

Abstract
PAX5 is a member of the paired box (PAX) family of transcription factors involved in B‐cell development. PAX5 P80R has recently been described as a distinct genetic B‐cell precursor (BCP) acute lymphoblastic leukemia (ALL) subtype with a favorable prognosis in adults. In contrast, an unfavorable outcome has been observed in children. Our aim was to determine the frequency of PAX5 P80R in childhood BCP‐ALL treated according to the Associazione Italiana Ematologia ed Oncologia Pediatrica‐Berlin‐Frankfurt‐Muenster (AIEOP‐BFM) ALL 2000 protocol and to evaluate its clinical significance within this study cohort. The analyses included 1237 patients with ALL treated in the AIEOP‐BFM ALL 2000 trial with complete information for copy number variations (CNVs) of IKZF1, PAX5, ETV6, RB1, BTG1, EBF1, CDKN2A, CDKN2B, and ERG. A customized TaqMan genotyping assay was used to screen for PAX5 P80R. Sanger sequencing was used to confirm PAX5 P80R‐positive results as well as to screen for second variants in PAX5. Agilent CGH + SNP arrays (e‐Array design 85 320; Agilent Technologies) were performed in PAX5 P80R‐positive patients to verify additional CNVs. Almost 2% (20/1028) of our BCP‐ALL cohort were PAX5 P80R‐positive. White blood cell counts higher than 50 000/μl as well as male sex were significantly (P < .05) associated with PAX5 P80R. Most of the PAX5 P80R‐positive cases were 10 years of age or older. PAX5 P80R‐positive samples were enriched for deletions affecting PAX5, IKZF1, CDKN2A, and CDKN2B. Compared to PAX5 P80R ‐wildtype BCP‐ALL, PAX5 P80R‐positive patients showed a significantly reduced 5‐year overall survival (P = .042). Further studies should evaluate the interaction of PAX5 P80R with other genetic aberrations to further stratify intermediate risk pediatric BCP‐ALL.
Keywords: acute lymphoblastic leukemia, AIEOP‐BFM ALL, B‐cell precursor ALL, PAX5, pediatric acute lymphoblastic leukemia
1. INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most common malignancy of childhood and adolescence. 1 Improvement of therapeutic strategies, including application of risk‐adapted treatment employing prognostic markers such as minimal residual disease (MRD), has increased long‐term survival rates of pediatric ALL to over 80%. 2 , 3 In addition to treatment response a broad spectrum of genetic aberrations is increasingly being used for patient stratification. 4 High‐resolution genome‐ and transcriptome‐wide analyses have led to the description of new biological subgroups and advanced treatment strategies in pediatric ALL. 5 , 6 Improved definitions of alterations or signatures associated with a high relapse risk may help to further improve patients' outcome. Potential targets for therapy de‐escalation might be able to reduce late therapeutic effects.
One of the genes most commonly affected by deletions, amplifications, translocations, or point mutations in ALL is PAX5. 7 , 8 , 9 PAX5 is a member of the paired box (PAX) family of transcription factor genes and encodes a B‐cell lineage‐specific activator protein, which controls the identity of B lymphocytes throughout B‐cell development from the pro‐B to the mature B‐cell. 10 A germline PAX5 variant conferring susceptibility to pre‐B‐cell ALL has lately been described 11 and intragenic amplification of PAX5 has been shown to be related to poor outcome and relapses. 12 A nucleotide exchange at aminoacid codon 80 (c.239C > G; NM_016734.2) of the PAX5 gene (PAX5 P80R) causes a proline to arginine exchange in the DNA‐binding domain of the PAX5 protein (Figure 1A). 13 This somatic PAX5 P80R variant has been observed with a frequency of 0.4% in standard ALL and 3.1% in high‐risk pediatric B‐cell precursor (BCP) ALL. 14 Importantly, in nearly all cases a biallelic alteration has been observed. 14 Due to its accumulation in the intermediate to poor outcome group, 14 we aimed to investigate if PAX5 P80R biallelic alteration represents a prognostic marker in pediatric ALL as observed in adult ALL. 15
FIGURE 1.
