

Abstract
An unprecedented one‐pot fully electrochemically driven Wittig olefination reaction system without employing a chemical reductant or sacrificial electrode material to regenerate triphenylphosphine (TPP) from triphenylphosphine oxide (TPPO) and base‐free in situ formation of Wittig ylides, is reported. Starting from TPPO, the initial step of the phosphoryl P=O bond activation proceeds through alkylation with RX (R=Me, Et; X=OSO2CF3 (OTf)), affording the corresponding [Ph3POR]+X− salts which undergo efficient electroreduction to TPP in the presence of a substoichiometric amount of the Sc(OTf)3 Lewis acid on a Ag‐electrode. Subsequent alkylation of TPP affords Ph3PR+ which enables a facile and efficient electrochemical in situ formation of the corresponding Wittig ylide under base‐free condition and their direct use for the olefination of various carbonyl compounds. The mechanism and, in particular, the intriguing role of Sc3+ as mediator in the TPPO electroreduction been uncovered by density functional theory calculations.
Keywords: bond activation, electrosynthesis, Lewis acid, phosphine oxide reduction, scandium complexes
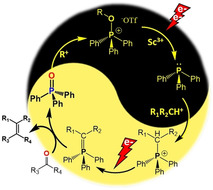
Fully electrified: For the first time, a electrochemical system for a one‐pot Wittig olefination reaction (WOR) is reported, which includes a very efficient recycling of triphenylphosphine from triphenylphosphine oxide waste and subsequent carbonyl olefinations through in situ base‐free Wittig ylide formation, avoiding chemical reductants or sacrificial electrodes.
Introduction
The Wittig olefination reaction (WOR) is one of the most common synthetic routes to produce functional alkenes.1 The perhaps most prominent case of WOR at industrial scale is the production of vitamin A.2 The latter affords a stoichiometric amount of triphenylphophine oxide (TPPO) resulting as a by‐product in several tons per year during the synthesis (Scheme 1 a). Henceforth, reduction of organophosphine oxides to the corresponding phosphine (e.g., Ph3P, TPP) is not only of fundamental but also of utmost industrial interest.3 In addition, the complete removal of TPPO from reaction mixtures is often not straightforward.4 Until now, deoxygenation to regenerate TPP can nearly exclusively be achieved using chemical reductant, for example, silanes,3b, 5 boranes3a, 6 and aluminium (hydrides)7 and therefore, the pursuit of other reducing agents8 or regeneration processes has gained attention in the last few years (Scheme 1 a).3a, 3b Considering that regenerating TPP with expensive reduction agents is not a viable solution, alternative approaches have been tested.9 Among them, electrochemical reduction of TPPO to TPP is a highly attractive aim, but remains cumbersome.10 Up to now, electroreduction of TPPO to TPP suffers from shortcomings that prevent from its recycling on a commercially feasible scale.11
Scheme 1.
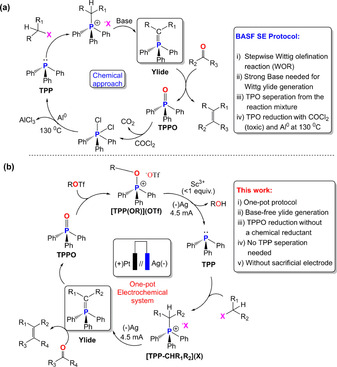
(a) Chemical route of Wittig olefination reactions (WOR) followed by chemical recycling of TPPO to TPP with phosgene (COCl2) and Al° at 130 °C applied by BASF SE for vitamin A production. (b) One‐pot electrochemical WOR protocol via cathodic recycling of TPPO without sacrificial electrode and subsequent Wittig ylide regeneration and carbonyl olefination.
Previous attempts towards selective electroreductive deoxygenation of TPPO remained unsatisfactory due to undesired C−P bond dissociation to Ph2P(O)H and benzene.10 The most promising approaches towards successful TPP regeneration from TPPO proceed through activation of the P=O bond with Lewis acids (LA) such as Me3SiCl,11a, 11b AlCl3 11a during electrolysis and/or formation of dichlorophophorane (Ph3PCl2).11c Recently, boron esters, [B(OAr)3], have also been demonstrated to act as suitable LAs to form a Ph3P=O→B(OAr)3 adduct which facilitates the rate‐determining phosphoryl P=O bond dissociation to form TPP and [{(ArO)3B}2O]2− diborate.11e However, its moderate overall efficiency along with side‐product formation and the difficulty to recover the boron ester from the diborate limits its suitability for TPP regeneration.11e An improvement in TPP regeneration has lately been achieved under mild electrochemical conditions, in the presence of AlCl3 as LA and tetramethylethylene diamine as an additive.11f However, the intrinsic dissolution of a sacrificial Al electrode by electro‐corrosion and subsequent formation of Al2O3, which has a high energy demand for recycling, limits this approach for sustainable TPP electro‐regeneration too.11a, 11c, 11d, 11f
On the other hand, and to the best of our knowledge, the direct use of electro‐regenerated TPP and base‐free electrochemical Ph3P=CR2 ylide formation and subsequent WOR in the presence of a carbonyl compound in a one‐pot protocol is currently unknown.
Herein, we describe a systems approach to a one‐pot fully electrochemically driven protocol for direct carbonyl olefination via WOR, combining the required subset of stoichiometric reactions. This includes the recycling of TPPO to TPP in excellent yields omitting any chemical reductant and subsequent in situ electrosynthesis of Wittig ylides in the absence of a base (Scheme 1 b).
Results and Discussion
The starting point is the phosphoryl P=O bond activation of TPPO via O‐alkylation to form the isolable [Ph3P(OR)](OTf) phosphonium salts (R=Me (1); Et (2)), using ROTf (OTf=OSO2CF3) as sources of R+. Structural (XRD), spectroscopic (31P NMR, IR) and cyclic voltammetry evidences in support of the P=O bond activation via alkylation is also provided. In the presence of a substoichiometric amounts of Sc(OTf)3 acting as a very effective redox‐innocent Lewis‐acid mediator, the electrochemical conversion of [Ph3P(OR)]+ to TPP could be achieved in 78 % (R=Me) and almost quantitative yield for R=Et, respectively, along with alcohol (ROH) formation. The mechanism and striking role of Sc3+ have been rationalized by means of density functional calculations (DFT) (see below).
Methyl triflate (MeOTf) was used as a facile source of Me+ to activate the P=O bond of TPPO, resulting in almost quantitative isolation of the methoxy‐phosphonium triflate, [Ph3P(OMe)](OTf) 1 (Figures S1–S4 in Supporting Information). The weakening of the P=O bond via methylation is evident by the large downfield shift in the 31P NMR signal of 1 vs. TPPO (Δδ=37.9 ppm) (Figure 1 a), which is further supported by FTIR spectroscopy with a bathochromic shift of the ν(PO) stretching vibration mode at 1191 cm−1 for TPPO (Figure S4). Accordingly, the P−O distance obtained from the X‐ray crystal structure analysis of 1 (Figure 1 b inset; Tables S1 and S2) is about 0.08(1) Å longer than that in TPPO.12 The activation of the P=O bond in TPPO via formation of 1 further leads to a drastic decrease of the redox potential as evident from the cyclic voltammograms recorded in acetonitrile solutions with 0.15 m tetrabutylammonium hexafluorophosphate (TBAPF6). A pseudo‐reversible one‐electron‐reduction of TPPO was observed at 2.92 V (vs. Fc/Fc+)10d, 11e, 11f (Figure 1 b, black curve). The methylation of P=O to P−OMe+ causes an anodic shift of 870 mV and irreversible reduction of 1 (Figure 1 b, red curve) which, in contrast, has not been previously achieved with TPPO→LA (LA: B(OAr)3 and AlCl3) adducts, apparently due to weaker association of TPPO and LA.11e, 11f However, the estimated free energy change (ΔG) determined by DFT calculations for the one‐electron‐reduction of [Ph3P(OMe)]+ (Figure S5) is about 63.7 kcal mol−1 preferable in comparison to TPPO, while that of the TPPO→AlCl3 adduct is stabilized by only 9–10 kcal mol−1, as reported recently.11f Additional four different phosphine oxides (R3PO) were selected bearing phenyl and/or alkyl substituents R attached to the P center and using the same methodology of phosphoryl P=O bond via alkylation, corresponding methoxy‐ and/or ethoxy‐phosphonium salts [R3P(OR)](OTf) were synthesized and isolated (see Supporting Information, Figures S6–S17). In all cases and the shifts of their 31P NMR signals and the redox potentials indicate a similar degree of P=O activation as observed for TPPO (Figures S6–S17, Table S3).
Figure 1.
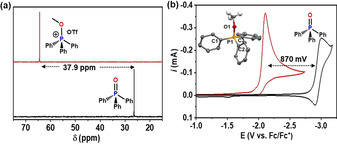
Activation of the P=O bond in TPPO via methylation with MeOTf to form 1. (a) Downfield shift of 37.9 ppm of the 31P NMR signal of TPO (bottom) vs. 1 (top). (b) Anodic shift (870 mv) of the first electron reduction of 1 with respect to TPO (electrochemical condition; 0.15 m TBAPF6 in CH3CN with a 0.1 Vs−1 scan rate, glassy carbon (GC) as working, Pt as a counter, Ag as a pseudo reference electrode). (b, inset) Molecular structure of the cation in 1 determined by a single‐crystal X‐ray diffraction analysis; hydrogen atoms of the phenyl rings are omitted for clarity.
The one‐pot electrochemical WOR was performed in a customized two‐compartment cell equipped with Pt foil as a counter electrode and Ag foil as a working electrode (Pt(+)//(−)Ag, geometric surface area 0.5×0.5 cm) separated by glass frit (G4 porosity) (Figure S18 in Supporting Information) to avoid further oxidation (at the counter electrode) using 1 (0.032 m) at a constant current of 4.5 mA (for 2 h, 32.4 C charge passed) followed by addition of MeOTf (0.048 m) and benzaldehyde (PhCHO, 0.048 m) and subsequent electrolysis of the mixture for additional 40 mins under similar electrochemical condition (entry 1 in Table 1). In the latter case, only a trace amount (<10 %, entry 1 in Table 1) of the desired olefin (styrene, PhCH=CH2) was produced (Figure S19a), presumably due to the moderate TPP regeneration from 1 (about 39 % yields; Figure S19b) before subsequent addition of MeOTf and PhCHO. DFT calculation of the free energy change associated with the electro‐reduction of 1 suggests a plausible pathway (Scheme S1) and predicts that a sequential two step electron uptake by the methoxyphosphonium cation in 1 to the corresponding anion via formation of the neutral mono‐radical is energetically favorable. The later steps consist of two parallel pathways, P‐O and C−O bond dissociation, with comparable free energy changes (ΔG), −50.3 and −45.4 kcal mol−1, respectively (Scheme S1 in Supporting Information). Most likely, the low selectivity towards TPP formation is presumably due to competitive P−O and C−O bond dissociation. Notably, alkene formation could not be achieved under an identical electrochemical condition only with TPPO which further highlights the significance of P=O bond activation via alkylation (Figure S20). To increase the TPP regeneration from 1, Sc(OTf)3 was employed as a redox‐innocent Lewis acid and MeO− acceptor to accelerate the rate‐determining P‐O dissociation step. Sc(OTf)3 has already been used in other LA‐mediated organic transformations13 and for the stabilization of high‐valent oxo‐transition‐metal intermediates.14 In fact, addition of a substoichiometric amount of Sc(OTf)3 to solutions of 1 and subsequent electrolysis under identical conditions (entry 2 in Table 1, in presence of MeOTf and PhCHO) resulted in a drastic increase in styrene formation to 63 % yields (Figures S21 and S22). The stoichiometry of Sc3+ (0.6 molar equivalents) was optimized by means of the highest yield of TPP electro‐regenerated from 1 (Figures S23–S26).
Table 1.
Screening of reaction conditions. (a) One‐pot reaction scheme for electrochemical WOR using [Ph2RP(OR1)]+ (R=Ph, Me, Et and R1=Me, Et) in the presence of substoichiometric amounts of Lewis acid (LA), (b) different [Ph2RP(OR1)]+, (c) alkyl electrophiles RX, (d) carbonyl compounds, and (e) olefins produced by electrochemical WOR.
|
| |||||||
|---|---|---|---|---|---|---|---|
|
Entry[a] |
WE[b] |
LA[c] |
[Ph2RP(OR1)]+[d] |
Alkyl electrophile[e] |
Carbonyl compound[f] |
Olefin |
Yield |
|
1 |
Ag |
– |
[Ph3P(OMe)]+ |
MeOTf |
PhCHO |
PhCH=CH2 |
<10 % |
|
2 |
Agg |
Sc3+ |
[Ph3P(OMe)]+ |
MeOTf |
PhCHO |
PhCH=CH2 |
63 % |
|
3 |
GCh |
Sc3+ |
[Ph3P(OMe)]+ |
MeOTf |
PhCHO |
PhCH=CH2 |
41 % |
|
4 |
Ag |
Yb3+i |
[Ph3P(OMe)]+ |
MeOTf |
PhCHO |
PhCH=CH2 |
33 % |
|
5 |
Ag |
Sc3+ |
[Ph2MeP(OMe)]+ |
MeOTf |
PhCHO |
PhCH=CH2 |
31 % |
|
6 |
Ag |
Sc3+ |
[Ph2EtP(OMe)]+j |
MeOTf |
PhCHO |
PhCH=CH2 |
– |
|
7 |
Ag |
Sc3+ |
[Ph3P(OEt)]+ |
EtOTf |
PhCHO |
PhCH=CHCH3 |
67 % |
|
8 |
Ag |
Sc3+ |
[Ph3P(OMe)]+ |
PhCH2Br |
PhCHO |
PhCH=CHPh |
43 % |
|
9 |
Ag |
Sc3+ |
[Ph3P(OMe)]+ |
MeOTf |
CyCHO |
CyCH=CH2 |
57 % |
|
10 |
Ag |
Sc3+ |
[Ph3P(OMe)]+ |
MeOTf |
FuCHO |
FuCH=CH2 |
46 % |
|
11 |
Ag |
Sc3+ |
[Ph3P(OMe)]+ |
MeOTf |
Ph2CO |
Ph2C=CH2 |
22 % |
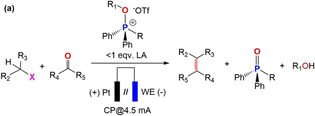
[a] Reaction conditions: all the experiments were conducted under N2 atmosphere (glove box), 0.175 m TBAPF6 as supporting electrolyte, dried CH3CN prior to use as solvent (total volume 3 mL), electrolysis in separated (by glass frit) two electrode cell setup at a constant current (chronopotentiometry; CP at 4.5 mA) using a Pt foil as counter electrode (0.5×0.5 cm working area). [b] working electrode (WE) (0.5×0.5 cm working area). [c] highest yield of TPP and WOR was achieved with 0.02 m (0.6 equiv) LA. [d] electrolysis (2 h) was conducted with 0.032 m [Ph2RP(OR1)]+ in the presence of 0.6 equiv LA and formation of TPP was confirmed analyzing the solution by 31P(1H) NMR. [e] 0.048 m alkyl halide was added to the working compartment after 2 h of electrolysis and stirred for additional 30 minutes to prepare the phosphonium salt in situ. [f] 0.048 m aldehyde and/or ketone added further to the working compartment. [g] Ag foil 0.1 mm thick connected to with copper wire and copper tape (under an identical condition Cu foil, Mg foil and Ni foam did not produce alkene), [h] glassy carbon rod (GC; 6 mm diameter). [i] 0.02 m Yb(OTf)3; other LA (Fe3+, Ni2+, Zn2+ and B(OPh)3) remain ineffective for one‐pot WOR. [j] [R3POMe](OTf) (R=nBu and n‐oct) did not produce any olefin due to poor TPP regeneration (Figures S40 and S41). 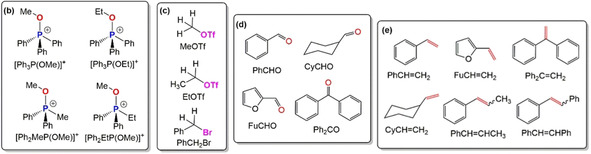
The efficiency of WOR was further tested with four different potential working electrode materials: Cu and Mg foils, Ni foam (NF, 0.5×0.5 cm, three‐dimensional) and glassy carbon rod (GC, 6 mm diameter). Under similar experimental conditions, GC carbon rod (entry 3 in Table 1) delivers a moderate yield (41 %) of styrene (Figure S27 and Figure S28) while Cu, Mg foils and Ni foam remain ineffective in the one‐pot WOR due to ineffective TPP formation and unwanted TPPO regeneration by C‐OPPh3 bond dissociation (Figures S29–S31 and Table S4). The highest efficiency on a Ag electrode surface could be ascribed to its higher conductivity which plays an important role in this electrochemical system.15 Other transition‐metal ions (Fe3+, Ni2+, Yb3+, Zn2+) were also probed as potential LAs for electrochemical WOR. In the presence of Yb3+, a conversion of ca. 33 % styrene was obtained (entry 4 in Table 1, Figures S32 and S33 in Supporting Information). Unfavorably, the desired electrochemical WOR could not occur in the presence of Fe3+, Ni2+, Zn2+ salts most likely due to the lower reduction potentials of Fe3+(E Fe3+/Fe=−0.04 V), Ni2+ (E Ni2+/Ni=−0.26 V) and Zn2+ (E Zn2+/Zn=−0.76 V)16 in comparison with 1 (one‐electron‐reduction at 2.04 V vs. Fc/Fc+) (Figures S34‐S35). Thus, the high reduction potential of Yb3+ (E Yb3+/Yb=−2.37 V) and Sc3+ (E Sc3+/Sc −2.03 V)16 makes them suitable LAs for electrochemical reduction of 1 to TPP and its disposal for a one‐pot WOR. In contrast, the addition of B(OPh)3 as LA does not result in olefin formation due to relatively low TPP regeneration (ca. 20 %) from 1 under the applied experimental conditions (Figure S36).
The displacement of Ph groups in 1 by alkyl substituents has also a significant influence on the WOR efficiency: using [Ph2MeP(OMe)](OTf), MeOTf and PhCHO lowers the yields of styrene to 31 % (entry 5 in Table 1, Figures S37 and S38), while monoethyl substitution on the P atom ([Ph2EtP(OMe)](OTf)) leads merely to unwanted (P)O−C bond scission and formation of Ph2EtP=O (entry 6 in, Table 1, and Figures S39–S41). Notably, due to the higher reduction potentials of fully alkyl‐substituted phosphine oxides (nBu3PO, nOct3PO), electro‐chemical and/or chemical reduction remain unfavorable under the here applied conditions.8b, 11f Although DFT calculations performed with [nBu3P(OMe)]+ revealed that the one‐electron reduction of the phosphonium center is energetically favorable, no minima was obtained in the potential energy surface for the two‐electron‐reduction to nBu3P (Scheme S2). Conversely, the computed ΔG value for the C−O bond dissociation of [nBu3P(OMe)]+ is energetically favorable and as a consequence, nBu3PO and nOct3PO were solely obtained during electroreduction of the respective methoxyphosphonium salts (Scheme S2) and, henceforth, alkyl‐substituted methoxy phosphonium salts are unsuitable for electrochemical WOR.
Starting from TPPO, the electrochemical WOR can also be achieved with other alkoxy‐phosphonium salts, [Ph3P(OR)]+ (R=Et) and using various carbonyl compounds (entry 7–11 in Table 1). Accordingly, electrolysis of solutions containing [Ph3P(OEt)](OTf) (2), EtOTf and PhCHO affords prop‐1‐enyl‐benzene in 67 % yields (PhCH=CHCH3, entry 7 in Table 1, inset c; Figures S42–S44). Very similarly, about 43 % stilbene (entry 8 in Table 1, Figures S45–S47), 57 % vinylcylohexane (CyCH=CH2, entry 9 in Table 1, Figure S48), 46 % 2‐vinalyfuran (FuCH=CH2 entry 10 in Table 1, Figure S49) and 22 % 1,1‐diphenylethene (Ph2C=CH2, entry 11 in Table 1, Figure S50) could be prepared. However, electrochemical WOR does not occur with unactivated TPPO in the presence of Sc3+ although a week interaction between “free” TPPO and Sc3+ is suggested by 31P(1H) NMR and also observed for the one‐electron redox potential of TPPO in the presence of Sc3+ in CV (Figure S51). This control experiment proves that both alkoxy‐phosphonium salt and Sc3+ are the two key components for successful one‐pot electrochemical WOR.
In the earlier reports of electroreduction of TPPO to TPP, the LAs such as B(OAr)3, AlCl3 and Me3SiCl act as sinks to trap O2−, producing the very stable [{(OAr)3B}2O]2− diborate,11e Al2O3,11f and (Me3Si)2O disiloxane,11b respectively. In contrast, the electro‐reductive P−O bond cleavage of 1 and 2 furnishes the respective alcohols CH3OH (from 1 in 68 % yields) and C2H5OH (from 2 in 75 % yields), respectively (see Figures S52–S54). Core level X‐ray photoelectron spectroscopic (XPS) analyses of crude solids isolated from concentrated electrolyzed solutions were performed to determine the valence state of scandium after electrolysis. The binding energies obtained for Sc 3p1/2 (407.8 eV) in these solids were identical to that of fresh Sc(OTf)3 (Figure S55), proving that Sc remains in the same oxidation state (+3) after the reaction.17 Although, TPPO can coordinate Sc3+ and forms a stable isolable complex,18 31P(1H) NMR and ESI‐mass spectra of a mixture of 1 and Sc(OTf)3 indicated the presence of “free” 1 in solution (Figures S56 and S57).
How does the electroreduction of the cation in 1, [1‐OTf]+, occur and what is the role of Sc3+ in this process? We propose a plausible mechanistic pathway based on results of DFT calculations as depicted in Figure 2. To explain the reduction of [1‐OTf]+ in the presence of Sc3+, geometry optimizations were carried out at the B3LYP19a, 19b, 19c, 19d‐D320 level of theory with Stuttgart RSC 1997 valence basis set and effective core potential21 for Sc and 6‐31G(d,p) basis set22 for all other atoms. To account for the solvent effect of acetonitrile, single point calculations of the optimized structures were performed using Polarizable Continuum Model (PCM). The calculations revealed that coordination of Sc3+ with [1‐OTf]+ is energetically disfavored, while ligation with acetonitrile takes place rather easy to form complex A. The latter can release one of the coordinating acetonitrile ligands to form the pentacoordinated Sc complex A’ but the reaction is slightly endergonic by 4.1 kcal mol−1 (Figure 2 a). Notably, the one‐electron‐reduction product of [1‐OTf]+, that is, the corresponding radical [1‐OTf] ., has a highly exergonic coordination affinity towards A’ with 57.9 kcal mol−1, affording the Sc‐arene π‐complex B (Figure 2 b); in fact, related scandium‐arene complexes have previously been reported.23 O‐ and P‐coordination of [1‐OTf]. to Sc could lead to the isomer B’ and B’’, respectively (Figure 2 b), but they are less stable than B. Upon formation of B, the methoxy group on phosphorus can be transferred to the electrophilic CN carbon atom of the ligated acetonitrile via the transition‐state TS(B‐C) to give the methoxy‐acetimidate‐substituted Sc complex C; the latter can undergo a one‐electron‐reduction to form the anionic complex D at ΔG=−159.0 kcal mol−1 (Figure 2 c). D releases a “free” phosphine via TS(D‐E) forming the complex E at ΔG=−170.4 kcal mol−1 in which the methoxy‐acetimidate oxygen atom coordinates to the Sc center. Cleavage of the NC−OMe bond of the methyl‐acetimidate group via TS(E–F) results in formation of the methoxy‐substituted scandium complex F at −187.4 kcal mol−1. Protonation of F can result in formation of methanol (Figure 2 d, as observed experimentally, see also Figure S52) along with regeneration of A.
Figure 2.
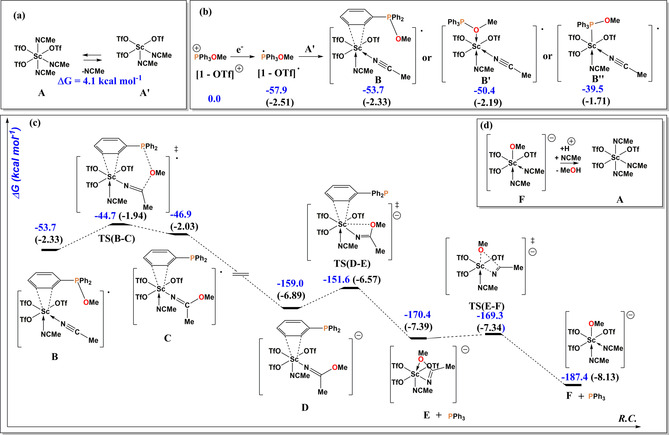
Calculated mechanism of electroreduction of 1 to triphenylphosphine (TPP) mediated by Sc3+. The values in eV are shown in parentheses.
After successful regeneration of TPP from 1 and 2, addition of alkyl electrophiles RX (R=Me, Et, benzyl; X=OTf, Br) resulted in the almost quantitative formation of the corresponding phosphonium salts, [TPP‐R](X), as evidenced by their downfield 31P chemical shifts from δ=−5.6 (TPP) to 21.4 ppm for [TPP‐Me]+ in the 31P(1H) NMR spectrum (Figure S58, see Figures S44 a for [TPP‐Et]+ and S45 for [TPP‐CH2Ph]+). Shano et al. proposed that a Wittig ylide can be formed in situ through one‐electron‐reduction of phosphonium cations.10b, 24 Up to now, no experimental evidence in support of ylide formation was reported. In fact, electroreduction of in situ regenerated methylphosphonium triflate, [TPP‐Me](OTf), from 1 (Figure S58) and/or independently prepared (Figure S59) under similar reaction conditions affords the corresponding Ph3P=CH2 Wittig ylide as proven by its characteristic 31P(1H) NMR spectrum (Figure S60); this confirms that the one‐pot olefination described herein occurs via in situ Wittig ylide formation (Scheme S4). After complete electrochemical WOR with Ph3P=CR’2 (CR’2=CH2, CHMe, CHPh) ca. 87 % TPPO is generated (Figure S61).
Conclusion
In summary, we reported a novel and facile one‐pot electrochemical strategy for Wittig olefination reactions directly recycling TPPO by means of P=O phosphoryl bond activation via alkylation with RX (R=Me, Et; X=OTf) to give [Ph3P(OR)](OTf), which proved to be a suitable pathway for electro‐recycling of TPP with high efficiency (80–98 %) in the presence of Sc(OTf)3 in acetonitrile solution. DFT calculations shed light on the crucial role of Sc3+ and acetonitrile which both act as mediators in the electroreduction of the P−OR bond in [Ph3P(OR)](OTf) (1 and 2) to form TPP. Interestingly, the formation of alcohol as a value‐added side product could be achieved (and explained by the DFT‐proposed mechanism), which distinguishes this approach from previous methods using chemical reductants (e.g., silanes, boranes) and sacrificial electrode material (e.g., Al). Moreover, a separated cell setup with Ag as a working electrode and Pt as a supporting electrode does not show anodic corrosion and/or electrode dissolution, representing a highly sustainable one‐pot approach for WOR. Furthermore, the electroreduction of [Ph3P(OR)]+ to TPP and subsequent olefination reaction via in situ electrochemical Wittig reagent formation could be realized in a one‐pot protocol with extended scope of substrates. The strategy presented herein could pave the way to bulk scale electrochemical WOR synthesis of more functionalized alkenes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funded by the Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2008–390540038—UniSysCat. Authors thank Dr. Xiaohoi Deng for his help in performing some preliminary experiments. Open access funding enabled and organized by Projekt DEAL.
B. Chakraborty, A. Kostenko, P. W. Menezes, M. Driess, Chem. Eur. J. 2020, 26, 11829.
References
- 1.
- 1a. Wittig G., Geissler G., Liebigs Ann. Chem. 1953, 580, 44–57; [Google Scholar]
- 1b. Wittig G., Schöllkopf U., Chem. Ber. 1954, 87, 1318–1330; [Google Scholar]
- 1c. Wittig G., Pommer H., Patent DBP954247, Germany, 1956;
- 1d. Maryanoff B. E., Reitz A. B., Chem. Rev. 1989, 89, 863–927; [Google Scholar]
- 1e. Gilheany D. G., Chem. Rev. 1994, 94, 1339–1374; [DOI] [PubMed] [Google Scholar]
- 1f. Byrne P. A., Gilheany D. G., Chem. Soc. Rev. 2013, 42, 6670–6696; [DOI] [PubMed] [Google Scholar]
- 1g. Hermeling D., Bassler P., Hammes P., Hugo R., Lechtken P., Siegel H., Patent 5527966, Germany, 1996.
- 2.
- 2a. Parker G. L., Smith L. K., Baxendale I. R., Tetrahedron 2016, 72, 1645–1652; [Google Scholar]
- 2b. Pommer H., Angew. Chem. 1960, 72, 811–819; [Google Scholar]
- 2c. Sarnecki W., Pommer H., Patent DE1060386B, Germany, 1957;
- 2d. Eggersdorfer M., Laudert D., Letinois U., McClymont T., Medlock J., Netcher T., Bonrath W., Angew. Chem. Int. Ed. 2012, 51, 12960–12990; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 13134–13165. [Google Scholar]
- 3.
- 3a. Hérault D., Nguyen D. H., Nuel D., Buono G., Chem. Soc. Rev. 2015, 44, 2508–2528; [DOI] [PubMed] [Google Scholar]
- 3b. Longwitz L., Werner T., Pure Appl. Chem. 2019, 91, 95; [Google Scholar]
- 3c. Li Y., Das S., Zhou S., Junge K., Beller M., J. Am. Chem. Soc. 2012, 134, 18325–18329; [DOI] [PubMed] [Google Scholar]
- 3d. Petit C., Favre-Re'guillon A., Albela B., Bonneviot L., Mignani G., Lemaire M., Green Chem. 2010, 12, 326–330; [Google Scholar]
- 3e. Horner L., Balzer W. D., Tetrahedron Lett. 1965, 6, 1157–1162; [Google Scholar]
- 3f. Bauld N. L., Farr F., J. Am. Chem. Soc. 1969, 91, 2788; [Google Scholar]
- 3g. Tang R., Mislow K., J. Am. Chem. Soc. 1969, 91, 5644–5655; [Google Scholar]
- 3h. Naumann K., Zon G., Mislow K., J. Am. Chem. Soc. 1969, 91, 7012–7023. [Google Scholar]
- 4.
- 4a. Batesky D. C., Goldfogel M. J., Weix D. J., J. Org. Chem. 2017, 82, 9931–9936; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Rose J. P., Lalancette R. A., Potenza J. A., Schugar H. J., Acta. Cryst. Sect. B 1980, 36, 2409–2411. [Google Scholar]
- 5.
- 5a. Coyle E. E., Doonan B. J., Holohan A. J., Walsh K. A., Lavigne F., Krenske E. H., O′Brien C. J., Angew. Chem. Int. Ed. 2014, 53, 12907–12911; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13121–13125; [Google Scholar]
- 5b. O′Brien C. J., Tellez J. L., Nixon Z. S., Kang L. J., Carter A. L., Kunkel S. R., Przeworski K. C., Chass G. A., Angew. Chem. Int. Ed. 2009, 48, 6836–6839; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6968–6971; [Google Scholar]
- 5c. Wang L., Sun M., Ding M.-W., Eur. J. Org. Chem. 2017, 2568–2578. [Google Scholar]
- 6. Provis-Evans C. B., Emanuelsson E. A. C., Webster R. L., Adv. Synth. Catal. 2018, 360, 3999–4004. [Google Scholar]
- 7. Imamoto T., Kikuchi S., Miura T., Wada Y., Org. Lett. 2001, 3, 87–90. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Li P., Wischert R., Métivier P., Angew. Chem. Int. Ed. 2017, 56, 15989–15992; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16205–16208; [Google Scholar]
- 8b. Stepen A. J., Bursch M., Grimme S., Stephan D. W., Paradies J., Angew. Chem. Int. Ed. 2018, 57, 15253–15256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15473–15476; [Google Scholar]
- 8c. Petit C., Poli E., Favre-Réguillon A., Khrou L., Denis-Quanquin S., Bonneviot L., Mignani G., Lemaire M., ACS Catal. 2013, 3, 1431–1438; [Google Scholar]
- 8d. Schirmer M. L., Jopp S., Holz J., Spannenberg A., Werner T., Adv. Synth. Catal. 2016, 358, 26–29. [Google Scholar]
- 9.
- 9a. Yanilkin V. V., Gromakov Y. S., Nigmadzyanov F. F., Russ. Chem. Bull. 1996, 45, 1257–1258; [Google Scholar]
- 9b. Smith J. M., Harwood S. J., Baran P. S., Acc. Chem. Res. 2018, 51, 1807–1817; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Yan M., Kawamata Y., Baran P. S., Angew. Chem. Int. Ed. 2018, 57, 4149–4155; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4219–4225. [Google Scholar]
- 10.
- 10a. Saveant J. M., Khac Binh S., Electrochim. Acta 1975, 20, 21–26; [Google Scholar]
- 10b. Saveant J. M., Su Khac B., J. Org. Chem. 1977, 42, 1242–1248; [Google Scholar]
- 10c. Saveant J. M., Binh S. K., J. Electroanal. Chem. 1978, 88, 27–41; [Google Scholar]
- 10d. Santhanam K. S. V., Bard A. J., J. Am. Chem. Soc. 1968, 90, 1118–1122. [Google Scholar]
- 11.
- 11a. Kuroboshi M., Yano T., Kamenoue S., Kawakubo H., Tanaka H., Tetrahedron 2011, 67, 5825–5831; [Google Scholar]
- 11b. Tanaka H., Yano T., Kobayashi K., Kamenoue S., Kuroboshi M., Kawakubo H., Synlett 2011, 2011, 582–584; [Google Scholar]
- 11c. Yano T., Kuroboshi M., Tanaka H., Tetrahedron Lett. 2010, 51, 698–701; [Google Scholar]
- 11d. Kawakubo H., Kuroboshi M., Yano T., Kobayashi K., Kamenoue S., Akagi T., Tanaka H., Synthesis 2011, 2011, 4091–4098; [Google Scholar]
- 11e. Elias J. S., Costentin C., Nocera D. G., J. Am. Chem. Soc. 2018, 140, 13711–13718; [DOI] [PubMed] [Google Scholar]
- 11f. Manabe S., Wong C. M., Sevov C. S., J. Am. Chem. Soc. 2020, 142, 3024–3031. [DOI] [PubMed] [Google Scholar]
- 12. Al-Farhan K. A., J. Crystallogr. Spectrosc. Res. 1992, 22, 687–689. [Google Scholar]
- 13. Kobayashi S., Eur. J. Org. Chem. 1999, 15–27. [Google Scholar]
- 14. Liu Y., Lau T.-C., J. Am. Chem. Soc. 2019, 141, 3755–3766. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Menezes P. W., Indra A., Zaharieva I., Walter C., Loos S., Hoffmann S., Schlögl R., Dau H., Driess M., Energy Environ. Sci. 2019, 12, 988–999; [Google Scholar]
- 15b. Hori Y., in Modern Aspects of Electrochemistry (Eds.: C. G. Vayenas, R. E. White, M. E. Gambo-Aldeco), Springer, New York, 2008; [Google Scholar]
- 15c. Kuhl K. P., Cave E. R., Abram D. N., Jaramilo T. E., Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar]
- 16. Huheey J. E., Keiter E. A., Keiter R. L., Inorganic Chemistry: Principples of Structure and Reactivity, 4th ed., Addison Wesley, 1993. [Google Scholar]
- 17. Biesinger M. C., Lau L. W. M., Gerson A. R., Smart R. S. C., Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar]
- 18. Fawcett J., Platt A. W. G., Russell D. R., Polyhedron 2002, 21, 287–293. [Google Scholar]
- 19.
- 19a. Becke A. D., J. Chem. Phys. 1993, 98, 5684–5652; [Google Scholar]
- 19b. Vosko S. H., Wilk L., Nusair M., Can. J. Phys. 1980, 58, 1200–1211; [Google Scholar]
- 19c. Stephens P. J., Devlin F. J., Chabalowski C. F., Frisch M. J., J. Phys. Chem. 1994, 98, 11623–11927; [Google Scholar]
- 19d. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465; [DOI] [PubMed] [Google Scholar]
- 20b. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 21. Andrae D., Haeussermann U., Dolg M., Stoll H., Preuss H., Theor. Chem. Acc. 1990, 77, 123. [Google Scholar]
- 22. Hariharan P. C., Pople J. A., J. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- 23. Cloke F. G. N., Khan K., Perutzb R. N., J. Chem. Soc. Chem. Commun. 1991, 1372–1373. [Google Scholar]
- 24. Shono T., Mitani M., J. Am. Chem. Soc. 1968, 90, 2728–2729. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary