest that Dex-BSA selectively activates membranous GR in HEKs and does not contribute to transcriptional regulation of genomic GR.
Membranous glucocorticoid receptor activates phospholipase C/protein kinase C/glycogen synthase kinase 3 beta pathway that induces c-myc
To assess if mbGR activates β-catenin and by which mechanism, we first examined the phosphorylation state of GSK-3β (Goode et al., 1992) by treating with either Dex or Dex-BSA for 30 minutes and immunoblotting against phosphorylated GSK-3β (Supplementary Figure S1d online). We observed a clear induction of GSK-3β (S9) phosphorylation in keratinocytes treated with Dex-BSA (comparable to Dex), indicating rapid GSK-3β phosphorylation mediated by the mbGR. Because inhibition of PKC by treatment with CC resulted in a significant reduction of nuclear β-catenin (see Figure 1), we focused on testing whether PKC pathway was essential for mbGR-mediated induction of GSK-3β phospho-inactivation and nuclear translocation of β-catenin. We treated keratinocytes with either Dex or Dex-BSA, and tested for activation of PKC and its upstream regulator PLCγ via immunoblotting. We observed a clear phosphorylation of both PKC (pan S660) and its upstream regulator PLCγ1 (Y783) (Supplementary Figure S1e online).
Next, we used a series of small enzymatic inhibitors of each step of the signaling pathway. HEKs were incubated with Dex-BSA alone or in combination with either GR-antagonist Ru486, PKC inhibitors CC or Go6976, and PLC inhibitor, genistein. Pretreatment of cells with Ru486 selectively inhibited the Dex-BSAemediated phosphorylation of GR (S211) shown by immunoblotting (Figure 2a). We observed that Dex-BSA induced phosphorylation of PLCγ1, whereas genistein pretreatment inhibited it (Figure 2b). Because genistein is a nonspecific tyrosine kinase inhibitor with multiple effects in the cell, we also performed experiments using a PLC-specific inhibitor, U73122, and found similar results (Supplementary Figure S1h online). Furthermore, Dex-BSA induced phosphorylation of PKC, which was ameliorated by pretreatment of cells with CC (Figure 2c). Lastly, we pretreated cells with either CC (general PKC inhibitor) or Go6976 (selective PKCα/β1 inhibitor) and assessed phosphorylation of GSK-3β (S9) by immunoblotting. We observed that Dex-BSA induced phosphorylation of GSK-3β; however, pretreatment of cells with either CC or Go6976 selectively inhibited Dex-BSAemediated phosphorylation of GSK-3β (Figure 2d). Taken together, these results show that GCs mediate GSK-3β phosphorylation by activating PLC/PKC pathway, thus resulting in nuclearization of β-catenin via a pathway that uses mbGR. To confirm that indeed Dex-BSAemediated activation of the PLC/PKC pathway results in transcriptionally active β-catenin, we tested the expression of c-myc (a known β-catenin target gene) (Stojadinovic et al., 2005) by incubating HEKs with Dex-BSA for 4 hours in the presence or absence of PKC inhibitor, CC. We confirmed that Dex-BSAemediated induction of c-myc expression is effectively blocked by CC (Figure 2e). Altogether, we conclude that activation of mbGR induces c-myc expression in a PLC/PKC-dependent manner.
Figure 2. mbGR stimulation activates PLC/PKC signaling cascade and induces phosphorylation and subsequent inactivation of GSK-3β resulting in c-myc induction.
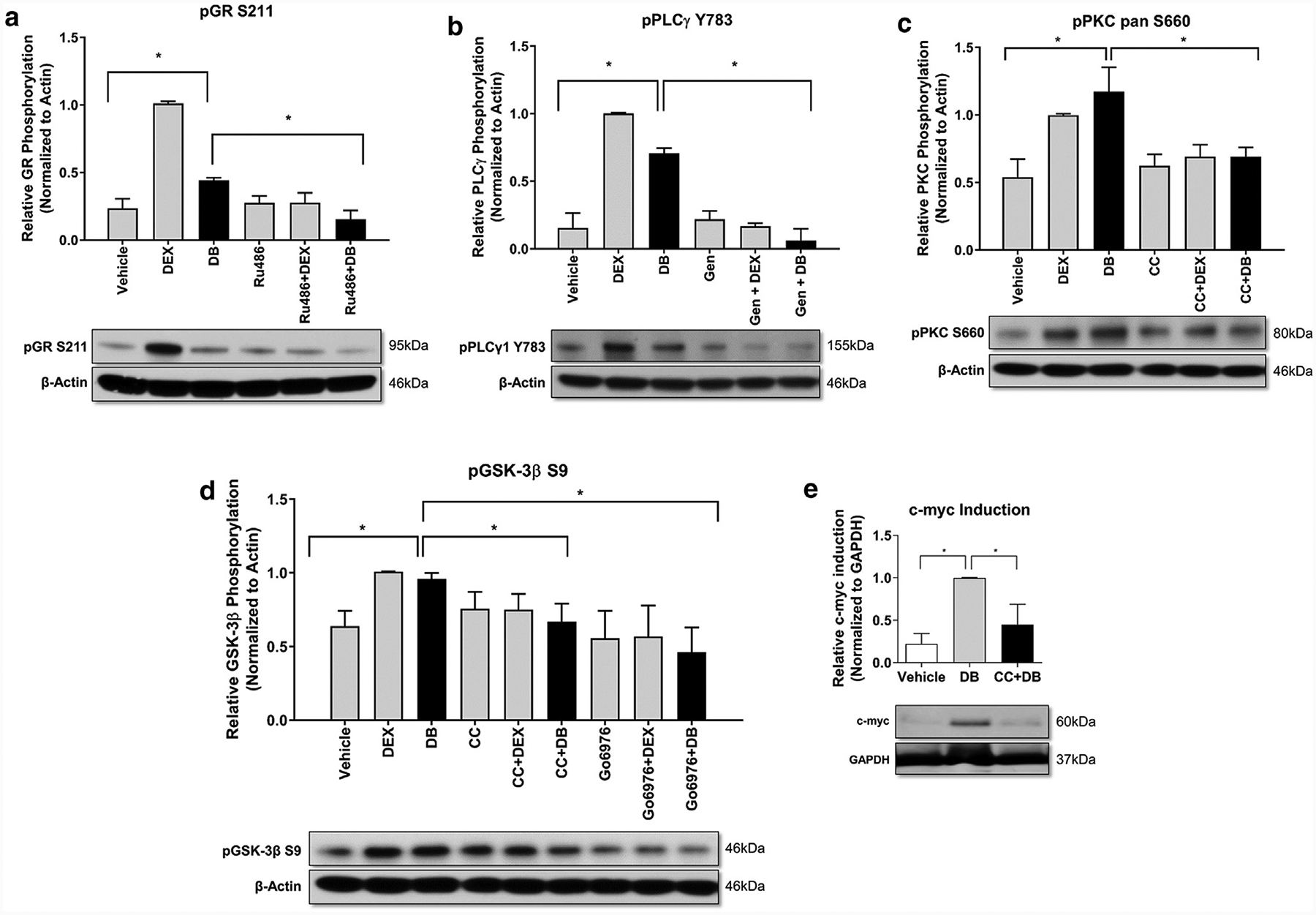
Cells were stimulated with vehicle (DMSO), 1 μM Dex, or 100 nM Dex-BSA (DB) for 30 minutes upon treatment with GR antagonist Ru486 (1 μM), PLCγ inhibitor, genistein (50 μM) or PKC inhibitors CC (200 nM) or Go6976 (4μM). Phosphorylation of GR (a), PLCγ (b), PKC (c), and GSK-3β (d) were assessed by western blot. (e) Cells were stimulated with vehicle (DMSO) or 100 nM Dex-BSA for 4 hours upon treatment with 200 nM CC, and induction of c-myc was assessed by western blotting. All quantifications were performed using ImageJ with error bars corresponding to standard deviation from n = 3. CC, calphostin C; Dex, dexamethasone; Dex-BSA, BSA-conjugated dexamethasone (DB); GR, glucocorticoid receptor; GSK-3β, glycogen synthase kinase 3 beta; mbGR, membranous glucocorticoid receptor; PKC, protein kinase C; PLC, phospholipase C. *P ≤ 0.05 (Student t test).
Membranous glucocorticoid receptor/phospholipase C/protein kinase C pathway is not restricted to skin or epithelial cells
Because we observed a novel mbGR-mediated signaling mechanism in HEKs, we then examined whether the observed mbGR/PLC/PKC/GSK-3β signaling pathway is restricted to skin epithelium or whether it is found in cells of other epithelial origin. We treated D407 human retinal epithelial cells (Figure 3a), undifferentiated or differentiated primary human bronchial epithelial cells (Figure 3b; Supplementary Figure S1f online) in presence or absence of 100 nM Dex-BSA for 30 minutes and assayed existence of the mbGR/PLC/PKC/GSK-3β signaling cascade by immunoblotting. We observed a clear induction in phosphorylation of GR (S211), PLCγ1 (Y783), PKC pan (S660), and GSK-3β (S9) in both eye and lung epithelia (Figure 3a and b; Supplementary Figure S1f) as well as the downstream target c-myc in D407 human retinal epithelial cells (Supplementary Figure S1g online). Interestingly, we also observed the presence of the same signaling cascade in primary human fibroblasts (Figure 3c), leading us to conclude that mbGR signaling cascade is conserved not only in various epithelial cell types but also in other cells of mesenchymal origin, like fibroblasts.
Figure 3. mbGR mediated activation of PLCγ/PKC/GSK-3β is present not only in cells of epithelial origin (eye and lung), but also in cells of mesenchymal origin (foot fibroblasts).
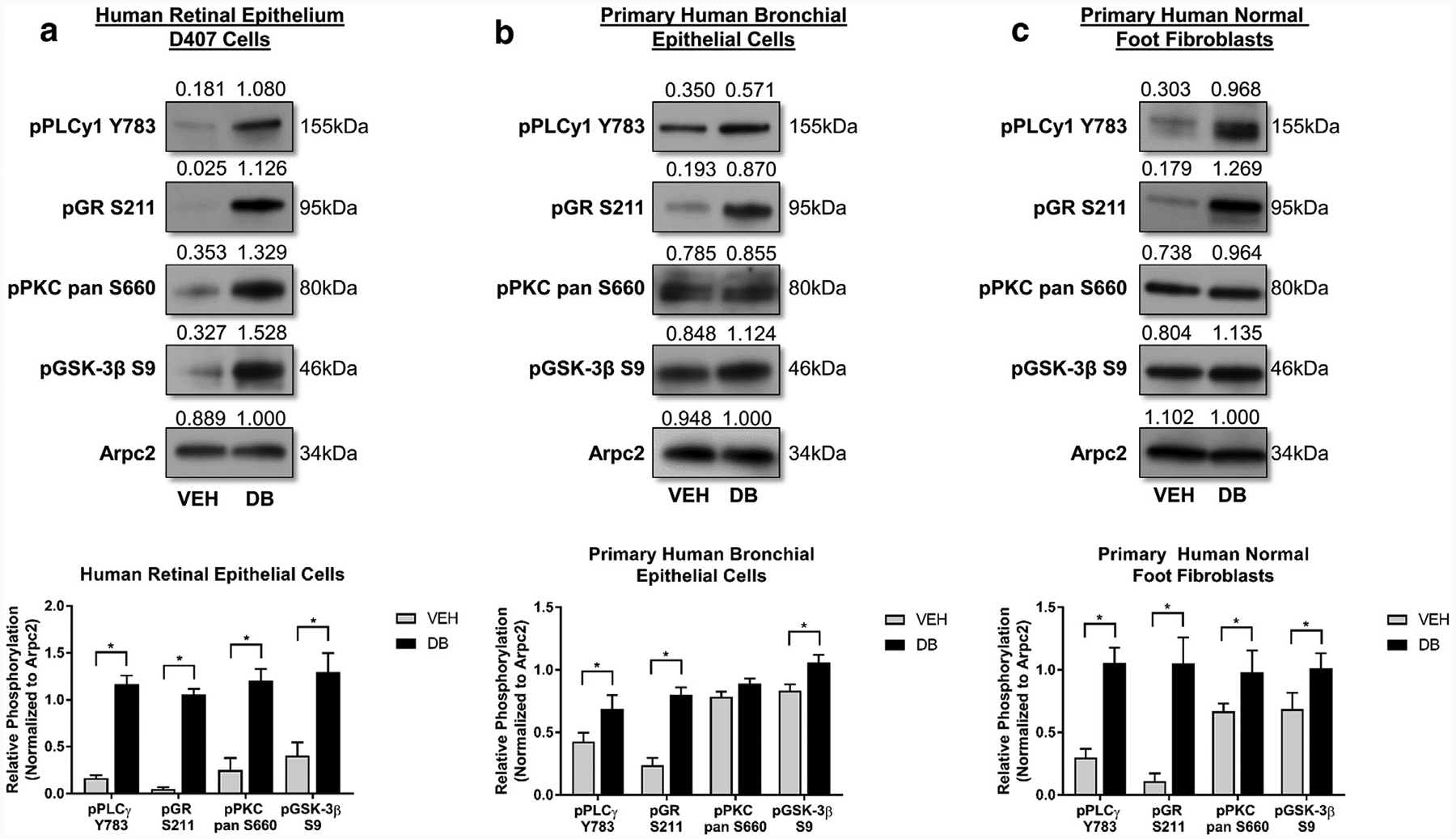
Presence of mbGR mediated activation of PLCy/PKC/GSK-3B signaling cascade was assessed in (a) D407-human retinal epithelial cells and (b) primary human bronchial epithelial cells, as well as in cells of nonepithelial origin like (c) primary human foot fibroblasts. Cells were stimulated with vehicle (DMSO) or 100 nM Dex-BSA (DB) for 30 minutes, and phosphorylation of PLCγ, GR, PKC pan, and GSK-3β were assessed by western blot, with Arpc2 serving as loading control. All quantifications were performed using ImageJ with error bars corresponding to standard deviation from n = 3. Dex-BSA, BSA-conjugated dexamethasone (DB); GR, glucocorticoid receptor; GSK-3β, glycogen synthase kinase 3 beta; mbGR, membranous glucocorticoid receptor; PKC, protein kinase C; PLC, phospholipase C. *P ≤ 0.05 (Student t test).