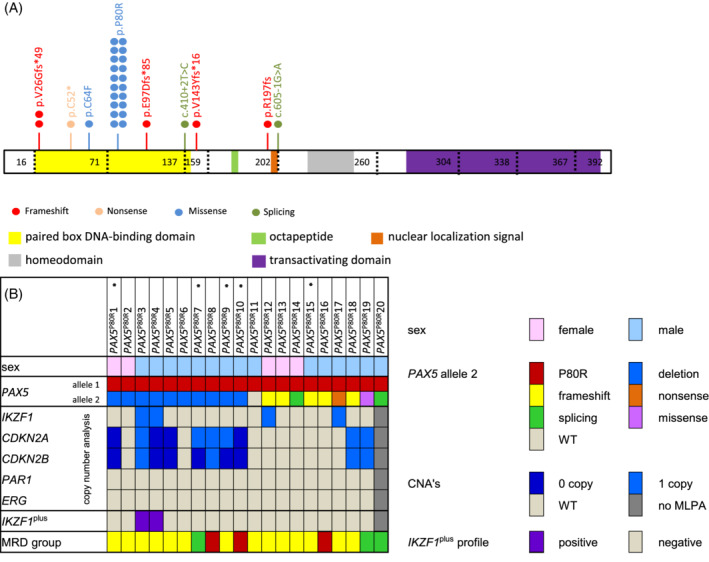
Summary of biallelic PAX5 alteration involving PAX5 P80R observed in a cohort of 1237 pediatric BCP‐ALL cases. A, Protein structure and variants of PAX5 P80R‐positive patients in the Associazione Italiana Ematologia ed Oncologia Pediatrica‐Berlin‐Frankfurt‐Muenster (AIEOP‐BFM) ALL 2000 trial. Each dot symbolizes one variant; patients can harbor more than one variant. Variants leading to frameshifts are shown in red, nonsense variants in rose, missense variants in light blue and splice‐site variants in dark green. B, Additional CNVs detected in PAX5 P80R‐positive patients. Each column represents one patient; the characteristics examined are listed on the left hand. Sex is shown in rose for female and light blue for male patients. The status of PAX5 is displayed in red for PAX5 P80R, blue for deletion of PAX5, yellow for variants leading to frameshift and orange for nonsense variants. Green represents splicing variants, pink missense variants and the WT is represented in light gray. The legend for all copy number analyses displays wildtype in light gray, loss of one copy in light blue and loss of two copies in dark blue. Copy numbers were detected via MLPA. Patients without MLPA data are depicted in dark gray. Patients that fulfill the IKZF1plus criteria are marked in purple, patients that do not fulfill the criteria are represented in light gray and patients without MLPA data in dark gray. The MRD group is labelled red for high risk patients, yellow for medium and green for standard risk patients. ALL, acute lymphoblastic leukemia; BCP, B‐cell precursor; MLPA, multiplex ligation‐dependent probe amplification; MRD, minimal residual disease; p.P80R, PAX5 P80R; WT, wildtype; *, patients that experienced an event and were analyzed by Agilent CGH + SNP arrays
2. MATERIALS AND METHODS
The present study included 1237 ALL patients diagnosed between August 1999 and May 2009 who have been enrolled in the international multicenter trial Associazione Italiana Ematologia ed Oncologia Pediatrica‐Berlin‐Frankfurt‐Muenster ALL 2000 on treatment of pediatric ALL in Germany and had complete information available for copy number alterations of IKZF1, PAX5, ETV6, RB1, BTG1, EBF1, CDKN2A, CDKN2B, Xp22.33/Yp11.31 (PAR1 region; CRLF2, CSF2RA, and IL3RA genes), and ERG. 16 In addition, included patients had to have diagnostic DNA available for further molecular analyses (1237 out of 1323). A customized TaqMan genotyping assay (PAX5_P80R.ANH6G97; ThermoFisher) was used to screen for PAX5 P80R and positive results were confirmed by Sanger sequencing (Supplementary Table S1).
3. RESULTS
We identified 20 PAX5 P80R‐positive cases (1.9% of 1028 BCP‐ALL, Supplementary Figure S1). As expected, none of the 197 patients with T‐ALL were PAX5 P80R‐positive (and none of the 12 patients with unknown immunophenotype as well [Supplementary Figure S2]). Only B‐ALL cases were considered for the following statistical analyses. For 11 of the 20 PAX5 P80R‐positive patients, cytogenetic examinations were available; all of these 11 patients belonged to the B‐other group, 17 , 18 , 19 none showed BRC‐ABL, t(4;11), TCF3‐PBX1 or ETV6‐RUNX1 fusions nor high hyperdiploidy or hypodiploidy. Yet, they were designated as B‐other throughout the remaining document. For the remaining nine patients who lacked information on ETV6/RUNX1, BCR/ABL1 and MLL/AF4 no clear statement can be made regarding their classification to the B‐other collective.
