

Abstract
Erosion of biodiversity generated by anthropogenic activities has been studied for decades and in many areas at the species level, using taxa monitoring. In contrast, genetic erosion within species has rarely been tracked, and is often studied by inferring past population dynamics from contemporaneous estimators. An alternative to such inferences is the direct examination of past genes, by analysing museum collection specimens. While providing direct access to genetic variation over time, historical DNA is usually not optimally preserved, and it is necessary to apply genotyping methods based on hybridization‐capture to unravel past genetic variation. In this study, we apply such a method (i.e., HyRAD), to large time series of two butterfly species in Finland, and present a new bioinformatic pipeline, namely PopHyRAD, that standardizes and optimizes the analysis of HyRAD data at the within‐species level. In the localities for which the data retrieved have sufficient power to accurately examine genetic dynamics through time, we show that genetic erosion has increased across the last 100 years, as revealed by signatures of allele extinctions and heterozygosity decreases, despite local variations. In one of the two butterflies (Erebia embla), isolation by distance also increased through time, revealing the effect of greater habitat fragmentation over time.
Keywords: HyRAD, Lepidoptera, museomics, past gene frequencies, population dynamics
1. INTRODUCTION
An increasing number of studies have revealed that a dramatic collapse of biodiversity has been ongoing since the early 20th century, and that decay rates have accelerated over the past 50 years (de Oliveira Roque et al., 2018; Johnson et al., 2017). There is no doubt that this global ongoing crisis of biodiversity loss is due to anthropogenic factors that have transformed species' habitats, and caused a decrease in population sizes, with potential long‐term consequences for local or global extinction. Many studies have highlighted the consequences of this extinction in terms of erosion of species diversity for decades (Ceballos, Ehrlich, & Dirzo, 2017; Ehrlich, 1995), providing the basis for international reports assessing the state of biodiversity and the ecosystem services it provides to society (e.g., the Intergovernmental Platform on Biodiversity and Ecosystem Services, IPBES; https://bit.ly/IPBESReport). However, it is only recently that observations and empirical approaches have shown that biomass and, by extension, population sizes have also declined significantly over the last 50 years, especially for insects, resulting in multitrophic cascades affecting the biomass of many insectivorous species (Karp et al., 2013). In particular, Hallmann et al. (2017) have measured insect biomass during 27 years at 63 German localities, and found a greater than three‐quarters decline in this short time span, which represents an average 3% drop per year. Similarly, Lister and Garcia (2018) reported a decrease of more than 90% of the terrestrial arthropod biomass and 80% of that of the canopy over the last 36 years in Puerto Rico, which has in turn strongly reduced the abundance of predator populations of lizards, frogs and birds. Anthropogenic factors are strong candidates for this recognized “insectageddon” including: pesticide and fertilizer use in agriculture, land‐use change (habitat destruction), climate change, chemical contamination (Whiteside & Marvin Herndon, 2018), light pollution, invasion by exogenous species (Sánchez‐Bayo & Wyckhuys, 2019; van Strien, van Swaay, van Strien‐van Liempt, Poot, & WallisDeVries, 2019) and wireless communication systems (Thielens et al., 2018).
These anthropic factors will impact the populations at different intensities, ranging from reduction of their size to local extinction. The reduction in population sizes will have several consequences. A reduction in effective population size (N e) will cause a loss of genetic diversity (i.e., the number of alleles in the populations), thus decreasing both neutral variation and adaptive potential. In addition, shrinking populations will experience increased allele extinction due to inbreeding that causes allele fixation by genetic drift. Also, inbred populations will tend to accumulate deleterious alleles and such a mechanism, referred to as inbreeding depression, will further decrease the average fitness of a population (Keller & Waller, 2002; Kristensen, Pedersen, Vermeulen, & Loeschcke, 2010). Furthermore, the extinction dynamics will weaken the connectivity network between remaining populations and thus reduce overall gene flow. This will increase their divergence and prevent their capacity to exchange potential adaptive alleles. The combination of these mechanisms will further decrease the population persistence likelihood over time (Bouzat, 2010).
However, empirical data providing time series of abundance data and genetic diversity remain scarce, which limits our ability to precisely infer the recent demographic trajectory of most populations. A variety of indirect proxies can help to overcome the general lack of long‐term abundance data across taxa to estimate demographics. For example, the genetic information present in museum specimens collected across the last few decades or even centuries provides a unique opportunity to obtain temporal snapshots of past population genetic diversity and quantify the extent and dynamics of the current biodiversity crisis (Jensen et al., 2018; Meineke, Davies, Daru, & Davis, 2018; Ryan et al., 2018). It is estimated that the number of museum specimens collected around the world exceeds 1 billion individuals, and covers ~1.2 million species (Pyke & Ehrlich, 2010). The molecular diversity present in these specimens can help assess population sizes through time but signatures can also help track adaptation to changing environments (Hoffmann, Sgrò, & Kristensen, 2017). These approaches can thus both help define conservation priorities and estimate the future resilience to ongoing environmental change.
However, analysing DNA contained in museum collections (hereafter referred to as historical DNA) is not devoid of difficulties owing to post‐mortem decay reactions fragmenting the DNA backbone and modifying the chemical nature of individual nucleotidic bases (Dabney, Meyer, & Pääbo, 2013). The extensive fragmentation of historical DNA molecules precludes widely used conventional genetic analyses, such as high‐density array genotyping (Decker et al., 2009) and Restriction Site‐Associated DNA Sequencing (RADseq) (Linck, Hanna, Sellas, & Dumbacher, 2017). Over the past decade, a number of alternative molecular methods have been developed to gather historical DNA data at population and genome‐wide scales (reviewed in Burrell, Disotell, & Bergey, 2015; Horn, 2012; Orlando, Gilbert, & Willerslev, 2015). These most often rely on the construction of next‐generation sequencing DNA libraries, and the capture of those DNA library templates annealing to short synthetic nucleic acid baits spread across predefined loci of interest. This rationale was applied in 2016 to target Genotyping‐By‐Sequencing (GBS) or RADseq loci, either through bench‐top synthesized probes (i.e., HyRAD; Suchan et al., 2016), or commercially produced synthetic GBS or RADseq oligonucleotides (Ali et al., 2016; Boucher, Casazza, Szövényi, & Conti, 2016; Hoffberg et al., 2016; Sánchez Barreiro et al., 2017). These methods generally improve the quality of the genotypic information retrieved by increasing the overall average depth‐of‐coverage and reducing the fraction of loci for which no data could be obtained (Suchan et al., 2016). The HyRAD method, which utilizes the standard double digest (dd) RAD protocol (Mastretta‐Yanes et al., 2015) to prepare a set of DNA probes from fresh samples, has proven especially versatile in its applications to ancient (Schmid et al., 2017) and historical DNA (Schmid et al., 2018; Suchan et al., 2016), and for obtaining genome‐wide data at the population scale in a cost‐effective manner.
The museum collections of Finland, in particular the Finnish Museum of Natural History in Helsinki (Luonnontieteellinen keskusmuseo, Luomus), contains ~22 million specimens collected over recent centuries (Tegelberg, Haapala, Mononen, Pajari, & Saarenmaa, 2012). This provides a fantastic opportunity to obtain molecular data across wide time series, in a range of species, including butterfly taxa that have declined over the past century. This is notably the case of Erebia embla—a northern European and eastern Palearctic butterfly species inhabiting bogs—which has experienced a strong decrease in southern Finland, following the extensive drainage of its habitat (Rassi, Hyvärinen, Juslén, & Mannerkoski, 2010). The same holds true for Lycaena helle, an arctic‐alpine Palearctic butterfly species inhabiting fresh, damp meadows, which shows a patchy distribution throughout Europe and has declined throughout most of its range, especially in Finland, where it remains present at only two sites, Kuivaniemi Simo and Kuusamo (Heino, Poykko, & Itames, 1998).
In this study, we aim to examine how the genetic structures and diversities of E. embla and L. helle have been impacted by recent environmental changes and human activities, relying on HyRAD data generated from 118 and 165 museum specimens from Finland, respectively, collected across the last century as well as from extant populations. To achieve this, we developed a pipeline, namely PopHyRAD, that (a) aligns each sequence read against the probe catalogue (including reference genomes when available, or probe sequences provided by users), (b) identifies and controls for deamination patterns (typical of historical DNA), (c) eliminates putative paralogues, PCR duplicates, low‐quality genotypes and indels, and (d) keeps only bi‐allelic loci for downstream analyses. We then investigated the spatial and temporal dynamics of genetic diversity indices as well as isolation by distance in the two above‐mentioned butterflies, both locally and regionally.
2. MATERIAL AND METHODS
2.1. Sampling
Historical samples of L. helle and E. embla were sampled among the Lepidoptera collection of the Finnish Museum of Natural History in 2014 and 2015. Fresh samples were collected following fieldwork in summer 2015, by capturing flying adults with a net, sampling and storing a single leg in 95% EtOH, before releasing individuals alive. The sampling details are given in Table 1, and illustrated in Figure 1.
TABLE 1.
Sample information comprising locality name, approximate GPS positions, year of collection and number of samples. The three time slices used for the analyses have been indicated
| Erebia embla | Lycaena helle | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Locality | Latitude | Longitude | Year | N | Locality | Latitude | Longitude | Year | N | |
| Before 1950 | Haminalahti | 62.8534641 | 27.5326783 | 1909 | 12 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 1928 | 10 |
| Pirkkala | 61.4654497 | 23.6456252 | 1909 | 1 | Paanajarvi | 66.4555006 | 28.9798017 | 1934 | 1 | |
| Pirkkala | 61.4654497 | 23.6456252 | 1925 | 2 | Paanajarvi | 66.4555006 | 28.9798017 | 1935 | 6 | |
| Pirkkala | 61.4654497 | 23.6456252 | 1930 | 5 | Ivalo | 68.6588185 | 27.5348114 | 1937 | 13 | |
| Pernio | 60.2050782 | 23.1235771 | 1932 | 1 | Mikkeli | 61.6877956 | 27.2726569 | 1938 | 3 | |
| Portom | 62.7100207 | 21.6163442 | 1937 | 13 | Harmoinen | 61.4852477 | 25.1409736 | 1940 | 8 | |
| Muonio | 67.9593397 | 23.6774037 | 1938 | 13 | Kannus | 63.9007773 | 23.9170363 | 1940 | 5 | |
| Nurmes | 63.5422079 | 29.1410100 | 1941 | 6 | Nurmes | 63.5422079 | 29.1410100 | 1941 | 13 | |
| Pernio | 60.2050782 | 23.1235771 | 1944 | 2 | Ruovesi | 61.9856303 | 24.0703481 | 1941 | 13 | |
| Pirkkala | 61.4654497 | 23.6456252 | 1945 | 2 | Mikkeli | 61.6877956 | 27.2726569 | 1942 | 9 | |
| Jakobstad Pietarsaari | 63.6666709 | 22.7000229 | 1947 | 1 | Haapavesi | 64.1378737 | 25.3658176 | 1946 | 5 | |
| Pelkosenniemi | 67.1095969 | 27.5118116 | 1947 | 9 | Pelkosenniemi | 67.1095969 | 27.5118116 | 1947 | 12 | |
| Jakobstad Pietarsaari | 63.6666709 | 22.7000229 | 1949 | 4 | Paltamo | 64.4068668 | 27.8335512 | 1949 | 10 | |
| Between 1950 and 2000 | Jakobstad Pietarsaari | 63.6666709 | 22.7000229 | 1951 | 2 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 1955 | 1 |
| Jakobstad Pietarsaari | 63.6666709 | 22.7000229 | 1953 | 6 | Tohmajarvi | 62.2259448 | 30.3335512 | 1957 | 1 | |
| Kuusamo | 65.9645637 | 29.1883283 | 1955 | 8 | Tohmajarvi | 62.2259448 | 30.3335512 | 1958 | 5 | |
| Tyrvanto | 61.1546112 | 24.3283168 | 1959 | 1 | Kuopio | 62.8241424 | 27.5945615 | 1959 | 4 | |
| Karttula | 62.8952013 | 26.9723784 | 1963 | 4 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 1959 | 1 | |
| Ikaalinen | 61.7701493 | 23.0633777 | 1965 | 5 | Kuopio | 62.8241424 | 27.5945615 | 1960 | 4 | |
| Ikaalinen | 61.7701493 | 23.0633777 | 1969 | 5 | Tohmajarvi | 62.2259448 | 30.3335512 | 1960 | 4 | |
| Tuulos | 61.1181656 | 24.8337064 | 1970 | 2 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 1962 | 2 | |
| Tuulos | 61.1181656 | 24.8337064 | 1973 | 2 | Kuopio | 62.8241424 | 27.5945615 | 1964 | 1 | |
| Tuulos | 61.1181656 | 24.8337064 | 1975 | 1 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 1975 | 3 | |
| Kuusamo | 65.9645637 | 29.1883283 | 1977 | 4 | Kuivaniemi Simo | 65.6040217 | 25.2038392 | 1980 | 6 | |
| Mikkeli | 61.6877956 | 27.2726569 | 1979 | 2 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 1985 | 12 | |
| Kuusamo | 65.9645637 | 29.1883283 | 1980 | 1 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 1991 | 13 | |
| Kuusamo | 65.9645637 | 29.1883283 | 1981 | 3 | ||||||
| Mikkeli | 61.6877956 | 27.2726569 | 1983 | 1 | ||||||
| 2015 | Ivalo | 68.6588185 | 27.5348114 | 2015 | 5 | Kuivaniemi Simo | 65.6040217 | 25.2038392 | 2015 | 9 |
| Kuusamo | 65.9645637 | 29.1883283 | 2015 | 9 | Kuusamo Liikasenvaara | 65.9645637 | 29.1883283 | 2015 | 23 | |
| Muonio | 67.9593397 | 23.6774037 | 2015 | 9 | ||||||
| Oulu | 65.0118734 | 25.4716809 | 2015 | 12 | ||||||
| Pelkosenniemi | 67.1095969 | 27.5118116 | 2015 | 8 | ||||||
| Rovaniemi | 66.4976214 | 25.7192101 | 2015 | 11 | ||||||
FIGURE 1.

(a, c) Pictures of the two focal species, Lycaena helle and Erebia embla. (b, d) Maps of Finland with the sampling localities. The different colours indicate whether the samples considered were historical, with a distinction before and after 1950, or modern (year 2015)
2.2. DNA extraction and HyRAD protocol
The HyRAD protocol was carried out according to Suchan et al. (2016). Briefly, for historical and fresh samples, DNA was extracted from a leg using the QIAamp DNA Micro kit (Qiagen). The probe precursors were prepared using a ddRAD protocol applied to six fresh samples from each focal species (Mastretta‐Yanes et al., 2015; Peterson, Weber, Kay, Fisher, & Hoekstra, 2012). Total genomic DNA was digested with the restriction enzymes SbfI and MseI, DNA adapters were ligated and the resulting library was purified, size‐selected with a range of 200–250 bp and amplified by PCR for 30 cycles. An aliquot of the final library was sequenced on one lane of an Illumina MiSeq 150‐bp paired‐end at the Lausanne Genomic Technology Facility (LGTF) in order to obtain a sequence catalogue of the loci represented in the ddRAD probes, and the rest of the library was converted into probes by removing adapter sequences.
Individual Illumina DNA libraries were prepared from the fresh and museum specimens based on a published protocol for degraded DNA samples (Tin, Economo, & Mikheyev, 2014). Hybridization‐capture and enrichment was performed as described in Schmid et al. (2017), using a dual‐indexing tagging (i.e., with different combinations of barcodes and indexes), allowing extensive multiplexing of samples on a single sequencing lane: in L. helle, nine and 25 indexes and barcodes were used, respectively; while 10 and 25 were used in E. embla, respectively. For each butterfly species, the final (capture‐hybridized) enriched libraries were sequenced on two lanes of a HiSeq 2500 instrument using a 100‐bp paired‐end protocol at the LGTF.
2.3. PopHyRAD bioinformatic pipeline
The sequence pairs generated from the ddRAD probe libraries were first cleaned and overlapping reads were collapsed using adapterremoval version 2 (Schubert, Lindgreen, & Orlando, 2016). Then a first data set reduction was performed following the first step of the ddocent pipeline (Puritz, Hollenbeck, & Gold, 2014), keeping only alleles covered by at least four reads and, thus, removing the vast majority of sequencing errors. We merged intra‐individual loci using cd‐hit‐est from the cd‐hit tool (Fu, Niu, Zhu, Wu, & Li, 2012) using a minimum identity threshold of 90%. This step was repeated across samples to produce a combined catalogue of all loci identified for a given species. Loci shared by at least half of the probe samples (i.e., three out of six) were kept to remove uninformative specific loci. The absence of contamination in the catalogue was evaluated using centrifuge (Kim, Song, Breitwieser, & Salzberg, 2016).
Reads from each historical sample were cleaned using trimmomatic (Bolger, Lohse, & Usadel, 2014) and individually mapped on the loci catalogue generated above using bwa aln (Li & Durbin, 2009). PCR duplicates were removed using markduplicates from the picard toolkit (http://broadinstitute.github.io/picard). Nucleotide misincorporation patterns were investigated using mapdamage 2.0 (Jónsson, Ginolhac, Schubert, Johnson, & Orlando, 2013), and base quality scores were rescaled according to their probability to represent a post‐mortem DNA deamination event in order to reduce the impact of DNA decay on downstream analyses. Finally, individual genotypes were called using freebayes (Garrison & Marth, 2012), and considered for further analyses when (a) showing qualities higher than 100, (b) shared by at least 80% of the samples and (c) biallelic (Figure 2). This complete pipeline has been implemented under the name PopHyRAD, with “Pop” standing for population genetics.
FIGURE 2.
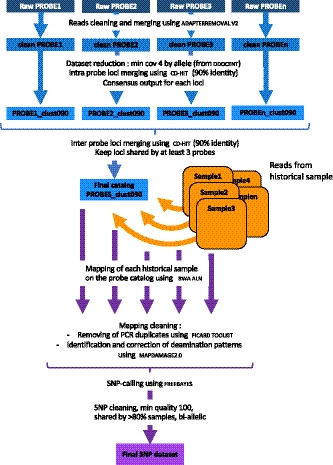
Schematic PopHyRAD pipeline including the creation of the catalogue from probes and the treatment of samples
Although bwa aln is the recommended tool for ancient DNA analysis (Schubert et al., 2012), we evaluated the performance of two other mapping tools, namely bwa mem (Li, 2013) and bowtie2 (Langmead & Salzberg, 2012). Similarly, we evaluated the possible impact of the genotyping method on downstream analyses using varscan (Koboldt et al., 2009).
2.4. Population genetics analyses
Allelic frequencies, observed gene diversity (H s) and inbreeding level (F IS) were estimated using the r package hierfstat (Goudet, 2005). The frequency of fixed alleles, which is a proxy for the frequency of allele extinction, was estimated using a custom script. For these estimations only localities with more than eight samples were kept. These estimations were performed after the samples were binned within temporal groups (or time slices). More precisely, we merged samples from each geographical location within three time slices: before 1950, between 1950 and 2000, and present samples (i.e., 2015; see details in Table 1). Additionally, we merged all samples from all locations within the same temporal bin to identify patterns at the scale of Finland.
Population genetic structure was investigated using a subset of SNPs, where only one SNP per locus was considered so as to minimize linkage disequilibrium (i.e., to minimize redundant information), as generally recommended (Falush, Stephens, & Pritchard, 2003). We used Bayesian admixture analysis implemented in structure (Pritchard, Stephens, & Donnelly, 2000) to estimate admixture proportions, that is, the proportion of each individual's genome inherited from each of K hypothetical source populations. We ran analyses with K from 1 to 5 with 10 independent Markov chains each, using 1,000,000 steps and including 50,000 burn‐in steps. We visually checked the results obtained in each run to assess whether the Markov chains were convergent. The most likely number of clusters was identified using Evanno's method (Evanno, Regnaut, & Goudet, 2005) implemented in structure harvester (Earl & vonHoldt, 2012). Then each sample was associated to the corresponding cluster and the proportion of the samples present in each cluster was drawn on the Finland map using pie charts.
Isolation‐by‐distance (IBD) was tested by examining the correlation between genetic differentiation, F ST/1 − F ST, using F ST estimations between population pairs performed with hierfstat (Goudet, 2005), and the log‐transformed distance, as suggested by Rousset (1997). Due to the ongoing debate in using Mantel tests for IBD examination (Diniz‐Filho et al., 2013), we used a simple Spearman's correlation. To corroborate the role of geographical distance as well as time in the differentiation between localities with another approach than a simple Spearman's correlation, we performed a distance‐based redundancy analysis (dbRDA) integrating geographical variables and year of collection, in an individual‐based approach, following recommendations from Laura Benestan (unpublished data; https://github.com/laurabenestan/db‐RDA‐and‐db‐MEM). In breif, we first created spatial variables using Moran Eigenvector's Maps (MEMs) implemented in the adespatial r package (Dray, Blanchet, Borcard, Guenard, & Jombart, 2016). Second, a principal coordinates analysis (PCoA) was performed on the Euclidean genetic distances based on genotypes of each sample. Finally a global dbRDA was applied by integrating the year of collection as an additional variable to spatial components, and an ANOVA with 1,000 permutations was performed to assess the significance of each variable within the model using the vegan r package (Oksanen, Kindt, Legendre, & O'Hara, 2016).
3. RESULTS
3.1. HyRAD efficiency
The HyRAD wetlab and PopHyRAD bioinformatic pipeline developed for this study were particularly efficient in retrieving DNA sequences of historical samples collected up to more than a century ago and stored in museum conditions. The analysis of ddRAD data used for designing the probes allowed us to obtain sufficient data at 3,826 loci for E. embla and 3,443 for L. helle, which allowed us to undertake population genomic analyses. In addition, the identification of deamination patterns at the extremities of the reads from the collection samples and not in the fresh samples confirmed the endogenous nature of DNA present in these historical samples (Figure S1).
3.2. Mapping and SNP calling comparison
We first aimed to test whether different read alignment procedures could impact on the amount of HyRAD data retrieved. bwa aln was able to align only a limited number of reads against the catalogue of HyRAD probes, representing a mean of 11.2% (SD 6.7) across all samples. This proportion was improved to 37.1% (SD 8.1) when using bwa mem (Table S2). bowtie2 showed intermediate efficiency with a mean value of 18.3% (SD 7.9) across all samples. These differences, however, decreased after quality filtering, with bwa aln showing 6.9% (SD 2.0) of mapped reads, bwa mem 12.1% (SD 4.8) and bowtie2 11.0% (SD 2.8).
We also observed broad differences between methodologies aimed at calling genotypes. We counted the total number of positions covered by at least 80% of the samples and showing biallelic SNP polymorphisms. This number was maximal when combining bwa mem alignments and varscan genotype calling, representing a total of 10,116 SNPs from 1,289 loci in E. embla and 11,534 SNPs from 1,241 loci for L. helle. The number was minimal when bwa aln alignments and freebayes genotype calls were combined, which led to the identification of 2,742 SNPs from 869 loci for E. embla and 2,549 SNPs from 1,015 loci for L. helle.
For each of these six combinations (three mappers and two SNP callers), we applied the complete pipeline and subsequently estimated genetic diversity statistics. We observed that these statistics were extremely variable according to the SNP calling tool used, especially the inbreeding coefficient (F IS) (Figure S2). With varscan, a large proportion of the SNPs demonstrate an inbreeding coefficient below zero with a non‐normal distribution, while the normal distribution obtained from freebayes is more consistent with biological expectations. Therefore, we decided to utilize the conservative data set obtained using freebayes on the mapping generated by bwa aln, despite being associated with an overall smaller number of SNPs; and we implemented freebayes and bwa aln as the standard caller*mapper combination in the PopHyRAD pipeline.
3.3. Patterns of population declines
To investigate genetic patterns associated with population size reduction, we focused on three descriptive statistics: (a) the observed gene diversity (H s), (b) the frequency of fixed alleles and (c) the inbreeding coefficient (F IS). Results are depicted in Figure 3.
FIGURE 3.
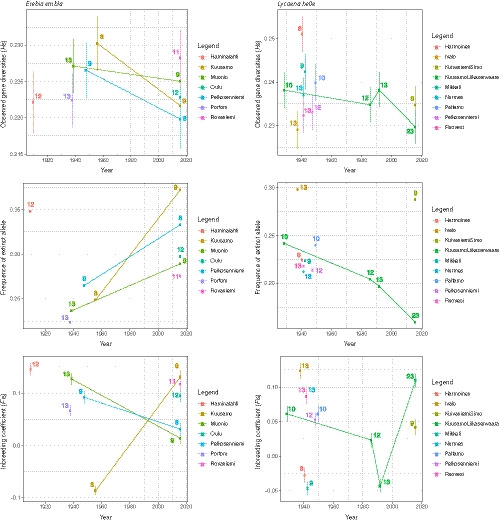
Observed gene diversity (H s), frequency of fixed alleles and inbreeding coefficient (F IS) estimated for each locality and years. For each point a number indicates the number of samples. Vertical lines represent the standard error across all SNPs (2,742 for Erebia embla and 2,549 for Lycaena helle). Localities sampled at different time points are linked by a line of the corresponding colour
For E. embla, sufficient temporal data could only be retrieved for three localities, namely Kuusamo, Muonio and Pelkosenniemi. These indicated reduction of the observed gene diversity (H s) and the frequency of fixed alleles through time. This overall reduction is, paradoxically, associated with a sharp increase of the inbreeding coefficient at only one locality, Kuusamo. For L. helle, only one locality, Kuusamo Liikasenvaara, was sampled at different time points. It showed a dynamic diversity trend in which the observed gene diversity (H s) and the frequency of fixed alleles were relatively stable between the 1920s and the 1990s, but declined after the 1990s. In contrast, the inbreeding coefficient (F IS) was found to decrease between the 1920s and the 1990s and to increase sharply since.
Grouping all samples within three main time slices (i.e., before 1950, between 1950 and 2000, and in 2015) increased the sample size of each locality and reinforced the observations made above (Figure 4). Indeed, we observed a decrease of the observed allelic richness (H s) over time in both species, concomitant with an increase in the frequency of fixed alleles.
FIGURE 4.
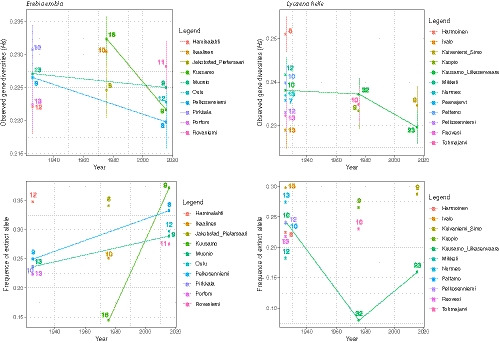
Observed gene diversity (H s) and frequency of fixed alleles over time (i.e., before 1950, between 1950 and 2000, and 2015), and for each locality. Vertical lines represent the standard error across all SNPs (2,742 for Erebia embla and 2,549 for Lycaena helle). Localities sampled at different time slices are linked by a line of the same colour
To investigate whether the patterns observed at the local level were retrieved at the global level (i.e., throughout Finland), we merged all the samples within the three time slices considered above, regardless of their geographical origin. This provided an opportunity to retrace the temporal trajectory of the Finland‐wide genetic diversity present in both focal species. We found that the observed gene diversity decreased through time and that the frequency of fixed alleles increased, especially in the most recent time period (Figure 5).
FIGURE 5.
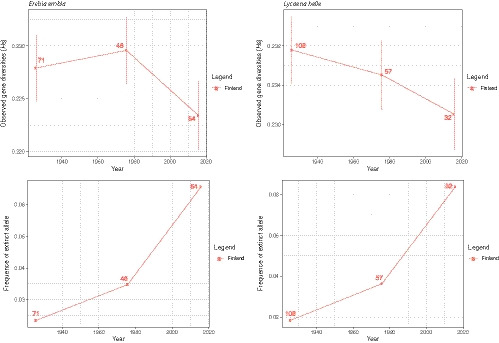
Observed gene diversity (H s) and frequency of fixed alleles estimated by time slices, before 1950, between 1950 and 2000, and 2015, over Finland as a whole after samples merging. Vertical lines represent the standard error across all SNPs (2,742 for Erebia embla and 2,549 for Lycaena helle)
3.4. Genetic structure and isolation by distance
To investigate the mechanisms that may explain the observed decrease in genetic diversity and the increase in the inbreeding coefficient (F IS), we studied the spatial genetic structure of populations, which only revealed a faint structuring (Figure 6). In contrast, a more marked pattern of spatial structuring was retrieved in the IBD analysis, revealing a varying correlation between genetic and geographical distances over time (Figure 7). Before 1950, no correlation between genetic and geographical distances was found in any of the two species investigated. Between the 1950s and the 2000s, a significant correlation between genetic and geographical distances was found for E. embla. In this species the level of correlation and the associated slope increased further when considering the modern time period (year 2015), suggesting an increasing spatial structuring from 1950 onwards, probably in relation to increased habitat fragmentation. No significant correlation could be retrieved in L. helle in any of the two historical time periods considered (this correlative analysis could not be carried out for modern times, as only two extant populations are known) (Figure 7). Despite an overall low level of genetic structuring through space and time (overall R 2 of 0.95% and 1.52%, based on one temporal and eight spatial axes, for E. embla and L. helle, respectively), the dbRDA approach indicated a significant impact of time (p = .002) to explain population differentiation, as well as a fainter effect of 3/8 spatial variables for E. embla and 2/8 spatial variables for L. helle, although with respective contributions of spatial variables to the overall R 2 remaining <0.5% (see Table S3).
FIGURE 6.
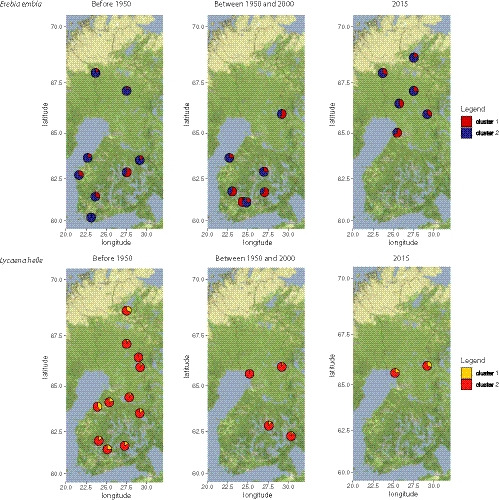
Population structure of all sampled localities according to the three studied periods. The pie charts indicate the proportion of samples associated with each cluster as identified by structure with K = 2
FIGURE 7.
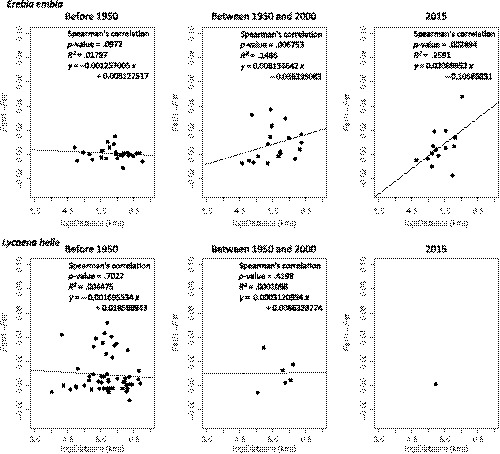
Isolation by distance was investigated for each time slice by computing the correlation between the genetic differentiation between populations (F ST/1 − F ST) and the log‐transformed geographical distance (log(distance)). Correlations were tested using Spearman's rank correlation, and coefficient of determination, intercept and slopes using a linear model are shown
4. DISCUSSION
4.1. A direct estimation of genetic variation across the past
The study of past population dynamics has received considerable attention in recent decades (Bi et al., 2019; Nadachowska‐Brzyska, Li, Smeds, Zhang, & Ellegren, 2015; Tallavaara, Luoto, Korhonen, Järvinen, & Seppä, 2015). Those studies classically identify the most likely demographic model underlying the allelic frequency spectrum measured in modern specimens (Csilléry, Blum, Gaggiotti, & François, 2010; Espíndola et al., 2012; François & Durand, 2010). These demographic inference approaches are, however, often limited as different models can produce similar allelic frequency spectrum and summary statistics, and cannot necessarily be discriminated (Lapierre, Lambert, & Achaz, 2017). In contrast, genomic data from historical specimens catch evolution red‐handed, and can help overcome such limitations by providing direct snapshots of the past genetic diversity present in a population.
In this study, we collected a large sample set of two butterfly species spread across Finland, and spanning the last 110 years. This sampling provided us with a unique opportunity to quantify the variation of the genetic diversity in both species at a time when their distribution drastically declined (Rassi et al., 2010). We have benefited from the HyRAD genome–complexity–reduction method to obtain genetic data from these valuable samples. HyRAD has been increasingly used in different laboratories, not only to identify genetic variation in historical material (Crates et al., 2019; Keighley, Heinsohn, Langmore, Murphy, & Peñalba, 2019; Linck et al., 2017; Linck, Freeman, & Dumbacher, 2019; Schmid et al., 2017), but also in ancient DNA (Schmid et al., 2017). Indeed, these methods based on hybridization capture allow us to retrieve even very small quantities of degraded DNA, which often remain unquantifiable before capture (Table S1). The amount of DNA in historical samples and the ability to extract, capture and sequence it depends on the history of the sample, the conditions of collection, sample preparation (drying, pinning etc.) and storage. Unfortunately, for most of our historical samples we do not have access to such information. However, in this study we were able to perform the entire process from historical specimen subsampling to SNP calling for ~75% of the samples analysed from both species, thus suggesting that it is compliant with most preparation histories.
For this study we developed a specific pipeline, PopHyRAD, to optimally exploit the genetic information contained in samples. For now, the PopHyRAD computational pipeline released here facilitates HyRAD sequence analysis at the within‐species level by automating the steps underlying read cleaning, trimming and merging, as well as read mapping, and probe clustering. This pipeline is versatile, and can be used to analyse any type of hybridization‐capture data, using either probes from ddRAD or another RADseq protocol, or any tool able to reduce genomic complexity such as selective extraction of organellar genomes or amplification of specific genes. Before this study, the catalogue definition and remaining analytical workflow using HyRAD data have essentially been empirically explored (or a posteriori chosen), considering the outputs, and thus the tools that provided the best geographical or phylogenetic structure (Schmid et al., 2018). Here, we take the opportunity to test more accurately the performance of aligners and SNP‐callers on HyRAD data, using different tool combinations, and using a realistic criterion from the point of view of population genetics, namely F IS. The results revealed large differences on the SNPs identified and on the estimation of genetic diversity and inbreeding coefficient and suggested the bwa aln read aligner and the freebayes SNP‐caller as the most conservative combination. The nonbiologically relevant F IS values obtained with other combinations are likely to be due, at least in part, to increased false positive alignment rates (e.g., misidentified paralogues) as well as to the oversplitting of loci (i.e., a locus separated in two loci in the catalogue). This type of difference has already been highlighted in analyses of standard RADseq data (Shafer et al., 2017) and calls for caution in downstream analyses. An analysis based on data simulation is outside the scope of this study but would probably clarify the specificity and sensitivity of the different aligners and SNP‐callers, and help each user to refine the most appropriate parameters for their analyses and model species.
4.2. Genetic diversity decline in butterfly populations across Finland
The HyRAD data gathered in this study supported an overall erosion of genetic diversity at the country‐wide level of Finland in both species (Figure 5). Although interpretation of variation in genetic diversity should be tempered due to our relatively reduced sampling size per locality and temporal binning, one should keep in mind that in the context of museomics, our sampling remains substantial. This pattern of genetic diversity reduction parallels those found by similar studies on butterflies in Northern Europe (Fountain et al., 2016; Ugelvig, Nielsen, Boomsma, & Nash, 2011) but also more broadly in other taxa (Dufresnes et al., 2018; Schmid et al., 2018). Our data also uncovered strong regional differences, with at least one locality (i.e., Kuusamo) showing a local increase in diversity at a given time point, potentially following migration linked to the persistence of their habitat in these specific localities (Habel, Meyer, & Schmitt, 2014) playing a role as refuges for individuals from other populations carrying genetic diversity (Craioveanu, Sitar, & Rákosy, 2014). However, estimations based on recent samples (i.e., collected in 2015) still show a decline in genetic diversity in this particular locality. The overall erosion of genetic diversity, both locally and country‐wide, is expected given that most Finnish populations of these two butterflies have gone extinct through the 20th century, as a result of a drastic reduction in habitat availability, with the remaining populations not being able to maintain genetic diversity to levels that once existed in an area of ~340,000 km2 a century or even a few decades ago.
The second striking result of this study is the increase in IBD over time, at least in one of the two species. Indeed, in E. embla, when considering time slices that divide the time frame of collected specimens into three periods, only the two last (i.e., 1950–2000, and 2015) are associated with significant IBD, with an increasing slope as we reach contemporaneous times. The effect of time was also retrieved in the dbRDA approach, although because this analysis is individual‐based, it was less representative of genetic variation per deme through space and time, thus explaining the low R 2 retrieved in the overall model.
Our main result of an overall increase in the slope of the IBD pattern is probably the consequence of an increase in habitat fragmentation, revealing a reduced number of migrants among demes, and thus an increase in the differentiation of populations, essentially due—given the short time span involved—to drift. This signature might be also found in L. helle, even if our sampling does not allow the estimation of IBD for the most recent period (i.e., only two populations are still extant today). This transition from a virtually countrywide panmictic system to a more marked structuring in space is indicative of the fact that despite acknowledged dispersal capabilities of these butterflies in Finland (Habel, Rödder, Schmitt, & Nève, 2011; Habel, Finger, Schmitt, & Nève, 2011), generally related to a colonization‐edge syndrome characteristic of populations found at the northern edge of a species' distribution (Duplouy, Wong, Corander, Lehtonen, & Hanski, 2017), the fragmentation of habitats has led to a decrease in these exchanges, and thus to local differentiation.
Through their impact on biodiversity, human activities are accelerating the extinction of populations and the differentiation of those that persist. This could be catalyzing lineage divergence, except that habitat destruction is an ongoing process, potentially hampered by geopolitical, but potentially ubiquitous, decisions. Our study of two species of butterflies in Finland indicates that not all species might respond identically to this fragmentation, and that comparative studies, involving a larger number of species represented by fresh but also historical specimens, are needed to understand how life history traits influence the species' population response to anthropogenic habitat disturbance and destruction. With the application of both the wetlab HyRAD protocol to historical and fresh specimens, and the PopHyRAD bioinformatic pipeline as described in this study, access to both past and extant genetic diversity should allow a better understanding and anticipation of the neutral response of populations to drastic habitat loss.
AUTHOR CONTRIBUTIONS
N.A. and M.P. designed the study. M.P. and L.K. performed sampling. M.P. performed laboratory work. J.G. analysed the molecular data, with contributions from S.N., M.P., N.A., L.O. and S.S. All authors took part in discussions concerning the analyses and interpretations. J.G. and N.A. wrote the paper, with contributions from all authors.
DATA ACCESSIBILITY
Sequence reads are archived at Zenodo: http://doi.org/10.5281/zenodo.3668644 for E. embla and http://doi.org/10.5281/zenodo.3668660 for L. helle. Scripts for the whole analytical process have been uploaded to Github (https://github.com/JeremyLGauthier/Scripts_Gauthier_et.al_2019_MER). The PopHyRAD pipeline is constantly under development and improvement. The current version can be found at https://github.com/JeremyLGauthier/PHyRAD).
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Christophe Dufresnes, Tomasz Suchan, Camille Pitteloud and Kimmo Saarinen for their invaluable help in the field, to Nils Arrigo for setting preliminary bioinformatic pipelines to process HyRAD data, to Stéphanie Manel for advice with distance‐based redundancy analysis, and to Jérôme Goudet for support during early development of the PopHyRAD pipeline and for commenting on this manuscript. We thank the Lausanne Genomic Technologies Facility (LGTF) for the sequencing service as well as two anonymous reviewers for their useful comments that led us to improve the manuscript.
Gauthier J, Pajkovic M, Neuenschwander S, et al. Museomics identifies genetic erosion in two butterfly species across the 20th century in Finland. Mol Ecol Resour. 2020;20:1191–1205. 10.1111/1755-0998.13167
Jérémy Gauthier and Mila Pajkovic contributed equally to this work and are considered as joint first authors.
Funding information
This research was funded by the Swiss National Science Foundation grants PP00P3_144870 and PP00P3_172899 awarded to Nadir Alvarez.
REFERENCES
- Ali, O. A. , O'Rourke, S. M. , Amish, S. J. , Meek, M. H. , Luikart, G. , Jeffres, C. , & Miller, M. R. (2016). RAD capture (rapture): Flexible and efficient sequence‐based genotyping. Genetics, 202, 389–400. 10.1534/genetics.115.183665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, K. , Linderoth, T. , Singhal, S. , Vanderpool, D. , Patton, J. L. , Nielsen, R. , … Good, J. M. (2019). Temporal genomic contrasts reveal rapid evolutionary responses in an alpine mammal during recent climate change. PLoS Genetics, 15, e1008119 10.1371/journal.pgen.1008119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher, F. C. , Casazza, G. , Szövényi, P. , & Conti, E. (2016). Sequence capture using RAD probes clarifies phylogenetic relationships and species boundaries in Primula sect. Auricula . Molecular Phylogenetics and Evolution, 104, 60–72. 10.1016/j.ympev.2016.08.003 [DOI] [PubMed] [Google Scholar]
- Bouzat, J. L. (2010). Conservation genetics of population bottlenecks: The role of chance, selection, and history. Conservation Genetics, 11, 463–478. 10.1007/s10592-010-0049-0 [DOI] [Google Scholar]
- Burrell, A. S. , Disotell, T. R. , & Bergey, C. M. (2015). The use of museum specimens with high‐throughput DNA sequencers. Journal of Human Evolution, 79, 35–44. 10.1016/j.jhevol.2014.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceballos, G. , Ehrlich, P. R. , & Dirzo, R. (2017). Biological annihilation via the ongoing sixth mass extinction signaled by vertebrate population losses and declines. Proceedings of the National Academy of Sciences USA, 114, E6089–E6096. 10.1073/pnas.1704949114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craioveanu, C. , Sitar, C. , & Rákosy, L. (2014). Mobility, behaviour and phenology of the Violet Copper Lycaena helle in North‐Western Romania In Habel J. C., Meyer M., & Schmitt T. (Eds.), Jewels in the mist. A synopsis on the endangered Violet Copper butterfly Lycaena helle (pp. 91–105). Sofia, Bulgaria; Moscow, Russia: Pensoft. [Google Scholar]
- Crates, R. , Olah, G. , Adamski, M. , Aitken, N. , Banks, S. , Ingwersen, D. , … Heinsohn, R. (2019). Genomic impact of severe population decline in a nomadic songbird. PLoS ONE, 14, e0223953 10.1371/journal.pone.0223953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csilléry, K. , Blum, M. G. B. , Gaggiotti, O. E. , & François, O. (2010). Approximate Bayesian Computation (ABC) in practice. Trends in Ecology & Evolution, 25, 410–418. 10.1016/j.tree.2010.04.001 [DOI] [PubMed] [Google Scholar]
- Dabney, J. , Meyer, M. , & Pääbo, S. (2013). Ancient DNA damage. Cold Spring Harbor Perspectives in Biology, 5, a012567 10.1101/cshperspect.a012567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Roque, F. , Menezes, J. F. S. , Northfield, T. , Ochoa‐Quintero, J. M. , Campbell, M. J. , & Laurance, W. F. (2018). Warning signals of biodiversity collapse across gradients of tropical forest loss. Scientific Reports, 8, 1622 10.1038/s41598-018-19985-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker, J. E. , Pires, J. C. , Conant, G. C. , McKay, S. D. , Heaton, M. P. , Chen, K. , … Taylor, J. F. (2009). Resolving the evolution of extant and extinct ruminants with high‐throughput phylogenomics. Proceedings of the National Academy of Sciences USA, 106, 18644–18649. 10.1073/pnas.0904691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz‐Filho, J. A. F. , Soares, T. N. , Lima, J. S. , Dobrovolski, R. , Landeiro, V. L. , de Campos Telles, M. , … Bini, L. M. (2013). Mantel test in population genetics. Genetics and Molecular Biology, 36, 475–485. 10.1590/S1415-47572013000400002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray, S. , Blanchet, G. , Borcard, D. , Guenard, G. , & Jombart, T. (2016). adespatial: Multivariate multiscale spatial analysis. R package version 0.0.3. [Google Scholar]
- Dufresnes, C. , Mazepa, G. , Rodrigues, N. , Brelsford, A. , Litvinchuk, S. N. , Sermier, R. , … Jeffries, D. L. (2018). Genomic evidence for cryptic speciation in tree frogs from the Apennine peninsula, with description of Hyla perrini sp. nov. Frontiers in Ecology and Evolution, 6, 144 10.3389/fevo.2018.00144 [DOI] [Google Scholar]
- Duplouy, A. , Wong, S. C. , Corander, J. , Lehtonen, R. , & Hanski, I. (2017). Genetic effects on life‐history traits in the Glanville fritillary butterfly. PeerJ, 5, e3371 10.7717/peerj.3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl, D. A. , & vonHoldt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4, 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- Ehrlich, P. R. (1995). The scale of human enterprise and biodiversity loss In Lawton H., & May R. M. (Eds.), Extinction rates (pp. 214–226). Oxford, UK: Oxford University Press. [Google Scholar]
- Espíndola, A. , Pellissier, L. , Maiorano, L. , Hordijk, W. , Guisan, A. , & Alvarez, N. (2012). Predicting present and future intra‐specific genetic structure through niche hindcasting across 24 millennia. Ecology Letters, 15, 649–657. 10.1111/j.1461-0248.2012.01779.x [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14, 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics, 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountain, T. , Nieminen, M. , Sirén, J. , Wong, S. C. , Lehtonen, R. , & Hanski, I. (2016). Predictable allele frequency changes due to habitat fragmentation in the Glanville fritillary butterfly. Proceedings of the National Academy of Sciences USA, 113, 2678–2683. 10.1073/pnas.1600951113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- François, O. , & Durand, E. (2010). Spatially explicit Bayesian clustering models in population genetics. Molecular Ecology Resources, 10, 773–784. 10.1111/j.1755-0998.2010.02868.x [DOI] [PubMed] [Google Scholar]
- Fu, L. , Niu, B. , Zhu, Z. , Wu, S. , & Li, W. (2012). CD‐HIT: Accelerated for clustering the next‐generation sequencing data. Bioinformatics, 28, 3150–3152. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison, E. , & Marth, G. (2012). Haplotype‐based variant detection from short‐read sequencing. Arxiv preprint Retrieved from http://arxiv.org/abs/1207.3907
- Goudet, J. (2005). Hierfstat, a package for R to compute and test hierarchical F‐statistics. Molecular Ecology Notes, 5, 184–186. 10.1111/j.1471-8286.2004.00828.x [DOI] [Google Scholar]
- Habel, J. C. , Rödder, D. , Schmitt, T. , & Nève, G. (2011). Global warming will affect genetic diversity and uniqueness of Lycaena helle populations. Global Change Biology, 17, 194–205. 10.1111/j.1365-2486.2010.02233.x [DOI] [Google Scholar]
- Habel, J. C. , Finger, A. , Schmitt, T. , & Nève, G. (2011). Survival of the endangered butterfly Lycaena helle in a fragmented environment: Genetic analyses over 15 years. Journal of Zoological Systematics and Evolutionary Research, 49, 25–31. 10.1111/j.1439-0469.2010.00575.x [DOI] [Google Scholar]
- Habel, J. C. , Meyer, M. , & Schmitt, T. (2014). Jewels in the mist: A synopsis on the endangered Violet Copper butterfly Lycaena helle. Sofia‐Moscow: Pensoft. [Google Scholar]
- Hallmann, C. A. , Sorg, M. , Jongejans, E. , Siepel, H. , Hofland, N. , Schwan, H. , … de Kroon, H. (2017). More than 75 percent decline over 27 years in total flying insect biomass in protected areas. PLoS ONE, 12, e0185809 10.1371/journal.pone.0185809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heino, J. , Poykko, H. , & Itames, J. (1998). Occurrence, biology and conservation possibilities of Lycaena helle in the area of Koillismaa, eastern Finland. Baptria, Helsinki, 23, 163–168. [Google Scholar]
- Hoffberg, S. L. , Kieran, T. J. , Catchen, J. M. , Devault, A. , Faircloth, B. C. , Mauricio, R. , & Glenn, T. C. (2016). RADcap: Sequence capture of dual‐digest RADseq libraries with identifiable duplicates and reduced missing data. Molecular Ecology Resources, 16, 1264–1278. 10.1111/1755-0998.12566 [DOI] [PubMed] [Google Scholar]
- Hoffmann, A. A. , Sgrò, C. M. , & Kristensen, T. N. (2017). Revisiting adaptive potential, population size, and conservation. Trends in Ecology & Evolution, 32, 506–517. 10.1016/j.tree.2017.03.012 [DOI] [PubMed] [Google Scholar]
- Horn, S. (2012). Target enrichment via DNA hybridization capture. Methods in Molecular Biology, 840, 177–188. 10.1007/978-1-61779-516-9_21 [DOI] [PubMed] [Google Scholar]
- Jensen, E. L. , Edwards, D. L. , Garrick, R. C. , Miller, J. M. , Gibbs, J. P. , Cayot, L. J. , … Russello, M. A. (2018). Population genomics through time provides insights into the consequences of decline and rapid demographic recovery through head‐starting in a Galapagos giant tortoise. Evolutionary Applications, 11, 1811–1821. 10.1111/eva.12682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, C. N. , Balmford, A. , Brook, B. W. , Buettel, J. C. , Galetti, M. , Guangchun, L. , & Wilmshurst, J. M. (2017). Biodiversity losses and conservation responses in the Anthropocene. Science, 356, 270–275. 10.1126/science.aam9317 [DOI] [PubMed] [Google Scholar]
- Jónsson, H. , Ginolhac, A. , Schubert, M. , Johnson, P. L. F. , & Orlando, L. (2013). mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics, 29, 1682–1684. 10.1093/bioinformatics/btt193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, D. S. , Mendenhall, C. D. , Sandí, R. F. , Chaumont, N. , Ehrlich, P. R. , Hadly, E. A. , & Daily, G. C. (2013). Forest bolsters bird abundance, pest control and coffee yield. Ecology Letters, 16, 1339–1347. 10.1111/ele.12173 [DOI] [PubMed] [Google Scholar]
- Keighley, M. V. , Heinsohn, R. , Langmore, N. E. , Murphy, S. A. , & Peñalba, J. V. (2019). Genomic population structure aligns with vocal dialects in Palm Cockatoos (Probosciger aterrimus); evidence for refugial late‐Quaternary distribution? Emu‐Austral Ornithology, 119, 24–37. 10.1080/01584197.2018.1483731 [DOI] [Google Scholar]
- Keller, L. F. , & Waller, D. M. (2002). Inbreeding effects in wild populations. Trends in Ecology & Evolution, 17, 230–241. 10.1016/S0169-5347(02)02489-8 [DOI] [Google Scholar]
- Kim, D. , Song, L. , Breitwieser, F. P. , & Salzberg, S. L. (2016). Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Research, 26, 1721–1729. 10.1101/gr.210641.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt, D. C. , Chen, K. , Wylie, T. , Larson, D. E. , McLellan, M. D. , Mardis, E. R. , … Ding, L. (2009). VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics, 25, 2283–2285. 10.1093/bioinformatics/btp373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen, T. N. , Pedersen, K. S. , Vermeulen, C. J. , & Loeschcke, V. (2010). Research on inbreeding in the “omic” era. Trends in Ecology & Evolution, 25, 44–52. 10.1016/j.tree.2009.06.014 [DOI] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre, M. , Lambert, A. , & Achaz, G. (2017). Accuracy of demographic inferences from the site frequency spectrum: The case of the Yoruba population. Genetics, 206, 439–449. 10.1534/genetics.116.192708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. Arxiv Preprint Retrieved from http://arxiv.org/abs/1303.3997
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linck, E. B. , Freeman, B. G. , & Dumbacher, J. P. (2019). Speciation with gene flow across an elevational gradient in New Guinea kingfishers. bioRxiv. https://www.biorxiv.org/content/10.1101/589044v2 [DOI] [PubMed] [Google Scholar]
- Linck, E. B. , Hanna, Z. R. , Sellas, A. , & Dumbacher, J. P. (2017). Evaluating hybridization capture with RAD probes as a tool for museum genomics with historical bird specimens. Ecology and Evolution, 7, 4755–4767. 10.1002/ece3.3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister, B. C. , & Garcia, A. (2018). Climate‐driven declines in arthropod abundance restructure a rainforest food web. Proceedings of the National Academy of Sciences USA, 115, E10397–E10406. 10.1073/pnas.1722477115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastretta‐Yanes, A. , Arrigo, N. , Alvarez, N. , Jorgensen, T. H. , Piñero, D. , & Emerson, B. C. (2015). Restriction site‐associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Molecular Ecology Resources, 15, 28–41. 10.1111/1755-0998.12291 [DOI] [PubMed] [Google Scholar]
- Meineke, E. K. , Davies, T. J. , Daru, B. H. , & Davis, C. C. (2018). Biological collections for understanding biodiversity in the Anthropocene. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 374 10.1098/rstb.2017.0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadachowska‐Brzyska, K. , Li, C. , Smeds, L. , Zhang, G. , & Ellegren, H. (2015). Temporal dynamics of avian populations during Pleistocene revealed by whole‐genome sequences. Current Biology, 25, 1375–1380. 10.1016/j.cub.2015.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Kindt, R. , Legendre, P. , & O'Hara, B. (2016). The vegan package. R package version 2.4‐1. [Google Scholar]
- Orlando, L. , Gilbert, M. T. P. , & Willerslev, E. (2015). Reconstructing ancient genomes and epigenomes. Nature Reviews Genetics, 16, 395–408. 10.1038/nrg3935 [DOI] [PubMed] [Google Scholar]
- Peterson, B. K. , Weber, J. N. , Kay, E. H. , Fisher, H. S. , & Hoekstra, H. E. (2012). Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non‐model species. PLoS ONE, 7, e37135 10.1371/journal.pone.0037135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puritz, J. B. , Hollenbeck, C. M. , & Gold, J. R. (2014). dDocent: A RADseq, variant‐calling pipeline designed for population genomics of non‐model organisms. PeerJ, 2 10.7717/peerj.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyke, G. H. , & Ehrlich, P. R. (2010). Biological collections and ecological/environmental research: A review, some observations and a look to the future. Biological Reviews of the Cambridge Philosophical Society, 85, 247–266. 10.1111/j.1469-185X.2009.00098.x [DOI] [PubMed] [Google Scholar]
- Rassi, P. , Hyvärinen, E. , Juslén, A. , & Mannerkoski, I. (2010). The 2010 Red List of Finnish species. Helsinki, Finland: Ministry of the Environment. Finnish Environment Institute. [Google Scholar]
- Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F‐statistics under isolation by distance. Genetics, 145, 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, S. F. , Deines, J. M. , Scriber, J. M. , Pfrender, M. E. , Jones, S. E. , Emrich, S. J. , & Hellmann, J. J. (2018). Climate‐mediated hybrid zone movement revealed with genomics, museum collection, and simulation modeling. Proceedings of the National Academy of Sciences USA, 115, E2284–E2291. 10.1073/pnas.1714950115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez Barreiro, F. , Vieira, F. G. , Martin, M. D. , Haile, J. , Gilbert, M. T. P. , & Wales, N. (2017). Characterizing restriction enzyme‐associated loci in historic ragweed (Ambrosia artemisiifolia) voucher specimens using custom‐designed RNA probes. Molecular Ecology Resources, 17, 209–220. 10.1111/1755-0998.12610 [DOI] [PubMed] [Google Scholar]
- Sánchez‐Bayo, F. , & Wyckhuys, K. A. G. (2019). Worldwide decline of the entomofauna: A review of its drivers. Biological Conservation, 232, 8–27. 10.1016/j.biocon.2019.01.020 [DOI] [Google Scholar]
- Schmid, S. , Genevest, R. , Gobet, E. , Suchan, T. , Sperisen, C. , Tinner, W. , & Alvarez, N. (2017). HyRAD‐X, a versatile method combining exome capture and RAD sequencing to extract genomic information from ancient DNA. Methods in Ecology and Evolution/British Ecological Society, 8, 1374–1388. 10.1111/2041-210X.12785 [DOI] [Google Scholar]
- Schmid, S. , Neuenschwander, S. , Pitteloud, C. , Heckel, G. , Pajkovic, M. , Arlettaz, R. , & Alvarez, N. (2018). Spatial and temporal genetic dynamics of the grasshopper Oedaleus decorus revealed by museum genomics. Ecology and Evolution, 8, 1480–1495. 10.1002/ece3.3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, M. , Ginolhac, A. , Lindgreen, S. , Thompson, J. F. , Al‐Rasheid, K. A. S. , Willerslev, E. , … Orlando, L. (2012). Improving ancient DNA read mapping against modern reference genomes. BMC Genomics, 13, 178 10.1186/1471-2164-13-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, M. , Lindgreen, S. , & Orlando, L. (2016). AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Research Notes, 9, 88 10.1186/s13104-016-1900-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer, A. B. A. , Peart, C. R. , Tusso, S. , Maayan, I. , Brelsford, A. , Wheat, C. W. , & Wolf, J. B. W. (2017). Bioinformatic processing of RAD‐seq data dramatically impacts downstream population genetic inference. Methods in Ecology and Evolution/British Ecological Society, 8, 907–917. 10.1111/2041-210X.12700 [DOI] [Google Scholar]
- Suchan, T. , Pitteloud, C. , Gerasimova, N. S. , Kostikova, A. , Schmid, S. , Arrigo, N. , … Alvarez, N. (2016). Hybridization capture using RAD probes (hyRAD), a new tool for performing genomic analyses on collection specimens. PLoS ONE, 11, e0151651 10.1371/journal.pone.0151651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallavaara, M. , Luoto, M. , Korhonen, N. , Järvinen, H. , & Seppä, H. (2015). Human population dynamics in Europe over the Last Glacial Maximum. Proceedings of the National Academy of Sciences USA, 112, 8232–8237. 10.1073/pnas.1503784112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegelberg, R. , Haapala, J. , Mononen, T. , Pajari, M. , & Saarenmaa, H. (2012). The development of a digitising service centre for natural history collections. ZooKeys, 209, 75–86. 10.3897/zookeys.209.3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielens, A. , Bell, D. , Mortimore, D. B. , Greco, M. K. , Martens, L. , & Joseph, W. (2018). Exposure of insects to radio‐frequency electromagnetic fields from 2 to 120 GHz. Scientific Reports, 8, 3924 10.1038/s41598-018-22271-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tin, M.‐M.‐Y. , Economo, E. P. , & Mikheyev, A. S. (2014). Sequencing degraded DNA from non‐destructively sampled museum specimens for RAD‐tagging and low‐coverage shotgun phylogenetics. PLoS ONE, 9, e96793 10.1371/journal.pone.0096793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugelvig, L. V. , Nielsen, P. S. , Boomsma, J. J. , & Nash, D. R. (2011). Reconstructing eight decades of genetic variation in an isolated Danish population of the large blue butterfly Maculinea arion . BMC Evolutionary Biology, 11, 201 10.1186/1471-2148-11-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Strien, A. J. , van Swaay, C. A. M. , van Strien‐van Liempt, W. T. F. H. , Poot, M. J. M. , & WallisDeVries, M. F. (2019). Over a century of data reveal more than 80% decline in butterflies in the Netherlands. Biological Conservation, 234, 116–122. 10.1016/j.biocon.2019.03.023 [DOI] [Google Scholar]
- Whiteside, M. , & Marvin Herndon, J. (2018). Previously unacknowledged potential factors in catastrophic bee and insect die‐off arising from coal fly ash geoengineering. Asian Journal of Biology, 6, 1–13. 10.9734/AJOB/2018/43268 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Sequence reads are archived at Zenodo: http://doi.org/10.5281/zenodo.3668644 for E. embla and http://doi.org/10.5281/zenodo.3668660 for L. helle. Scripts for the whole analytical process have been uploaded to Github (https://github.com/JeremyLGauthier/Scripts_Gauthier_et.al_2019_MER). The PopHyRAD pipeline is constantly under development and improvement. The current version can be found at https://github.com/JeremyLGauthier/PHyRAD).