Inhibition of protein kinase C restores keratinocyte migration in vitro and accelerates wound closure ex vivo
We found previously that activated GR inhibits wound healing by disrupting migration of keratinocytes (Lee et al., 2005; Stojadinovic et al., 2007). To functionally test whether activation of the PLC/PKC pathway by mbGR inhibits epithelialization and wound closure, migration of keratinocytes was quantified by a wound-scratch assay at 0 hours, 24 hours, and 48 hours. Keratinocytes were treated with Dex or Dex-BSA in the presence or absence of either inhibitors of GR (Ru486), PLCγ (genistein), and PKC (CC) or EGF, a known stimulator. We found that Dex-BSA inhibits keratinocyte migration similarly to Dex, suggesting that mbGR regulates inhibition of migration in HEK (Figure 4; Video 1). Furthermore, inhibition of either GR, PLCγ, or PKC (by Ru486, genistein, and CC, respectively) reversed the Dex-BSAemediated inhibition (Figure 4; Video 1). Similar effects can be observed as early as 4 hours after treatment with Dex-BSA (data not shown). We conclude that sustained stimulation of mbGR by GCs elicits activation of mbGR/PLCγ/PKC signaling cascade and leads to inhibition of keratinocyte migration.
Figure 4. Selective activation of the mbGR results in inhibition of keratinocyte migration.
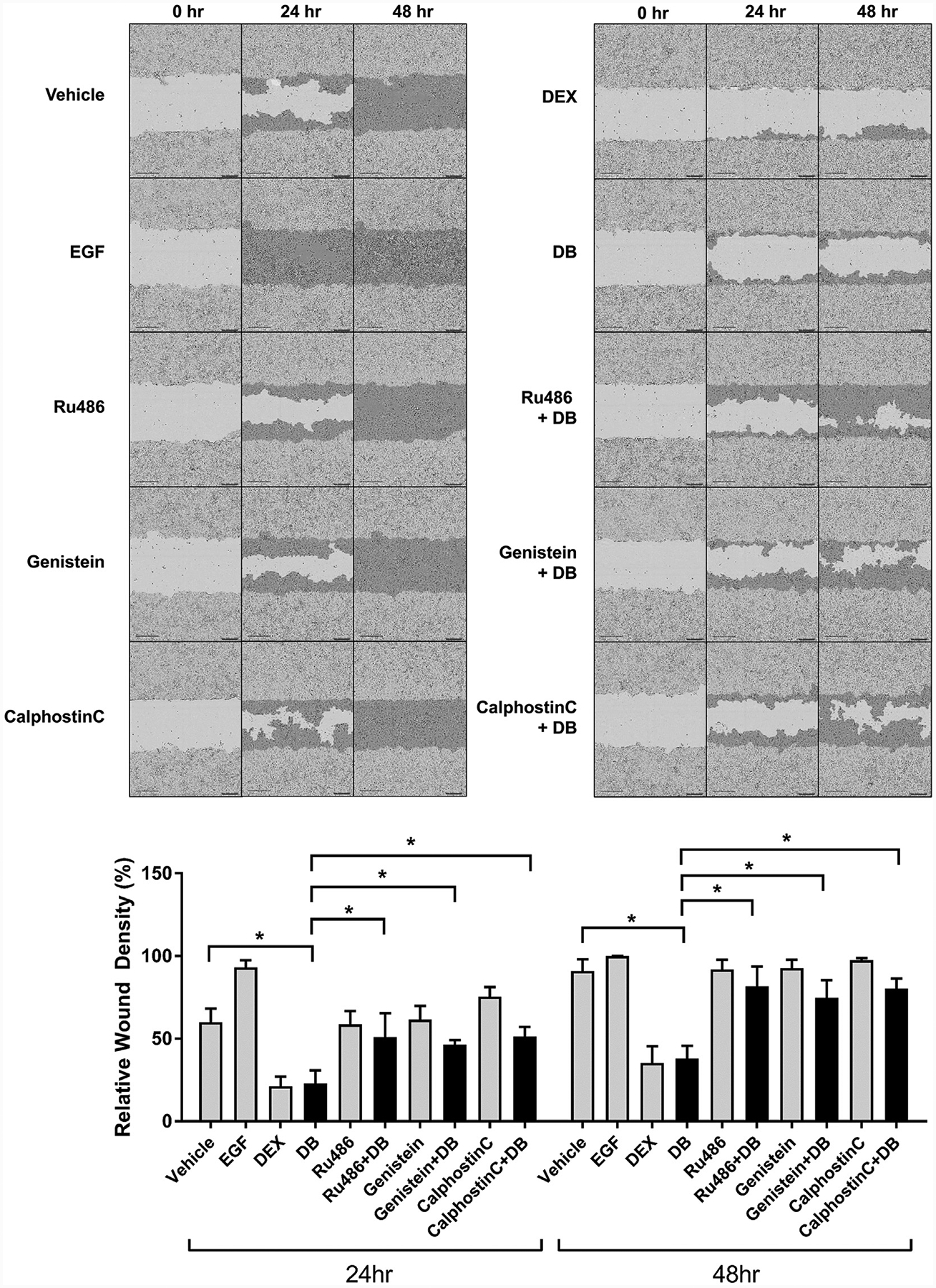
Primary human keratinocytes were pretreated with 4 μg/ml mitomycin-C and stimulated with vehicle (DMSO), 25 ng/ml EGF (positive control), 1 μM Dex or 100 nM Dex-BSA (DB). Cells were wounded by a scratch and their migration was assessed at the time of the scratch (0 h) every 2 hours for 48 hours. Representative images at 0, 24, and 48 hours after the initial scratch were used to quantify migration by using Cell Migration Analysis software module (Essen Bioscience) comparing relative wound density, with light gray corresponding to initial wound scratch and dark gray corresponding to repopulation of the wound over time. Error bars correspond to standard deviation from n = 16. *P ≤ 0.05 (Student t test). Dex, dexamethasone; Dex-BSA, BSA-conjugated dexamethasone (DB); mbGR, membranous glucocorticoid receptor.
To test functional implications of the mbGR nongenomic pathway, we used two established models: human skin ex vivo and organotypic culture wound model (Ojeh et al., 2014; Stojadinovic and Tomic-Canic, 2013). In both models, skin was wounded by 3-mm punch biopsies and treated with Dex, Dex-BSA, and EGF as positive control. Histologic assessments at day 4 postwounding show that Dex-BSA treatment inhibited epithelialization similarly to Dex in both models (Figure 5; Supplementary Figure S2 online). Furthermore, pretreatment of skin with CC prevented Dex-BSAemediated inhibition of wound healing. When present alone, CC accelerated wound closure (Figure 5). This acceleration of wound closure by CC can be contributed by blocking of activation of mbGR by endogenously synthesized cortisol (Vukelic et al, 2011).
Figure 5. Selective targeting of mbGR by Dex-BSA impedes epithelialization and can be ameliorated by inhibiting PKC in an ex vivo model of wound closure.
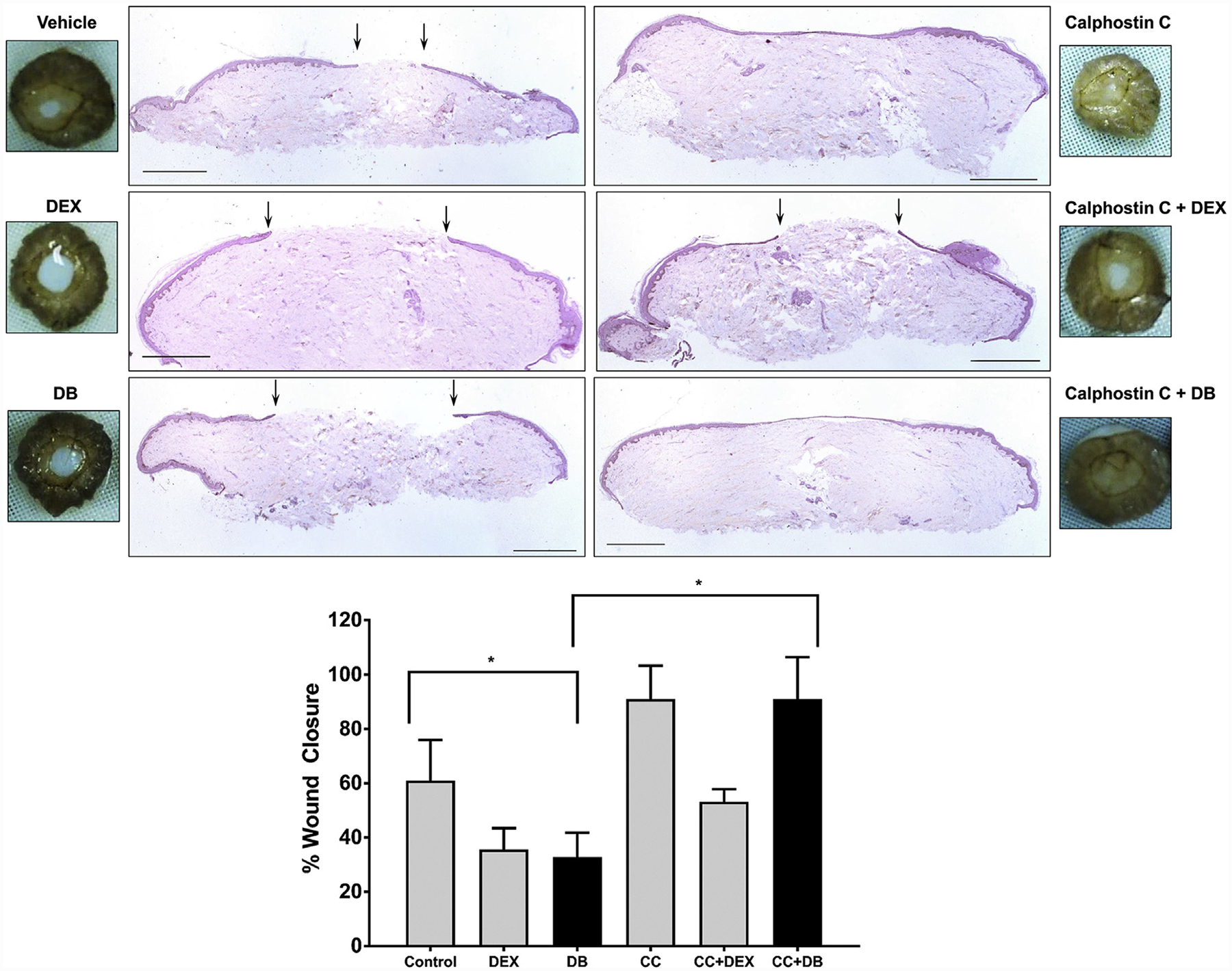
Normal human skin was wounded using a 3-mm biopsy punch and maintained at the air-liquid interface in presence or absence of 1 μM Dex, 100 nM Dex-BSA (DB), or PKC inhibitor CC (0.2 μM). Wound healing was assessed at day 4 after wounding, a time when exponential epithelialization occurs. Wound closure was quantified by histology using ImageJ software. Gross photos show visual signs of closure and correspond to the histology assessments with black arrows pointing to epithelial tongue location at day 4. Error bars correspond to standard deviation from n = 4. *P ≤ 0.05 (Student t test). Scale bar 1 = mm. Dex, dexamethasone; Dex-BSA, BSA-conjugated dexamethasone (DB); mbGR, membranous glucocorticoid receptor; PKC, protein kinase C.
Taken together, our data show how a new molecular mechanism of activation of the PLC/PKC by GCs via the mbGR results in inhibition of keratinocyte migration and epithelialization, thus contributing to wound healing impairment. Conversely, inhibition of this pathway accelerates wound closure, providing new therapeutic approaches to stimulate wound healing.
DISCUSSION
Here we show a novel mechanism that converts incoming stress signal, mediated by mbGR, into growth signals similar to those of Wnt, by activation of the β-catenin (Figure 6). We also show that stress mediators, GCs, trigger fast-acting nongenomic effects via mbGR that activate the PLC/PKC/β-catenin pathway, induce c-myc, and contribute to impairment of healing. Furthermore, we show that inhibition of PKC restores keratinocyte migration and accelerates epithelialization. Importantly, this novel signaling pathway is functional in many cell types and tissues, suggesting a more general impact. The nongenomic GC signaling that activates PKC and merges multiple major pathways may have significant impacts on understanding molecular mechanisms that govern homeostasis or pathophysiology of cutaneous (and other) diseases. The potential implications of such a concept are immense, as components of the Wnt signaling are key regulators of cell differentiation and proliferation and have, thus, been found to be deregulated in many diseases ranging from skin, cardiovascular, and neuronal disorders to various forms of cancer (Abrahamsson et al., 2009; Clements et al., 2003; Tian et al., 2003; van Gijn et al., 2002). It also underscores the potential role of stress-related signaling in these diseases. However, the contribution of GC-mediated activation of β-catenin in these processes remains to be elucidated.
Figure 6. Proposed mechanism by which GC-mediated activation of mbGR contributes to inhibition of keratinocyte migration and wound healing.
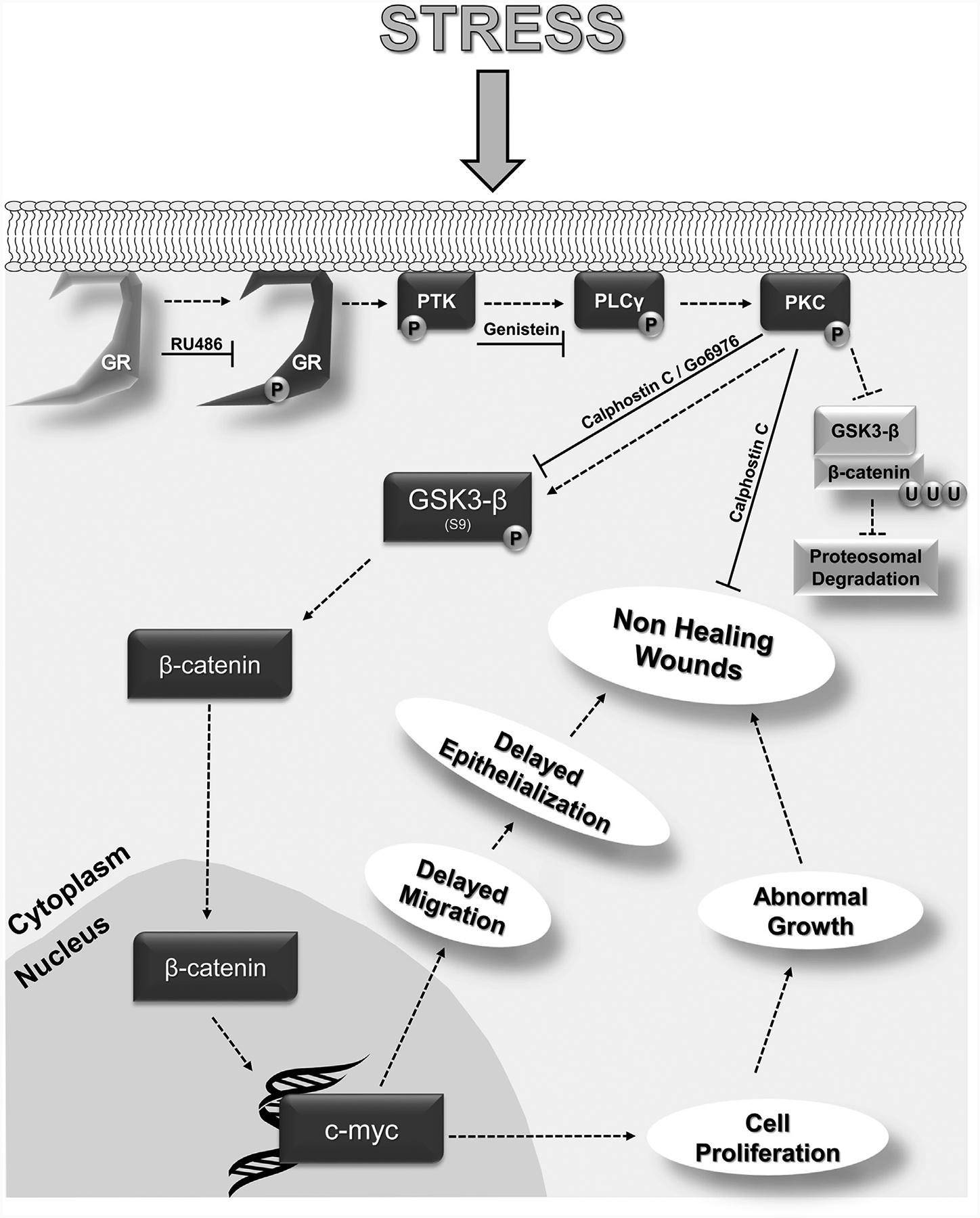
A diagram summarizing findings from this study is shown. Upon binding to the membranous fraction of GR, GCs induce a phosphorylation and activation of GR, followed by activation PLCγ (via PTK), PKC, and subsequent phospho-inactivation of GSK-3β. This in turn liberates β-catenin from the inactivation complex thereby allowing it to translocate into the nucleus and induce c-myc expression, thus inducing cell proliferation while delaying keratinocyte migration and subsequent epithelialization. GC, glucocorticoid; GR, glucocorticoid receptor; GSK-3β, glycogen synthase kinase 3 beta; mbGR, membranous glucocorticoid receptor; P, phosphorylation; PKC, protein kinase C; PLC, phospholipase C; PTK, protein tyrosine kinase; U, ubiquitination.
In the context of skin, Wnt signaling controls development, stem cell cycling, hair follicle expansion, and terminal differentiation of the hair lineage (Fuchs et al., 2004; Zhou et al., 1995). Although β-catenin promotes the recruitment of fibroblast to the wound site, uncontrolled Wnt signaling may lead to aggressive fibromatosis and resemble a hyperactive wound. In most types of chronic wounds, impairment of epithelialization arises from aberrant signaling that governs activity of keratinocytes in response to injury (Brem et al., 2007; Brem et al., 2003; Pastar et al., 2014; Stojadinovic et al., 2005). A hyperproliferative epidermis is a hallmark of the nonhealing epidermal edge of chronic wounds (Nunan et al., 2014; Stojadinovic et al., 2008) that was documented by activation of Wnt pathway and overexpression of c-myc (Eming et al., 2014; Stojadinovic et al., 2005). In spite of the activation of these growth signals, keratinocytes in chronic wounds are not epithelializing because of impairment in migration. In this report, we show a similar paradox. Although stimulation of the membranous fraction of GR results in activation of the same growth signals (β-catenin nuclearization and subsequent induction of c-myc), it results in inhibition of wound closure. All these findings suggest that GCs, in part via mbGR, may play an important role in inhibition of epithelialization and pathophysiology of chronic wounds.
The identification of PKC as a downstream target of mbGR has multiple implications, especially if one takes into account the relative abundance of PKC signaling in tissues and systemic circulating presence of GC (Czerwinski et al., 2005; Helfrich et al., 2007; Mellor and Parker, 1998; Nishizuka, 1988; Selbie et al., 1993). The PKC family is involved in signals that control cellular survival, proliferation, and differentiation (Denning et al., 1998; Farese et al., 1992; Nishizuka, 1988; Nishizuka, 1995; Wertheimer et al., 2001), and therefore plays a major role in development of diseases (Braiman et al., 1999; Dlugosz and Yuspa, 1993; Gordge et al., 1996; Idris et al., 2001; Prevostel et al., 1995; Standaert et al., 1997; Wallace et al., 2014). Our discovery of PKC activation by mbGR provides new insights into mechanisms by which systemic and local corticosteroids may contribute to PKC-mediated effects. Although we are currently investigating whether mbGR exhibits selective activation of PKC isoenzymes in both skin and other epithelia, further understanding of greater physiologic/pathologic implications of this mechanism that extend beyond skin will provide important insights into systemic GC action and its contribution to various diseases.
Deregulation of physiologic pathways underlies mechanisms of how psychosocial stressors are hypothesized to influence biology of chronic disease. Psychosocial stressors have been known to influence the hypothalamic-pituitary-adrenal axis, one of the major physiologic pathways controlling neuroendrocrine response. GCs are also commonly used as antiemetics in various forms of cancer therapy (Keith, 2008; Mitre-Aguilar et al., 2015; Rutz, 2002). Moreover, cancer patients frequently exhibit alterations in cortisol regulation (Sephton et al., 2000). The discovery that a physiologically relevant dose of glucocorticoids in epithelial and mesenchymal cells stimulates a Wnt-like signaling cascade, which culminates in elevated expression of c-myc, a regulator of cell cycle, may have profound effects on how to potentially mitigate adverse effects of both endogenous and exogenous GCs in cancer patients and opens up many avenues to pharmaceutically target mbGR in conjunction with already established inducers of tumor cell death.
Selective targeting of mbGR, as we have shown above, could prove invaluable in not only understanding various disease development but also targeted steroid therapies and their side effects. This is particularly important in the context of the endogenous constitutive synthesis of cortisol by keratinocytes (Slominski et al., 2015; Slominski et al., 2013). Topical CC, by blocking mbGR activation triggered by endogenously synthesized cortisol, resulted in accelerated epithelialization and subsequent wound healing, suggesting that selective targeting of subsets of GR, such as mbGR, may have therapeutic effects or may alleviate multiple side effects of prolonged GC therapy. It is not surprising that CC shows less-effective rescue of Dex-mediated wound healing inhibition when compared with Dex-BSA. Unlike Dex-BSA, which acts only on mbGR, Dex can stimulate multiple fractions of the receptor and simultaneously activates non-genomic and genomic GR effects.
We identified a novel molecular mechanism by which mbGR exerts a rapid, nongenomic effect via PLC/PKC cascade, leading to activation of β-catenin and c-myc. A functional consequence of mbGR/PLC/PKC pathway is inhibition of keratinocyte migration and wound closure. This mechanism may have major effects on the understanding of how GC signaling regulates homeostasis and its role in development of skin diseases that possibly extends beyond to other tissues.
MATERIALS AND METHODS
Immunofluorescence
Immunofluorescence protocol was followed as previously described (Jozic et al., 2012). Cells were grown to 65% confluency. After washing with phosphate buffered saline, fixing in 5% formalin for 5 minutes, and being permeabilized with 0.1% Triton X-100 (Sigma, St. Louis, MO) for 10 minutes, cells were blocked with 1% BSA in phosphate buffered saline for 30 minutes and then incubated in β-catenin Ab in 1% BSA for 24 hours. This was followed by phosphate buffered saline washes and incubation with Alexa Flour Ab (Thermo Fisher Scientific, Waltham, MA) in 1% BSA for 1 hour. Cells were mounted and nuclei were visualized with propidium iodide.
Immunoblotting
Cell lysates for immunoblotting and cell fractionation were prepared from a subconfluent 10-cm plate of HEKs following published protocol (Jozic et al., 2011; Jozic et al., 2012). Protein from each sample was resolved on 4 to 20% gradient Tris-Glycine gels (Bio-Rad Laboratories, Hercules, CA) and transferred onto polyvinylidene difluoride membranes. A list of antibodies used is provided in Supplementary Materials.
Quantitative PCR
RNA isolation and purification were performed as previously described (Ramirez et al., 2015). A total of 1.0 μg of total RNA from HEK was reverse transcribed using a qScript cDNA kit and real-time PCR was performed in triplicate using the Bio-Rad CFX Connect thermal cycler and detection system (Bio-Rad Laboratories) and a PerfeCTa SYBR Green Supermix (QuantaBio, Beverly, MA). Relative expression was normalized for levels of HPRT1. The primer sequences are provided in Supplementary Materials. Statistical comparisons were performed using Student t test.
Wound migration assay
HEK were grown to confluence in a 96-well ImageLock plates (Essen Bioscience, Ann Arbor, MI), treated with 4cμg/ml mitomycin-C, and wounded by scratch with a 96-pin wound making tool (Wound-Maker, Essen Bioscience). Cells were incubated for 48 hours, and two representative images from each well of relative migration were taken every 2 hours after the initial wound using IncuCyte Zoom system (Essen Bioscience) and quantified using Cell Migration Analysis software module (Essen Bioscience).
Ex vivo wound closure
Human skin specimens from reduction surgery were used to generate acute wounds as previously described (Ojeh et al., 2014; Stojadinovic and Tomic-Canic, 2013). Briefly, a 3-mm biopsy punch was used to create acute wounds (n = 9 per treatment), which were treated at the time of wounding in presence, absence, or combination of 1 μM Dex, 0.1 μM Dex-BSA, and 0.2 μM CC and treated daily for 4 days on air-liquid interface with DMEM, 1% antibiotic-antimycotic (Invitrogen) and 10% fetal bovine serum (Lonza, Basel, Switzerland) at 37°C, 5% CO2, and 95% relative humidity. Ex vivo acute wound specimens were frozen in optimal cutting temperature compound, and rate of healing was analyzed for epithelialization by histology assessment using a Nikon eclipse E400 microscope and NIS Elements software.
Statistics
Results from quantitative experiments are expressed as mean ± standard error of the mean. For pairwise comparisons we used the Student t test, and significance was accepted at a P < 0.05. Where appropriate, two-way analysis of variance was also used via the Bonferroni multiple comparison procedure (GraphPad Prism; GraphPad Software, La Jolla, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank Anthony Barrientos, Gregory Plano, Marcia Boulina, Gregory Conner, Monica G. Valencia, Wei Li, Michelle E. Leblanc, Camillo Ricordi, and Marta Garcia-Contreras for their generosity in sharing laboratory resources and equipment. We also thank Agata Krzyzanowska and Ashley M. Rosa for technical assistance and members of M.T.-C. laboratory for overall support and critical evaluation of the manuscript. This work was funded in part by 3M Healthcare Fellowship granted by Wound Healing Foundation (IJ), SAC Award SAC-2013-19 (MTC), NR008029 (MTC), and University of Miami Department of Dermatology and Cutaneous Surgery.
Abbreviations:
- CC
calphostin C
- Dex
dexamethasone
- Dex-BSA
BSA-conjugated dexamethasone
- GC
glucocorticoid
- GR
glucocorticoid receptor
- GILZ
glucocorticoid response element-induced leucine zipper
- GSK-3β
glycogen synthase kinase 3 beta
- HEK
human keratinocyte
- mbGR
membranous glucocorticoid receptor
- PKC
protein kinase C
- PLC
phospholipase C
Footnotes
CONFLICT OF INTEREST
Dr. Tomic-Canic is listed as an inventor of a patent PCT/US2010/062361 “Composition and methods for promoting epithelialization and wound closure” issued to the New York University based on the data presented, in part, in the study and stands to potentially gain royalties from future commercialization.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at www.jidonline.org, and at http://dx.doi.org/10.1016/j.jid.2016.11.036.
REFERENCES
- Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG, et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA 2009;106:3925–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almawi WY, Melemedjian OK. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J Leukoc Biol 2002;71:9–15. [PubMed] [Google Scholar]
- Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J 2004;18: 70–80. [DOI] [PubMed] [Google Scholar]
- Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, et al. Protein kinase Cdelta mediates insulin-induced glucose transport in primary cultures of rat skeletal muscle. Mol Endocrinol 1999;13:2002–12. [DOI] [PubMed] [Google Scholar]
- Brem H, Stojadinovic O, Diegelmann RF, Entero H, Lee B, Pastar I, et al. Molecular markers in patients with chronic wounds to guide surgical debridement. Mol Med 2007;13:30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem H, Tomic-Canic M, Tarnovskaya A, Ehrlich HP, Baskin-Bey E, Gill K, et al. Healing of elderly patients with diabetic foot ulcers, venous stasis ulcers, and pressure ulcers. Surg Technol Int 2003;11:161–7. [PubMed] [Google Scholar]
- Chen HC, Farese RV. Steroid hormones: interactions with membrane-bound receptors. Curr Biol 1999;9:R478–81. [DOI] [PubMed] [Google Scholar]
- Christian SL, Sims PV, Gold MR. The B cell antigen receptor regulates the transcriptional activator beta-catenin via protein kinase C-mediated inhibition of glycogen synthase kinase-3. J Immunol 2002;169:758–69. [DOI] [PubMed] [Google Scholar]
- Clements WM, Lowy AM, Groden J. Adenomatous polyposis coli/beta-catenin interaction and downstream targets: altered gene expression in gastrointestinal tumors. Clin Colorectal Cancer 2003;3(2):113–20. [DOI] [PubMed] [Google Scholar]
- Czerwinski R, Aulabaugh A, Greco RM, Olland S, Malakian K, Wolfrom S, et al. Characterization of protein kinase C theta activation loop auto-phosphorylation and the kinase domain catalytic mechanism. Biochemistry 2005;44:9563–73. [DOI] [PubMed] [Google Scholar]
- Denning MF, Wang Y, Nickoloff BJ, Wrone-Smith T. Protein kinase Cdelta is activated by caspase-dependent proteolysis during ultraviolet radiation-induced apoptosis of human keratinocytes. J Biol Chem 1998;273: 29995–30002. [DOI] [PubMed] [Google Scholar]
- Dlugosz AA, Yuspa SH. Coordinate changes in gene expression which mark the spinous to granular cell transition in epidermis are regulated by protein kinase C. J Cell Biol 1993;120:217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming SA, Martin P, Tomic-Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med 2014;6:265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farese RV, Standaert ML, Arnold T, Yu B, Ishizuka T, Hoffman J, et al. The role of protein kinase C in insulin action. Cell Signal 1992;4:133–43. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell 2004;116:769–78. [DOI] [PubMed] [Google Scholar]
- Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem 1992;267:16878–82. [PubMed] [Google Scholar]
- Gordge PC, Hulme MJ, Clegg RA, Miller WR. Elevation of protein kinase A and protein kinase C activities in malignant as compared with normal human breast tissue. Eur J Cancer 1996;32A:2120–6. [DOI] [PubMed] [Google Scholar]
- Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. Down-regulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol 1998;8: 573–81. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509–12. [DOI] [PubMed] [Google Scholar]
- Helfrich I, Schmitz A, Zigrino P, Michels C, Haase I, le Bivic A, et al. Role of aPKC isoforms and their binding partners Par3 and Par6 in epidermal barrier formation. J Invest Dermatol 2007;127:782–91. [DOI] [PubMed] [Google Scholar]
- Hinoi T, Yamamoto H, Kishida M, Takada S, Kishida S, Kikuchi A. Complex formation of adenomatous polyposis coli gene product and axin facilitates glycogen synthase kinase-3 beta-dependent phosphorylation of beta-catenin and down-regulates beta-catenin. J Biol Chem 2000;275:34399–406. [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang M, Czeh B, Flugge G, Zhang W. Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 2010;35:1693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idris I, Gray S, Donnelly R. Protein kinase C activation: isozyme-specific effects on metabolism and cardiovascular complications in diabetes. Diabetologia 2001;44:659–73. [DOI] [PubMed] [Google Scholar]
- Jozic I, Blanco G, Barbieri MA. Inhibition of Rab5 activation during insulin receptor-mediated endocytosis. Curr Cell Biochem 2011;1:20–32. [PMC free article] [PubMed] [Google Scholar]
- Jozic I, Saliba SC, Barbieri MA. Effect of EGF-receptor tyrosine kinase inhibitor on Rab5 function during endocytosis. Arch Biochem Biophys 2012;525:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jozic I, Stojadinovic O, Kirsner RS, Tomic-Canic M. Stressing the steroids in skin: paradox or fine-tuning? J Invest Dermatol 2014;134:2869–72. [DOI] [PubMed] [Google Scholar]
- Jozic I, Stojadinovic O, Kirsner RS, Tomic-Canic M. Skin under the (spot)-light: cross-talk with the central hypothalamic-pituitary-adrenal (HPA) Axis. J Invest Dermatol 2015;135:1469–71. [DOI] [PubMed] [Google Scholar]
- Keith BD. Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 2008;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Vouthounis C, Stojadinovic O, Brem H, Im M, Tomic-Canic M. From an enhanceosome to a repressosome: molecular antagonism between glucocorticoids and EGF leads to inhibition of wound healing. J Mol Biol 2005;345:1083–97. [DOI] [PubMed] [Google Scholar]
- Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J 1998;332(Pt 2):281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitre-Aguilar IB, Cabrera-Quintero AJ, Zentella-Dehesa A. Genomic and non-genomic effects of glucocorticoids: implications for breast cancer. Int J Clin Exp Pathol 2015;8:1–10. [PMC free article] [PubMed] [Google Scholar]
- Nahar J, Haam J, Chen C, Jiang Z, Glatzer NR, Muglia LJ, et al. Rapid non-genomic glucocorticoid actions in male mouse hypothalamic neuroendocrine cells are dependent on the nuclear glucocorticoid receptor. Endocrinology 2015;156:2831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, et al. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells 1998;3:395–403. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature 1988;334:661–5. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 1995;9:484–96. [PubMed] [Google Scholar]
- Nunan R, Harding KG, Martin P. Clinical challenges of chronic wounds: searching for an optimal animal model to recapitulate their complexity. Dis Model Mech 2014;7:1205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara T, Negishi T, Chik CL, Ho AK. Differential effects of two protein kinase C inhibitors, calphostin C and Go6976, on pineal cyclic nucleotide accumulation. J Neurochem 1998;71(4):1405–12. [DOI] [PubMed] [Google Scholar]
- Ojeh N, Stojadinovic O, Pastar I, Sawaya A, Yin N, Tomic-Canic M. The effects of caffeine on wound healing. Int Wound J 2014;13:605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, et al. Epithelialization in wound healing: a comprehensive review. Adv Wound Care (New Rochelle) 2014;3:445–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevostel C, Alvaro V, de Boisvilliers F, Martin A, Jaffiol C, Joubert D. The natural protein kinase C alpha mutant is present in human thyroid neoplasms. Oncogene 1995;11:669–74. [PubMed] [Google Scholar]
- Radoja N, Komine M, Jho SH, Blumenberg M, Tomic-Canic M. Novel mechanism of steroid action in skin through glucocorticoid receptor monomers. Mol Cell Biol 2000;20:4328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez HA, Liang L, Pastar I, Rosa AM, Stojadinovic O, Zwick TG, et al. Comparative genomic, mircoRNA, and tissue analyses reveal subtle differences between non-diabetic and diabetic foot skin. PLoS One 2015;10: e0137133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutz HP. Effects of corticosteroid use on treatment of solid tumours. Lancet 2002;360:1969e70. [DOI] [PubMed] [Google Scholar]
- Samarasinghe RA, Witchell SF, DeFranco DB. Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle 2012;11:2819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbie LA, Schmitz-Peiffer C, Sheng Y, Biden TJ. Molecular cloning and characterization of PKC iota, an atypical isoform of protein kinase C derived from insulin-secreting cells. J Biol Chem 1993;268:24296–302. [PubMed] [Google Scholar]
- Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D. Diurnal cortisol rhythm as a predictor of breast cancer survival. J Natl Cancer Inst 2000;92: 994–1000. [DOI] [PubMed] [Google Scholar]
- Skobowiat C, Slominski AT. UVB activates hypothalamic-pituitary-adrenal axis in C57BL/6 mice. J Invest Dermatol 2015;135:1638–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski A, Wortsman J, Tuckey RC, Paus R. Differential expression of HPA axis homolog in the skin. Mol Cell Endocrinol 2007;265–266:143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski A, Zbytek B, Semak I, Sweatman T, Wortsman J. CRH stimulates POMC activity and corticosterone production in dermal fibroblasts. J Neuroimmunol 2005a;162:97–102. [DOI] [PubMed] [Google Scholar]
- Slominski A, Zbytek B, Szczesniewski A, Semak I, Kaminski J, Sweatman T, et al. CRH stimulation of corticosteroids production in melanocytes is mediated by ACTH. Am J Physiol Endocrinol Metab 2005b;288:E701–6. [DOI] [PubMed] [Google Scholar]
- Slominski A, Zjawiony J, Wortsman J, Semak I, Stewart J, Pisarchik A, et al. A novel pathway for sequential transformation of 7-dehydrocholesterol and expression of the P450scc system in mammalian skin. Eur J Biochem 2004;271:4178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski AT, Manna PR, Tuckey RC. On the role of skin in the regulation of local and systemic steroidogenic activities. Steroids 2015;103:72–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski AT, Zmijewski MA, Zbytek B, Tobin DJ, Theoharides TC, Rivier J. Key role of CRF in the skin stress response system. Endocr Rev 2013;34: 827–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert ML, Galloway L, Karnam P, Bandyopadhyay G, Moscat J, Farese RV. Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J Biol Chem 1997;272:30075–82. [DOI] [PubMed] [Google Scholar]
- Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, et al. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol 2005;167:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Gordon KA, Lebrun E, Tomic-Canic M. Stress-induced hormones cortisol and epinephrine impair wound epithelization. Adv Wound Care (New Rochelle) 2012;1:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Lee B, Vouthounis C, Vukelic S, Pastar I, Blumenberg M, et al. Novel genomic effects of glucocorticoids in epidermal keratinocytes: inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J Biol Chem 2007;282:4021–34. [DOI] [PubMed] [Google Scholar]
- Stojadinovic O, Pastar I, Vukelic S, Mahoney MG, Brennan D, Krzyzanowska A, et al. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med 2008;12:2675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Sawaya A, Pastar I, Tomic-Canic M. Glucocorticoid receptor localizes to adherens junctions at the plasma membrane of keratinocytes. PLoS One 2013;8:e63453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojadinovic O, Tomic-Canic M. Human ex vivo wound healing model. Methods Mol Biol 2013;1037:255–64. [DOI] [PubMed] [Google Scholar]
- Strehl C, Buttgereit F. Unraveling the functions of the membrane-bound glucocorticoid receptors: first clues on origin and functional activity. Ann N Y Acad Sci 2014;1318:1–6. [DOI] [PubMed] [Google Scholar]
- Strehl C, Gaber T, Lowenberg M, Hommes DW, Verhaar AP, Schellmann S, et al. Origin and functional activity of the membrane-bound glucocorticoid receptor. Arthritis Rheum 2011;63:3779–88. [DOI] [PubMed] [Google Scholar]
- Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med 2003;349:2483–94. [DOI] [PubMed] [Google Scholar]
- van Gijn ME, Daemen MJ, Smits JF, Blankesteijn WM. The wnt-frizzled cascade in cardiovascular disease. Cardiovasc Res 2002;55:16–24. [DOI] [PubMed] [Google Scholar]
- Vernocchi S, Battello N, Schmitz S, Revets D, Billing AM, Turner JD, et al. Membrane glucocorticoid receptor activation induces proteomic changes aligning with classical glucocorticoid effects. Mol Cell Proteomics 2013;12:1764–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukelic S, Stojadinovic O, Pastar I, Rabach M, Krzyzanowska A, Lebrun E, et al. Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J Biol Chem 2011;286:10265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JA, Pitarresi JR, Sharma N, Palettas M, Cuitino MC, Sizemore ST, et al. Protein kinase C Beta in the tumor microenvironment promotes mammary tumorigenesis. Front Oncol 2014;4:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem 2002;277: 26573–80. [DOI] [PubMed] [Google Scholar]
- Wertheimer E, Spravchikov N, Trebicz M, Gartsbein M, Accili D, Avinoah I, et al. The regulation of skin proliferation and differentiation in the IR null mouse: implications for skin complications of diabetes. Endocrinology 2001;142:1234–41. [DOI] [PubMed] [Google Scholar]
- Zhou P, Byrne C, Jacobs J, Fuchs E. Lymphoid enhancer factor 1 directs hair follicle patterning and epithelial cell fate. Genes Dev 1995;9: 700–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.