Next, we were interested to determine whether additional mutational events were present in the 20 PAX5 P80R‐positive samples. MLPA data indicated a deletion of one PAX5 allele in nine patients, leading to loss of heterozygosity. In confirmation with an allelic loss, the TaqMan assay showed a hemizygous PAX5 P80R genotype in eight cases (one case was addressed as heterozygous). Sequencing of the entire gene in the remaining 11 samples with no loss showed a second nucleotide variant in 10 cases. One of them appeared to be homozygous for PAX5 P80R and nine showed a second PAX5 variant suggesting compound heterozygosity. Of those nine additional variants, five resulted in a frameshift and two were splice site variants. In addition, we identified one missense and one nonsense variant (Figure 1A). Four of the additional variants have previously been described as somatically acquired variants, 14 the other five have not been previously described. All variants were classified as loss of function variants. In summary, only one PAX5 P80R‐positive patient had no detectable second variant, whereas 19 presented a second loss of function variant leading to PAX5 biallelic alteration (and most likely complete loss of function) (Figure 1B and Supplementary Figure S2). For 18 of the PAX5 P80R‐positive patients, follow‐up material was collected during remission, which was used as a germline surrogate. In none of the available remission samples PAX5 P80R was detectable, showing that PAX5 P80R is not a germline variant but rather somatically acquired. This confirms previous observations. 15
MLPA data were used to further analyze the copy number profile of the PAX5 P80R‐positive patients (Figure 1B). As mentioned before, nine PAX5 P80R‐positive samples showed an additional PAX5 deletion due to 9p deletion including CDKN2A/B. CDKN2A was deleted biallelically in three and CDKN2B deleted biallelically in five cases. In addition, four samples had a monoallelic, intragenic IKZF1 deletion (located at 7p12.2). Two of the four IKZF1 deleted patients fulfilled the recently described IKZF1 plus criteria. 20 The IKZF1 plus criteria are fulfilled if in addition to an IKZF1 deletion at least one additional deletion in CDKN2A and/or CDKN2B (homozygous deletion only), a deletion in PAX5 or the PAR1 region is present and an ERG deletion (ERG del) is absent. 20 ETV6 was deleted in one patient, as was BTG1. There were no alterations in ERG, CRLF2, EBF1, and RB1 in PAX5 P80R‐positive patients.
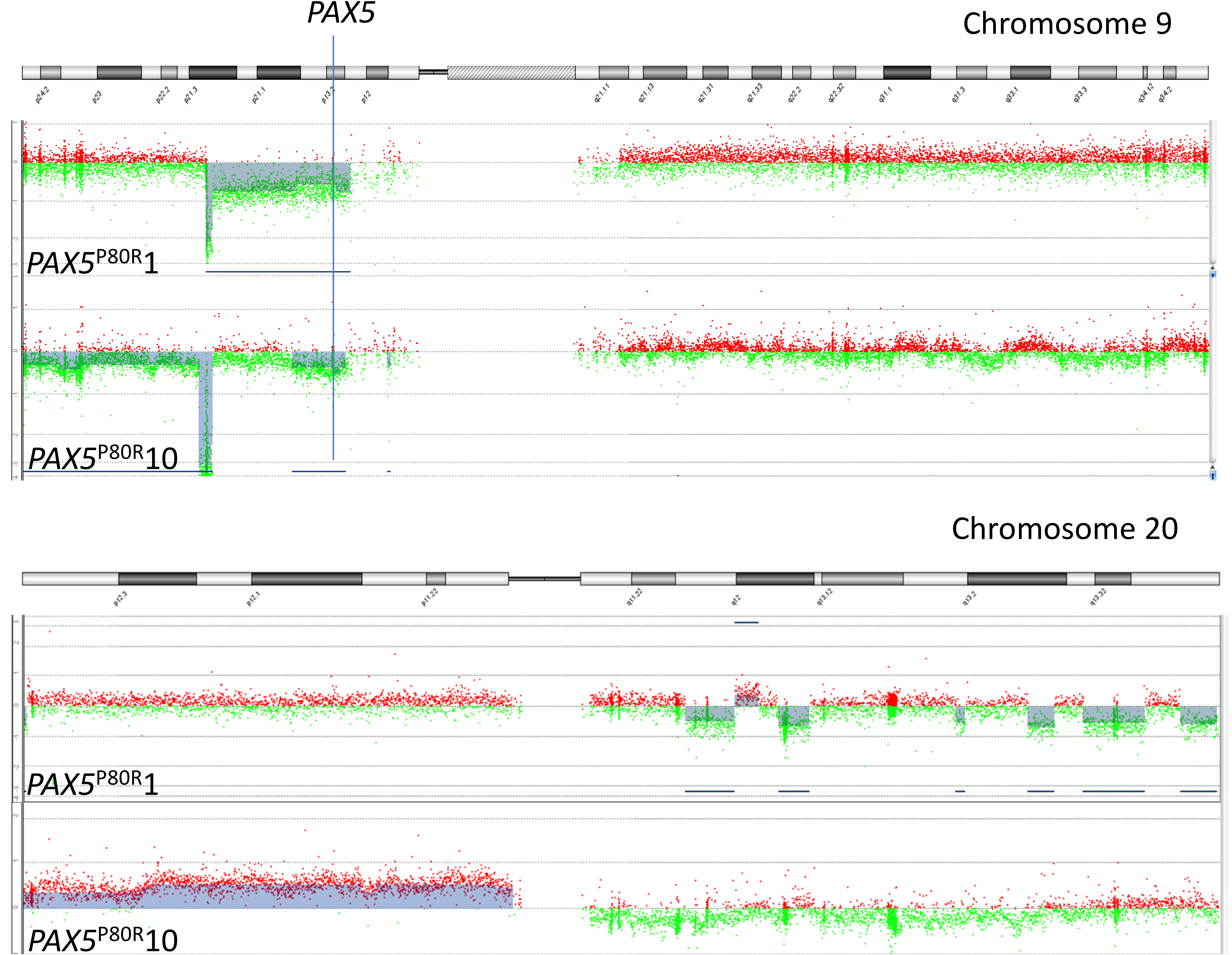
Agilent CGH + SNP arrays (e‐Array design 85 320; Agilent Technologies, Supplementary Table S2) were used to screen for additional copy number alterations in five PAX5 P80R patients, who experienced an event (Figure 2A). This included the patient that appeared to be homozygous for PAX5 P80R at first (#10). Importantly, array analyses showed a 9p deletion leading to hemizygosity of PAX5 in this case, too (Supplementary Figure S3). Additionally, in this case and in another one, alterations at chromosome 20 (Supplementary Figure S3) were present. All but one of the five patients had a biallelic CDKN2A/B deletion, which was in line with the MLPA data (Supplementary Table S2).
FIGURE 2.
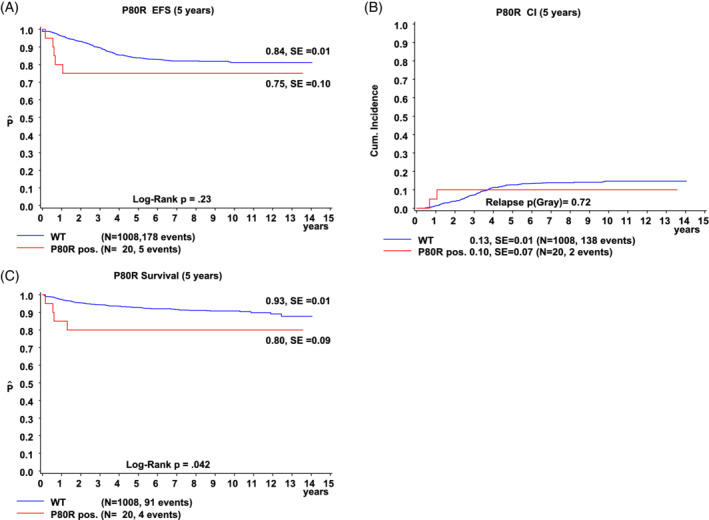
Long‐term outcome data according to PAX5 P80R status. PAX5 P80R‐WT patients are represented in blue, PAX5 P80R‐positive patients in red. A, EFS, B, CI, and C, OS. CI, cumulative incidence; EFS, event‐free survival; OS, overall survival; P80R, PAX5 P80R; WT, wildtype
In our cohort, white blood cell counts higher than 50 000/μl as well as male sex were significantly (P < .05) associated with the presence of PAX5 P80R. In addition, most of the PAX5 P80R cases were 10 years of age or older and only 15% were MRD‐negative (Supplementary Table S3). Furthermore, 70% of our PAX5 P80R‐positive cases were initially stratified to the intermediate risk (IR) group (Figure 1B), a group of patients still positive for MRD after induction. 3 IR patients neither fulfill criteria for treatment intensification, nor for therapy de‐escalation and may benefit from improved treatment stratification. 3
Compared to PAX5 P80R ‐wildtype BCP‐ALL, PAX5 P80R‐positive patients showed no significant differences in event‐free‐survival (Figure 2A) and the cumulative incidence of relapse (Figure 2B). During the 5 years of follow‐up, events were recorded for five patients: three died treatment‐related, one deceased of a relapse and one relapsed but survived (Figure 2A). PAX5 P80R‐positive patients had a significantly poorer 5‐year overall survival (P = .042, Figure 2C). Although the overall survival was significantly different in univariate analysis, this was not observed in multivariate analysis (Supplementary Table S4).
4. DISCUSSION
PAX5 P80R has been the focus of interest of several other studies. 13 , 14 , 15 , 21 While our research was focused on children, others have examined adults 13 or both 14 , 15 with opposing conclusions regarding outcome. In our BCP‐ALL cohort 1.9% of the patients were positive for PAX5 P80R, whereas other trials have shown higher prevalence (ie, 2.4%, 14 5.3%, 13 and 6.4% 15 of BCP‐ALL). Most of our PAX5 P80R‐positive patients were 10 years of age or older (P < .0001) matching the findings of increased prevalence with increasing age 14 and in young adults. 15 Bastian et al have described an equal sex distribution, 15 whereas Gu et al have observed a larger proportion of males (65.9%), 14 as we did in our cohort (75%, P < .05). As described previously, PAX5 P80R‐positive samples were enriched for deletions of PAX5, IKZF1, CDKN2A, 13 , 14 , 15 and CDKN2B. 15 Bastian et al have recently described PAX5 plus, a subgroup defined by these enrichments, the frequent occurrence of RAS pathway mutations and the lack of assignability to a previously known group. 15 As formerly described none of our patients belonged to one of the known groups as well. However, we cannot confirm the favorable outcome of PAX5 P80R described in adults. 13 , 15 In fact, we observed rather intermediate to poor outcome 14 for patients harboring PAX5 P80R, which is in line with Gu et al (Figure 2).
With a rate of 1.9% in our study and the absence of established genetic risk markers, PAX5 P80R‐positive patients might represent a novel subgroup in BCP‐ALL, especially in adolescents and young adults. This subgroup seems to be further characterized by enrichment of copy number variations (CNVs) in specific genes other than PAX5 and by an intermediate to poor outcome. Although PAX5 P80R is not a germline variant it may represent an initiating event in the development of B‐ALL, 14 and subsequent biallelic loss of PAX5 function further contributes to neoplastic progression. 22 One important limitation of our study is the rare occurrence of PAX5 P80R in children, which resulted in a moderate case number despite the large cohort. Nevertheless, our data underline the importance of PAX5 P80R in pediatric BCP‐ALL and that international collaborative approaches with larger cohorts are warranted to further evaluate the interaction of PAX5 P80R with other genetic aberrations (eg, additional deletions in 9p, or complex aberrations of chromosome 20) and its impact on the patient's outcome.
Supporting information
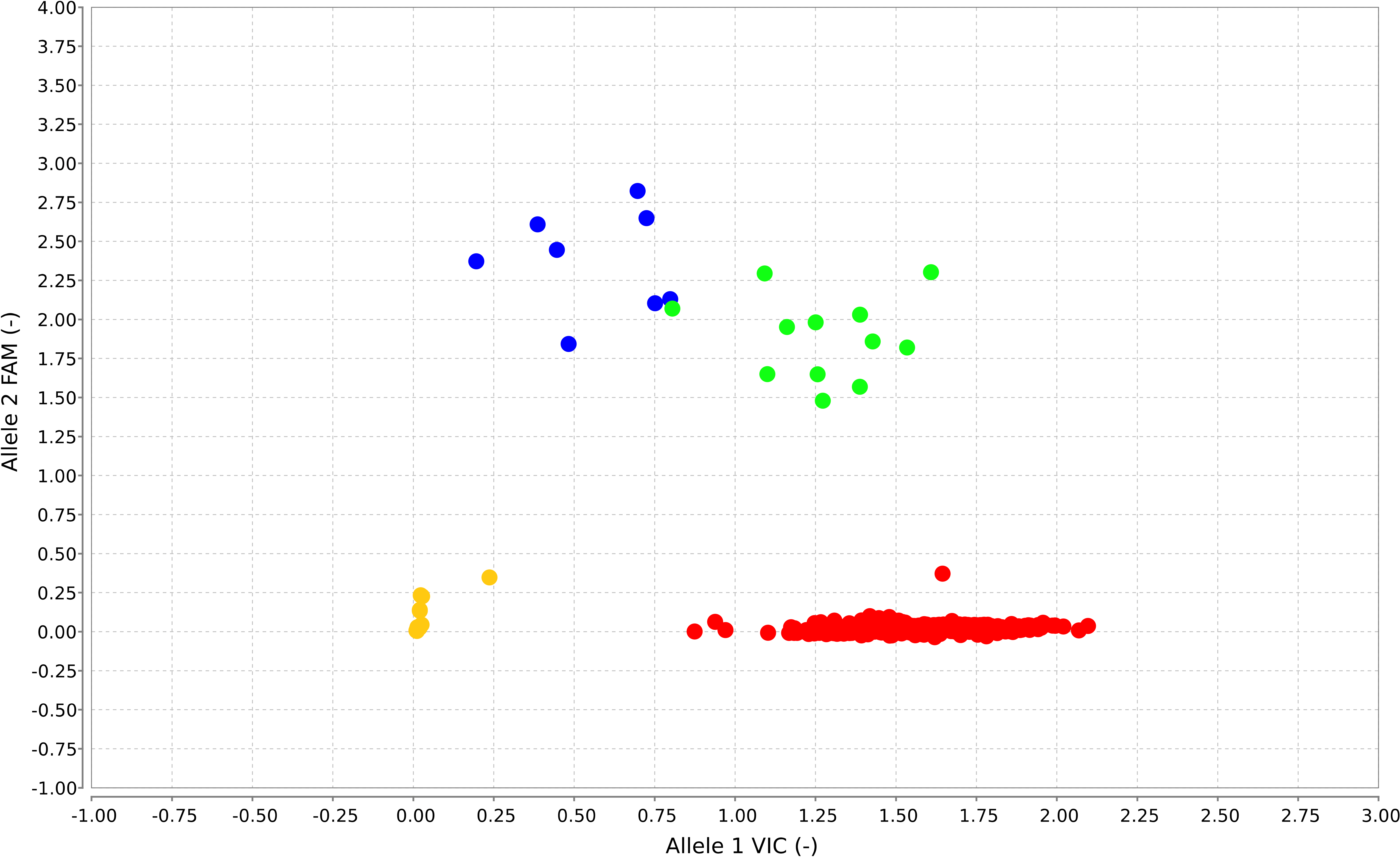
Supplementary Figure S1 TaqMan assay results of 1237 patients in trial AIEOP‐BFM (Associazione Italiana Ematologia ed Oncologia Pediatrica‐ Berlin ‐Frankfurt‐Muenster) ALL 2000. Allele discrimination plot: Each dot represents one patient or one non‐template control. Non‐template controls are depicted in yellow; PAX5 P80R‐negative patients are shown in red (WT/WT). Green and blue dots display hetero‐ or hemizygous PAX5 P80R‐positive patients (WT/P80R or P80R/− respectively) with the hemizygous cluster (blue) close to the heterozygous cluster (green). Green dots represent heterozygous patients with two different alleles (P80R/WT (12)), whereas blue dots represent those cases with P80R allele only (P80R/del (8)). Abbreviations: ALL, acute lymphoblastic leukemia.
{kind=link}
Supplementary Figure S2 Flow chart of identification of PAX5 P80R‐positive patients in trial AIEOP‐BFM (Associazione Italiana Ematologia ed Oncologia Pediatrica‐ Berlin‐Frankfurt‐Muenster) ALL 2000.
Abbreviations: AIEOP‐BFM, Associazione Italiana Ematologia ed Oncologia Pediatrica‐ Berlin‐Frankfurt‐Muenster; ALL, acute lymphoblastic leukemia, B‐ALL, B acute lymphoblastic leukemia; MLPA, multiplex ligation‐dependent probe amplification; T‐ALL, T‐cell acute lymphoblastic leukemia.
{kind=link}
Supplementary Figure S3 Chromosomal alterations in two PAX5 P80R ‐positive patients (patients 1 and 10) examined by Agilent CGH + SNP arrays (e‐Array design 85 320; Agilent Technologies). Abbreviations: 9, chromosome 9; 20, chromosome 20.
{kind=link}
Supplementary Table S1 PAX5 primers and nested primers used for Sanger sequencing of PAX5.
Supplementary Table S2 Agilent CGH + SNP array results.
Supplementary Table S3 Positive correlations of PAX5 P80R.
Supplementary Table S4 Estimated HRs from the Multivariable Cox Proportional Hazards Model on Event‐Free Survival and Overall Survival in 842 patients with B‐Cell precursor ALL from trial AIEOP‐BFM ALL 2000.
Jung M, Schieck M, Hofmann W, et al. Frequency and prognostic impact of PAX5 p.P80R in pediatric acute lymphoblastic leukemia patients treated on an AIEOP‐BFM acute lymphoblastic leukemia protocol. Genes Chromosomes Cancer. 2020;59:667–671. 10.1002/gcc.22882
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Stanulla M, Cavé H, Moorman AV. IKZF1 deletions in pediatric acute lymphoblastic leukemia: still a poor prognostic marker? Blood. 2020;135:252‐260. 10.1182/blood.2019000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schrappe M, Valsecchi MG, Bartram CR, et al. Late MRD response determines relapse risk overall and in subsets of childhood T‐cell ALL: results of the AIEOP‐BFM‐ALL 2000 study. Blood. 2011;118(8):2077‐2084. 10.1182/blood-2011-03-338707. [DOI] [PubMed] [Google Scholar]
- 3. Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B‐cell precursor acute lymphoblastic leukemia: results in 3184 patients of the AIEOP‐BFMALL 2000 study. Blood. 2010;115:3206‐3214. 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 4. Pui C‐H, Yang JJ, Hunger SP, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. 2015;33(27):2938‐2948. 10.1200/JCO.2014.59.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schwab C, Harrison CJ. Advances in B‐cell precursor acute lymphoblastic leukemia genomics. HemaSphere. 2018;2(4):e53 10.1097/hs9.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu YF, Wang BY, Zhang WN, et al. Genomic profiling of adult and pediatric B‐cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173‐183. 10.1016/j.ebiom.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mullighan CG, Goorha S, Radtke I, et al. Genome‐wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758‐764. 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 8. Nebral K, Denk D, Attarbaschi A, et al. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23(1):134‐143. 10.1038/leu.2008.306. [DOI] [PubMed] [Google Scholar]
- 9. Cazzaniga G, Daniotti M, Tosi S, et al. The paired box domain gene PAX5 is fused to ETV6/TEL in an acute lymphoblastic leukemia case. Cancer Res. 2001;61(12):4666‐4670. http://www.ncbi.nlm.nih.gov/pubmed/11406533 Accessed November 25, 2019. [PubMed] [Google Scholar]
- 10. Cobaleda C, Schebesta A, Delogu A, Busslinger M. Pax5: the guardian of B cell identity and function. Nat Immunol. 2007;8(5):463‐470. 10.1038/ni1454. [DOI] [PubMed] [Google Scholar]
- 11. Shah S, Schrader KA, Waanders E, et al. A recurrent germline PAX5 mutation confers susceptibility to pre‐B cell acute lymphoblastic leukemia. Nat Genet. 2013;45(10):1226‐1231. 10.1038/ng.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwab C, Nebral K, Chilton L, et al. Intragenic amplification of PAX5: a novel subgroup in B‐cell precursor acute lymphoblastic leukemia? Blood Adv. 2017;1(19):1473‐1477. 10.1182/bloodadvances.2017006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Passet M, Boissel N, Sigaux F, et al. PAX5 P80R mutation identifies a novel subtype of B‐cell precursor acute lymphoblastic leukemia with favorable outcome. Blood. 2019;133(3):280‐284. 10.1182/BLOOD-2018-10-882142. [DOI] [PubMed] [Google Scholar]
- 14. Gu Z, Churchman ML, Roberts KG, et al. PAX5‐driven subtypes of B‐progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296‐307. 10.1038/s41588-018-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bastian L, Schroeder MP, Eckert C, et al. PAX5 biallelic genomic alterations define a novel subgroup of B‐cell precursor acute lymphoblastic leukemia. Leukemia. 2019;33(8):1895‐1909. 10.1038/s41375-019-0430-z. [DOI] [PubMed] [Google Scholar]
- 16. Zaliova M, Zimmermannova O, Dörge P, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. 2014;28:182‐185. 10.1038/leu.2013.282. [DOI] [PubMed] [Google Scholar]
- 17. den Boer ML, van Slegtenhorst M, de Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome‐wide classification study. Lancet Oncol. 2009;10(2):125‐134. 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roberts KG, Morin RD, Zhang J, et al. Genetic alterations activating kinase and cytokine receptor signaling in high‐risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153‐166. 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roberts KG, Li Y, Payne‐Turner D, et al. Targetable kinase‐activating lesions in Ph‐like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005‐1015. 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stanulla M, Dagdan E, Zaliova M, et al. IKZF1plus defines a new minimal residual disease‐dependent very‐poor prognostic profile in pediatric B‐cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36(12):1240‐1249. 10.1200/JCO.2017.74.3617. [DOI] [PubMed] [Google Scholar]
- 21. Li J‐F, Dai Y‐T, Lilljebjörn H, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A. 2018;115(50):E11711‐E11720. 10.1073/pnas.1814397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dang J, Wei L, de Ridder J, et al. PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood. 2015;125(23):3609‐3617. 10.1182/blood-2015-02-626127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 TaqMan assay results of 1237 patients in trial AIEOP‐BFM (Associazione Italiana Ematologia ed Oncologia Pediatrica‐ Berlin ‐Frankfurt‐Muenster) ALL 2000. Allele discrimination plot: Each dot represents one patient or one non‐template control. Non‐template controls are depicted in yellow; PAX5 P80R‐negative patients are shown in red (WT/WT). Green and blue dots display hetero‐ or hemizygous PAX5 P80R‐positive patients (WT/P80R or P80R/− respectively) with the hemizygous cluster (blue) close to the heterozygous cluster (green). Green dots represent heterozygous patients with two different alleles (P80R/WT (12)), whereas blue dots represent those cases with P80R allele only (P80R/del (8)). Abbreviations: ALL, acute lymphoblastic leukemia.
Supplementary Figure S2 Flow chart of identification of PAX5 P80R‐positive patients in trial AIEOP‐BFM (Associazione Italiana Ematologia ed Oncologia Pediatrica‐ Berlin‐Frankfurt‐Muenster) ALL 2000.
Abbreviations: AIEOP‐BFM, Associazione Italiana Ematologia ed Oncologia Pediatrica‐ Berlin‐Frankfurt‐Muenster; ALL, acute lymphoblastic leukemia, B‐ALL, B acute lymphoblastic leukemia; MLPA, multiplex ligation‐dependent probe amplification; T‐ALL, T‐cell acute lymphoblastic leukemia.
Supplementary Figure S3 Chromosomal alterations in two PAX5 P80R ‐positive patients (patients 1 and 10) examined by Agilent CGH + SNP arrays (e‐Array design 85 320; Agilent Technologies). Abbreviations: 9, chromosome 9; 20, chromosome 20.
Supplementary Table S1 PAX5 primers and nested primers used for Sanger sequencing of PAX5.
Supplementary Table S2 Agilent CGH + SNP array results.
Supplementary Table S3 Positive correlations of PAX5 P80R.
Supplementary Table S4 Estimated HRs from the Multivariable Cox Proportional Hazards Model on Event‐Free Survival and Overall Survival in 842 patients with B‐Cell precursor ALL from trial AIEOP‐BFM ALL 2000.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.