

Abstract
Objective
Conventional genetic tests (quantitative fluorescent‐PCR [QF‐PCR] and single nucleotide polymorphism‐array) only diagnose ~40% of fetuses showing ultrasound abnormalities. Rapid exome sequencing (rES) may improve this diagnostic yield, but includes challenges such as uncertainties in fetal phenotyping, variant interpretation, incidental unsolicited findings, and rapid turnaround times. In this study, we implemented rES in prenatal care to increase diagnostic yield.
Methods
We prospectively studied 55 fetuses. Inclusion criteria were: (a) two or more independent major fetal anomalies, (b) hydrops fetalis or bilateral renal cysts alone, or (c) one major fetal anomaly and a first‐degree relative with the same anomaly. In addition to conventional genetic tests, we performed trio rES analysis using a custom virtual gene panel of ~3850 Online Mendelian Inheritance in Man (OMIM) genes.
Results
We established a genetic rES‐based diagnosis in 8 out of 23 fetuses (35%) without QF‐PCR or array abnormalities. Diagnoses included MIRAGE (SAMD9), Zellweger (PEX1), Walker‐Warburg (POMGNT1), Noonan (PTNP11), Kabuki (KMT2D), and CHARGE (CHD7) syndrome and two cases of Osteogenesis Imperfecta type 2 (COL1A1). In six cases, rES diagnosis aided perinatal management. The median turnaround time was 14 (range 8‐20) days.
Conclusion
Implementing rES as a routine test in the prenatal setting is challenging but technically feasible, with a promising diagnostic yield and significant clinical relevance.
What's already known about this topic?
Conventional genetic tests (quantitative fluorescent‐PCR and single nucleotide polymorphism‐array) in fetuses with ultrasound anomalies only yield a diagnosis in ~40% of cases.
Exome sequencing is a promising tool to improve this diagnostic yield.
Exome sequencing includes challenges for implementation such as uncertainties in fetal phenotyping, variant interpretation, incidental unsolicited findings, and rapid turnaround times.
What does this study add?
This prospective study confirms that it is feasible to implement rapid exome sequencing during pregnancy for unselected multiple fetal anomalies with a turnaround time of 2 weeks .
Exome sequencing adds 18% to the diagnostic yield.
1. INTRODUCTION
Congenital anomalies occur in 2% to 3% of children in the Northern Netherlands. 1 Second trimester ultrasound can detect congenital anomalies and reveals structural anomalies in ~1% of fetuses. 2 Detecting the underlying cause of ultrasound anomalies is important for a number of reasons. In the short‐term, it may provide a prognosis that allows parents to make better informed choices about continuing or terminating the pregnancy (termination is legally possible in the Netherlands until 24 weeks of gestation and is possible after this time under exceptional circumstances). In the medium‐term, a prognosis can help the obstetrician‐gynecologist determine the best obstetric management (eg, delivery mode) and assist neonatologists in optimizing neonatal care. In the long‐term, identifying the underlying genetic cause for an ultrasound anomaly is crucial for determining recurrence risk.
Current genetic diagnostic tests only detect numerical chromosomal anomalies, copy number variations, and uniparental disomies. These tests reveal the underlying causes in ~40% of ultrasound anomaly cases. 3 However, this means that ~60% of cases remain undiagnosed during the pregnancy, and a proportion of these may be monogenic diseases caused by single nucleotide variants or small indels. Prenatally, exome sequencing (ES) is a promising tool for detecting monogenic causes. Adding rapid ES (rES) to the standard diagnostic procedures can reveal an additional 6% to 80% of causes, with a higher yield in fetuses with multiple congenital anomalies or clinical suspicion of a syndrome. 3 , 4
However, although ES may improve diagnostic yield, implementing prenatal ES is challenging due to uncertainties about the fetal phenotype, ethical counseling issues, the difficulty of variant interpretation, the need to make choices about reporting variants of unknown clinical significance, and the need for short turnaround times. 3 , 5 , 6 In this prospective study, we offered rES as a diagnostic test in standard prenatal care in addition to quantitative fluorescent‐polymerase chain reaction (QF‐PCR) and single nucleotide polymorphism‐array (SNP‐array) during pregnancy. We present the diagnostic yield, pregnancy outcomes, and clinical experiences to demonstrate the impact of implementing this test in routine care.
2. METHODS
2.1. Study design and inclusion of patients
This prospective cohort study was performed between March 2018 and January 2019 in the Departments of Genetics and Prenatal Diagnostics of the UMCG, Groningen, the Netherlands, and the Department of Obstetrics of the Isala Hospital, Zwolle, the Netherlands. We initially included only one case per week to limit pressure on the laboratory. After inclusion of the first four cases, we optimized procedures during a four‐week break in study inclusion, after which all eligible cases were included.
All pregnant women who opted for invasive prenatal diagnostics due to suspected fetal anomalies were counseled by a clinical geneticist. The inclusion criteria for rES were: (a) two or more independent major fetal anomalies, (b) either hydrops fetalis or bilateral renal cysts alone, or (c) one major fetal anomaly and a first‐degree relative with the same anomaly. We excluded fetuses diagnosed prenatally of having an anomaly for which no underlying genetic defect is known, for example, a body stalk anomaly, limb body wall complex or OEIS complex (OMIM 258040). Fetal DNA was derived from chorionic villi or amniotic fluid. Parental DNA of the biological father and mother was required for trio‐analysis.
The study was approved by the local medical ethics committee (GEN14.0117, UMCG research register code 201700782).
2.2. Parental counseling and informed consent
Parents received our standard, extensive pre‐test counseling about QF‐PCR and SNP‐array and additional counseling about the potential value and limitations of rES. Amongst other topics, unsolicited findings and the possibility of discovering non‐paternity were discussed, in accordance with the joint position statement on ES. 5 Parents were informed that rES cannot identify or rule out all genetic conditions and that our knowledge of genetic diseases and variant interpretations is continuously evolving. As part of the informed consent process, we asked for parental consent to follow‐up and data collection about the pregnancy and the health and genetic diagnosis of the child postnatally and to recontact if new, clinically significant information related to the diagnosis of their child becomes available. Both parents were asked to sign an informed consent form as a condition for rES.
2.3. Logistics
Upon inclusion, patients followed routine prenatal diagnostics starting with QF‐PCR for detection of trisomies 13, 18, and 21, monosomy X and triploidy. Following normal QF‐PCR results, routine‐care SNP‐array and rES were started in parallel (Figure 1 for study workflow).
FIGURE 1.
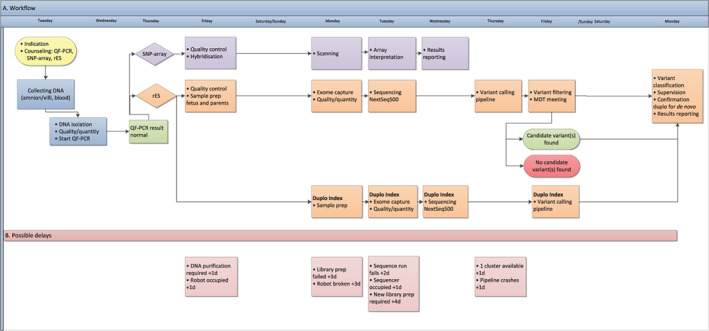
Logistics of rapid prenatal diagnostic testing in the study setting. Panel A (top) shows a realistic, common scenario of inclusion for a fetus with ultrasound anomalies. The collection of fetal material via chorionic villus sampling/amniocentesis is typically scheduled on fixed days of the week, which allows the subsequent genetic diagnostic workflow to be scheduled, including QF‐PCR, SNP‐array, and rES. Panel B (bottom) shows the observed delays in the workflow with the additional time spent on such delays in days. MDT, multidisciplinary team; QF‐PCR, quantitative fluorescent‐polymerase chain reaction; rES, rapid exome sequencing; SNP‐array, single nucleotide polymorphism‐array [Colour figure can be viewed at wileyonlinelibrary.com]
Fetal DNA was extracted from chorionic villi or amniotic fluid and parental DNA from peripheral blood using standard diagnostic procedures. Identification of the sample and exclusion of maternal cell contamination in the fetal DNA was carried out using QF‐PCR data of the fetus and the mother. Fetal and parental DNA were prepared for rES using SureSelect Human All Exon V6 (Agilent) target enrichment, according to standard procedures, on Bravo automated liquid handling robots (Agilent), and then sequenced on an Illumina NextSeq500 sequencer aiming for 20x coverage for 95% of the target genes. Fetal DNA was prepared for rES in duplo and sequenced in two separate runs for validation purposes in order to avoid time‐consuming Sanger sequencing validation. Two automated DNA isolation systems (Maxwell, Promega), two automated library and enrichment robots (Bravo, Agilent), two TapeStation systems (Agilent) for measuring DNA concentrations, two Nextseq500 sequencers (Illumina) and two data analysis clusters ensured redundancy in case of failure or malfunctions.
2.4. Data analysis
Raw rES data was processed according to standardized protocols as described in Data S1. Sequence variants were filtered using Alissa NGS‐Bench Lab software (Alissa, Agilent) using an automated filtering tree. We generated a virtual gene panel of monogenic diseases based on approximately 3850 genes from the Clinical Genomics Database and Online Mendelian Inheritance in Man (OMIM), and removed genes associated with late‐onset diseases as described previously. 7 , 8 , 9 The full gene list can be found in Table S1.
We analyzed the variants in the gene panel using GAVIN (Gene‐Aware Variant Interpretation), minor allele frequencies from GnomAD, subsequent annotation with OMIM terms, Combined Annotation Dependent Depletion (CADD) scores and reported modes of inheritance using MOLGENIS. 10 , 11 , 12 , 13 , 14 , 15 Variants remaining after these filtering steps were evaluated by a multidisciplinary team comprised of, at minimum, the operating technician, a clinical geneticist, and a genetic laboratory specialist in matching the fetal phenotype. A detailed description of the variant‐filtering process can be found in Data S1.
2.5. Reporting of variants
All variants that might explain the fetal phenotype and all unsolicited findings were classified using Alamut software according to standardized guidelines based on Richards et al, 16 while taking into account the Human Gene Mutation Database, CADD score, and population frequency. 17 , 18 Only variants classified as pathogenic and likely pathogenic were communicated to parents. Variants of unknown significance (VUS) were not communicated, as is recommended in the joint position statement on genome‐wide sequencing in fetuses. 5
Unsolicited findings were divided into three categories:
(likely) pathogenic variants matching the inheritance pattern of the associated, actionable disease, knowledge of which could lead to health benefits for the child and/or parents;
(likely) pathogenic variants in genes associated with developmental delay and/or intellectual disability unrelated to the fetal phenotype;
(likely) pathogenic variants in autosomal recessive disease genes with a carrier frequency above 1:60 in the general Dutch population, or both parents carrying a heterozygous variant in the same autosomal recessive disease gene.
2.6. Approach for postnatal follow‐up
We collected data from medical files in participating hospitals or from the patient's healthcare providers. We studied the phenotype seen on additional ultrasounds, the pregnancy outcome, the phenotype after birth (including autopsy report if applicable) and the results of additional genetic tests (if requested), as well as other factors.
2.7. Primary endpoints
We measured diagnostic yield, turnaround times and the clinical consequences of a rapid genetic diagnostic approach using ES. Turnaround time was measured from the moment of the invasive procedure until the definitive report of rES results.
3. RESULTS
3.1. Inclusion of patients
Of the 55 pregnancies with invasive procedures that fulfilled the inclusion criteria, 28 were excluded. Exclusion was mainly due to abnormal QF‐PCR (N = 18), while two couples were in the process of considering rES when a cause for the fetal anomalies was identified by SNP‐array. Only one couple declined rES (Figure 2 and Data S1).
FIGURE 2.
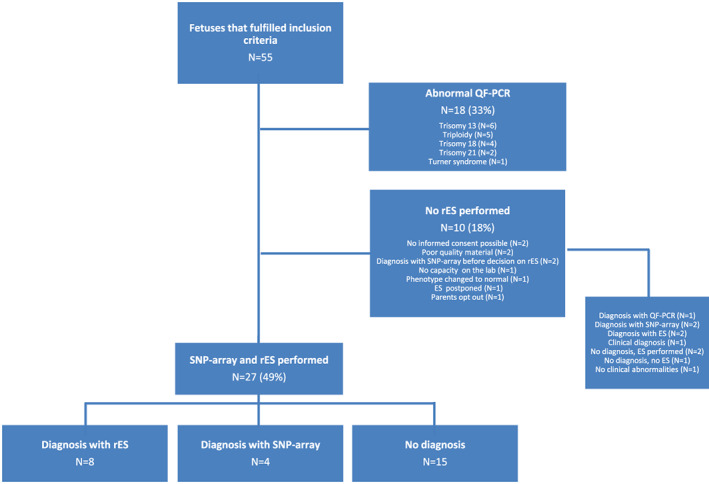
Inclusion of fetuses with ultrasound anomalies for rES. ES, exome sequencing; QF‐PCR, quantitative fluorescent‐polymerase chain reaction; rES, rapid exome sequencing; SNP‐array, single nucleotide polymorphism‐array [Colour figure can be viewed at wileyonlinelibrary.com]
In total, 27 cases underwent testing with both SNP‐array and rES (for indications, see Figure 2 and Table 1). Gestational ages were between 11 weeks and 34 weeks and 4 days, with a median of 20 weeks and 3 days. The most common indications for rES were major anomalies of the brain, heart and skeleton, and/or hydrops. Six fetuses had a first‐degree relative with a congenital anomaly related to the fetal anomaly. In four of these cases, the congenital anomaly of the first‐degree relative was necessary to fulfill the inclusion criteria for this study. In 24 cases, rES was requested in parallel with SNP‐array. In three cases, rES was requested after SNP‐array because the phenotype changed when additional anomalies were detected after the initial ultrasound.
TABLE 1.
Characteristics of fetuses with ultrasound anomalies at the moment of inclusion in the study that underwent rES and the outcomes of the pregnancies
| ID | Gestational age at | Criteria for inclusion: Ultrasound anomalies and/or family history | Genetic diagnosis | Pregnancy outcome | Did WES result affect decision? | |
|---|---|---|---|---|---|---|
| inclusion | result rES | |||||
| 1 | 20w6d | 24w1d | VSD, ASD, bilateral talipes equinovarus, oligohydramnios | Yes, rES | Termination a | Yes |
| 2 | 19w5d | 21w5d |
Severe hydrocephaly Previous pregnancy of fetus with severe hydrocephaly |
Yes, rES | Termination | No |
| 3 | 19w1d | 21w2d |
Cerebellar vermis hypoplasia, hydronephrosis Previous pregnancy of fetus with cerebellar vermis hypoplasia, cystic hygroma and bilateral talipes equinovarus |
Yes, rES | Termination | Yes |
| 4 | 19w3d | 22w2d | Decreased skull ossification, abnormality of the ribs, bowed forearm bones, bowed humerus | Yes, rES | Termination | No |
| 5 | 16w2d | 18w1d | Hydrops fetalis, pleural effusion | Yes, rES | Termination | No |
| 6 | 20w1d | 22w2d |
Bilateral CLP, abnormal heart morphology Parent: CL |
Yes, rES | Termination | No |
| 7 | 19w5d | 22w1d | CP, bilateral CL, abnormal heart morphology | Yes, rES | Termination | No |
| 8 | 28w4d | 32w5d | Abnormality of the skull, hypotelorism, proptosis, thoracic hypoplasia, abnormality of the ribs, severe limb shortening, talipes equinovarus, bowed humerus, femoral bowing, flexion contracture | Yes, rES | Continued, neonatal death | No |
| 9 | 31w4d | 33w6d | Absent septum pellucidum, abnormal heart morphology, severe IUGR, oligohydramnios | Yes, SNP‐array | Continued | No |
| 10 | 21w3d | 23w3d | Abnormality of the cerebellum, hypoplasia of the nasal bone, bilateral CL, IUGR | Yes, SNP‐array | Termination | No |
| 11 | 21w5d | 23w6d | Enlarged cisterna magna, dilated third ventricle, absence of stomach bubble, growth abnormality | Yes, SNP‐array | Termination | No |
| 12 | 20w5d | 23w3d | Abnormality of the skull, dextrocardia, abnormal lung morphology, unilateral renal agenesis, unilateral oligodactyly, SUA | Yes, SNP‐array | Termination | No |
| 13 | 12w2d | 15w5d | Hydrops fetalis, SUA | No b | Termination | No |
| 14 | 19w4d | 21w4d | Multicystic kidney dysplasia, renal agenesis unilateral, absence of stomach bubble on fetal sonography, IUGR, oligohydramnios, SUA | No c | Termination | No |
| 15 | 14w2d | 16w1d | Retrognathia, fetal cystic hygroma, short forearm, deviation of the hand | No d | Termination | No |
| 16 | 14w0d | 16w2d | Hydrops fetalis, hypoplastic right heart, increased NT | No | Termination | No |
| 17 | 21w2d | 23w6d | VSD, ectopic kidney | No | Continued | Yes |
| 18 | 11w0d | 13w4d |
Hydrops fetalis Previous pregnancy of fetus with hydrops fetalis |
No | Miscarriage before rES result | No |
| 19 | 21w5d | 23w5d | Abnormal heart morphology, omphalocele | No | Continued, fetal death | Yes |
| 20 | 19w2d | 21w2d | Hydrocephalus, ACC, cerebellar hypoplasia | No | Termination | No |
| 21 | 34w4d | 37w0d | Ventriculomegaly, short fetal femur length, short fetal humerus length | No | Continued | No |
| 22 | 21w5d | 23w6d |
BAV, echogenic fetal bowel, possible duplicated collecting system (urinary tract) Parent with VSD |
No | Continued | Yes |
| 23 | 18w6d | 21w5d |
Abnormality of the myocardium, abnormal tricuspid valve morphology, possible VSD Previous child with VSD |
No | Continued | Unknown |
| 24 | 21w0d | 23w0d | Anomaly of the posterior cranial fossa, thickened nuchal skin fold | No | Continued | Unknown |
| 25 | 14w6d | 16w1d | Cerebellar malformation, increased NT, abnormal heart morphology, hyperechogenic kidneys, spina bifida, hydrops fetalis | No | Termination | No |
| 26 | 20w1d | 22w2d | Frontal bossing, overlapping fingers, hydrops fetalis | No | Fetal death before rES result | No |
| 27 | 20w3d | 22w2d | Macrocephaly, enlarged cerebellum, ambiguous genitalia, contracture joints upper limb and lower limb, rocker bottom feet | No | Termination | No |
Note: Ultrasound anomalies are described using the terminology of the human phenotype ontology (HPO).
Abbreviations: ACC, agenesis of corpus callosum; ASD, atrial septal defect; BAV, bicuspid aortic valve; CL, cleft lip; CLP, cleft lip and palate; CP, Cleft palate IUGR, intrauterine growth retardation; NT, nuchal translucency; rES, rapid exome sequencing; SUA, single umbilical artery; VSD, ventricular septal defect; w, weeks; d, days.
Termination after 24 weeks is only possible in severe lethal disorders.
Maternal likely pathogenic variant in MYH7 identified with rES. An effect of this variant on the phenotype cannot be excluded.
Mosaicism of trisomy 15 identified with SNP‐array in DNA extracted from chorionic villi. SNP‐array in cord blood and pericardium DNA revealed no mosaicism.
Postnatally diagnosed with Nager syndrome, based on clinical features.
3.2. Turnaround time and impact on laboratory workflow
We decided to limit inclusion to one case per week in the initial 4 weeks of the study in order to streamline procedures. Indeed, when the study commenced, we experienced unfamiliarity with logistics, difficulties with transfer of knowledge, and automation errors in the bioinformatics pipeline. Importantly, laboratory personnel and physicians also experienced time pressure, mainly due to the short turnaround time and the legal limit for terminating the pregnancy. However, after the four‐week run‐in period, the clinicians and laboratory personnel adapted to the procedures, and we managed to obtain a median turnaround time of 14 days (range 8‐20 days). However, even though our laboratory is equipped with all essential equipment in redundancy, delays in our workflow still occurred due to occupied robots, failed sample preparations and bioinformatics delays due to cluster maintenance, amongst other factors (Figure 1, panel B).
3.3. Diagnostic yield
Of the 55 fetuses included in this study (Figure 2), a genetic diagnosis was made in 35 (64%). In 28 fetuses, no rES was performed. In 18 cases, this was because of an abnormal QF‐PCR. In the other 10 no rES was performed for various other reasons, but five fetuses did receive a genetic diagnosis (Figure 2 and Data S1).
Of the 27 fetuses included for rES in combination with SNP‐array after a normal QF‐PCR result, a genetic diagnosis was made in 12 (44%): four using SNP‐array and eight using rES (Figure 2 and Tables 1 and 2).
TABLE 2.
Overview of genetic diagnoses made for fetus with ultrasound anomalies who underwent rapid exome sequencing (rES)
| rES result | ||||||
|---|---|---|---|---|---|---|
| ID | Gene | Causative variant | Transcript ID | Inheritance | SNP‐array result | Syndrome |
| 1 | SAMD9 | c.2920G>A, p.Glu974Lys | NM_017654.3 | AD de novo | MIRAGE syndrome | |
| 2 | POMGNT1 | c.636C>T, (=) AND c.630G>T, p.Trp210Cys | NM_001243766.1 | AR mother and father | Walker‐Warburg syndrome | |
| 3 | PEX1 | c.2097dupT, p.Ile700Tyrfs*42 | NM_000466.2 | AR mother and father | Zellweger syndrome | |
| 4 | COL1A1 | c.3150_3158del, p.Ala1053_Gly1055del | NM_000088.3 | AD de novo | Osteogenesis imperfecta, lethal type | |
| 5 | PTPN11 | c.227A>T, p.Glu76Val | NM_002834.3 | AD de novo | Noonan syndrome 1 | |
| 6 | CHD7 | c.3301T>C, p.Cys1101Arg | NM_017780.2 | AD de novo | CHARGE syndrome | |
| 7 | KMT2D | c.696dupT, p.Glu233* | NM_003482.3 | AD de novo | Kabuki syndrome | |
| 8 | COL1A1 | c.2102G>T, p.Gly701Val | NM_000088.3 | AD de novo | Osteogenesis Imperfecta, lethal type | |
| 9 | Trisomy Chr22 in villi and amnion | |||||
| 10 | Deletion 4p16.3p15.31 | Wolf‐Hirschhorn syndrome | ||||
| 11 | Duplication 11q23.3q25 and duplication 22q11.1q11.21 | Emanuel syndrome | ||||
| 12 | Mosaic trisomy 16 (arr[16]x2 ~ 3 dn) | |||||
Abbreviations: AD, autosomal dominant; AR, autosomal recessive.
3.4. Unsolicited findings
We communicated 12 unsolicited findings to 11 couples, as shown in Table 3. In two parental couples, a likely pathogenic mutation was identified that was communicated because of possible health benefits. The parent carrying the likely pathogenic variant in ABCC9 or MYH7 was screened by a cardiologist, and advice for follow‐up was given. In addition, one likely benign variant (MYLK) was communicated due to misclassification.
TABLE 3.
Unsolicited findings reported to parents
| IF category | Gene | Variant | Transcript ID | Classification | Phenotype |
|---|---|---|---|---|---|
| Gene associated with developmental delay and/or intellectual disability | NAA15 |
c.1826_1830del, p.Glu609Glyfs*20 (fetal de novo) |
NM_057175.3 | P | Intellectual disability |
| Variant(s) matching inheritance pattern of the actionable disease | ABCC9 | c.4537G>A, p.Ala1513Thr (paternal) | NM_005691.2 | LP | Atrial fibrillation |
| MYH7 | c.976G>C, p.Ala326Pro (maternal) | NM_000257.2 | LP | Cardiomyopathy | |
| MYLK | c.493delA, p.Thr165Profs*72(maternal) | NM_053025.3 | LP ➔ LB* | Aortic aneurysm | |
| Autosomal recessive carrier (>1:60) or both parents carry a heterozygous variant in the same gene | PIGV | c.494C>A, p.Ala165Glu (maternal, paternal) | NM_001202554.1 | LP | Mabry syndrome |
| ATP7B | c.3207C>A, p.His1069Gln (maternal) | NM_000053.3 | P | Wilson disease | |
| CYP21A2 | c.949C>T, p.Arg317* (paternal) | NM_000500.5 | P | Congenital adrenal hyperplasia | |
| CEP290 a | c.3175dupA, p.Ile1059Asnfs*11 (paternal) | NM_025114.3 | P | Ciliopathy | |
| HFE b | c.845G>A, p.Cys282Tyr (maternal) | NM_000410.3 | P | Hereditary Hemochromatosis | |
| SERPIN1A | c.1096G>A, p.Glu366Lys (maternal) | NM_001127701.1 | P | Alfa‐1‐antitrypsin deficiency | |
| WNT10A | c.682T>A, p.Phe228Ile (paternal) | NM_025216.2 | P | Odontoonychodermal dysplasia | |
| WNT10A | c.383G>A, p.Arg128Gln (maternal) | NM_025216.2 | LP | Odontoonychodermal dysplasia |
Variant was reported because phenotype matched, even though carrier frequency was below 1:60. This is our postnatal standard operating procedure. Extensive screening did not identify any second variant in the WES data or SNP array affecting this gene. In a prenatal setting, this variant should not have been reported. An abnormal SNP array result was identified as the cause of the ultrasound anomalies in this case, excluding the CEP290 variant as the main cause.
High carrier frequency in the general population, and reporting policy was adjusted to non‐reporting during the study, because of low clinical impact.
One variant was identified in a gene known to cause a variable developmental delay. In this case, the fetal anomalies seen on ultrasound were already explained by another variant.
Most (likely) pathogenic variants in autosomal recessive disease genes were only identified in the fetus and one of the parents. The whole gene of the other parent was re‐analyzed, but no (likely) pathogenic variants were identified. In one couple, both the parents and the fetus were heterozygous for a likely pathogenic variant in an autosomal recessive disease gene and therefore at increased risk of having a child with this autosomal recessive disorder.
3.5. Effects of rES and SNP array results on pregnancy outcome, parental choices and medical intervention
Ten out of 12 pregnancies with a genetic diagnosis were terminated. Conversations with patients during the consultations at which the abnormal rES and SNP‐array results were communicated suggested that the genetic results played a crucial role in the decision to terminate the pregnancy in five cases (Table 1). In two other cases, the severity of the genetic diagnosis supported the choice to pursue an obstetric non‐intervention policy and comfort care after birth. In the remaining 15 pregnancies without a genetic diagnosis, having a result in which no clearly pathogenic variant was detected supported at least four couples in their decision to continue their pregnancy. In two cases, the results were important for decision making surrounding active obstetric and neonatal management. Pregnancy outcome data is available for 14 out of 15 cases without a prenatal genetic diagnosis. Seven pregnancies were terminated based on the severity of the ultrasound abnormalities. One case ended in a miscarriage around 13 weeks of gestation. There were two cases of intra‐uterine demise at 21 and 32 weeks of gestation. Four children were born alive.
3.6. Follow‐up after rES
Most ultrasound anomalies were confirmed postnatally or postmortem. However, one fetus suspected of having an inlet ventricular septal defect based on prenatal ultrasound had a normal cardiac ultrasound after birth. In other fetuses, additional anomalies were seen after birth beyond those detected on ultrasound. These included dysmorphic facial features, seizures, position anomalies of limbs and syndactyly.
In 8 of the 15 cases without a prenatal genetic diagnosis, additional genetic investigations such as specific gene panels or open exome analysis were performed postnatally or postmortem. However, no definitive genetic diagnosis was made in these cases.
A clinical diagnosis of Nager syndrome was made postmortem in case 15 based on micrognathia, radial aplasia and absent thumbs, but no variant in the SF3B4 gene (OMIM 605593) was identified. In case 14, a mosaicism of trisomy 15 was identified in DNA extracted from chorionic villi. SNP‐array in fetal DNA after termination of pregnancy revealed no mosaicism. The growth restriction of this fetus is thus probably explained by confined placental mosaicism, but the cause of the other anomalies (Table 1) remains unexplained. In case 13 with hydrops, a maternal likely pathogenic variant in MYH7 (OMIM 160760) was identified using rES. Pathogenic variants in MYH7 are a cause of cardiomyopathy, and anomalies of the heart may cause fetal hydrops. A possible effect of this variant on the fetal phenotype cannot therefore be excluded.
4. DISCUSSION
In this prospective study, we implemented rES as a diagnostic genetic test in prenatal care, in addition to QF‐PCR and SNP‐array. We made a genetic diagnosis in 64% of all included cases, with ES contributing 18% of this diagnostic yield. Our rES yield is comparable to the yields of 15% to 19% reported by two other large prospective studies of fetuses with multiple congenital anomalies. 19 , 20 While QF‐PCR still yield the majority of diagnoses, ES identified more causal variants than SNP‐array in our study sample (18% vs 11%). These data support a policy of starting with QF‐PCR and then adding SNP‐array and rES, performed in parallel due to time limitations, when necessary.
Our strict inclusion criteria were formulated in light of limited capacity and an anticipated increase in workload, in combination with the anticipated greater yield, as had been seen previously. 19 , 20 , 21 , 22 In addition, several studies have shown that more rES‐based diagnoses are made in specific subcategories of anomalies (eg, skeletal dysplasia) than others (eg, increased nuchal translucency). 19 , 20 , 21 , 22 This could be explained by the fact that anomalies in these organ systems are more likely to be caused by single gene variants, but a higher yield in a specific organ system could also be the result of greater knowledge about genetic variants associated with anomalies in these organ systems in the prenatal setting. 20 Our dataset is too small to draw conclusions on this matter and, since other studies are inconsistent, more data are needed to estimate the expected prenatal diagnostic yield per anomaly, or combination thereof.
Recognizing the fetal phenotype on ultrasound is challenging. Ultrasound images are a digital representation of the anatomy of the fetus, and not every anomaly will be detected by ultrasound, with dysmorphic features being especially difficult to identify. For example, for rES diagnostics, we excluded established fetal anomaly associations without a known genetic background, such as limb body wall complex; however, this diagnosis was made postmortem in one fetus in our cohort.
Detailed information on the phenotype is still essential for accurate interpretation of genetic data. Uncertainty about the fetal phenotype and lack of knowledge on the fetal phenotype in relation to disease‐causing genes are possible, but unavoidable, obstacles to correct data interpretation. 23 On the other hand, suspected diagnoses might turn out to be false. For example, the expected diagnosis in Case 3 was Joubert syndrome based on the combination of cerebellar anomalies seen in this fetus, including suspected molar tooth sign and hydronephrosis, and the cerebellar anomalies, cystic hygroma, echogenic kidneys, and talipes seen in a previous fetus of the parents. However, rES established a homozygous pathogenic variant in PEX1 (OMIM 602136), which is known to cause Zellweger syndrome (OMIM 214100). Cerebellar anomalies are a rare feature of Zellweger syndrome, while the other features of the syndrome did match the fetal phenotype. This case shows the value of using broad virtual gene panels rather than specific gene panels like that used for ciliopathies. Not only do we lack knowledge about the prenatal phenotype of disease‐causing genes, variants in genes that cause lethal phenotypes in the embryonic, fetal, or perinatal period may have escaped detection until now. Increased use of ES in the prenatal period will probably reveal more of these lethal variants. Indeed, Meier et al (2019) identified new disease genes in a cohort of severely affected fetuses. 24
Prenatal ES can have a significant impact on parental decision‐making and pre‐ and perinatal management. In our study, the molecular diagnosis was a deciding factor for terminating the pregnancy in five cases. However, it is equally important to recognize that a result in which no clearly pathogenic variant was detected supported the parental decision to continue the pregnancy in at least four cases. These observations are based on conversations with parents during genetic counseling. To gain deeper insights into parental decision‐making in our study group, parents were also asked to participate in a mixed methods patient perception study, the results of which will soon be submitted.
We chose not to report any VUS identified using rES during pregnancy because this was expected to hamper parental decision‐making and increase stress. We also minimized the number of unsolicited findings by excluding late‐onset disease genes not known to cause a phenotype at younger ages. We did detect an unsolicited finding of two likely pathogenic variants in the autosomal dominant genes ABCC9 and MYH7 in both a fetus and one of the parents (Table 3). ABCC9 and MYH7 are both cardiomyopathy genes, warranting a cardiology exam in the parent who carried the likely pathogenic variant, which led to distress without medical benefits at the time.
Another example of an unsolicited finding is a variant in MYLK that was first classified as likely pathogenic. Pathogenic MYLK variants cause familial aorta pathology, an “actionable” condition, and the variant was therefore communicated to the parents. However, after reevaluation, the variant was re‐classified as likely benign since pathogenic MYLK variants have only been reported in one particular transcript, and the variant we identified was not within this transcript. This kind of situation might cause unwarranted distress and inconvenience to parents.
Based on these and other examples, we adapted our filtering strategy to minimize unsolicited findings, especially those of autosomal recessive disease variants detectable in one parent. We did this by only including homozygous and compound heterozygous variants in recessive genes, autosomal dominant de novo variants and X‐linked hemizygous variants. Another way to address this issue would be to use a tiered process in which variants related to the fetal phenotype are reported within 10 working days, whereas unsolicited “actionable” findings are reported with a longer turnaround time. This, however, has the disadvantage of not supporting completely informed decision‐making regarding the termination or continuation of pregnancy. More research on how to deal with unsolicited findings in the prenatal setting that includes both health care professionals and parents is therefore needed.
To conclude, our study clearly demonstrates the added value of rES in terms of diagnostic yield in comparison to the current standard of QF‐PCR and SNP‐array for fetuses with ultrasound anomalies. An underlying genetic cause was identified in 64% of cases, with 18% identified via rES. Despite its high diagnostic yield, rES remains challenging in a prenatal context, particularly with respect to the short turnaround times required, uncertainties surrounding the prenatal phenotype and classification of variants and unavoidable unsolicited findings.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Data S1. Supporting Information
Table S1. Gene list.
ACKNOWLEDGMENTS
We want to thank the patients, technicians, laboratory specialists, and clinical geneticists of the Genetics department of the University Medical Center Groningen (UMCG) for their contribution and expertise in the multidisciplinary team meetings when discussing rES results. We thank the gynecologists of the UMCG and Isala for referring patients. We thank Kate McIntyre for editorial assistance. We want to thank the UMCG for its financial support via a healthy ageing pilot grant (HAP; CDO17.0028/2017‐2/321).
Corsten‐Janssen N, Bouman K, Diphoorn JCD, et al. A prospective study on rapid exome sequencing as a diagnostic test for multiple congenital anomalies on fetal ultrasound. Prenatal Diagnosis. 2020;40:1300–1309. 10.1002/pd.5781
Birgit Sikkema‐Raddatz and Helga Westers shared authorship.
Funding information University Medical Center Groningen Healthy Ageing Pilot grant, Grant/Award Number: CDO17.0028/2017‐2/321
DATA AVAILABILITY STATEMENT
Absence of shared data.
REFERENCES
- 1.Eurocat update: short report congenital anomalies Northern Netherlands 1981–2017 (update); 2019. www.eurocat.umcg.nl
- 2. Fleurke‐Rozema JH, Bakker MK, Snijders RJM, Bilardo CM. Uptake of the 20‐week scan and the detection rate of selected sonomarkers and structural congenital anomalies (Chapter 8 thesis) In: Fleurke‐Rozema JH, ed. Impact of the 20‐week scan. Groningen, The Netherlands: Rijksuniversiteit Groningen; 2017. https://www.rug.nl/research/portal/files/46538014/Chapter_8.pdf. [Google Scholar]
- 3. Best S, Wou K, Vora N, van der Veyver IB, Wapner R, Chitty LS. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn. 2018;38(1):10‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Leung GKC, Mak CCY, Fung JLF, et al. Identifying the genetic causes for prenatally diagnosed structural congenital anomalies (SCAs) by whole‐exome sequencing (WES). BMC Med Genomics. 2018;11(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. The International Society for Prenatal Diagnosis , The Society for Maternal and Fetal Medicine , The Perinatal Quality Foundation . Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome‐wide sequencing for fetal diagnosis. Prenatal Diagn. 2018;38(1):6‐9. [DOI] [PubMed] [Google Scholar]
- 6. Horn R, Parker M. Opening Pandora's box?: ethical issues in prenatal whole genome and exome sequencing. Prenat Diagn. 2018;38(1):20‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Diemen CC, Kerstjens‐Frederikse WS, Bergman KA, et al. Rapid targeted genomics in critically ill Newborns. Pediatrics. 2017;140(4):e20162854. [DOI] [PubMed] [Google Scholar]
- 8. Solomon BD, Nguyen AD, Bear KA, Wolfsberg TG. Clinical genomic database. Proc Natl Acad Sci U S A. 2013;110(24):9851‐9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Johns Hopkins University . Online Mendelian Inheritance in Man, OMIM®. McKusick‐Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD. www.omim.com.
- 10. van der Velde KJ, de Boer EN, van Diemen CC, et al. GAVIN: gene‐aware variant INterpretation for medical sequencing. Genome Biol. 2017;18(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv. 2019; 531210 10.1101/531210. [DOI] [Google Scholar]
- 12. Online Mendelian Inheritance in Man (OMIM) , Morbid Map; 2014. Downloaded from OMIM FTP.
- 13.CADD scores. (Release 1.0); 2013. http://cadd.gs.washington.edu/download. Accessed September 26, 2013
- 14. Swertz MA, Dijkstra M, Adamusiak T, et al. The MOLGENIS toolkit: rapid prototyping of biosoftware at the push of a button. BMC Bioinformatics. 2010;11(suppl 12):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. MOLGENIS . Current version in use is 1.4.0.
- 16. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids res. 2019;47(D1):D886‐D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stenson PD, Mort M, Ball EV, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Hum Genet. 2017;136(6):665‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lord J, Mcmullan DJ, Eberhardt RY, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393(10173):747‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petrovski S, Aggarwal V, Giordano JL, et al. Whole‐exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. The Lancet. 2019;393(10173):758‐767. [DOI] [PubMed] [Google Scholar]
- 21. Normand EA, Braxton A, Nassef S, et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018;10(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chandler N, Best S, Hayward J, et al. Rapid prenatal diagnosis using targeted exome sequencing: a cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet Med. 2018;20(11):1430‐1437. [DOI] [PubMed] [Google Scholar]
- 23. Gray KJ, Wilkins‐Haug LE, Herrig NJ, Vora NL. Fetal phenotypes emerge as genetic technologies become robust. Prenat Diagn. 2019;39(9):811‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meier N, Bruder E, Lapaire O, et al. Exome sequencing of fetal anomaly syndromes: novel phenotype‐genotype discoveries. EJHG. 2019;27(5):730‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Table S1. Gene list.
Data Availability Statement
Absence of shared data.