

Abstract
It is known that co‐administration of CYP3A inducers may decrease the effectiveness of oral contraceptives containing progestins as mono‐preparations or combined with ethinylestradiol. In a randomized clinical drug‐drug interaction study, we investigated the effects of CYP3A induction on the pharmacokinetics of commonly used progestins and ethinylestradiol. Rifampicin was used to induce CYP3A. The progestins chosen as victim drugs were levonorgestrel, norethindrone, desogestrel, and dienogest as mono‐products, and drospirenone combined with ethinylestradiol. Postmenopausal women (n = 12–14 per treatment group) received, in fixed sequence, a single dose of the victim drug plus midazolam without rifampicin, with rifampicin 10 mg/day (weak induction), and with rifampicin 600 mg/day (strong induction). The effects on progestin exposure were compared with the effects on midazolam exposure (as a benchmark). Unbound concentrations were evaluated for drugs binding to sex hormone binding globulin. Weak CYP3A induction, as confirmed by a mean decrease in midazolam exposure by 46%, resulted in minor changes in progestin exposure (mean decreases: 15–37%). Strong CYP3A induction, in contrast, resulted in mean decreases by 57–90% (mean decrease in midazolam exposure: 86%). Namely, the magnitude of the observed induction effects varied from weak to strong. Our data might provide an impetus to revisit the currently applied clinical recommendations for oral contraceptives, especially for levonorgestrel and norethindrone‐containing products, and they might give an indication as to which progestin could be used, if requested, by women taking weak CYP3A inducers—although it is acknowledged that the exact exposure‐response relationship for contraceptive efficacy is currently unclear for most progestins.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ CYP3A inducers may decrease the effectiveness of hormonal contraceptives. However, comparative clinical data on the effects of CYP3A induction on different progestins are absent.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Are all progestins equally vulnerable to CYP3A induction and what is the relationship between changes in midazolam exposure and the exposure of different progestins after CYP3A induction?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ It showed that CYP3A induction affects progestins in varying degrees and provided a ranking of the vulnerability of different progestins to CYP3A induction, where the perpetrator’s effect on midazolam exposure served as a benchmark. Further, it showed that unbound drug concentrations should be considered for progestins binding to sex hormone binding globulin.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The study results allow predicting the inductive effect of new drugs on different progestins based on clinical screening data obtained with midazolam as an index substrate. This might result in fewer and more focused interaction studies with oral contraceptives and might help counseling women taking CYP3A inducers who seek contraception.
Oral contraceptives (OC) (i.e., progestin‐only products or products containing a progestin combined with an estrogen, usually ethinylestradiol (EE)), are widely used and highly effective. Their perfect‐use Pearl indices are below 1; however, typical‐use Pearl indices are higher. 1 Leaving the possibility of incorrect or inconsistent use aside, pharmacokinetic (PK) drug‐drug interactions (DDIs) between the OC and concomitantly used drugs that decrease the systemic exposure of the OC might be a key factor responsible for this discrepancy in failure rates. Of particular concern are interactions mediated by CYP3A enzymes, which are induced by many therapeutic drugs (e.g., rifampicin, phenytoin, carbamazepine, and efavirenz). Thus, women taking such drugs who are seeking contraception need counseling on which contraceptive to choose and their gynecologists need qualified information on the relative interaction potential of different OCs.
For many of the commonly used OCs, it is known from in vitro investigations as well as from clinical studies that they are eliminated by metabolic processes mediated by CYP3A enzymes. However, the quantitative contribution of this elimination pathway, the fraction metabolized, is unknown for most OCs 2 and there are no comparative clinical studies available addressing the question of whether the exposure of different progestins is equally vulnerable to CYP3A induction. In a recent meeting hosted by the US Food and Drug Administration (FDA) it was pointed out that it is challenging to extrapolate the results of a study with one specific OC to another OC due to possible differences in the compounds’ metabolism and transport pathways 3 ; see also Zhang et al. 2 and Ramsden et al. 4 This makes advising women in need of treatment with CYP3A inducers on adequate contraception very difficult. The same applies to the question of which OCs should be tested as DDI victim drugs during the development of a new drug with the potential to induce CYP3A. Prescribing information for OCs do not solve these problems. The FDA guidance on labeling for combined hormonal contraceptives 5 contains a “class labeling” applicable to all OCs. It also includes a comprehensive list of CYP3A inducers with high diverging induction potential (Supplementary Material s, Table S1 ) but the statement “Enzyme inducers (CYP3A4): May decrease the effectiveness” of the OC is applicable to any inducer/OC combination.
Therefore, we considered a head‐to‐head comparison very useful to assess the sensitivity of different progestins toward a graded induction of CYP3A. The focus of our study was on progestins, which are the main driver of the contraceptive efficacy of modern OCs. 6 , 7 Rifampicin (RIF), which is described as an index perpetrator in the FDA DDI guidance, 8 was used at a low dosage and a high dosage to study the impact of weak and of strong CYP3A induction on the exposure of selected progestins and EE. The progestins chosen as DDI victims for this study were levonorgestrel (LNG), norethindrone (NET), desogestrel (DSG), and dienogest (DNG) as mono‐products and drospirenone (DRSP) in a fixed combination with EE. The total global market share of these 5 contraceptive progestins amounted to 81% in 2016, with LNG having the largest share globally, in Europe, and in Latin America (39%, 37%, and 42%, respectively), whereas NET had the largest market share in the United States (36%) (details in Figure S1 ). To calibrate the impact of CYP3A induction on the investigated hormones, a subtherapeutic dose of midazolam (MDZ), a sensitive and selective CYP3A index substrate with a known high fraction metabolized via CYP3A (~90%), 9 , 10 was co‐administered with each dose of hormone. Additional aspects studied were: (i) RIF’s impact on the concentrations of sex hormone binding globulin (SHBG), a main binding protein for several progestins, among them LNG, NET, and DSG’s active metabolite etonogestrel (ENG); and (ii) the association among changes in progestin, EE, and MDZ exposure and changes in the plasma concentrations of 4ß‐hydroxycholesterol (4ß‐HC), an endogenous biomarker of hepatic CYP3A activity. 11 , 12 , 13
METHODS
The protocol for this study (EudraCT 2017‐002792‐26; clinicaltrials.gov NCT03353857) was approved by the relevant independent ethics committee before the start of the study (Ethics Committee of Nordrhein; reference number 101/17‐03 RN). All subjects gave their written informed consent before entry into the study.
Study design and treatments
This was a randomized, open‐label, fixed‐sequence, cross‐over study with five parallel treatment groups conducted in two centers in Germany. It was designed to investigate the impact of two different degrees of CYP3A induction by RIF on the PKs of commonly used progestins and EE. A subtherapeutic dose of MDZ was co‐administered with the hormones as a benchmark for the induction effect achieved.
The study comprised a screening and randomization phase, three treatment periods, and a follow‐up visit for each subject (Figure 1 ). The first treatment period served as a control period. During this period, subjects received a single dose of the randomly assigned test drug (hormone(s) plus MDZ) without prior medication. In periods 2 and 3, administration of the test drug was preceded and followed by once daily administration of RIF 10 or 600 mg, respectively, resulting in two 11‐day induction phases. On the days of test drug administration, RIF was administered 12 hours after the test drug to minimize competitive inhibition of the clearance of the test drug. 14 Daily administration of RIF was continued after test drug administration to maintain the CYP3A activity at the induced level for the duration of the PK sampling period. A fixed sequence of dosing with RIF was chosen because a randomized sequence crossover design would have required long washout periods to ensure the return of enzyme activity to baseline.
Figure 1.
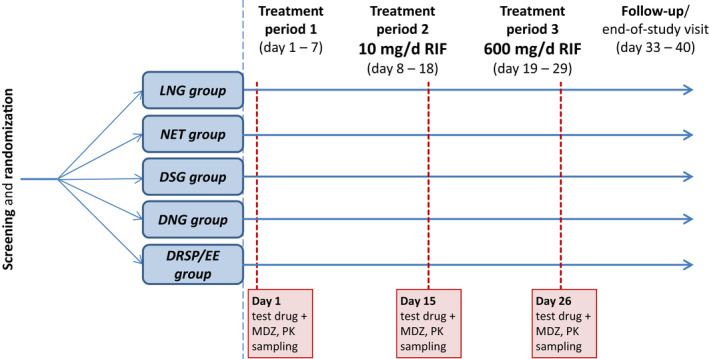
Study design. Treatment period 1 (control period): single administration of the randomly assigned test drug plus MDZ on day 1. Treatment period 2 (weak‐CYP3A‐induction period): administration of the randomly assigned test drug plus MDZ on day 15 was preceded and followed by once daily administration of rifampicin 10 mg for 7 and 4 days, respectively. Treatment period 3 (strong‐CYP3A‐induction period): administration of the randomly assigned test drug plus MDZ on day 26 was preceded and followed by once daily administration rifampicin 600 mg for 7 and 4 days, respectively. On days 15 and 26, rifampicin was administered 12 hours after the contraceptive and MDZ. There was no washout between low‐dose and high‐dose administration of rifampicin. The following test drugs (hormones) were co‐administered with MDZ 1 mg: LNG 0.03 mg; NET 0.35 mg; DSG 0.075 mg; DNG 2 mg; and DRSP 3 mg/EE 0.03 mg. Each treatment group included 12–14 subjects. DNG, dienogest; DRSP, drospirenone; DSG, desogestrel; EE, ethinylestradiol; LNG, levonorgestrel; MDZ, midazolam; NET, norethindrone; PK, pharmacokinetic; RIF, Rifampicin.
The hormones administered as single doses in this study were LNG 0.03 mg (MICROLUT; Jenapharm, Jena, Germany), NET 0.35 mg (NORIDAY; Pfizer, Sandwich, UK), DSG 0.075 mg (CERAZETTE; Merck Sharp & Dohme, Haar, Germany), DNG 2 mg (28 MINI (Visanne); Jenapharm, Jena, Germany), and a fixed combination of DRSP 3 mg and EE 0.03 mg (YASMIN; Jenapharm, Jena, Germany). MDZ 1 mg (the CYP3A probe substrate) was co‐administered with the hormones in the form of an oral solution (MIDAZOLAM‐RATIOPHARM; 2 mg/mL, Ratiopharm, Ulm, Germany). For weak induction of CYP3A in period 2, a RIF‐containing suspension was used (EREMFAT SIRUP; Riemser Pharma, Greifswald, Germany; 10 mg in 0.5 mL). For strong induction of CYP3A in period 3, RIF tablets were used (EREMFAT 600 mg ; Riemser Pharma, Greifswald, Germany). All study medications were taken by mouth. RIF was taken at home, except on PK sampling days. The intake of other drugs or herbal remedies, in particular those potentially inhibiting or inducing CYP3A (e.g., St. John’s wort), and the consumption of food and beverages containing furanocoumarin derivatives (e.g., grapefruits) was not allowed during the study.
Study population
This was a study in healthy postmenopausal women. To be eligible, subjects had to be between 45 and 70 years of age (inclusive), be nonsmokers, and to have a body weight of ≥ 50 kg and a body mass index ≥ 18.5 and ≤ 30 kg/m2. Their postmenopausal state had to be confirmed by medical history (natural menopause ≥ 12 months before first dose of study medication or bilateral ovariectomy ≥ 3 months before first dose) or, for women younger than 60 years by hormone measurements (follicle stimulating hormone > 40 IU/L and estradiol ≤ 20 pg/mL). Women with specific risks or with conditions or habits suspected to have an impact on the aims of the study were excluded from participation.
Assessments and variables
Blood samples (PK samples) for the determination of LNG, NET, ENG, DNG, DRSP, EE, MDZ, and 1′‐OH‐MDZ, the metabolite of MDZ formed by CYP3A, were taken up to 144 hours postdose in period 1 and up to 96 hours in periods 2 and 3, assuming that clearance would be faster with co‐administration of RIF. Plasma samples were taken predose, and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 12, 15, 24, 48, 72, 96, 120, and 144 hours postdose (for MDZ only until 24 hours postdose). Plasma samples to determine RIF trough concentrations and 4ß‐HC concentrations were taken on days 8, 10, 13, 15, 17, 19, and 28 before RIF intake and on day 30. Serum samples to determine SHBG by immunoassay were taken on days 1, 8, 15, 19, and 26.
High‐performance liquid chromatography–tandem mass spectrometry and fluoroimmunometric methods, which were validated according to the relevant European and US guidelines and a pertinent reflection paper of the European Medicines Agency (EMA), 15 , 16 , 17 were used to determine the concentrations of the above‐mentioned analytes in individual plasma samples (detailed description in Methods S1). The fractions unbound of LNG, NET, and ENG in human plasma were determined using ultrafiltration after spiking predose plasma samples with the corresponding 3H‐labeled tracer. Because LNG, NET, and ENG bind to SHBG, which increased during RIF treatment, the unbound concentrations of these progestins were calculated by multiplying the obtained fractions unbound with the corresponding total plasma concentrations.
The following noncompartmental single dose PK parameters were determined for LNG, LNGunbound, NET, NETunbound, ENG, ENGunbound, DNG, DRSP, EE, MDZ, and 1′‐OH MDZ without and with concomitant administration of RIF 10 or 600 mg/day using the software WinNonlin version 5.3 (Certara, Princeton, NJ): area under the concentration‐time curve (AUC) and the maximum concentration (Cmax). Further, the metabolic ratio for MDZ, AUC(0−t last)1−OH−MDZ/AUC(0–t last)MDZ, was determined using the area under the plasma concentration‐time curve from zero to the time of the last quantifiable concentration (AUC(0−t last)).
Statistical analyses
For each cohort and analyte, a parametric mixed‐effects analysis of variance model was fitted to the natural log‐transformed AUC and Cmax values assuming normal distributed data. Analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC). The ratio of geometric least‐squares means for test vs. reference treatment periods was calculated, as well as the associated 90% confidence interval (CI).
The magnitude of the induction effects was classified into the following categories based on the with RIF(low or high dose)/no RIF AUC or Cmax geometric mean ratios of the respective analyte: weak induction (ratio > 0.5 and ≤ 0.8), moderate induction (ratio > 0.2 and ≤ 0.5) and strong induction (ratio ≤ 0.2); cf the FDA and EMA DDI guidelines. 8 , 18 The standard bioequivalence criterion, geometric mean ratio and 90% CI within the acceptance interval of 0.8–1.25, was applied for the classification “no effect.”
Determination of sample size
Based on published data from DDI studies with OCs or MDZ as the victim drug and bioequivalence studies with combined OCs, it was assumed that the within‐subject coefficient of variation for PK parameters is 0.30. Further, it was assumed that the PK parameters are log‐normally distributed. With 12 valid subjects per group, the 90% CI for the mean of a prespecified progestin induction ratio was expected to lie within the ranges (0.87 × µ; 1.14 × µ) with 90% probability. Here, µ denotes the observed mean induction ratio. This sample size was determined by a simulation approach using SAS version 9.4. To adjust for dropouts, it was planned to randomize 14 subjects to each treatment group.
RESULTS
Supplementary information accompanies this paper on the Clinical Pharmacology and Therapeutics website (www.cpt‐journal.com). Furthermore, individual de‐identified PK data from the study will be made publicly available at the Open Systems Pharmacology GitHub repository (https://github.com/Open‐Systems‐Pharmacology).
Study population
In total, 68 healthy postmenopausal women between 51 and 70 years of age were enrolled in the study and received study medication. Sixty‐five of these subjects were included in the PK analysis set (12–14 subjects/group). Three subjects were excluded from PK analysis: one subject because of obvious incompliance (progestin not quantifiable in plasma in period 3), another because of violation of inclusion/exclusion criteria, and a third subject withdrew prematurely. The subject characteristics were well balanced with no notable differences between treatment groups (Table S2 ).
Impact of rifampicin administration on the PKs of midazolam
Co‐administration of RIF (i.e., prior and concomitant intake of RIF; trough concentrations shown in Figure S2 ), was consistently associated with the expected, dose‐dependent decrease in the concentrations of MDZ (and also 1'‐OH‐MDZ) in plasma (Figure 2 a,b; Figure S3 ). In total, co‐administration of RIF 10 mg/day led to a decrease in the exposure (AUC) of MDZ by 46% and coadministration of RIF 600 mg/day to a decrease by 86% compared with MDZ administration alone (Table 1 ; Table S3 ), confirming that RIF 10 mg/day acted as a weak inducer of CYP3A and RIF 600 mg/day as a strong inducer according to the classification provided in the DDI guidelines. 8 , 18 The effect of RIF on MDZ exposure was similar in all treatment groups with the same RIF dose (Figure S3 ; i.e., it was not affected by the co‐administered progestins or EE). The relationship between individual RIF trough concentrations and the corresponding decrease in MDZ exposure is shown in Figure S4 A.
Figure 2.
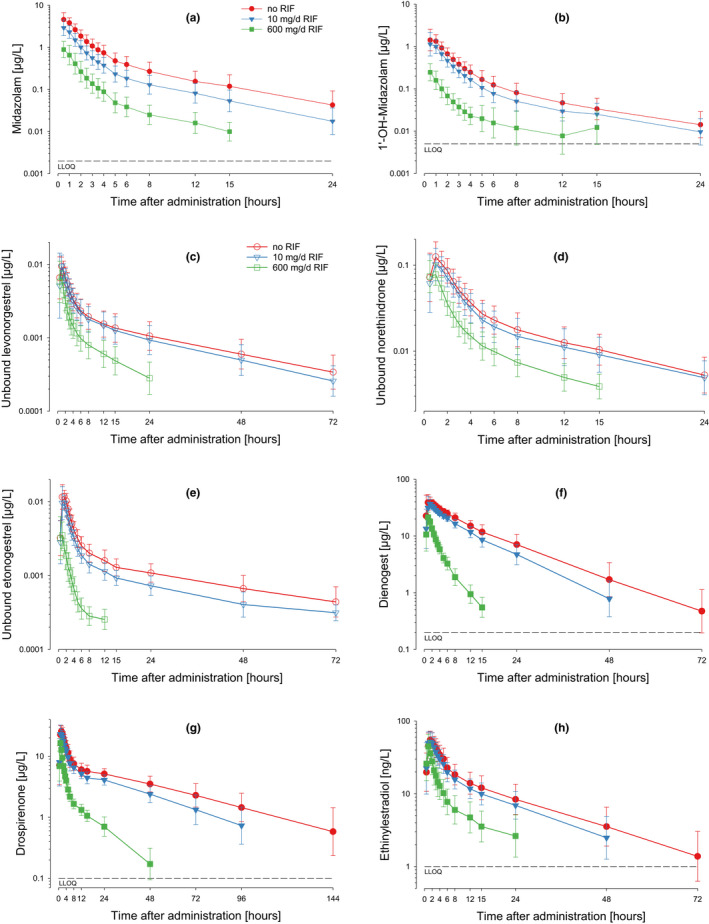
Midazolam, 1′‐OH‐midazolam, progestin, and EE concentrations in plasma after single dose administration of the test drug with and without co‐administration of rifampicin. Geometric means and standard deviations. (a) Midazolam concentration vs. time; (b) 1′‐OH‐midazolam concentration vs. time; (c) unbound levonorgestrel concentration vs. time; (d) unbound norethindrone concentration vs. time; (e) unbound etonogestrel (active metabolite of desogestrel) concentration vs. time; (f) dienogest concentration vs. time; (g) drospirenone concentration vs. time; (h) ethinylestradiol concentration vs. time. LLOQ, lower limit of quantitation; RIF, rifampicin.
Table 1.
Geometric mean AUC ratios (with RIF/no RIF) of different progestins, ethinylestradiol, and midazolam following 10 and 600 mg/day RIF
| Strong‐induction phase (RIF 600 mg/day) | Weak‐induction phase (RIF 10 mg/day) | N | |||
|---|---|---|---|---|---|
| GMRAUC | 90% CI | GMRAUC | 90% CI | ||
| MDZ | 0.137 | (0.124–0.150) | 0.539 | (0.491–0.592) | 65 |
| ENGunbound (DSG) | 0.105 | (0.091–0.120) | 0.627 | (0.547–0.719) | 12 |
| ENG (DSG) | 0.128 | (0.113–0.145) | 0.625 | (0.553–0.708) | 12 |
| DNG | 0.130 | (0.116–0.144) | 0.722 | (0.648–0.804) | 12 |
| DRSP | 0.139 | (0.130–0.149) | 0.701 | (0.657–0.749) | 14 |
| LNGunbound | 0.292 | (0.255–0.335) | 0.853 | (0.744–0.978) | 13 |
| LNG | 0.427 | (0.395–0.460) | 0.832 | (0.771–0.897) | 13 |
| NETunbound | 0.430 | (0.392–0.471) | 0.850 | (0.775–0.932) | 14 |
| NET | 0.539 | (0.489–0.595) | 0.875 | (0.794–0.965) | 14 |
| EE | 0.358 | (0.320–0.400) | 0.817 | (0.730–0.915) | 14 |
Progestins are sorted in ascending order by the GMRAUC (RIF 600 mg per day/no RIF) value.
AUC, area under the concentration‐time curve extrapolated to infinity; CI, confidence interval; DNG, dienogest; DRSP, drospirenone; DSG, desogestrel; EE, ethinylestradiol; ENG, etonogestrel (active metabolite of DSG); GMR, geometric mean ratio (with RIF/no RIF); LNG, levonorgestrel; MDZ, midazolam; NET, norethindrone; RIF, rifampicin.
Impact of rifampicin co‐administration on the PKs of the progestins and ethinylestradiol in comparison to MDZ
Co‐administration of RIF 600 mg/day led to substantial decreases in the exposure of the 5 progestins and EE (Table 1 ; Figure 2 c–h). The largest decreases were observed for ENG, DNG, and DRSP. Smaller decreases were observed for LNG and NET. For LNG, NET, and ENG, the effects of RIF co‐administration were more pronounced when unbound drug concentrations were considered; see also Figure 3 c,d. Compared with co‐administration of RIF 600 mg/day, co‐administration of RIF 10 mg/day led to less pronounced decreases in the exposure of the 5 progestins and EE, but the ranking of the effects was similar.
Figure 3.
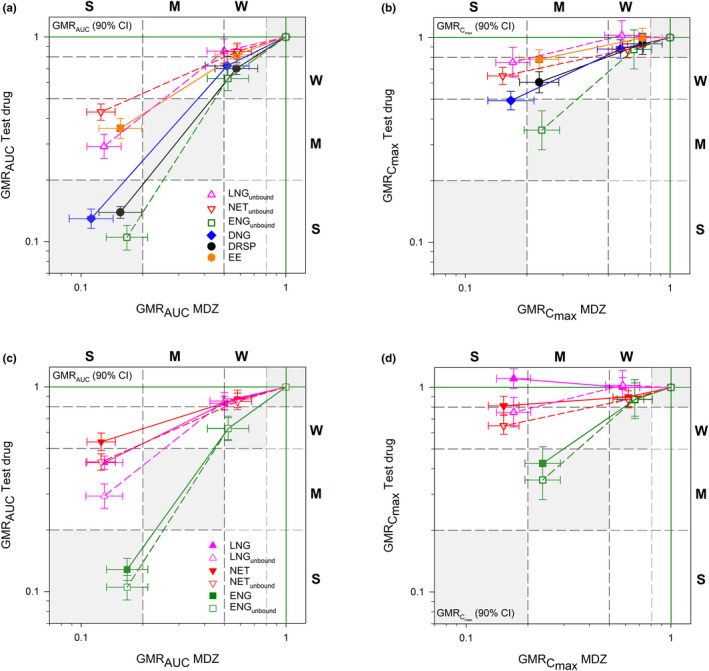
Rifampicin‐dependent induction relationship between midazolam and different progestins and EE. Induction effects on AUC (a), Cmax (b) and unbound vs. total AUC (c) and Cmax (d). The magnitude of the induction effect is classified into three categories: weak induction (W, GMR > 0.5 and ≤ 0.8), moderate induction (M, GMR > 0.2 and ≤ 0.5) and strong induction (S, GMR ≤ 0.2). Gray shaded areas indicate parity of induction effects. AUC, area under the concentration‐time curve extrapolated to infinity; Cmax, observed maximum concentration; CI, confidence interval; DNG, dienogest; DRSP, drospirenone; EE, ethinylestradiol; ENG(unbound), (unbound) etonogestrel (active metabolite of desogestrel); GMR, geometric mean ratio (with RIF/no RIF); LNG(unbound), (unbound) levonorgestrel; MDZ, midazolam; NET(unbound), (unbound) norethindrone.
The geometric mean AUC ratios (and 90% CIs) presented in Table 1 and Figure 3 a show that co‐administration with RIF 10 mg/day had a weak effect on (in descending order) MDZ, ENGunbound, DRSP, and DNG, and almost no effect on EE, LNGunbound, and NETunbound. The effects of RIF 600 mg/day were stronger: there were moderate effects on LNG unbound, EE, and NETunbound observed and strong effects on ENGunbound, DNG, MDZ, and DRSP. Hence, parity between the effect on the progestin and the effect on MDZ was observed only for ENGunbound, DNG, and DRSP (Figure 3 a, gray shades areas). The effect on LNGunbound, EE, and NETunbound, in contrast, was smaller than the effect on MDZ. The effects of RIF co‐administration on the Cmax values of the progestins and EE were weak at most and lower than the effects on Cmax of MDZ (Table S3 ; Figure 3 b).
Impact of rifampicin administration on SHBG concentrations
The mean concentrations of SHBG in serum substantially increased in all treatment groups from day 19 to day 26 by ~ 100%, confirming that RIF at a dosage of 600 mg/day induces SHBG in serum (Figure 4 ). The subjects in the DRSP/EE group showed—probably EE‐related—higher SHBG concentrations than the other subjects, with a first peak still present 8 days after the first dose of EE (+DRSP; i.e., without any concomitant RIF administration), and a steep increase 4 days after the second dose (i.e., at the 10 mg/day RIF dose, which was shown not to induce SHBG).
Figure 4.
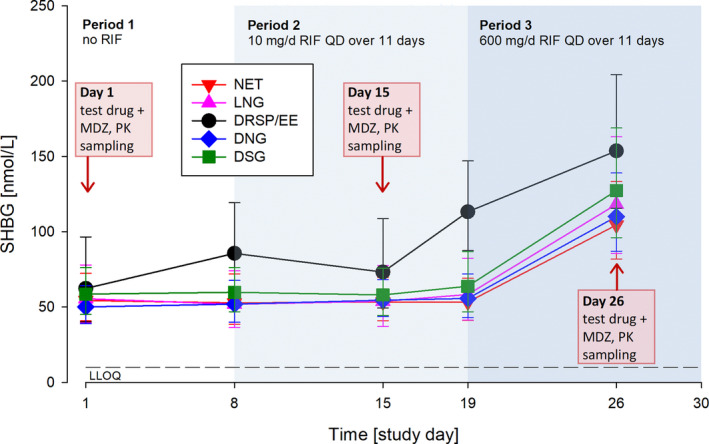
Sex hormone binding globulin concentrations in serum prior to and during repeated administration of rifampicin. Geometric means with geometric SDs; N = 12 to 14 per group. DNG, dienogest; DRSP, drospirenone; DSG, desogestrel; EE, ethinylestradiol; LLOQ, lower level of quantification; LNG, levonorgestrel; MDZ, midazolam; NET, norethindrone; PK, pharmacokinetic; RIF, rifampicin; SHBG, sex hormone binding globulin.
Impact of rifampicin administration on 4ß‐HCs
Across all treatment groups, the mean concentrations of 4β‐HC in plasma substantially increased during administration of 600 mg/day RIF (day 26: increase by 215% on day 8); the 10 mg/day dose, in contrast, had almost no effect (day 15: 14% increase on day 8) (Table S3 ; Figure 5 ). The 4β‐HC concentrations increased continuously during the two 11‐day RIF administrations without reaching steady‐state. Visual inspection of the scatter plot of RIF concentrations vs. changes in 4β‐HC concentrations (4β‐HC ratios (day 19/day 8 and day 30/day 8); Figure S4 B) revealed a correlation between the two parameters and a good differentiation between low‐dose and high‐dose RIF. The 4β‐HC ratios correlated also with the ratios of MDZ (Figure 6 a; Figure S4 C) and the ratios of the progestins and EE (Figure 6 b), showing the same pattern of effects as the CYP3A index substrate MDZ (Figure 3 a).
Figure 5.
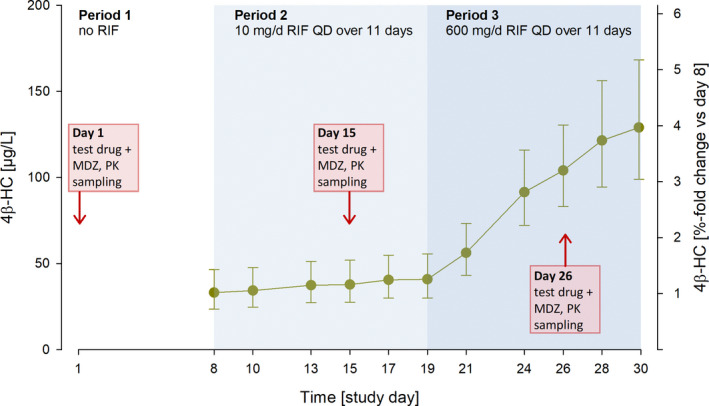
The 4β‐hydroxycholesterol concentrations in plasma before and during repeated oral administration of rifampicin. Geometric means with geometric SDs; N = 65. 4β‐HC, 4β‐hydroxycholesterol, MDZ, midazolam; PK, pharmacokinetic; RIF, rifampicin.
Figure 6.
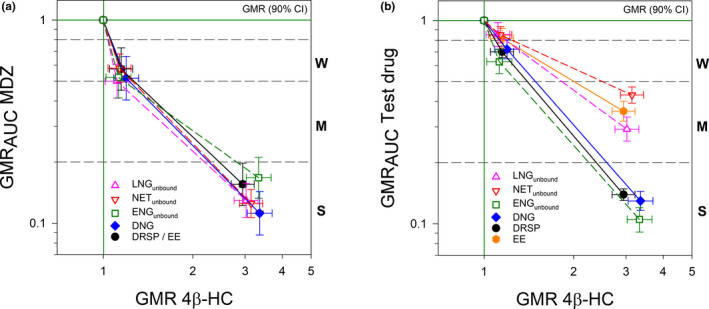
Rifampicin‐dependent induction relationship between 4ß‐hydroxycholesterol and midazolam, different progestins, and ethinylestradiol. The magnitude of the induction effect on the progestins and MDZ is classified into three categories: weak induction (W, > 0.5 and ≤ 0.8), moderate induction (M, > 0.2 and ≤ 0.5) and strong induction (S, ≤0.2). 4ß‐OH, 4ß‐hydroxycholesterol; AUC, area under the concentration‐time curve extrapolated to infinity; CI, confidence interval; DNG, dienogest; DRSP, drospirenone; EE, ethinylestradiol; ENGunbound, unbound etonogestrel (active metabolite of desogestrel); GMR, geometric mean ratio; LNGunbound, unbound levonorgestrel; MDZ, midazolam; NETunbound, unbound norethindrone.
DISCUSSION
This study is the first head‐to‐head comparison of the effects of CYP3A induction on different progestins commonly used in OCs. The results of this study clearly showed that the progestin components of OCs are vulnerable to CYP3A induction to varying degrees and that the results obtained in a study with one OC cannot be directly extrapolated to another OC. The effects of high‐dose RIF administration (i.e., the effects of strong CYP3A induction as judged by the concomitantly gauged changes in MDZ exposure) were substantial for all five progestins studied, but they varied considerably in size. The exposures of DRSP, DNG, and the DSG‐metabolite ENGunbound were decreased by > 80% when the drugs were administered in combination with high‐dose RIF compared with their administration alone. The exposure of NETunbound, in contrast, was reduced by < 60%. The effects of low‐dose RIF co‐administration were weak (ENGunbound, DRSP, and DNG) or virtually absent (LNGunbound and NETunbound). The decrease in the exposure of EE was moderate with co‐administration of high‐dose RIF and negligible with low‐dose RIF. In summary, the effects of strong CYP3A induction seen in our study show a clear ranking (Figure 3 a): On the one hand, there were decreases in drug (or metabolite in case of ENG) exposure by > 80% observed for DNG, DRSP, and ENGunbound and, on the other hand, there were decreases in exposure of between 50% and < 80% observed for LNGunbound and NETunbound and also for EE. The decreases in the exposure of NET and LNG were less pronounced than the simultaneously assessed decreases in the exposure of MDZ (cf Ramsden et al. 4 ), whereas the decreases in DNG, DRSP, and ENG exposure were in the same order of magnitude (the fact that postmenopausal instead of premenopausal women (the target population of OCs) were included in our study does not diminish the validity of our findings. Previous studies have shown that age does not influence the extent of intestinal and hepatic CYP3A induction as determined by the oral and systemic clearance of midazolam 19 ). These differences in the vulnerability of the different progestins tested toward induction of CYP3A metabolizing enzymes by rifampicin suggest that the fractions metabolized via CYP3A are higher for DNG, DRSP, and ENG than for LNG, NET, and EE (Table S4 ).
Progestins commonly used in OCs and EE are known to be extensively metabolized via phase I and II biotransformation pathways (with a very low amount of unchanged excretion indicating that metabolic clearance is the main elimination pathway) whereas active transport plays a minor role. CYP3A is one of the main contributing enzymes for the primary metabolic pathway (Table S4 ). Most progestins and EE are highly permeable compounds. Although some of them are in vitro substrates of certain transporters (e.g., P‐gp), 20 the contribution of active transport to overall elimination is considered insubstantial.
Rifampicin is a ligand of pregnane xenobiotic receptor that is inducing the transcription not only of CYP3A but also of other drug metabolizing enzymes and a few drug transporters. Based on the current knowledge, a major contribution of other than CYP3A elimination pathways and active transport is, however, considered of minor importance for progestins and EE. Additionally, it was recently shown that the clinical effect of rifampicin on other than CYP3A enzymes as well as drug transporters is one category less than CYP3A induction (e.g., strong on CYP3A, moderate on CYP2C9 and P‐gp). 21 Therefore, we assume that the observed clinical effects in this study are mainly due to induction of CYP3A by rifampicin. Furthermore, the effect of rifampicin on progestins and EE may be translatable to other CYP3A inducers acting via the same pregnane xenobiotic receptor‐mediated mechanism taking the potency of the inducer into account. 22
Overall, the effects of RIF co‐administration on progestins observed in our study were in the same range as the effects reported for prior DDI studies with RIF and NET or DNG. 23 , 24 , 25 Comparable studies with LNG, ENG, and DRSP do not seem to be available. The effects of low‐dose and high‐dose RIF on MDZ were also in the same range as previously reported. 11 , 14 , 21 , 26 , 27 , 28 In this context, it might be worth mentioning that most of the published DDI studies with OCs were conducted with LNG or NET, two progestins with a large market share in the United States (Figure S1 ) and—based on our study—the two progestins with the lowest vulnerability toward CYP3A induction. Further, it is worth mentioning that none of the studies published so far provides a comparison between different OCs or a direct comparison with a CYP3A index substrate.
Based on such comparisons, practical guidance can be given as to which OCs should be tested as DDI victims during the development of a new drug. The results of our study suggest that LNG or NET should be used as victim drugs for studying the drug‐OC interaction potential of new drug candidates in a best‐case scenario (i.e., when the focus of the study is on the general suitability of different OC options as concomitant medications). For predicting the drug candidate’s effects on OCs in a worst‐case scenario (i.e., when the focus is on the potential maximum effect on progestin exposure), the results obtained in a DDI study with MDZ can be used. Knowledge of a drug’s effects on both highly vulnerable and less vulnerable OCs will also be helpful for prescribers counseling patients using this drug. However, the comparative data obtained in our study are valuable also from another point of view. As the effects of high‐dose RIF on ENGunbound, DNG, DRSP, and MDZ were very similar, it might be possible to estimate the effect on another CYP3A substrate with similar fm,CYP3A by a DDI induction study using DSG, DNG, or DRSP.
Furthermore, as the exposures of LNG and NET (and EE) were less impacted by CYP3A induction compared with the index substrate midazolam, the currently applied clinical recommendations for OCs might be revisited especially for the combination of LNG or NET containing OCs and weak inducers (e.g., topiramate and rufinamide; Table S1 ). However, we are aware that the detailed knowledge of the exposure‐response relationship required for qualified guidance on what degree of exposure reduction would still be acceptable is currently very limited for most progestins. (A promising project that might help to establish exposure thresholds for the efficacious and safe use of OC has recently been described by Lesko et al. 29 ). Nevertheless, we assume that our data provide an indication as to which OCs could be used, if requested, by a woman taking a weak CYP3A inducer. Furthermore, our data imply that taking a higher dose of progestin would be reasonable in this case, if available (e.g., 150 mg LNG and 0.03 mg EE instead of 100 mg and 0.02 mg EE).
An additional objective of our study was to investigate the impact of RIF administration on the concentrations of SHBG because LNG, ENG, and NET bind with high affinity to SHBG. 30 During treatment with high‐dose RIF (600 mg/day), but not during treatment with low‐dose RIF (10 mg/day), we observed a marked increase in SHBG concentrations in human plasma, as previously reported. 31 , 32 , 33 Accordingly, the decrease in the exposure after co‐administration with RIF calculated as unbound LNG, ENG, and NET was more pronounced than that calculated based on total concentrations (Figure 3 c). These findings indicate that not considering the concentrations of unbound drug in these cases would have underestimated the true pharmacologically relevant effect of CYP3A induction in the progestin exposure. Therefore, concentrations of unbound hormones should always be considered when evaluating the risk of interactions between a progestin that binds to SHBG and a perpetrator expected to increase SHBG concentration in plasma, such as rifampicin, or other strong enzyme inducers, such as phenytoin, phenobarbital, or carbamazepine. 32 , 34 , 35 Furthermore, EE even after a single dose should not be ignored as an SHBG inducer either. The subjects in the DRSP/EE group showed an increase of SHBG concentrations already after a single dose of DRSP/EE and a further increase after the second dose. Similar effects on SHBG have been observed after a single dose of NET/EE by Boyd et al. 36 and in a more manifested manner (e.g., by Fotherby 30 ) after multiple administration of EE.
Further, we studied the association among changes in progestin, EE, and MDZ exposure and changes in the plasma concentrations of 4ß‐HC, an endogenous CYP3A substrate and biomarker of CYP3A activity. The results are promising: 4β‐HC concentration in plasma was substantially increased after 11‐day intake of high‐dose RIF compared to baseline (no RIF) and the changes in 4β‐HC concentrations showed the same pattern of effects as the CYP3A index substrate MDZ, substantiating 4ß‐HC as a robust biomarker reflecting the hepatic component of strong CYP3A induction. 13 Especially in studies were a MDZ microdose is not feasible (e.g., patient studies), it might be possible to use this endogenous marker instead of the exogenous probe MDZ for calibrating, but proper reference ranges still have to be established.
In conclusion, our study showed that CYP3A induction affects progestins in varying degrees and provided a ranking of the vulnerability of the exposure of different progestins to CYP3A induction, where the perpetrator’s effect on MDZ exposure served as a benchmark. The magnitude of the observed induction effects varied from weak to strong. Our data might provide an impetus to revisit the currently applied clinical recommendations for OCs, especially for LNG and NET containing products, and they might give an indication as to which progestin could be used, if requested, by women taking weak CYP3A inducers—although it is acknowledged that the exact exposure‐response relationship for contraceptive efficacy is currently unclear for most progestins.
Funding
The study was sponsored by Bayer AG, Berlin, Germany.
Conflict of Interest
All authors are employees of Bayer AG.
Author Contributions
C.F., I.G., J.H., S.K., A.R., K.R., and H.W. wrote the manuscript. M.B.‐K., C.F., I.G., J.H., S.K., A.R., and H.W. designed the research. M.B.‐K., R.F., B.N., and K.R. performed the research. I.G., S.K., and H.W. analyzed the data.
Supporting information
Acknowledgments
The clinical part of the study was conducted at the research units of the CRS Clinical Research Services group in Mönchengladbach and Mannheim, Germany. B.N. (now at Bayer AG, Wuppertal, Germany) was principal investigator in the study. Bioanalytical measurements (LNG, NET, ENG, DNG, DRSP, EE, RIF, MDZ, and 1′‐OH‐MDZ) were carried out at InVentive Health Clinique (now Syneos Health), Einstein, Québec, Canada. Measurements of SHBG were performed at Syrinx Bioanalytics, Turku, Finland; 4ß‐hydroxycholesterol was determined at Covance Laboratories Ltd, Madison, WI. The f u of LNG, ENG, and NET was determined at Bayer AG, Wuppertal using 3H‐labeled tracer synthesized at Pharmaron, Cardiff, UK. The authors would like to thank the following colleagues from Bayer AG, Berlin, Germany, for their contributions to this manuscript: Antonia Kohnke for preparing the figures, Nina Besche and Jochen Vornbäumen for statistical evaluations, Tatjana Gust and Helen Gutsch for study management, Michaela Bairlein and Thomas Wendl for helpful discussions during study preparation, and Thorsten Poethko for supervising the tracer synthesis. Medical writing assistance for this manuscript was provided by C. Hilka Wauschkuhn, Bonn, Germany, on behalf of Bayer AG, Berlin, Germany.
References
- 1. Trussell, J. Contraceptive failure in the United States. Contraception 83, 397–404 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang, N. et al Role of CYP3A in oral contraceptives clearance. Clin. Transl. Sci. 11, 251–260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akbar, M. et al Public meeting report on "Drug interactions with hormonal contraceptives: public health and drug development implications". J. Clin. Pharmacol. 58, 1655–1665 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramsden, D. et al Perspectives from the Innovation and Quality Consortium Induction Working Group on factors impacting clinical drug‐drug interactions resulting from induction: focus on cytochrome 3A substrates. Drug Metab. Dispos. 47, 1206–1221 (2019). [DOI] [PubMed] [Google Scholar]
- 5. U.S. Department of Health and Human Services , Food and Drug Administration (FDA) , Center for Drug Evaluation and Research (CDER) . Labeling for combined hormonal contraceptives ‐ Guidance for industry. Draft guidance <https://www.fda.gov/media/110050/download> (2017). Accessed December 19, 2019.
- 6. Erkkola, R. & Landgren, B.‐M. Role of progestins in contraception. Acta Obstet. Gynecol. Scand. 84, 207–216 (2005). [DOI] [PubMed] [Google Scholar]
- 7. Reimers, A. , Brodtkorb, E. & Sabers, A. Interactions between hormonal contraception and antiepileptic drugs: clinical and mechanistic considerations. Seizure 28, 66–70 (2015). [DOI] [PubMed] [Google Scholar]
- 8. U.S. Department of Health and Human Services , Food and Drug Administration (FDA) , Center for Drug Evaluation and Research (CDER) . Clinical drug interaction studies ‐ Study design, data analysis, and clinical implications ‐ Guidance for industry <https://www.fda.gov/media/134581/download> (2020). Accessed March 16, 2020.
- 9. Fahmi, O.A. , Maurer, T.S. , Kish, M. , Cardenas, E. , Boldt, S. & Nettleton, D. A combined model for predicting CYP3A4 clinical net drug‐drug interaction based on CYP3A4 inhibition, inactivation, and induction determined in vitro. Drug Metab. Dispos. 36, 1698–1708 (2008). [DOI] [PubMed] [Google Scholar]
- 10. Gertz, M. , Harrison, A. , Houston, J.B. & Galetin, A. Prediction of human intestinal first‐pass metabolism of 25 CYP3A substrates from in vitro clearance and permeability data. Drug Metab. Dispos. 38, 1147–1158 (2010). [DOI] [PubMed] [Google Scholar]
- 11. Bjorkhem‐Bergman, L. et al Comparison of endogenous 4beta‐hydroxycholesterol with midazolam as markers for CYP3A4 induction by rifampicin. Drug Metab. Dispos. 41, 1488–1493 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Penzak, S.R. & Rojas‐Fernandez, C. 4beta‐Hydroxycholesterol as an endogenous biomarker for CYP3A activity: literature review and critical evaluation. J. Clin. Pharmacol. 59, 611–624 (2019). [DOI] [PubMed] [Google Scholar]
- 13. Mao, J. et al Perspective: 4beta‐hydroxycholesterol as an emerging endogenous biomarker of hepatic CYP3A. Drug Metab. Rev. 49, 18–34 (2017). [DOI] [PubMed] [Google Scholar]
- 14. Chattopadhyay, N. et al CYP3A4‐mediated effects of rifampicin on the pharmacokinetics of vilaprisan and its UGT1A1‐mediated effects on bilirubin glucuronidation in humans. Br. J. Clin. Pharmacol. 84, 2857–2866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. European Medicines Agency (EMA) , Committee for Medicinal Products for Human Use (CHMP) . Guideline on bioanalytical method validation <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf> (2011). Accessed December 17, 2019.
- 16. U.S. Department of Health and Human Services , Food and Drug Administration (FDA) , Center for Drug Evaluation and Research (CDER) , Center for Veterinary Medicine (CVM) . Bioanalytical method validation ‐ Guidance for industry <https://www.fda.gov/media/70858/download> (2018). Accessed December 17, 2019.
- 17. European Medicines Agency (EMA) , GCP Inspectors Working Group . Reflection paper for laboratories that perform the analysis or evaluation of clinical trial samples. GCP Inspectors Working Group. EMA/INS/GCP/532137/2010 <https://www.ema.europa.eu/en/documents/regulatory‐procedural‐guideline/reflection‐paper‐laboratories‐perform‐analysis‐evaluation‐clinical‐trial‐samples_en.pdf> (2012). Accessed December 17, 2019.
- 18. European Medicines Agency (EMA) , Committee for Human Medicinal Products (CHMP) . Guideline on the investigation of drug interactions <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf> (2012). Accessed December 17, 2019.
- 19. Gorski, J.C. et al The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin. Pharmacol. Ther. 74, 275–287 (2003). [DOI] [PubMed] [Google Scholar]
- 20. Kim, W.Y. & Benet, L.Z. P‐glycoprotein (P‐gp/MDR1)‐mediated efflux of sex‐steroid hormones and modulation of P‐gp expression in vitro. Pharm. Res. 21, 1284–1293 (2004). [DOI] [PubMed] [Google Scholar]
- 21. Lutz, J.D. et al Cytochrome P450 3A induction predicts P‐glycoprotein induction; Part 1: establishing induction relationships using ascending dose rifampin. Clin. Pharmacol. Ther. 104, 1182–1190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lutz, J.D. et al Cytochrome P450 3A induction predicts P‐glycoprotein induction; part 2: prediction of decreased substrate exposure after rifabutin or carbamazepine. Clin. Pharmacol. Ther. 104, 1191–1198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. LeBel, M. et al Effects of rifabutin and rifampicin on the pharmacokinetics of ethinylestradiol and norethindrone. J. Clin. Pharmacol. 38, 1042–1050 (1998). [DOI] [PubMed] [Google Scholar]
- 24. Barditch‐Crovo, P. et al The effects of rifampin and rifabutin on the pharmacokinetics and pharmacodynamics of a combination oral contraceptive. Clin. Pharmacol. Ther. 65, 428–438 (1999). [DOI] [PubMed] [Google Scholar]
- 25. Blode, H. et al Evaluation of the effects of rifampicin, ketoconazole and erythromycin on the steady‐state pharmacokinetics of the components of a novel oral contraceptive containing estradiol valerate and dienogest in healthy postmenopausal women. Contraception 86, 337–344 (2012). [DOI] [PubMed] [Google Scholar]
- 26. Backman, J.T. , Olkkola, K.T. & Neuvonen, P.J. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin. Pharmacol. Ther. 59, 7–13 (1996). [DOI] [PubMed] [Google Scholar]
- 27. Kharasch, E.D. , Francis, A. , London, A. , Frey, K. , Kim, T. & Blood, J. Sensitivity of intravenous and oral alfentanil and pupillary miosis as minimal and noninvasive probes for hepatic and first‐pass CYP3A induction. Clin. Pharmacol. Ther. 90, 100–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanke, N. et al PBPK Models for CYP3A4 and P‐gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacometrics Syst. Pharmacol. 7, 647–659 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lesko, L.J. et al Establishing a multidisciplinary framework to study drug‐drug interactions of hormonal contraceptives: an invitation to collaborate. CPT Pharmacometrics Syst. Pharmacol. 7, 706–708 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fotherby, K. Interactions of contraceptive steroids with binding proteins and the clinical implications. Ann. NY Acad. Sci. 538, 313–320 (1988). [DOI] [PubMed] [Google Scholar]
- 31. Back, D.J. et al The effect of rifampicin on the pharmacokinetics of ethynylestradiol in women. Contraception 21, 135–143 (1980). [DOI] [PubMed] [Google Scholar]
- 32. Brodie, M.J. , Boobis, A.R. , Gill, M. & Mashiter, K. Does rifampicin increase serum levels of testosterone and oestradiol by inducing sex hormone binding globulin capacity? Br. J. Clin. Pharmacol. 12, 431–433 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lonning, P.E. , Bakke, P. , Thorsen, T. , Olsen, B. & Gulsvik, A. Plasma levels of estradiol, estrone, estrone sulfate and sex hormone binding globulin in patients receiving rifampicin. J. Steroid Biochem. 33, 631–635 (1989). [DOI] [PubMed] [Google Scholar]
- 34. Victor, A. , Lundberg, P.O. & Johansson, E.D. Induction of sex hormone binding globulin by phenytoin. Br. Med. J. 2, 934–935 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Connell, J.M. , Rapeport, W.G. , Beastall, G.H. & Brodie, M.J. Changes in circulating androgens during short term carbamazepine therapy. Br. J. Clin. Pharmacol. 17, 347–351 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boyd, R.A. , Zegarac, E.A. , Eldon, M. & Koup, J.R. Induction of sex hormone binding globulin (SHBG) by a single dose of norethindrone acetate/ethinyl estradiol (NA/EE). Clin. Pharmacol. Ther. 61, 171 (1997). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials