

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder affecting primarily the motor system, but in which extra‐motor manifestations are increasingly recognized. The loss of upper and lower motor neurons in the motor cortex, the brain stem nuclei and the anterior horn of the spinal cord gives rise to progressive muscle weakness and wasting. ALS often has a focal onset but subsequently spreads to different body regions, where failure of respiratory muscles typically limits survival to 2–5 years after disease onset. In up to 50% of cases, there are extra‐motor manifestations such as changes in behaviour, executive dysfunction and language problems. In 10%–15% of patients, these problems are severe enough to meet the clinical criteria of frontotemporal dementia (FTD). In 10% of ALS patients, the family history suggests an autosomal dominant inheritance pattern. The remaining 90% have no affected family members and are classified as sporadic ALS. The causes of ALS appear to be heterogeneous and are only partially understood. To date, more than 20 genes have been associated with ALS. The most common genetic cause is a hexanucleotide repeat expansion in the C9orf72 gene, responsible for 30%–50% of familial ALS and 7% of sporadic ALS. These expansions are also a frequent cause of frontotemporal dementia, emphasizing the molecular overlap between ALS and FTD. To this day there is no cure or effective treatment for ALS and the cornerstone of treatment remains multidisciplinary care, including nutritional and respiratory support and symptom management. In this review, different aspects of ALS are discussed, including epidemiology, aetiology, pathogenesis, clinical features, differential diagnosis, investigations, treatment and future prospects.
Keywords: amyotrophic lateral sclerosis, sporadic and familial ALS, TDP‐43 pathology
Introduction
Amyotrophic lateral sclerosis (ALS) was originally defined as a pure motor neuron disease by Jean‐Martin Charcot in 1869 but is now recognized as a multisystem neurodegenerative disorder, with disease heterogeneity at the clinical, genetic and neuropathological level [1, 2, 3].
The clinical presentation of ALS typically consists of adult onset focal muscle weakness and wasting, which has a tendency to spread with disease progression. The weakness most commonly starts in the limb muscles, more often in distal muscles than in proximal muscles. In about 25%–30% of cases there is a bulbar onset of the disease, presenting with dysarthria, dysphagia, dysphonia, or more rarely with masseter weakness. There is a high degree of variability in the age at onset, the site of onset and the disease progression rate of ALS. The disease is relentlessly progressive in most patients, with a median survival of about 3 years after symptom onset, where death is mostly attributed to respiratory failure. About 50% of patients will suffer from extra‐motor manifestations to some degree in addition to their motor problems. In 10%–15% of cases, an additional diagnosis of frontotemporal dementia (FTD) can be made [4], whilst 35%–40% of patients will have mild behavioural and/or cognitive changes. FTD is characterized by the degeneration of frontal and anterior temporal lobes and presents clinically by behavioural changes, impairment of executive functioning and/or language impairment [5]. ALS and FTD are now considered to be two ends of a spectrum due to the overlap in molecular mechanisms underlying both neurodegenerative disorders [6].
At the genetic level there is considerable disease heterogeneity as well, with more than 20 genes that have been associated with ALS. The five most common genetic causes are hexanucleotide expansions in chromosome 9 open reading frame 72 (C9orf72) and mutations in superoxide dismutase 1 (SOD1), TAR DNA‐binding protein 43 (TARDBP), fused in sarcoma (FUS) and TANK‐binding kinase 1 (TBK1). Together, they explain about 15% of all patients [1, 2, 3].
The most common neuropathological signature of ALS is cytoplasmic aggregation of TDP‐43, a protein encoded by TARDBP, which is found in more than 95% of ALS cases [7]. TDP‐43 inclusions are not unique to patients with mutations in TARDBP, but are also present in patients with C9orf72 expansions or with TBK1 mutations and in patients with sporadic ALS (sALS). TDP‐43 is predominantly localized to the nucleus under basal conditions, but in ALS it mislocalizes to the cytoplasm to form aggregates and become phosphorylated. Other aggregating proteins, such as SOD1 and FUS, are found in patients bearing SOD1 and FUS mutations, respectively. Patients with C9orf72 hexanucleotide repeat expansions have accumulations of dipeptide repeat proteins which are translated from the GGGGCC repeats, although this repeat is located in a non‐coding region of the gene.
The diagnosis of ALS remains a clinical diagnosis and is based on the presence of both upper motor neuron (UMN) and lower motor neuron (LMN) signs, in patients with progressive muscle weakness in whom no alternative explanation can be found. Most clinicians do not rely on the available revised El Escorial criteria [8] or the Awaji algorithm [9], as these criteria lack sensitivity, rather capturing disease progression and only indirectly diagnostic certainty [10]. Moreover, these criteria have been developed for research purposes to select patients for participation in clinical trials. There is a high need for clinical diagnostic criteria of ALS and related subtypes of motor neuron disease, to reduce the diagnostic delay, which is unfortunately still often up to a year after disease onset. Recently, new simplified diagnostic criteria for ALS have been proposed, requiring only combined UMN and LMN dysfunction in one body region, or LMN dysfunction in at least two regions [11]. Whether this will reduce the diagnostic delay requires further study.
The only European Medicines Agency approved drug to treat ALS is riluzole, a glutamate antagonist, which has a small but significant effect on survival in ALS [12]. Despite the ever‐growing knowledge about the causes and disease mechanisms underlying ALS, more than 40 randomized clinical trials have been negative [13]. There are many potential reasons for this lack of success, but treating ALS as one disease regardless of the underlying cause or disease mechanisms involved may be one of them.
Epidemiology
Amyotrophic lateral sclerosis has an estimated incidence of 1.75–3 per 100 000 persons per year and a prevalence of 10–12 per 100 000 in Europe, but significant geographical differences exist [14, 15, 16]. The incidence amounts to 4–8 per 100 000 persons per year in the age group with the highest risk of developing ALS (45–75 years). Mean age at onset of symptoms is variable: 58–63 years for sALS and 40–60 years for familial ALS (fALS) [14]. An estimation of the cumulative lifetime risk for developing ALS is 1:350 in men and 1:400 in women [17, 18]. Men have a higher risk of developing sporadic limb onset ALS compared to women; the global sex ratio is 1.2–1.5 [19].
Aetiology
Similar to other neurodegenerative conditions, ALS is thought to be caused by a combination of genetic factors, environmental factors and aging‐related dysfunction. At the genetic level, more than 20 genes have been linked with the disease to date, and it is anticipated that more genetic factors will be discovered. The genetic architecture of ALS appears complex, where monogenetic mutations with high effect size currently explain about 15% of patients, but where common and rare genetic variants with low and moderate effect size seem to contribute to the risk of developing ALS as well. The overall heritability of ALS is high; in patients with sALS the heritability is estimated to be 30%–60% [18, 20]. The risk of developing ALS doubles in first degree relatives of ALS patients [20].
Autosomal dominant causes of ALS
In 1993, the first ALS‐related gene was discovered: SOD1, responsible for 20% of fALS and 1%–2% of sALS [21]. Mutations in this gene do not cause ALS by loss of SOD1 function but rather by rendering the protein prone to aggregation, which disturbs multiple important cellular functions.
In 2008 and 2009, mutations in TARDBP and FUS, the genes encoding the RNA‐binding proteins TDP‐43 and FUS, were discovered. These mutations are responsible for 3%–5% of fALS and for <1% of sALS [22, 23, 24, 25]. In 2011, C9orf72 was discovered, responsible for 30%–50% of fALS and for 7%–10% of sALS [26, 27]. Patients with hexanucleotide repeat expansions in C9orf72 are more likely to get bulbar onset ALS and to have cognitive and behavioural impairment as well.
Mutations in TBK1 are most probably the fifth most common cause of autosomal dominant ALS, responsible for about 1% of patients [28, 29] but up to 10% of patients with ALS‐FTD [30].
Although most SOD1 mutations have a high penetrance, the other genes mentioned are known to have a reduced penetrance, which complicates genetic counselling. Rarely, patients carry mutations in more than one of these genes, suggesting that ALS can be oligogenic in origin [31].
Using next‐generation sequencing, several rare variants in additional genes have been identified [1, 2, 3]. Whilst mutations in many of these genes are rarely identified as the cause of ALS, they appear to cluster in some emerging disease pathways (Fig. 1).
Figure 1.
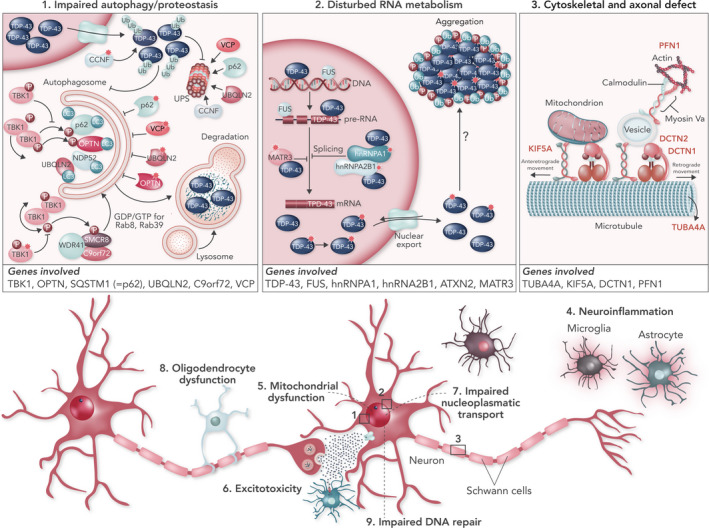
Clustering of ALS genes in pathogenic pathways. (1) Mutations in TBK‐1, OPTN, SQSTM1 (= p62), UBQLN2, C9orf72 and VCP affect the protein degradation pathways and may contribute to TDP‐43 accumulation. (2) Mutations in TARDBP, FUS, MATR3, TIA1, hnRNPA1, hnRNA2B1 and ATXN2 may all affect RNA metabolism. (3) Mutations in TUBA4A, PFN1, KIF5A and DCTN1 alter cytoskeletal dynamics and axonal transport.
Amyotrophic lateral sclerosis risk factors
Only few genetic risk factors for ALS have been identified. An at risk genotype is UNC13A [32], and intermediate repeat expansions in ATXN2 increase the risk of getting ALS [33, 34].
Apart from genetic factors, age and male sex increase the risk for ALS. Several studies have suggested environmental risk factors for ALS, such as smoking, body mass index, physical exercise, occupational and environmental exposures to metals, pesticides, β‐methylamino‐l‐alanine, head injury and viral infections [35, 36, 37]. However, the causal relationship of these factors with ALS remains to be established.
Pathogenesis
The neuropathological signature of ALS is characterized by loss of the neuromuscular connection, axonal retraction and subsequent cell death of UMNs and LMNs, surrounded by astrogliosis and microgliosis, with ubiquitin‐positive inclusions being observed in surviving neurons. TDP‐43 is the main component of these inclusions in more than 95% of ALS patients [7]. TDP‐43 is an RNA‐ and DNA‐binding protein involved in multiple processes such as transcription, splicing, micro RNA maturation, RNA transport and stress granule formation. In line with its nuclear and cytoplasmic functions, TDP‐43 can shuttle between the nucleus and the cytoplasm, but its localization is mainly nuclear. Mislocalization to the cytoplasm, leading to nuclear depletion of TDP‐43 along with cytoplasmic protein aggregation, is a hallmark of ALS [38].
Multiple molecular pathways have been implicated in the pathogenesis of ALS, such as failure of proteostasis, excitotoxicity, neuroinflammation, mitochondrial dysfunction and oxidative stress, oligodendrocyte dysfunction, cytoskeletal disturbances and axonal transport defects, disturbed RNA metabolism, nucleocytoplasmic transport deficits and impaired DNA repair [2, 39]. Interestingly, many of the genes associated with ALS appear to cluster in key pathways: protein quality control and degradation, RNA metabolism, and cytoskeletal and axonal transport (Fig. 1).
Failure of proteostasis
Protein aggregates or, more likely, their oligomeric complex precursors disturb normal protein homeostasis and induce cellular stress. Molecular chaperones can aid in refolding misfolded proteins, but when the cell is overloaded with misfolded proteins they will be targeted for degradation after ubiquitination via the ubiquitin–proteasome system. Alternatively, protein aggregates can also undergo lysosomal degradation by the autophagy pathway after binding to p62 (sequestosome 1).
Multiple ALS‐related genes support an important role for protein aggregation and impaired degradation as key factors in ALS pathogenesis. Indeed, ubiquilin‐2 (UBQLN2) has a role in the delivery of ubiquitinated proteins to the proteasome [40]. Several other mutations are found in genes involved in cargo recognition for the autophagy pathway, as they encode proteins that interact with the ubiquitinated cargo and the phagophore membrane: SQSTM1 (encoding the protein p62, which targets ubiquitinated proteins to the phagophore) [41], optineurin (OPTN, functioning as a receptor for autophagy) [42], TBK1 (activates OPTN by phosphorylation) [29], valosin‐containing protein (VCP) [43] and the C9orf72 protein [44].
Disturbed RNA metabolism
A remarkable number of RNA‐binding proteins are involved in the pathogenesis of ALS. Identification of mutations in the genes of two related RNA‐binding proteins TDP‐43 and FUS has introduced the mechanism of dysregulation of RNA metabolism to ALS [45]. Additional mutations in other RNA‐binding proteins such as angiogenin (ANG), senataxin (STX), matrin‐3 (MATR3), heterogeneous nuclear ribonucleoproteins A1 (hnRNPA1) and A2B1 (hnRNPA2B1), and ataxin‐2 (ATXN2) further support the notion that disrupted RNA metabolism probably plays an important role in ALS [46]. Under normal conditions, these proteins reside predominantly in the nucleus, where they serve important functions in transcription, splicing, non‐coding RNA metabolism and micro RNA biogenesis. Hence, nuclear depletion can be detrimental and induce gross transcriptome abnormalities. Mislocalization to the cytoplasm with aggregation may induce toxicity as well.
Cytoskeletal disturbances and axonal transport defects
Several genetic factors in ALS point toward the importance of cytoskeletal integrity and axonal transport [47]: profilin‐1 (PFN1) and tubulin alpha‐4A (TUBA4A) mutations only rarely cause ALS but were found to destabilize the tubulin network and cause axonal transport deficits. The dynactin complex is an important activator of the dynein motor that stabilizes the binding of cargoes and modulates motor function. Point mutations in the gene encoding the dynactin1 (DCTN1) subunit of the dynactin complex may cause ALS or FTD [48, 49]. Mutations in the C‐terminus of kinesin‐1, encoded by kinesin heavy chain isoform 5A (KIF5A), may impair the anterograde transport of cargoes along the microtubules [50, 51].
Clinical features
Clinical presentation
The hallmark of ALS is progressive muscle weakness, accompanied by muscle atrophy, fasciculations, muscle cramps and slowness of movements with muscle stiffness. The onset of muscle weakness in ALS is usually focal and typically spreads to adjacent body regions. This pattern is compatible with spreading of disease pathology within the motor system, with neuroanatomic propagation within the spinal cord segments and the motor cortex [52].
The disease usually presents with unilateral distal muscle weakness and atrophy in upper or lower limb muscles (spinal ALS, roughly in two‐thirds of patients) or in bulbar muscles (bulbar ALS, in about one‐third of patients). Upper limb onset is most commonly in the dominant hand [53], with thenar muscles being more affected than hypothenar muscles (which is referred to as the split‐hand syndrome) [54], with early involvement of the first interosseous muscle and finger extensors more affected than finger flexors [55]. In the lower limb the anterior tibial muscle is typically affected earlier in the disease course than the gastrocnemius muscle, the hamstrings before the quadriceps muscles [56].
Bulbar onset ALS presents most commonly with dysarthria or dysphagia, less commonly with dysphonia, or reduced mouth closure or chewing problems. Axial muscle weakness with head drop and problems with posture are common in later stages of the disease, but rarely can be the presenting symptom. In about one‐third of patients, there can be bouts of uncontrolled laughing or crying (referred to as a pseudobulbar affect) [57].
In some patients, the muscle weakness is preceded by a period in which fasciculations, muscle cramps or mild weight loss has been noted.
On neurological examination, a combination of signs of UMN and LMN involvement is found in patients with classic ALS. Signs of LMN involvement include muscle weakness, atrophy, fasciculations and reduced muscle tone. Signs of UMN involvement to look for include hyperreflexia (or retained reflexes in atrophic muscles), increased muscle tone (especially in upper limb flexors and lower limb extensors) and slowness of movements (e.g. of tongue movement).
Although the majority of patients can be labelled as having a classic ALS phenotype with spinal or bulbar onset, it is increasingly recognized that ALS is clinically a heterogeneous syndrome with distinct motor and extra‐motor manifestations (Fig. 2). There is considerable heterogeneity within the motor manifestations of the disease itself and the motor manifestations can be accompanied by variable degrees of frontotemporal involvement. This results in different phenotypic presentations of the disease which have different disease trajectories. Although no widely accepted clinical criteria for the different ALS phenotypes exist, there is a growing need for a new classification system using universally accepted terms to account for the disease heterogeneity in ALS [58].
Figure 2.
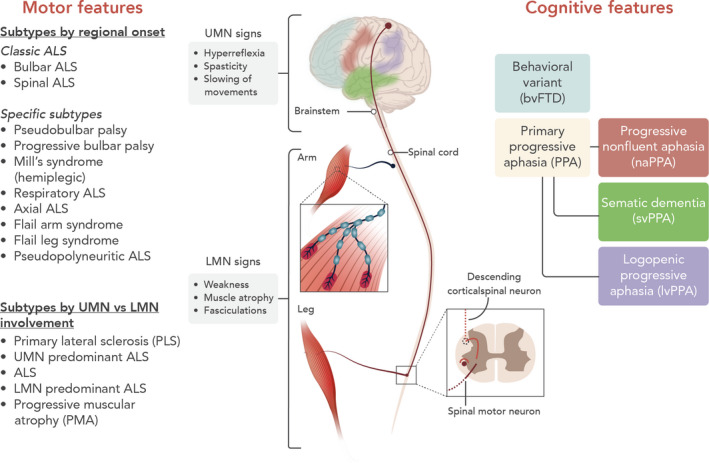
Phenotypic presentations of ALS. Motor features of ALS vary in regional distribution and relative UMN versus LMN involvement. Cognitive and behavioural features are detectable in up to 50% of patients.
Amyotrophic lateral sclerosis phenotypes
Many different motor phenotypes of ALS exist and they are mainly classified based on the relative UMN versus LMN involvement and the regional distribution of involvement (Fig. 2) [3, 58]. It is important to recognize the different motor phenotypes, as life expectancy varies considerably between subtypes of ALS [59]. In addition, variable degrees of cognitive and behavioural impairment can be present.
Subtypes of ALS based on relative UMN versus LMN involvement
In classic ALS, signs of combined UMN and LMN loss are present in one or more body regions and most patients presenting with a motor neuron disease can be labelled as classic ALS.
Primary lateral sclerosis (PLS) is characterized by progressive spasticity and slowing of movements with isolated UMN signs on clinical examination. There should be no muscle atrophy or visible fasciculations, and no signs of denervation on electromyography (EMG) 4 years from symptom onset [60]. Most commonly, the symptoms begin symmetrically in the lower limbs but can begin in the bulbar region as well. PLS represents 3%–5% of all motor neuron diseases. PLS can evolve into ALS, typically within 3–4 years after disease onset. The median survival of PLS patients is more than 20 years. Patients with UMN predominant ALS display some features of LMN involvement but much less pronounced than the UMN features. They have a shorter survival compared to PLS, but a slower disease progression compared to classic ALS.
Lower motor neuron predominant ALS patients have very limited UMN signs and can have different rates of progression. Progressive muscular atrophy is characterized by progressive isolated LMN signs without clinical evidence of UMN dysfunction, although up to 30% of progressive muscular atrophy patients will develop UMN signs during follow‐up.
Subtypes of motor neuron disease based on regional distribution of involvement
Bulbar ALS is a devastating variant of ALS, characterized by a rapid decline and a median survival of 2 years from disease onset. Bulbar UMN dysfunction results in spastic dysarthria, which is characterized by slow, laboured and distorted speech. Bulbar LMN dysfunction is characterized by tongue wasting and fasciculation, accompanied by flaccid dysarthria and dysphagia. Whilst only approximately 30% of patients present with bulbar symptoms, the majority of ALS cases eventually suffer from speech and swallowing difficulties.
Pseudobulbar palsy is characterized by absent facial expressions (expressionless face), spastic dysarthria, and difficulty in chewing, dysphagia and tongue protrusion due to spasticity, but no tongue fasciculation or wasting [61]. As this concerns UMN involvement, the jaw jerk is exaggerated or clonic. This disorder should be differentiated from progressive bulbar palsy, where the LMNs are affected, although there is no consensus on this syndrome in the literature.
Mill’s syndrome (hemiplegic variant) describes a hemiplegic or asymmetrical pattern of involvement. The symptoms are gradually progressive, and the progression is frequently more ascending than descending; the palsy can also involve the facial muscles. Pyramidal signs are usually predominant at the side of hemiplegia.
About 3% of patients present with diaphragm weakness (e.g. dyspnoea at exertion, dyspnoea at rest or orthopnoea) as the initial problem (respiratory ALS). The patients with respiratory onset have a poor prognosis. In axial variant ALS, the disease starts in paravertebral muscles, with stooped posture as a presenting symptom.
Flail arm ALS (brachial amyotrophic diplegia, man‐in‐the‐barrel syndrome or Vulpian–Bernhardt syndrome) is a progressive predominantly LMN pattern of weakness in the upper limbs, a mostly symmetrical pattern of weakness that typically begins in proximal muscles with progression to distal involvement. Bulbar symptoms develop in up to 77%. There is a high male preponderance (male to female ratio 3:1) [62]. Flail leg ALS is a progressive, asymmetrical, predominantly LMN pattern of weakness with distal‐onset weakness and wasting of the lower limbs. There is no significant weakness or wasting in the upper limbs and bulbar region within 12 months after onset, and progression is slightly slower compared to classic ALS. Pseudopolyneuritic ALS is characterized by distal weakness of the lower limbs and absence of Achilles tendon reflex, and should be distinguished from peripheral neuropathy.
Subtypes of ALS based on additional frontotemporal involvement
After Alzheimer’s disease, FTD is the most common cause of dementia in patients <65 years of age. In about 50% of ALS patients, the degenerative process can extend to the frontal and anterior temporal lobes, giving rise to a variable degree of executive dysfunction, language impairments or behavioural changes (Fig. 2). If not specifically sought for, these changes can go unnoticed. The Edinburgh cognitive and behavioural ALS screen is a useful screening assay to identify frontotemporal dysfunction [63]. About 50% of patients will have normal cognition but in about 10%–15% of patients a diagnosis of ALS‐FTD can be made, when the criteria for behavioural variant FTD or criteria for primary progressive aphasia are fulfilled (Table 1). ALS‐behavioural impairment only requires two of six criteria for behavioural variant FTD. ALS without cognitive or behavioural impairment is associated with dysfunction in two non‐executive domains (memory or visuospatial functions), whilst ALS‐cognitive impairment is associated with impairment on two tests for executive function [64].
Table 1.
Criteria for FTD
| Disorder | Variants | Clinical diagnosis | Imaging (18F FDG PET/CT of the brain) |
|---|---|---|---|
| Primary progressive aphasia (PPA) | Non‐fluent agrammatic variant primary progressive aphasia (naPPA) |
At least one:
|
Atrophy of anterior perisylvian atrophy involving inferior, opercular and insular portions of the left frontal lobe |
| Semantic variant of primary progressive aphasia (svPPA) |
|
Atrophy of left anterior temporal atrophy affecting lateral and ventral surfaces as well as the anterior hippocampus and the amygdala | |
| Logopenic variant primary progressive aphasia (lv‐PPA) |
|
Atrophy of left posterior perisylvian or parietal lobe | |
| Behavioural variant frontotemporal dementia (bvFTD) |
At least three:
|
Prefrontal or anterior temporal cortex loss, particularly in the right hemisphere |
FTD, frontotemporal dementia; 18F FDG PET/CT, 18F‐fluorodeoxyglucose positron emission tomography/computed tomography.
Prediction of prognosis
Life expectancy in ALS is extremely variable. Many different clinical features, already present at first disease presentation, are known to be associated with a shorter survival. They include a bulbar onset, a short diagnostic delay, a fast functional decline [e.g. as measured by the revised ALS Functional Rating Scale (ALSFRS‐R) decline], a pronounced loss of weight (or body mass index), the presence of FTD, an older age at the onset of symptoms and a low forced vital capacity. Moreover, genetic factors also influence survival. Some monogenetic causes are associated with a shorter survival (Ala5Val mutation in SOD1, C9orf72 repeat expansion, P525L mutation in FUS), but common and rare variants with effects on survival have been described as well. For example, homozygosity for the C allele of rs12608932 in UNC13a is associated with a shorter survival [65].
The first personalized prediction models have been developed which can estimate the survival outcome in individual patients based on a combination of clinical parameters [66]. Such tools are valuable for patient selection or stratification in clinical trials and may become important for personalized risk estimation and planning of care.
Important differential diagnoses
The diagnosis of ALS in patients with a typical disease presentation is relatively straightforward and is based on the recognition of signs of UMN and LMN degeneration, in the presence of a progressively worsening spread of symptoms or signs within a region or to other regions (Fig. 2). However, in patients with very early disease presentations, with slow disease progression or with concurrent central or peripheral nervous system disorders, the diagnosis can be challenging. The probability of misdiagnosis, the so‐called ‘ALS mimicking syndromes’, is about 7%–8% [67]. The ALS mimicking syndromes should be ruled out as delay in treatment may have an unfavourable effect on outcome.
For patients with predominant UMN or LMN involvement, the differential diagnosis becomes broader. In patients with predominant UMN involvement, a cervical radiculomyelopathy, hereditary spastic paraplegia, adrenomyeloneuropathy and cerebrotendinous xanthomatosis should be considered. In the case of pure LMN features, the diagnosis of plexopathy, peripheral neuropathy (e.g. multifocal motor neuropathy with conduction block, chronic inflammatory demyelinating polyneuropathy, infectious neuropathy) or myopathies (e.g. inclusion body myositis) should be ruled out. Flail arm ALS should to be distinguished from mimics such as spinal muscular atrophy, Kennedy’s disease, multifocal motor neuropathy and monomelic amyotrophy. In the case of focal onset of neck extensor weakness, myasthenia gravis and focal myopathy should be considered. Muscle‐specific tyrosine kinase (MuSK) myasthenia can be accompanied by tongue weakness and atrophy and be mistaken for bulbar ALS [68].
Investigations
The diagnosis of ALS relies on the medical history, physical examination, electrodiagnostic testing (with needle EMG) and neuroimaging. EMG remains a very useful diagnostic tool to confirm LMN involvement in clinically affected and non‐affected muscles (with fibrillation potentials, sharp waves, fasciculation potentials in relaxed muscles and chronic neurogenic changes upon contraction) [8, 69].
Biomarkers can play a crucial role in diagnostic, prognostic or predictive research studies. They could potentially become important for stratification of patients and monitoring treatment effects in clinical trials. Although not yet integrated into standard clinical practice, several biomarkers such as cerebrospinal fluid neurofilament levels (especially phosphorylated neurofilament heavy subunit) are useful in supporting the diagnosis [70, 71, 72], particularly in patients with very recent onset of muscle weakness, without clear signs of UMN involvement, or with concomitant neuropathy/plexopathy/cervical myelopathy.
Brain and spinal cord magnetic resonance imaging are often performed to exclude structural lesions affecting the motor system [73]. Furthermore, 18F‐fluorodeoxyglucose (18F‐FDG) positron emission tomography, if readily available, can reveal a typical pattern of hypometabolism in Rolandic brain regions and frontotemporal involvement [74, 75].
Genetic testing of the five most prevalent genes found to be mutated in ALS is routinely offered to patients with a positive family history (C9orf72, SOD1, TDP‐43, FUS, TBK‐1). Although there is no consensus on genetic testing for patients with sALS, there is a trend to offer it to all patients [76, 77]. However, genetic testing should only be performed if genetic counselling can be provided in the event that a pathogenic gene mutation is identified. Gene panels also including rarer ALS‐related genes are emerging, but the diagnostic yield on top of the five most prevalently mutated genes remains low.
Treatment/management
Over the last decades, more than 40 randomized controlled trials in patients with ALS failed to show a beneficial effect on disease progression or on survival, illustrating the complexity of the disease [13]. In most European countries, riluzole remains the only approved disease‐modifying drug. Riluzole 50 mg twice daily has antiglutamatergic effects and prolongs the mean patient survival by 3–6 months [12, 78, 79]. The most common side effects include nausea, diarrhoea, fatigue, dizziness and liver problems.
More recently, the free radical scavenger edaravone has been studied in ALS. A phase III randomized double‐blind study of intravenous edaravone 60 mg/day for 2 weeks per month in selected ALS patients showed a significantly smaller decline of the scores on the ALSFRS‐R after 6 months of treatment [80]. The study has been criticized because of the small study size, the short study duration, the selection of patients and the lack of data on survival [81]. To date, edaravone has been approved for the treatment of ALS in the USA, Canada, Japan, South Korea and Switzerland, but not in the European Union.
Another therapy under investigation is masitinib, an oral tyrosine kinase inhibitor. A randomized controlled trial using 4.5 mg/kg/day of masitinib as an add‐on therapy to riluzole suggested a positive effect on the decline of ALSFRS‐R, at least in patients with a typical disease progression [82], an effect that will be further explored in a confirmatory study.
The cornerstone of disease management for ALS patients remains multidisciplinary care which has a positive effect on patient satisfaction and outcome [83]. Several discomforting symptoms of ALS can be managed by symptomatic treatment options, including pharmacological and non‐pharmacological interventions [83]. For instance, spasticity can be treated with baclofen, tizanidine, cannabinoids and muscle stretching, and sialorrhea can be treated with anticholinergic medications (amitriptyline, glycopyrronium bromide and oxybutynin) and botulin toxin injections into the salivatory glands. Muscle cramps may respond to magnesium supplements, quinine sulfate, gabapentin or carbamazepine. A selective serotonin reuptake inhibitors, amitriptyline, benzodiazepines and dextromethorphan hydrobromide/quinidine sulfate, can be used in the case of emotional lability. Dietary changes can help to improve nutrition and a gastrostomy tube is an option if the caloric intake is insufficient or when swallowing becomes hazardous. Speech therapy is frequently necessary and assisted communication (customized software) can also be used. Non‐invasive ventilation is the preferred life‐prolonging treatment for respiratory insufficiency. At all disease stages, the patient’s individual wishes should be taken into account and advance care planning should be initiated early.
Future prospects
Over the last years, the first steps in the direction of a precision medicine approach for ALS have been taken. For several genetic subtypes of ALS, therapies that target the upstream genetic cause are being developed. One of these therapeutic approaches uses antisense oligonucleotides (ASOs), which are short single‐stranded nucleotide sequences that bind pre‐mRNA and mRNA to modulate gene expression or to alter splicing. ASOs have been used successfully in several pre‐clinical models of ALS caused by SOD1 mutations and C9orf72 repeat expansions [84, 85]. Clinical studies with intrathecal administration of ASOs targeted against SOD1 and C9orf72 are currently ongoing and the results are anxiously awaited. Stem cell treatments, such as granulocyte‐colony stimulating factor‐induced peripheral blood stem cells, bone marrow mesenchymal stem cells, non‐neural progenitor cells have been proved to be safe and well tolerated; however, the effects on disease progression are not yet known. Several phase II and III clinical trials are ongoing [86, 87, 88]. Overall, there is hope that a better categorization of cases based on pathogenic mechanisms will allow for targeted therapies with beneficial effects in selected ALS subgroups and that ALS will become a treatable condition in the future.
Disclosure of conflicts of interest
The authors declare no financial or other conflicts of interest.
Acknowledgements
The authors are supported by grants from KU Leuven (C1‐C14‐17‐107), Opening the Future Fund (KU Leuven), the Fund for Scientific Research Flanders (FWO‐Flanders), the ALS Liga Belgium, the KU Leuven funds ‘Een Hart voor ALS’, ‘Laeversfonds voor ALS Onderzoek’ and the ‘Valéry Perrier Race against ALS Fund’, the Alzheimer Research Foundation (SAO‐FRA 2017/023), the Flemish Government initiated Flanders Impulse Program on Networks for Dementia Research (VIND 135043), Flanders Innovation and Enterpreneurship (IWT grants Project MinE and iPSCAF), the Belgian National Lottery, the Latran Foundation, the European Union’s Horizon 2020 research and innovation programme (755094) and the European Union’s ERA‐Net for Research Programmes on Rare Diseases (INTEGRALS). PVD holds a senior clinical investigatorship of FWO‐Vlaanderen and is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders. Dr V. Bercier is thanked for carefully reading the manuscript and for giving constructive comments which substantially helped to improve the quality of this review.
References
- 1. Hardiman O, Al‐Chalabi A, Chio A, et al Amyotrophic lateral sclerosis. Nat Rev Dis Primers 2017; 3: 17085. [DOI] [PubMed] [Google Scholar]
- 2. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med 2017; 377: 162–172. [DOI] [PubMed] [Google Scholar]
- 3. van Es MA, Hardiman O, Chio A, et al Amyotrophic lateral sclerosis. Lancet 2017; 390: 2084–2098. [DOI] [PubMed] [Google Scholar]
- 4. Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol 2007; 6: 994–1003. [DOI] [PubMed] [Google Scholar]
- 5. Neary D, Snowden JS, Gustafson L, et al Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546–1554. [DOI] [PubMed] [Google Scholar]
- 6. Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain 2011; 134: 2582–2594. [DOI] [PubMed] [Google Scholar]
- 7. Neumann M, Sampathu DM, Kwong LK, et al Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314: 130–133. [DOI] [PubMed] [Google Scholar]
- 8. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1: 293–299. [DOI] [PubMed] [Google Scholar]
- 9. de Carvalho M, Dengler R, Eisen A, et al Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol 2008; 119: 497–503. [DOI] [PubMed] [Google Scholar]
- 10. Schrooten M, Smetcoren C, Robberecht W, Van Damme P. Benefit of the Awaji diagnostic algorithm for amyotrophic lateral sclerosis: a prospective study. Ann Neurol 2011; 70: 79–83. [DOI] [PubMed] [Google Scholar]
- 11. Shefner JM, Al‐Chalabi A, Baker MR, et al A proposal for new diagnostic criteria for ALS. Clin Neurophysiol 2020; 131: 1975–1978. [DOI] [PubMed] [Google Scholar]
- 12. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994; 330: 585–591. [DOI] [PubMed] [Google Scholar]
- 13. Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 2014; 13: 1127–1138. [DOI] [PubMed] [Google Scholar]
- 14. Logroscino G, Traynor BJ, Hardiman O, et al Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatry 2010; 81: 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collaborators GBDMND . Global, regional, and national burden of motor neuron diseases 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2018; 17: 1083–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marin B, Boumediene F, Logroscino G, et al Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta‐analysis. Int J Epidemiol 2017; 46: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnston CA, Stanton BR, Turner MR, et al Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol 2006; 253: 1642–1643. [DOI] [PubMed] [Google Scholar]
- 18. Ryan M, Heverin M, McLaughlin RL, Hardiman O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol 2019; 76: 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manjaly ZR, Scott KM, Abhinav K, et al The sex ratio in amyotrophic lateral sclerosis: a population based study. Amyotroph Lateral Scler 2010; 11: 439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Al‐Chalabi A, Fang F, Hanby MF, et al An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry 2010; 81: 1324–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosen DR, Siddique T, Patterson D, et al Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362: 59–62. [DOI] [PubMed] [Google Scholar]
- 22. Sreedharan J, Blair IP, Tripathi VB, et al TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008; 319: 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabashi E, Valdmanis PN, Dion P, et al TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 2008; 40: 572–574. [DOI] [PubMed] [Google Scholar]
- 24. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009; 323: 1205–1208. [DOI] [PubMed] [Google Scholar]
- 25. Vance C, Rogelj B, Hortobagyi T, et al Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009; 323: 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 2011; 72: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Renton AE, Majounie E, Waite A, et al A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 2011; 72: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cirulli ET, Lasseigne BN, Petrovski S, et al Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015; 347: 1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Freischmidt A, Wieland T, Richter B, et al Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nat Neurosci 2015; 18: 631–636. [DOI] [PubMed] [Google Scholar]
- 30. Le Ber I, De Septenville A, Millecamps S, et al TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging 2015; 36: 3116.e5–3116.e8. [DOI] [PubMed] [Google Scholar]
- 31. van Blitterswijk M, van Es MA, Hennekam EA, et al Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012; 21: 3776–3784. [DOI] [PubMed] [Google Scholar]
- 32. van Es MA, Veldink JH, Saris CG, et al Genome‐wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 2009; 41: 1083–1087. [DOI] [PubMed] [Google Scholar]
- 33. Elden AC, Kim HJ, Hart MP, et al Ataxin‐2 intermediate‐length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van Damme P, Veldink JH, van Blitterswijk M, et al Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology 2011; 76: 2066–2072. [DOI] [PubMed] [Google Scholar]
- 35. Pupillo E, Poloni M, Bianchi E, et al Trauma and amyotrophic lateral sclerosis: a European population‐based case–control study from the EURALS consortium. Amyotroph Lateral Scler Frontotemporal Degener 2018; 19: 118–125. [DOI] [PubMed] [Google Scholar]
- 36. Al‐Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 2013; 9: 617–628. [DOI] [PubMed] [Google Scholar]
- 37. Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol 2015; 7: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mackenzie IR, Rademakers R, Neumann M. TDP‐43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010; 9: 995–1007. [DOI] [PubMed] [Google Scholar]
- 39. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature 2016; 539: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deng HX, Chen W, Hong ST, et al Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 2011; 477: 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fecto F, Yan J, Vemula SP, et al SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011; 68: 1440–1446. [DOI] [PubMed] [Google Scholar]
- 42. Maruyama H, Morino H, Ito H, et al Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010; 465: 223–226. [DOI] [PubMed] [Google Scholar]
- 43. Johnson JO, Mandrioli J, Benatar M, et al Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010; 68: 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Webster CP, Smith EF, Bauer CS, et al The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 2016; 35: 1656–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F. Nuclear factor TDP‐43 can affect selected microRNA levels. FEBS J 2010; 277: 2268–2281. [DOI] [PubMed] [Google Scholar]
- 46. Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol 2016; 132: 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De Vos KJ, Hafezparast M. Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research? Neurobiol Dis 2017; 105: 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bercier V, Hubbard JM, Fidelin K, et al Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol Neurodegener 2019; 14: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Munch C, Sedlmeier R, Meyer T, et al Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004; 63: 724–726. [DOI] [PubMed] [Google Scholar]
- 50. Brenner D, Yilmaz R, Muller K, et al Hot‐spot KIF5A mutations cause familial ALS. Brain 2018; 141: 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nicolas A, Kenna KP, Renton AE, et al Genome‐wide analyses identify KIF5A as a novel ALS gene. Neuron 2018; 97: 1268–1283 e1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 2009; 73: 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Turner MR, Wicks P, Brownstein CA, et al Concordance between site of onset and limb dominance in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2011; 82: 853–854. [DOI] [PubMed] [Google Scholar]
- 54. Simon NG, Lomen‐Hoerth C, Kiernan MC. Patterns of clinical and electrodiagnostic abnormalities in early amyotrophic lateral sclerosis. Muscle Nerve 2014; 50: 894–899. [DOI] [PubMed] [Google Scholar]
- 55. Motor Neurone Disease: Assessment and Management. London, UK: National Clinical Guideline Centre, 2016. https://www.ncbi.nlm.nih.gov/books/NBK349620/ [Google Scholar]
- 56. Jenkins TM, Alix JJP, Fingret J, et al Correction to: Longitudinal multi‐modal muscle‐based biomarker assessment in motor neuron disease. J Neurol 2020; 267: 257–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parvizi J, Anderson SW, Martin CO, Damasio H, Damasio AR. Pathological laughter and crying: a link to the cerebellum. Brain 2001; 124: 1708–1719. [DOI] [PubMed] [Google Scholar]
- 58. Al‐Chalabi A, Hardiman O, Kiernan MC, Chio A, Rix‐Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol 2016; 15: 1182–1194. [DOI] [PubMed] [Google Scholar]
- 59. Chio A, Calvo A, Moglia C, Mazzini L, Mora G. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry 2011; 82: 740–746. [DOI] [PubMed] [Google Scholar]
- 60. Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain 1992; 115: 495–520. [DOI] [PubMed] [Google Scholar]
- 61. Finegan E, Chipika RH, Li Hi Shing S, Hardiman O, Bede P. Pathological crying and laughing in motor neuron disease: pathobiology, screening, intervention. Front Neurol 2019; 10: 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wijesekera LC, Mathers S, Talman P, et al Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology 2009; 72: 1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Niven E, Newton J, Foley J, et al Validation of the Edinburgh cognitive and behavioural amyotrophic lateral sclerosis screen (ECAS): a cognitive tool for motor disorders. Amyotroph Lateral Scler Frontotemporal Degener 2015; 16: 172–179. [DOI] [PubMed] [Google Scholar]
- 64. Strong MJ, Abrahams S, Goldstein LH, et al Amyotrophic lateral sclerosis – frontotemporal spectrum disorder (ALS‐FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017; 18: 153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Diekstra FP, van Vught PW, van Rheenen W, et al UNC13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging 2012; 33: 630.e3–630.e8. [DOI] [PubMed] [Google Scholar]
- 66. Westeneng HJ, Debray TPA, Visser AE, et al Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol 2018; 17: 423–433. [DOI] [PubMed] [Google Scholar]
- 67. Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman O. Amyotrophic lateral sclerosis mimic syndromes: a population‐based study. Arch Neurol 2000; 57: 109–113. [DOI] [PubMed] [Google Scholar]
- 68. Huijbers MG, Niks EH, Klooster R, et al Myasthenia gravis with muscle specific kinase antibodies mimicking amyotrophic lateral sclerosis. Neuromuscul Disord 2016; 26: 350–353. [DOI] [PubMed] [Google Scholar]
- 69. Mills KR. Detecting fasciculations in amyotrophic lateral sclerosis: duration of observation required. J Neurol Neurosurg Psychiatry 2011; 82: 549–551. [DOI] [PubMed] [Google Scholar]
- 70. De Schaepdryver M, Jeromin A, Gille B, et al Comparison of elevated phosphorylated neurofilament heavy chains in serum and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2018; 89: 367–373. [DOI] [PubMed] [Google Scholar]
- 71. Steinacker P, Feneberg E, Weishaupt J, et al Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry 2016; 87: 12–20. [DOI] [PubMed] [Google Scholar]
- 72. Poesen K, De Schaepdryver M, Stubendorff B, et al Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology 2017; 88: 2302–2309. [DOI] [PubMed] [Google Scholar]
- 73. Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol 2011; 7: 639–649. [DOI] [PubMed] [Google Scholar]
- 74. Pagani M, Chio A, Valentini MC, et al Functional pattern of brain FDG‐PET in amyotrophic lateral sclerosis. Neurology 2014; 83: 1067–1074. [DOI] [PubMed] [Google Scholar]
- 75. Van Laere K, Vanhee A, Verschueren J, et al Value of 18fluorodeoxyglucose‐positron‐emission tomography in amyotrophic lateral sclerosis: a prospective study. JAMA Neurol 2014; 71: 553–561. [DOI] [PubMed] [Google Scholar]
- 76. Roggenbuck J, Quick A, Kolb SJ. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians. Genet Med 2017; 19: 267–274. [DOI] [PubMed] [Google Scholar]
- 77. Vajda A, McLaughlin RL, Heverin M, et al Genetic testing in ALS: a survey of current practices. Neurology 2017; 88: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose‐ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996; 347: 1425–1431. [DOI] [PubMed] [Google Scholar]
- 79. Hinchcliffe M, Smith A. Riluzole: real‐world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener Neurol Neuromuscul Dis 2017; 7: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2017; 16: 505–512. [DOI] [PubMed] [Google Scholar]
- 81. Al‐Chalabi A, Andersen PM, Chandran S, et al July 2017 ENCALS statement on edaravone. Amyotroph Lateral Scler Frontotemporal Degener 2017; 18: 471–474. [DOI] [PubMed] [Google Scholar]
- 82. Mora JS, Genge A, Chio A, et al Masitinib as an add‐on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Frontotemporal Degener 2020; 21: 5–14. [DOI] [PubMed] [Google Scholar]
- 83. Andersen PM, Abrahams S, Borasio GD, et al EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS) – revised report of an EFNS task force. Eur J Neurol 2012; 19: 360–375. [DOI] [PubMed] [Google Scholar]
- 84. Smith RA, Miller TM, Yamanaka K, et al Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest 2006; 116: 2290–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jiang J, Zhu Q, Gendron TF, et al Gain of toxicity from ALS/FTD‐linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC‐containing RNAs. Neuron 2016; 90: 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mazzini L, Mareschi K, Ferrero I, et al Stem cell treatment in amyotrophic lateral sclerosis. J Neurol Sci 2008; 265: 78–83. [DOI] [PubMed] [Google Scholar]
- 87. Deda H, Inci MC, Kurekci AE, et al Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow‐derived hematopoietic stem cell transplantation: a 1‐year follow‐up. Cytotherapy 2009; 11: 18–25. [DOI] [PubMed] [Google Scholar]
- 88. Moviglia GA, Moviglia‐Brandolino MT, Varela GS, et al Feasibility, safety, and preliminary proof of principles of autologous neural stem cell treatment combined with T‐cell vaccination for ALS patients. Cell Transplant 2012; 21: S57–S63. [DOI] [PubMed] [Google Scholar]