

Abstract
Metal‐ligand cooperativity (MLC) had a remarkable impact on transition metal chemistry and catalysis. By use of the calix[4]pyrrolato aluminate, [1]−, which features a square‐planar AlIII, we transfer this concept into the p‐block and fully elucidate its mechanisms by experiment and theory. Complementary to transition metal‐based MLC (aromatization upon substrate binding), substrate binding in [1]− occurs by dearomatization of the ligand. The aluminate trapps carbonyls by the formation of C−C and Al−O bonds, but the products maintain full reversibility and outstanding dynamic exchange rates. Remarkably, the C−C bonds can be formed or cleaved by the addition or removal of lithium cations, permitting unprecedented control over the system's constitutional state. Moreover, the metal‐ligand cooperative substrate interaction allows to twist the kinetics of catalytic hydroboration reactions in a unique sense. Ultimately, this work describes the evolution of an anti‐van't Hoff/Le Bel species from their being as a structural curiosity to their application as a reagent and catalyst.
Keywords: aluminum, calix[4]pyrrole, dynamic covalent chemistry, metal-ligand cooperativity, structural constraint
The calix[4]pyrrolato aluminate enables a transfer of metal–ligand cooperativity from the d‐ into the p‐block. The compound traps carbonyls and delivers products that are showing highly dynamic–covalent behavior. Lithium ions provide full control over the formation and cleavage of C−C bonds. The adjustability in substrate activation enables a unique concept for rate control in catalysis.
Introduction
Metal‐ligand cooperativity (MLC) within transition metal complexes has become an indispensable tool for bond activation chemistry and homogeneous catalysis.1 Due to enormous research efforts, e.g., by the Milstein group, a variety of atom‐economic and waste‐free catalytic processes have been developed.2 A particular impetus was provided by the aromatization/dearomatization strategy, which operates by reversible aromatization of the ligand backbone upon interaction with the substrates.2c The herein often encountered scenario involves the synergistic binding of unsaturated substrates (carbonyls, CO2, or nitriles) to the metal center and ligand backbone (Figure 1 A). Although this type of reactivity does not require oxidation‐state changes, consistently, redox‐active metals of the d‐block, such as manganese,3 molybdenum,4 ruthenium,5 rhodium,6 iridium7 or rhenium8 were used. Surprisingly, a transfer of the aromatization/dearomatization concept to the p‐block is still at its beginnings.9 Even the much more general field of MLC with p‐block elements is developing hesitantly only.9b, 10 A possible reason for this limited success might be the low inherent reactivity of p‐block elements in their conventional oxidation and saturated valence states. Structural constraint offers a promising alternative to amplify reactivity by exposing space for substrate coordination and alteration of the frontier molecular orbital energies.11
Figure 1.
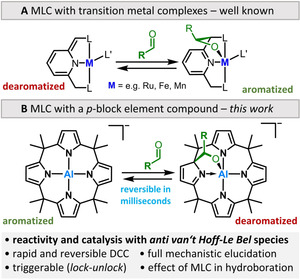
A) MLC by the aromatization/dearomatization strategy within transition‐metal complexes (exemplified for aldehyde binding). B) Complementary MLC reactivity of the calix[4]pyrrolato aluminate [1]− described in this work (DCC=dynamic covalent chemistry).
We recently reported the anti‐van't Hoff/Le Bel‐configurated meso‐octamethylcalix[4]pyrrolato aluminate ([1]−) with a square‐planar coordinated aluminum(III) atom (Figure 1 B).12 Planarization not only provokes a LUMO energy lowering in [1]− but also raises the energy of the ligand‐based HOMO in comparison to the tetrahedral counterpart Al(pyrrolato)4 −.12 Thus, [1]− provides a small HOMO–LUMO gap with an electron‐deficient metal center and a nearby electron‐rich pyrrole moiety—ideally setting the scene for MLC reactivity. Herein, we report the MLC binding of C=O‐containing substrates at [1]−, the highly dynamic covalent features of the products and its implications for catalysis.
Results and Discussion
Exposing [PPh4][1] to 2 bar CO2 in dichloromethane at room temperature resulted in a color change from colorless to yellow within seconds. The 1H NMR spectrum revealed a clean and quantitative reaction. Strongly downfield‐shifted signals of two pyrrolic protons indicate the dearomatization of one pyrrole unit (Figure 2 A). The initial D 2d‐symmetry of [1]− is lowered to C1, resulting in eight chemically inequivalent pyrrolic and methyl positions. Yellow crystals suitable for SCXRD analysis were obtained from a dichloromethane/toluene/THF solution at room temperature. The measurements illustrate the structure of [PPh4][1*‐CO2] (Figure 2 C, in the following, * designates a dearomatized state of one pyrrole unit in the ligand). Binding of CO2 occurs in a bidentate fashion by the formation of a C−C bond (155.8(6) pm) and Al−O bond (184.8(3) pm). The Al−O bond is in the range of other higher coordinated aluminum centres,13 whereas the C1‐C29 bond is elongated in comparison to common sp3‐sp2 C−C single bonds.14 The shortened C4‐N1 and C2‐C3 bonds verify the dearomatization of the pyrrole ring. This MLC‐type 1,2‐addition process is in striking contrast to the commonly observed insertion of CO2 into polar metal‐nitrogen bonds,15 or reported reactivities of Al‐porphyrin complexes,16 but manifests the electron‐rich ligand as nucleophilic reaction side.
Figure 2.
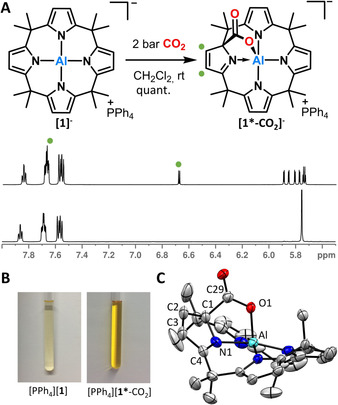
A) Synthesis of [PPh4][1*‐CO2] with in situ 1H NMR spectra of the pyrrole region. B) The observed color change. C) SCXRD structure of [1*‐CO2]− (cation and hydrogen atoms omitted for clarity, ellipsoids are drawn at the 50 % probability level) Selected bond lengths [pm]: Al–O1 184.8(3), C1–C29 155.8(6), N1–Al 197.0(3), C29–O1 129.7(5), C29–O2 121.8(4), N1–C4 129.3(5), C2–C3 135.1(6).24
Although calix[4]pyrroles have been extensively studied as ligands for transition metals, this mode of reactivity was observed only with TiIV and has not been followed any further.17 It strongly resembles the MLC binding of CO2 with transition metal complexes.3–,5c, 5d, 5e, 5f, 6, 7, 8
Appealed by the ease and purity of this reaction, [PPh4][1] was subjected to cyclohexane carboxaldehyde (CyA), para‐methylbenzaldehyde (pMe‐BA) and para‐nitrobenzaldehyde (pNO2‐BA) under analog conditions as for CO2. Quantitative conversions to the 1,2‐addition products were detected by 1H NMR spectroscopy immediately after aldehyde addition showing similar spectroscopic features of a dearomatized ligand pyrrole unit (Figure 3 A). Suitable yellow crystals of [PPh4][1*‐CyA], [PPh4][1*‐pNO2‐BA] and [PPh4][1*‐pMe‐BA] for SCXRD were obtained from saturated dichloromethane/toluene solutions at room temperature. Generally, those structures illustrate the same 1,2‐carboalumination pattern as for CO2 (Figure 3 B). Again, the formed Al−O bond lengths are in the range of common Al‐O‐bonds (Al‐O distances [pm]: [PPh4][1*‐CyA]: 176.7(2), [PPh4][1*‐pMeBA]: 179.0(2), [PPh4][1*‐pNO2BA]: 180.0(1)) and the C−C bonds slightly elongated (C−C distances [pm]: [PPh4][1*‐CyA]: 159.9(3), [PPh4][1*‐pMeBA]: 159.6(4), [PPh4][1*‐pNO2BA]: 160.1(2)). All adducts exhibit Al‐N distances in a range of 189–196 pm for the aromatic pyrroles but elongated dative Al−N bonds (Al‐Ndearom.=197–199 pm) for the dearomatized pyrrole rings. During the addition process, the formation of diastereomers was observed. All stereochemical aspects are discussed in section 12.2 of the supporting information.
Figure 3.
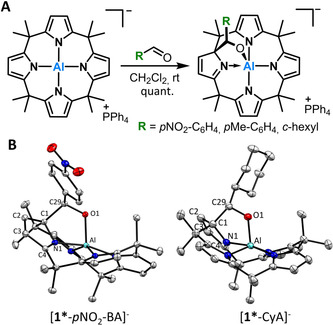
A) Synthesis of the aldehyde 1,2‐adducts. B) Representative SCXRD structures of 1,2‐adducts (cation and hydrogens omitted for clarity; ellipsoids are drawn at the 50 % probability level). Selected bond lengths [pm] for [1*‐pNO2‐BA]−: Al–O1 180.0(1), C1–C29 160.1(2), N1–Al 199.1(1), C29–O1 139.6(2), N1–C4 129.0(2), C2–C3 133.9(2). [1*‐CyA]−: Al–O1 176.7(2), C1–C29 159.9(3).24
Given the formation of enthalpically strong C−C and Al−O bonds and the liberation of a square‐planar aluminum center from its unique coordination environment, one would expect tight and irreversible binding of the substrates considered so far. However, in all cases, full reversibility of the addition process was observed. Adding 2 bar of CO2 to a 13C‐labeled adduct [Li(thf)n][1*‐13CO2] in dichloromethane resulted in a smooth exchange of 13CO2 over 18 h at room temperature, again in close similarity with PNN‐ or PNP complexes of RuII or ReI (Figure 4 A).5a, 5e, 8 Moreover, CO2 was readily replaced by the addition of most aldehydes to [1*‐CO2]−, yielding the aldehyde adducts in a remarkable purity (Figure 4 B). Even the aldehyde binding products maintain dynamic covalent bonding. For example, pNO2‐BA quantitatively replaces pMe‐BA from [1*‐pMe‐BA]− (for a full list of substrate replacement assays, see Table S1). Generally, aldehydes with lower resonance stabilization energy of the C=O bonds (electron‐poor aromatics)18 readily replace aldehydes with more stabilized C=O bonds—in perfect agreement with the computational results (vide infra). In line with this trend, the more electron‐rich para‐dimethylaminobenzaldehyde (pDMA‐BA) binds weaker to [1]−. Upon mixing [PPh4][1] and pDMA‐BA in dichloromethane at room temperature, broad signals were observed in the 1H NMR spectrum, pointing on a fast exchange on NMR timescale. Only by cooling this solution to −25 °C, signals for the 1,2‐adduct developed (Figure S4). Besides, other adduct forms were observed at low temperature, that will become of importance during the mechanistic studies of the process (vide infra).
Figure 4.
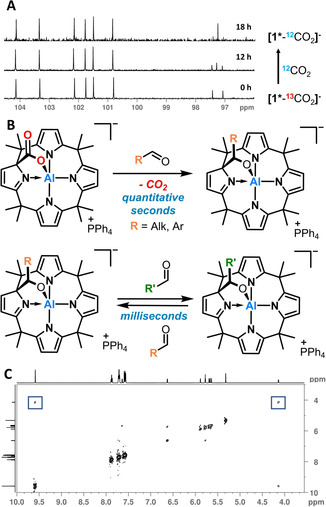
A) 13C{1H} NMR spectra of 13/12CO2 exchange in CD2Cl2. B) CO2 replacement by aldehydes and aldehyde cross‐exchange. C) Selected 1H EXSY cross signals for a mixture of [PPh4][1*‐CyA] and CyA; mixing time 400 ms, 295 K.
Next, we inspected the kinetics of the aldehyde‐aldehyde exchange process in detail. The substitution of pMe‐BA by adding pNO2‐BA to [1*‐pMe‐BA]− proceeded in <1 ms at −80 °C in dichloromethane and its kinetics were not even measurable with a stopped‐flow UV/Vis‐setup. However, such high reaction rates enable studies by dynamic NMR spectroscopy.
Indeed, a mixture of CyA and [PPh4][1*‐CyA] revealed self‐exchange detectable by phase‐sensitive EXSY‐NMR spectroscopy in CD2Cl2 (Figure 4 C). Based on the measured rate constant k=0.08 s−1 at 298.15 K, a value for ΔG ≠=79.4 kJ mol−1 was obtained and found in good agreement with DFT calculations of the rate‐determining step for the overall process (see computational section). An even lower barrier is demarcated by the broad signals and fast exchange regime with pDMA‐BA at room temperature (ΔG ≠<50 kJ mol−1), as described above. EXSY‐measurements with varying aldehyde concentrations revealed no effect on the reaction rate (Figure S13). This is an important finding, as it allowed us to characterize the process as dissociative and legitimates the neglect of associative pathways in the computational evaluation. Overall, these systems undergo the fastest reversible carbon‐carbon bond formation reactions ever determined, surpassing even the most rapid Diels Alder reactions with highly activated diene/dienophile combinations.19 Given the exceptional rates and the high selectivity of this process, the ideal basis for applications in the field of dynamic covalent chemistry (DCC) is at hand.20
After having obtained the fundamental thermodynamic and kinetic parameters for the binding process, further mechanistic insights were collected. The 1,2‐adduct formation can be conceived either as a step‐wise Al−O=C−R σ‐adduct formation/C−C bond formation or as a concerted process. First, the low‐temperature NMR spectroscopic results of the mixture of [PPh4][1] and pDMA‐BA were reconsidered. As mentioned above, besides the 1,2‐adduct, other species were observed at −25 °C. The apparent solution state symmetry of the second principal component indicated a local C 2v symmetry at the [1]− entity, known from our previous studies with pure σ‐donors (Figure S5).12 The observation of a clear EXSY exchange peak between the 1,2‐adduct and this σ‐adduct further substantiated its putative role as intermediate in the reaction sequence (Figure S8). The use of substrates with less reactive C=O bonds provided further insights. The reaction of [PPh4][1] with acetone led to broad 1H NMR signals, similar to the pDMA‐BA case (Figure S3). The characteristic signals of a σ‐adduct and the formation of the 1,2‐adducts occurred at a lower temperature (−50 °C). Noteworthy, this latter reaction disclosed that the reversible carbonyl binding is not restricted to aldehydes but holds likewise for ketones. By using formamide or dimethylformamide as substrates with even larger resonance stabilization energy of the C=O bond, the σ‐adduct formation was readily observed by 1H NMR spectroscopy (Figure S20), but the 1,2‐adduct formation did not occur. Having collected sufficient spectroscopic evidence, we were pleased to obtain unambiguous proof for the σ‐adducts also by SCXRD analysis.
Computational Section
After having spotted the participation of σ‐adducts, quantum‐theoretical studies gained a further understanding. For all substrates, the full reaction path, including σ‐adduct, the final 1,2‐adduct, and the corresponding transition states, were identified at the PW6B95‐D3(BJ), COSMO‐RS(CH2Cl2)/def2‐QZVPP//PBEh‐3c level of theory (Figure 5 A). First, the reaction process is illustrated for acetone, for which the σ‐adduct and the 1,2‐adduct have been observed by 1H NMR spectroscopy. In agreement with the experiment, the σ‐adduct between acetone and [1]− was found as a global minimum at (ΔG=−5.9 kJ mol−1), with the 1,2‐adduct lying higher in energy (ΔG=23.1 kJ mol−1). The reaction barrier to the σ‐adduct is caused by the bending of the calix[4]pyrrolato ligand during the entrance of the carbonyl in the coordination sphere of the planar aluminate [1]−. The formation of the C−C bond and the dearomatization of one pyrrole unit provokes the second reaction barrier. Intrinsic reaction coordinate (IRC) calculations were performed, and the intrinsic bond orbitals (IBOs)21 were calculated along the path (Figure 5 B).
Figure 5.
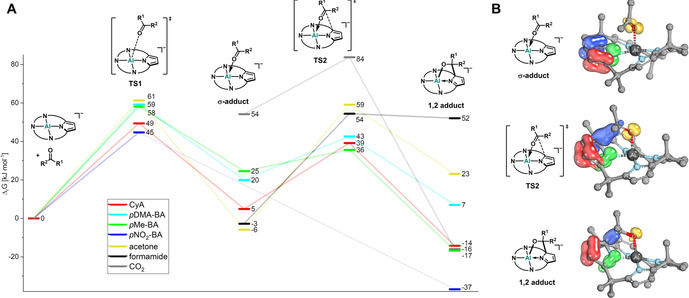
A) Computed Gibbs free energies (298.15 K) of σ‐adducts‐B, 1,2‐adducts‐B and transition states (PW6B95‐D3(BJ), COSMO‐RS(CH2Cl2)/def2‐QZVPP//PBEh‐3c). B) Intrinsic bond orbital representation of the 1,2‐adduct formation process for acetone with emphasis on the dearomatization.
They nicely illustrate the transformation of the aromatic pyrrole bond (blue IBO) into the C−C σ‐bond and the dearomatization of the pyrrole ring. A comparison of the thermodynamics and reaction barriers for all substrates was accomplished next (Figure 5 A). The more electron‐deficient the carbonyl functionality and the less sterically hindered, the more favorable is the 1,2‐adduct formation over the σ‐adduct—in agreement with the experimental findings. For the most electron‐deficient aldehyde pNO2‐BA, no stable σ‐adduct was found on the PES. The 1,2‐adducts of CyA and pMe‐BA are almost thermoneutral to each other. In line with that, it was observed experimentally that a mixture of [PPh4][1*‐pMe‐BA] and CyA immediately developed a dynamic mixture of [PPh4][1*‐CyA], [PPh4][1*‐pMe‐BA], free CyA and free pMe‐BA (Figure S39). Generally, the first reaction barriers for the σ‐adduct formation (ΔG ≠ TS1) do not differ significantly between the various substrates. Surprisingly, they are higher as for the C−C bond formation step (ΔG ≠ TS2), confirming the experimental non‐observability of the σ‐adduct intermediates for most cases. Importantly, for all aldehydes, two diastereomeric σ‐adducts and 1,2‐adducts have to be considered (a full discussion can be found in the supporting information, section 12.2). To conclude, the computed thermodynamics of the reaction products and intermediates ideally match with the experimental product distributions and the calculated reaction barriers are in good agreement with the experimentally derived values (e.g., for CyA: ΔG ≠ exp=79.4 kJ mol−1/ΔG ≠ comp=63.7 kJ mol−1).
Influence of the Countercation—Providing a Trigger for C−C Bond Formation and Catalytic Reaction Control
The previous sections clarified the binding of carbonyls with the [PPh4][1] system. Now, the influence of the counteraction to [1]− was studied. Cases in which the σ‐adduct formation with [PPh4][1] was favored were of particular interest. The addition of lithium iodide to the σ‐adduct of pDMA‐BA with [PPh4][1] in [D2]dichloromethane induced the quantitative transformation to the 1,2‐adducts (Figure 6 A and Figures S15). The process can be followed by the naked eye, as both adduct forms differ in their coloration.
Figure 6.
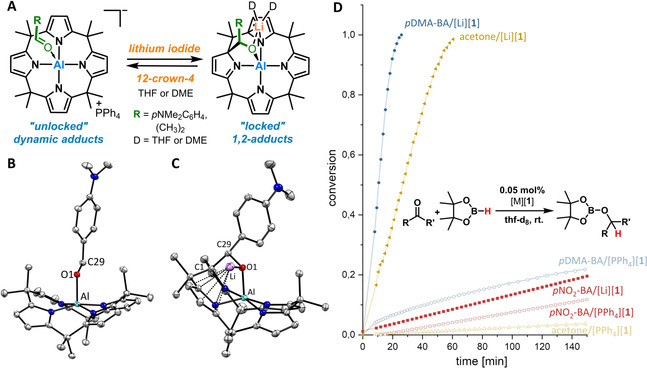
A) Locking of the 1,2‐adducts by addition of a soluble Li+ salt and unlocking by addition of 12‐crown‐4. B,C) SCXRD analysis of σ‐adducts of pDMA‐BA and [Li(thf)2][1*‐pDMA‐BA]; (cations and hydrogen atoms omitted for clarity, ellipsoids are drawn at the 50 % probability level), selected bond lengths [pm]: σ‐pDMA‐BA‐[1]−: Al‐O1 189.8(2), C29‐O1 125.8(2); Al‐N: 194.1(2)–194.7(2). [Li(thf)2][1*‐pDMA‐BA] (the two THF units at Li omitted for clarity): Al‐O1 182.2(1), C1‐C29 158.5(2), Al‐N1 196.7(1), C19‐Li 263.6(4), C18‐Li 266.0(4), N4‐Li 275.5(4), Li‐O1 198.3(3).24 D) Dependence of catalytic hydroboration rates for three substrates with 0.05 % mol catalyst loading of either [PPh4][1] or [Li(thf)4][1‐thf] (abbreviated as [Li][1]). In the pNO2‐BA curves, the slow background reaction was subtracted.
In addition to NMR spectroscopy, this behavior was validated by SCXRD analysis of [Li(thf)2][1*‐pDMA‐BA] (Figure 6 C). Herein the Li+ is η5‐coordinated to an aromatic pyrrole ring and bridges to the oxygen atom of the bound CO moiety. The Al−O and C−C bond lengths are similar to the spontaneously formed 1,2‐adducts, shown in Figure 3. The same sequence is found with the σ‐adduct of [PPh4][1] and acetone or cyclopentanone upon the addition of lithium iodide or use of [Li‐(thf)4][1‐thf] (for SCXRD see SI and Figures S13–S16). Strikingly, the lithium ion‐induced C−C bond formation can be reversed. After the addition of 12‐crown‐4 as a complexation agent for Li+, the original broad signals of the σ‐adducts reappear in the 1H NMR spectrum. By the addition of further lithium iodide, the process can be repeated (Figure S21). Quantum chemical calculations fully supported our observations. The coordination of a [Li‐dme]+ fragment to the [1*‐carbonyl] adducts leads to a computed stabilization of the 1,2‐adduct by 20–70 kJ mol−1 (Figure S104). In the case of acetone and pDMA‐BA, this effect indeed turns the process from endergonic to exergonic. Hence, the addition of a lithium cation offers quantitative yet reversible control over the formation of a C−C bond. Such a straightforward and mild trigger is, to the best of our knowledge, unprecedented. Given the very clean reaction outcome, achievable within seconds by merely combining easy to handle crystalline solids, it constitutes a triggerable reversible click reaction.22 It offers a stimulus to steer the constitutional state and covalent dynamics of systems, attractive for the controlled release of conjugated cargo, or as a temporary protecting group. The possibility for reversal permits to “lock‐and‐unlock” the constitutional dynamics of the system at will. Given the strongly altered physicochemical properties of the σ‐ vs. the 1,2‐adduct (e.g., dipole moment, light absorption), it might allow introducing features like visual feedback, responsiveness, and self‐healing into new functional materials.
Eager to identify a model catalytic application that is influenced by the MLC binding mode of [1]− , the hydroboration of aldehydes with pinacolborane was probed (Figure 6 D). The most activated aldehyde pNO2‐BA undergoes slow hydroboration already in the absence of a catalyst (Figure S46), but the other considered C=O containing substrates were unreactive without a catalyst under the given conditions (Figures S48/51). However, in the presence of 0.05 mol % of [PPh4][1], all compounds were catalytically hydroborated. Although not competing with the most efficient hydroboration catalysts,23 it is more the effect of MLC on the kinetics that makes this system interesting from a conceptual point of view.
With 0.05 mol % of [PPh4][1] in [D8]THF, pDMA‐BA is hydroborated only slowly (20 % conversion after 2 h, Figure 6 D, light blue curve). However, with the lithium salt [Li(thf)4][1‐thf] as the catalyst, the efficiency is boosted dramatically, and the reaction went to completion within 20 min under the same conditions (dark blue curve). The role of lithium cations as the catalytically active species was ruled out by employing 0.1 mol % LiOTf or LiI (without [1]−), in which case no reaction occurred (Figure S54). For acetone, a more than 50‐fold increase in reaction rate was observed between [PPh4][1] (dotted yellow curve) and [Li(thf)4][1‐thf] (thick yellow curve). Thus, with 0.05 mol % of the [Li(thf)4][1‐thf] catalyst system in [D8]THF, remarkable TOFs of >4500 h−1 for pDMA‐BA and 2000 h−1 for acetone were achieved. In [D2]dichloromethane as a solvent, even a >100‐fold rate increase was noted (Figure S42). In stark contrast, a much less pronounced increase of the reaction rate for the catalyzed hydroboration of pNO2‐BA happened with [Li(thf)4][1‐thf] in comparison to [PPh4][1] (Figure 6 D, red curves). Noteworthily, pNO2‐BA forms the 1,2‐adduct with both salts of [1]−. Since the 1,2‐bound states in d‐block MLC‐catalysis represent resting states,5d, 5g it was concluded that also in the present case, the 1,2‐form represents a deactivated state in the hydroboration cycle. However, the partial release of the 1,2‐mode into the σ‐adduct in the case of acetone and pDMA‐BA delivers a highly reactive form that is activated both by the Lewis acidic aluminum center and the nearby lithium cation. Hence, the considerable rate‐increase for only two substrates (pDMA‐BA and acetone) and the relatively small effect for the most activated substrate (pNO2‐BA) showcases the unique impact of MLC on the reaction kinetics. Usually, strongly activated substrates show high turnover frequencies with conventional catalysts (such as with Lewis acid or Lewis base catalysts).23 However, in the case of [1]− as the catalyst, such substrates (pNO2‐BA in our case) get effectively blocked by the MLC 1,2‐binding mode and allow the less activated substrates such as pDMA‐BA and acetone to outpace. In other words, the MLC‐dependent mode of substrate interaction allows to twist common selectivity trends and might enable a novel mode to steer regioselectivities in more complex substrates.
Conclusion
A successful transfer of the d‐block MLC‐concept into the p‐block would allow to couple the abundance of metals such as aluminum with the vast range of established d‐block MLC catalytic transformations. Understanding the mechanistic details of MLC with main group elements is vital for this transfer. The herein presented work provides critical steps in such a direction. The calix[4]pyrrolato aluminate offers exceptionally high rates in the capture and release of C=O containing substrates. These features promptly raise the question on the driving force, that facilitates rapid transformations by cleavage of a C−C and Al−O bond. In contrast to the aromatization/dearomatization process observed in transition metal complexes, [1]− operates by a reversed dearomatization/aromatization sequence. Thus, qualitatively, the regain of aromaticity in a pyrrole ring governs the behavior in the substrate bound state. The full reversibility paired with the substantial alteration of the substrates’ electronic structures upon binding provides an ideal starting point for catalytic applications. By now, it permits unique control over reaction kinetics if applied as a hydroboration catalyst. We believe that the hereby developed perspectives will serve exemplary for exciting progress and retrofit the fields of MLC and DCC with the beneficial earth abundance of aluminum.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. H.‐J. Himmel for his support and Prof. M. Enders for discussion. Financial support was provided by the FCI (Liebig fellowship, L.G.), the DFG (Emmy‐Noether program, GR 5007/2‐1, L.G.) and the Foundation of German Business (sdw, F.E.). The BWFor/BWUniCluster are acknowledged for computational resources, funded by the DFG. Open access funding enabled and organized by Projekt DEAL.
F. Ebner, L. M. Sigmund, L. Greb, Angew. Chem. Int. Ed. 2020, 59, 17118.
References
- 1.
- 1a. Khusnutdinova J. R., Milstein D., Angew. Chem. Int. Ed. 2015, 54, 12236–12273; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12406–12445; [Google Scholar]
- 1b. Higashi T., Kusumoto S., Nozaki K., Chem. Rev. 2019, 119, 10393–10402. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Gunanathan C., Milstein D., Acc. Chem. Res. 2011, 44, 588–602; [DOI] [PubMed] [Google Scholar]
- 2b. Gunanathan C., Milstein D., Science 2013, 341, 1229712; [DOI] [PubMed] [Google Scholar]
- 2c. Milstein D., Philos. Trans. R. Soc. London Ser. A 2015, 373, 20140189; [Google Scholar]
- 2d. Zell T., Milstein D., Acc. Chem. Res. 2015, 48, 1979–1994. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Mukherjee A., Nerush A., Leitus G., Shimon L. J. W., Ben David Y., Espinosa Jalapa N. A., Milstein D., J. Am. Chem. Soc. 2016, 138, 4298–4301; [DOI] [PubMed] [Google Scholar]
- 3b. Kumar A., Daw P., Espinosa-Jalapa N. A., Leitus G., Shimon L. J. W., Ben-David Y., Milstein D., Dalton Trans. 2019, 48, 14580–14584. [DOI] [PubMed] [Google Scholar]
- 4. Sieh D., Lacy D. C., Peters J. C., Kubiak C. P., Chem. Eur. J. 2015, 21, 8497–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Vogt M., Gargir M., Iron M. A., Diskin-Posner Y., Ben-David Y., Milstein D., Chem. Eur. J. 2012, 18, 9194–9197; [DOI] [PubMed] [Google Scholar]
- 5b. Montag M., Zhang J., Milstein D., J. Am. Chem. Soc. 2012, 134, 10325–10328; [DOI] [PubMed] [Google Scholar]
- 5c. Huff C. A., Kampf J. W., Sanford M. S., Organometallics 2012, 31, 4643–4645; [Google Scholar]
- 5d. Filonenko G. A., Conley M. P., Copéret C., Lutz M., Hensen E. J. M., Pidko E. A., ACS Catal. 2013, 3, 2522–2526; [Google Scholar]
- 5e. Huff C. A., Sanford M. S., ACS Catal. 2013, 3, 2412–2416; [Google Scholar]
- 5f. Huff C. A., Kampf J. W., Sanford M. S., Chem. Commun. 2013, 49, 7147–7149; [DOI] [PubMed] [Google Scholar]
- 5g. Bruffaerts J., von Wolff N., Diskin-Posner Y., Ben-David Y., Milstein D., J. Am. Chem. Soc. 2019, 141, 16486–16493. [DOI] [PubMed] [Google Scholar]
- 6. Heuermann I., Heitmann B., Stichauer R., Duvinage D., Vogt M., Organometallics 2019, 38, 1787–1799. [Google Scholar]
- 7.
- 7a. Feller M., Gellrich U., Anaby A., Diskin-Posner Y., Milstein D., J. Am. Chem. Soc. 2016, 138, 6445–6454; [DOI] [PubMed] [Google Scholar]
- 7b. Feller M., Ben-Ari E., Diskin-Posner Y., Milstein D., J. Coord. Chem. 2018, 71, 1679–1689; [Google Scholar]
- 7c. Langer J., Hamza A., Pápai I., Angew. Chem. Int. Ed. 2018, 57, 2455–2458; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2480–2483. [Google Scholar]
- 8.
- 8a. Vogt M., Nerush A., Diskin-Posner Y., Ben-David Y., Milstein D., Chem. Sci. 2014, 5, 2043–2051; [Google Scholar]
- 8b. Stichauer R., Helmers A., Bremer J., Rohdenburg M., Wark A., Lork E., Vogt M., Organometallics 2017, 36, 839–848. [Google Scholar]
- 9.
- 9a. Gellrich U., Diskin-Posner Y., Shimon L. J. W., Milstein D., J. Am. Chem. Soc. 2016, 138, 13307–13313; [DOI] [PubMed] [Google Scholar]
- 9b. Gellrich U., Angew. Chem. Int. Ed. 2018, 57, 4779–4782; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4869–4872. [Google Scholar]
- 10.
- 10a. Myers T. W., Berben L. A., J. Am. Chem. Soc. 2013, 135, 9988–9990; [DOI] [PubMed] [Google Scholar]
- 10b. Fedushkin I. L., Nikipelov A. S., Lyssenko K. A., J. Am. Chem. Soc. 2010, 132, 7874–7875; [DOI] [PubMed] [Google Scholar]
- 10c. Fedushkin I. L., Moskalev M. V., Lukoyanov A. N., Tishkina A. N., Baranov E. V., Abakumov G. A., Chem. Eur. J. 2012, 18, 11264–11276; [DOI] [PubMed] [Google Scholar]
- 10d. Abdalla J. A. B., Riddlestone I. M., Tirfoin R., Aldridge S., Angew. Chem. Int. Ed. 2015, 54, 5098–5102; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5187–5191. [Google Scholar]
- 11.
- 11a. Culley S. A., Arduengo A. J., J. Am. Chem. Soc. 1984, 106, 1164–1165; [Google Scholar]
- 11b. Dunn N. L., Ha M., Radosevich A. T., J. Am. Chem. Soc. 2012, 134, 11330–11333; [DOI] [PubMed] [Google Scholar]
- 11c. Robinson T. P., De Rosa D. M., Aldridge S., Goicoechea J. M., Angew. Chem. Int. Ed. 2015, 54, 13758–13763; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13962–13967; [Google Scholar]
- 11d. Gilhula J. C., Radosevich A. T., Chem. Sci. 2019, 10, 7177–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ebner F., Wadepohl H., Greb L., J. Am. Chem. Soc. 2019, 141, 18009–18012. [DOI] [PubMed] [Google Scholar]
- 13. Cruickshank M. C., Glasser L. S. D., J. Chem. Soc. Chem. Commun. 1985, 84–85. [Google Scholar]
- 14. Allen F. H., Kennard O., Watson D. G., Brammer L., Orpen A. G., Taylor R., J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar]
- 15.
- 15a. Lappert M. F., Prokai B. in Advances in Organometallic Chemistry, Vol. 5 (Eds.: F. G. A. Stone, W. Robert), Academic Press, New York, 1967, pp. 225–319; [Google Scholar]
- 15b. Yin X., Moss J. R., Coord. Chem. Rev. 1999, 181, 27–59; [Google Scholar]
- 15c. Habereder T., Nöth H., Paine R. T., Eur. J. Inorg. Chem. 2007, 4298–4305; [Google Scholar]
- 15d. Hengesbach F., Jin X., Hepp A., Wibbeling B., Würthwein E.-U., Uhl W., Chem. Eur. J. 2013, 19, 13901–13909; [DOI] [PubMed] [Google Scholar]
- 15e. Kraushaar K., Schmidt D., Schwarzer A., Kroke E. in Advances in Inorganic Chemistry, Vol. 66 (Eds.: M. Aresta, R. van Eldik), Academic Press, New York, 2014, pp. 117–162; [Google Scholar]
- 15f. Zijlstra H. S., Pahl J., Penafiel J., Harder S., Dalton Trans. 2017, 46, 3601–3610. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Aida T., Inoue S., J. Am. Chem. Soc. 1983, 105, 1304–1309; [Google Scholar]
- 16b. Kojima F., Aida T., Inoue S., J. Am. Chem. Soc. 1986, 108, 391–395. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Floriani C., Pure Appl. Chem. 1996, 68, 1–8; [Google Scholar]
- 17b. Solari G., Solari E., Lemercier G., Floriani C., Chiesi-Villa A., Rizzoli C., Inorg. Chem. 1997, 36, 2691–2695; [Google Scholar]
- 17c. Cuesta L., Sessler J. L., Chem. Soc. Rev. 2009, 38, 2716–2729. [DOI] [PubMed] [Google Scholar]
- 18. Hansch C., Leo A., Taft R. W., Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- 19.
- 19a. Chen X., Dam M. A., Ono K., Mal A., Shen H., Nutt S. R., Sheran K., Wudl F., Science 2002, 295, 1698–1702; [DOI] [PubMed] [Google Scholar]
- 19b. Boutelle R. C., Northrop B. H., J. Org. Chem. 2011, 76, 7994–8002; [DOI] [PubMed] [Google Scholar]
- 19c. Roy N., Lehn J.-M., Chem. Asian J. 2011, 6, 2419–2425. [DOI] [PubMed] [Google Scholar]
- 20. Rowan S. J., Cantrill S. J., Cousins G. R. L., Sanders J. K. M., Stoddart J. F., Angew. Chem. Int. Ed. 2002, 41, 898–952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 938–993. [Google Scholar]
- 21.
- 21a. Knizia G., J. Chem. Theory Comput. 2013, 9, 4834–4843; [DOI] [PubMed] [Google Scholar]
- 21b. Knizia G., Klein J. E. M. N., Angew. Chem. Int. Ed. 2015, 54, 5518–5522; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5609–5613. [Google Scholar]
- 22.
- 22a. Billiet S., De Bruycker K., Driessen F., Goossens H., Van Speybroeck V., Winne J. M., Du Prez F. E., Nat. Chem. 2014, 6, 815–821; [DOI] [PubMed] [Google Scholar]
- 22b. Diehl K. L., Kolesnichenko I. V., Robotham S. A., Bachman J. L., Zhong Y., Brodbelt J. S., Anslyn E. V., Nat. Chem. 2016, 8, 968–973. [Google Scholar]
- 23. Shegavi M. L., Bose S. K., Catal. Sci. Technol. 2019, 9, 3307–3336. [Google Scholar]
- 24. Deposition Numbers 1989029, 1989030, 1989031, 1989032, 1989033, 1989034, 1989035, 1989036 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary