

Abstract
Background
Drug repurposing enables the discovery of potential cancer treatments using publically available data from over 4000 published Food and Drug Administration approved and experimental drugs. However, the ability to effectively evaluate the drug's efficacy remains a challenge. Impediments to broad applicability include inaccuracies in many of the computational drug‐target algorithms and a lack of clinically relevant biologic modeling systems to validate the computational data for subsequent translation.
Methods
We have integrated our computational proteochemometric systems network pharmacology platform, DrugGenEx‐Net, with primary, continuous cultures of conditionally reprogrammed (CR) normal and prostate cancer (PCa) cells derived from treatment‐naive patients with primary PCa.
Results
Using the transcriptomic data from two matched pairs of benign and tumor‐derived CR cells, we constructed drug networks to describe the biological perturbation associated with each prostate cell subtype at multiple levels of biological action. We prioritized the drugs by analyzing these networks for statistical coincidence with the drug action networks originating from known and predicted drug‐protein targets. Prioritized drugs shared between the two patients’ PCa cells included carfilzomib (CFZ), bortezomib (BTZ), sulforaphane, and phenethyl isothiocyanate. The effects of these compounds were then tested in the CR cells, in vitro. We observed that the IC50 values of the normal PCa CR cells for CFZ and BTZ were higher than their matched tumor CR cells. Transcriptomic analysis of CFZ‐treated CR cells revealed that genes involved in cell proliferation, proteases, and downstream targets of serine proteases were inhibited while KLK7 and KLK8 were induced in the tumor‐derived CR cells.
Conclusions
Given that the drugs in the database are extremely well‐characterized and that the patient‐derived cells are easily scalable for high throughput drug screening, this combined in vitro and in silico approach may significantly advance personalized PCa treatment and for other cancer applications.
Keywords: conditionally reprogrammed cells, drug repurposing, primary prostate, systems network pharmacology, tissue
Abbreviations
- BTZ
bortezomib
- CFZ
carfilzomib
- CM
conditioned media
- CR cells
conditionally reprogrammed cells
- CRPC
castrate resistant prostrate cancer
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- DEGs
differentially expressed genes
- GSH
glutathione
- IPA
Ingenuity Pathways Analysis
- PCa
prostate cancer
- PSA
prostate specific antigen
- PTC
phenethyl isothiocyanate
- SFN
sulphoraphane
- TMFS
train match fit streamline
1. INTRODUCTION
Prostate cancer (PCa) remains the second most common cancer in men, with ∼1 in 6 men being diagnosed with PCa in their lifetime. Once diagnosed with PCa, most men undergo curative therapy, normally radiation, or surgery. While the 5‐year median survival rate for localized PCa approaches 100%, ∼30% of patients will have a biochemical recurrence and androgen deprivation therapy (ADT) remains the standard of care for these patients.
The death rate in patients with Gleason score 8 PCa has been reported as almost double that of Gleason score 6. 1 , 2 Men who progress to castrate‐resistant prostate cancer (CRPC) following ADT face a limited treatment repertoire, including docetaxel chemotherapy, next‐generation androgen/androgen receptor blockading drugs, sipuleucel‐T (autologous immunotherapy), and radium‐223. 3 , 4 , 5 The overall survival rates and quality of life for patients who have progressed to CRPC remains problematic due to the inability to effectively treat CRPC. In addition to better treatments for CRPC, there is an intense and justifiable need to identify or develop new therapeutics that more effectively prevent the need for ADT and the subsequent progression to CRPC.
To help achieve the goal of preventing PCa progression to CRPC, robust in silico approaches that identify approved drugs that may be cytotoxic to PCa cells, followed by validation of the predicted drug‐target interactions in biologically accurate model systems, are required. The ability to accurately predict drug efficacy at the gene, cellular, and organismal level is extremely challenging. We developed a novel proteochemometric drug repurpose method called “Train, Match, Fit, and Streamline” (TMFS) and a subsequent, refined version called RePurposeVS to predict alternative drug targets for all Food and Drug Administration (FDA)‐approved and experimental drugs. 6 Most recently, we integrated our repurposing methodology with network pharmacology called DrugGenEx‐Net and applied these approaches for use in melanoma and triple‐negative breast cancer (DrugGenEx‐TNBC). 7 , 8 As an example, our proteochemometric method successfully identified mebendazole as having the structural potential to inhibit kinases, especially BRAF (both wild type and mutant [BRAFV600]), and is a potential therapeutic option for melanoma patients in combination with trametinib. 9 Coupling network pharmacology with drug repurposing offers solutions to the problems encountered when a target is not well characterized structurally and/or is not known to be druggable, 7 as these targets are likely to be members of a pathway/network that contains other molecules that may already be druggable.
In this study, we applied the RePurpose network pharmacology platform and DrugGenEx‐Net and GenEx‐TNBC 7 , 8 to the transcriptomic data from our published Gleason 6 and Gleason 8 conditionally reprogrammed (CR) malignant prostate cells and their patient‐matched benign cells. 10 , 11 , 12 , 13 , 14 Using this approach, we identified drugs that were already in clinical trials for PCa, including carfilzomib (CFZ), 15 sulforaphane (SFN), 16 and others that are not currently in clinical trials, such as phenethyl isothiocyanate (PTC), SB220025, and glutathione (GSH). We show that the tumor‐derived prostate CR cells had enhanced sensitivity to bortezomib (BTZ) and CFZ, a first‐line therapy for multiple myeloma, 17 and that CFZ treatment resulted in the preferential regulation of proteasome and protease genes and of genes involved in cell cycle progression and proliferative signaling. Collectively, our data suggest that relative ease of use of the patient‐derived CR primary prostate cells, combined with computational modeling and in vitro efficacy testing, provides the feasibility to begin scale these approaches into larger preclinical studies and eventually into personalized clinical trials.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
Human radical prostatectomy samples were collected under the auspices and approval of the Georgetown University and Massachusetts General Hospital Institutional Review Boards. Following detailed pathological analyses that documented that the tissue sections collected were more than 70% tumor cells, the specimens were processed via protease dissociation as previously described. 11 , 18 Primary cultures were established at Georgetown University using the CR method, and drug senstivity experiments were performed in conditioned media (CM) as previously described. 19 Genetic profiles, obtained by performing metaphase spreads for the normal and tumor‐derived CR cells were performed. The Gleason 6 and 8 PCa CR cells remained diploid but show increasing incidences of chromosomal aberrations, with the Gleason 8 cells from patient 2 exhibiting numerous translocations and markers while the normal prostate CR cells are diploid 46XY with no known translocations. 14
2.2. Development of drug‐target biological associations
Experimentally validated drug‐protein interactions, as well as known agonism or antagonism, were curated from publicly available databases into a master set of direct biological targets for approved and experimental drugs. These data were obtained from the Drug‐Gene Interaction Database 20 and the Comparative Toxicogenomics Database. 21 Drug‐protein interactions were marked as agonistic, antagonistic, or unknown. Interactions with proteins whose genes were not assessed in our gene expression analysis were discarded. The resulting drug‐target set contains a total of 8054 drugs and 21, 353 interactions. In addition, we utilized a modified version of our TMFS method called RePurposeVS for generating reliable binding signature predictions. 6 , 22
2.3. Development of multi‐level drug‐target biological associations
Higher‐level drug‐biological interactions, including drug‐pathway and drug‐Gene Ontology (GO) function (drug‐function), were inferred from the protein targets for a given drug. Protein‐pathway and protein‐function annotations were obtained from ConsensusPathDB 23 and Database for Annotation, Visualization, and Integrated Discovery (DAVID) Functional Annotation Tool (FAT), 24 respectively. Pathways and functions whose component gene products were modulated in a known direction by a drug were considered to be modulated in that same direction. Thus for a given drug, a pathway or function could be marked as either upregulated, downregulated, unknown, or both up‐ and downregulated.
2.4. Differential gene expression and functional annotation using patient‐derived cancer and normal tissue samples
To determine differentially expressed genes (DEGs), a fold change cutoff for the cancer sample relative to the normal sample of greater than 2.5 or less than −2.5 was implemented from 47 315 total genes with no greater than 1500 upregulated and 1500 downregulated genes of the largest absolute fold change.
For each patient gene expression data set, upregulated and downregulated gene lists separately underwent functional enrichment analysis to determine those pathways and functions that were overrepresented in the set of genes differentially expressed by malignant prostatic tissue. Functional enrichment was carried out using the web interface tools with a P‐value cutoff of < .05. These tools include ConsensusPathDB 23 for pathway annotation and DAVID FAT 24 for function annotation.
FAT was also used to annotate pathways and functions for a given drug via its predicted direct and indirect targets through protein‐protein interactions (PPIs) using False Discovery Rate (FDR) less than 0.25. The final tier for biological characterization of DEGs is based on PPIs, which were curated from the STRING database using a “high confidence” score cutoff of more than 0.7. 25 Any PPI pairs where one of the partners did not exist in our protein target data set were excluded. A gene list comprised of the drugs' predicted and known direct targets and interacting partners was subjected to the DAVID annotation. For example, if Drug A was predicted to interact with Target A and Target B, and Target A also interacted with Protein C while Target B interacted with Protein D and Protein E, then the gene list for Drug A would consist of the following: Target A, Target B, Protein C, Protein D, and Protein E. The master set of PPIs were narrowed down to a disease‐PPI network which only included those gene products (A) whose genes were assessed in our patient‐derived gene expression analysis and (B) where at least one gene product was differentially expressed in our analysis.
2.5. Statistical overrepresentation analysis between drug and disease networks using a causal analysis algorithm
The above computational strategies resulted in drug‐biological and disease‐biological molecular association networks that were assessed for statistical co‐occurrence for each drug at every biological tier. This was done using an algorithmic strategy first implemented in the GenEx‐TNBC platform, 6 , 8 in which drug‐biologic interactions of an antagonistic nature matched with upregulated disease components, and vice versa. When the nature of the drug interaction is unknown or undefined, it can match with either up‐ or downregulated disease‐biological components. These disease and drug‐biological components were matched in this way at each level of biological activity, including genes, pathways, functions, and PPIs.
Statistical analysis of the significance of the interaction between matching disease and drug components was carried out using the hypergeometric test in R. Calculated probabilities for each tier were log‐normalized and summated to rank drugs according to potential therapeutic efficacy, as we described previously. 6 , 8 Briefly, each drug's Z‐score (Zi), which represents the final quantification of the drug‐PCa patient sample association, was calculated for a ranking using the following equation
| (1) |
where, A, B, C, and D correspond to the normalized hypergeometric test P‐values for drug‐gene product, pathway, function, and PPI associations, respectively. a, b, c, and d represent coefficient values of 2, 1, 1, and 1 with respect to each biological tier. As we previously described, coefficient values were determined to best prioritize direct drug‐gene product interactions over indirect interactions at higher‐order biological tiers while also allowing for the prioritization of drugs that do not necessarily have direct interactions but may be therapeutic through indirect mechanisms. 7 Using the final calculated Z‐score, drugs are ranked in descending order (eg, the drug with the highest Z‐score is considered the number one top‐ranked drug for a particular PCa patient sample. Thus, a high Z‐score indicates a drug's polypharmacological and multitiered potential to serve as a therapeutic for a given PCa patient sample.
2.6. Drug sensitivity assays
Drug sensitivity assay experiments were performed as described previously. 18 Briefly, 3000 prostate CR cells were seeded per well of 96 well plates in CM. Each CR ‐ line was seeded in triplicate. CM was added to a final volume of 100 μL. After 24 hour of seeding, CM containing drugs SFN, PTC, CFZ, BTZ, SB220025, or GSH was added. Cell survival was assessed after 72 hour using the WST‐1 reagent (Sigma #11644807001). Data were analyzed using Prism5 (GraphPad software, Inc, ver 5.0a). Each experiment was performed in triplicate.
2.7. RNA sequencing analysis
RNA from CR cells cultures grown in J2 CM was isolated from cell pellets of malignant and normal cell lines using an RNA Easy Kit (Qiagen Cat No. 74104). Expression analysis was performed in the Georgetown‐Lombardi Genomics and Epigenomics Shared Resource using the HumanHT‐12 v4 Expression BeadChip. Briefly, normalized data were imported into the R computing environment and analyzed using the Linear Models for Microarray Data package (LIMMA, 3.30.13) 26 as part of the larger Bioconductor project (www.Bioconductor.org). 27 A linear model was fit for the normalized log ratios of every gene using the “lmFit” function within LIMMA to estimate all systematic variability in the data. Using functions in LIMMA, pairwise comparisons were performed between tumor and normal cells cultured in CM to compute moderated t statistics, log‐odds ratios of differential expression (based on empirical Bayes for shrinkage of standard errors), and adjusted P‐values using the Benjamini‐Hochberg method. 28
2.7.1. Pathway analysis
Gene interaction networks, biofunctions, and pathway analysis were performed using Ingenuity Pathway Analysis (IPA) (Ingenuity Systems; Mountain View, CA) and GO Panther tools. 29 The DEGs were mapped to molecular functions and genetic networks available from the Ingenuity database. For protein class enrichment, the DEGs were uploaded into Panther tools and were mapped to the category of “protein class.” Venn diagrams (Venny 2.1.0) 30 yielded the list of genes exclusively regulated in tumor or normal CR cells. For canonical pathway analyses, z‐scores as a statistical measure of the match between expected relationship direction and observed gene expression were used, as previously described. 14 , 18
3. RESULTS
3.1. Microarray analysis and identification of drugs by network pharmacology
Two matched pairs of normal and tumor PCa samples were collected under the approval of the Institutional Review Boards of the Massachusetts General Hospital and Georgetown University Medical Center. The samples came from patients who underwent radical prostatectomy for Gleason 6 (patient 1) and Gleason 8 (patient 2) PCa and have been previously described. 10 , 14 , 31 Chromosomal analyses revealed that both of the tumor‐derived CR cells remained diploid, however, increased incidences of chromosomal aberrations were observed in the tumor‐derived cells from patient 2, which exhibited numerous chromosomal translocations and markers. 14
Gene transcription microarrays were performed by Illumina bead array on the matched normal and tumor cells cultured in CM as previously described. 14 , 18 , 19 The data were analyzed by IPA based on differential gene expression between the normal CR cells and the matched tumor‐derived CR cells as described. 14 , 18 Gene expression analysis identified a total of 122 DEGs in the tumor vs normal cells (fold change [FC] 1.5, P ≤ .09) in patient 1 and 203 genes (FC 1.5, P ≤ .09) in patient 2 (Table S1). GO analysis of the array data from the tumor vs normal CR cells revealed that hallmarks of cancer, including increased cancer, tumorigenesis, cancer cell invasion, and proliferation of epithelial cells, were more prominent in the PCa CR cells from patient 2 (Gleason 8) as compared with patient 1 (Gleason 6) (Figure 1A). Furthermore, the GO analysis revealed reductions in genes associated with cellular homeostasis and apoptosis in the tumor‐derived CR cells from patient 2 (Figure 1A).
Figure 1.
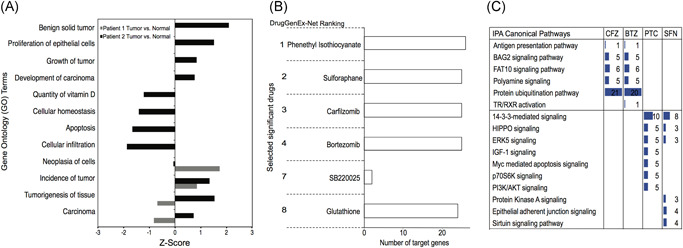
Comparison of tumor vs normal‐derived cells. A, Gene ontology analysis for the microarray expression profile of patient 1 and patient 2 tumor vs benign, normal CR cells cultured in J2‐conditioned media (FC 1.5, P < .09). Negative Z‐scores denote lower expression in tumor cells and positive Z‐scores indicate induced genes. B, Ranked FDA approved chemical or drugs as predicted by DrugGenEx‐Net for patient 1 and patient 2. C, Ingenuity Pathway Analysis (IPA) of the predicted target genes sets for carfilzomib (CFZ), bortezomib (BTZ), phenethyl isothiocyanate (PTC), and sulforaphane (SFN) as predicted by DrugGenEx‐Net. CR, conditionally reprogrammed; FC, fold change; FDA, Food and Drug Administration [Color figure can be viewed at wileyonlinelibrary.com]
The transcriptomic data were then processed by our previously published computational platform to build drug‐biological and disease‐biological molecular association networks and to prioritize thousands of approved and experimental drugs for therapeutic potential in PCa. 6 , 8 , 22 We constructed networks to define the biological perturbations that distinguish the PCa CR cells from their patient‐matched normal cells. These networks were analyzed for statistical coincidence with drug action networks stemming from known drug‐protein targets, while accounting for the direction of disease modulation, to prioritize the drugs for testing in the patient's cells. We identified more than 10 FDA approved and experimental drugs for each of the PCa CR cells as possible inhibitors of key cancer‐associated genes and pathways. A complete list of drugs and the associated genes and pathways is provided for patient 1 and patient 2 (Tables S2 and S3).
3.2. Target genes and pathways
The top scoring “hits” are shown (Figure 1B). Those that are currently undergoing clinical trials include SFN, CFZ, and BTZ, while GSH, PTC, and the p38 inhibitor, SB220025, are not currently in trials (Figure 1B).
IPA and DrugGenEx‐Net and GenEx‐TNBC 7 , 8 pathway analyses performed on the predicted genes showed that the proteasome inhibitors (BTZ and CFZ) and the isothiocyanates (SFN and PTC), target distinct signaling pathways (Figure 1C). With the exception of thyroid hormone receptor‐α within the TR/RXR activation pathway, which was a unique candidate target of BTZ, both CFZ and BTZ were predicted to target similar sets of genes (Tables S2 and S3). The highest impact of both proteasome inhibitors was predicted to be on the protein ubiquitination pathway, while for both SFN and PTC, the 14‐3‐3, HIPPO and ERK5 signaling pathways were predicted to be highly impacted pathways (Figure 1C). The full list of targeted genes and pathways is provided (Tables S2 and S3).
3.3. Drug sensitivity profiling
To validate the integrated drug repurpose network pharmacology platform predictions, the IC50 values of the normal and tumor CR cells from both patients were established for GSH, SB220025, SFN, PTC, CFZ, and BTZ. The dose‐response curves were performed on cells in CM as previously described. 21 The calculated IC50's of the matched normal and tumor CR cells from patient 1 and 2 are shown (Figures 2A,B and S1). Patient 1's tumor‐derived CR cells were more sensitive to SFN, CFZ, and BTZ than the benign CR cells (P < .05). Patient 2's tumor cells also showed significant responses to CFZ and BTZ (P < .01; Figure 2A), however, no differences in sensitivity between the normal and tumor‐derived CR cells from patient 2 were seen for either SFN or PTC (Figure 2B). No significant differential responses were found for SB00225 or GSH for either patient's cells (Figure S1). The full dose‐response curves for CFZ and BTZ for both patients are shown (Figure 2C‐F).
Figure 2.
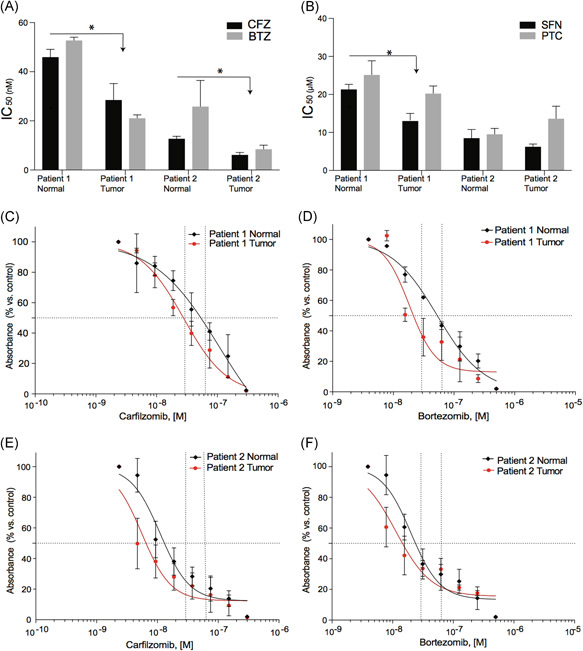
Comparison of IC50 for both patient's tumor and normal CR cells exposed to drugs ranked by DrugGenEx‐Net. IC50 calculations performed on the CR cells exposed to (A) CFZ and BTZ or (B) SFN or PTC for 72 hour. 32 : molar. (C and D) Detailed dose‐response curves of the CR cells from patient 1 treated with CFZ or BTZ, respectively. (E and F) Detailed dose‐response curves of the CR cells from patient 2 treated with CFZ or BTZ, respectively All values are the average ± SD of three separate experiments performed in triplicate. BTZ, bortezomib; CFZ, carfilzomib; CR, conditionally reprogrammed; PTC, phenethyl isothiocyanate; SFN, sulforaphane [Color figure can be viewed at wileyonlinelibrary.com]
3.4. Analyses of tumor‐derived CR cells to identify genes associated with increased sensitivity
Because of the heightened sensitivity of the PCa‐derived CR cells to CFZ and BTZ (Figure 2), the transcriptomic data of the untreated tumor and normal CR cells was next used to classify DEGs into protein classes using protein Panther (analysis through evolutionary relationships) tools 29 (Figure 3A). The hydrolases and enzyme modulators were the most enriched protein class in the tumor‐derived CR cells. Furthermore, subclassification of the hydrolases revealed that the proteases were the most enriched protein class in the tumor‐derived CR cells (Figure 3B), however, notable differences between the CR cells were seen. For example, lipases and esterases were enriched in the tumor CR cells from patient 1 while phosphodiesterases and deaminases were more highly enriched in the tumor CR cells from patient 2 (Figure 3B).
Figure 3.
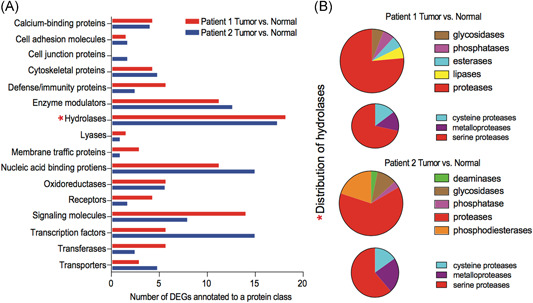
Differentially regulated genes. A, The list of differentially expressed genes, annotated to protein class using Panther tools. Hydrolases were identified as the most enriched protein class in tumor versus normal cells in both patients (*). B, Analyses of the subcategories of proteases differentially regulated in the tumor cells from both patients. DEGs, differentially expressed genes [Color figure can be viewed at wileyonlinelibrary.com]
3.5. Responses to drug treatment
Next, tumor and benign CR cells from both patients were treated with CFZ at the respective tumor CR cells IC50 (28 nM for patient 1 and 6.5 nM for patient 2) or with dimethyl sulfoxide (DMSO), followed by RNA sequencing (RNA‐seq) analysis. The RNA‐Seq data from CFZ‐treated patient 1 tumor and normal CR cells, compared with DMSO‐treated tumor and normal CR cells, identified 1689 and 1248 DEGs, respectively (FDR < 0.05 and FC 1.5). For patient 2, CFZ treatment of the tumor and normal CR cells, compared with DMSO‐treated cells, led to 9199 and 9562 DEGs, respectively. The list of all DEGs for both patient CR cells is provided (Table S4). These RNA‐seq data were then used to explore the effects of CFZ on proteasome and protease genes.
In the CR cells from the patient 1, the expression levels of a broad array of genes that comprise components of the 26S proteasome were more significantly repressed by CFZ in the tumor‐derived vs normal cells (blue squares) with a small subset being induced in both (red squares; Figure 4A). The total and differentially regulated serine‐ and metalloprotease genes impacted by CFZ are shown (Figure 4B). CFZ treatment resulted in significant differences in expression between the normal and tumor CR cells ‐ for PRSS21, PRSS22, MMP9, MMP11, MMP13, MMP16, MMP25, TMPRSS3, and TMPRSS4.
Figure 4.
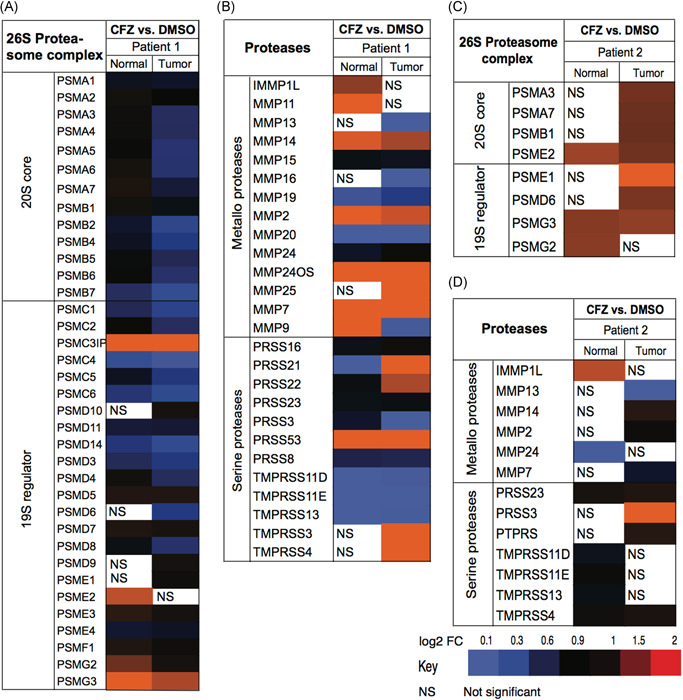
Differential effects of CFZ treatment on gene expression. A, Impact of CFZ on proteasomal subunit genes in the CR cells from patient 1. B, Impact of CFZ on serine proteases and metalloproteases in CR cells from patient 1. Differential effects of CFZ treatment on (C) proteasomal subunits and (D) serine proteases and metalloproteases in CR cells from patient 2. CFZ, carfilzomib; CR, conditionally reprogrammed; DMSO, dimethyl sulfoxide; FC, fold change [Color figure can be viewed at wileyonlinelibrary.com]
In the tumor CR cells from patient 2, CFZ modestly induced a small number of proteasomal genes that were repressed in the tumor CR cells from patient 1. Differences in responses to CFZ of the serine‐ and metalloprotease genes were also observed between patient 1 and patient 2, with only MMP13 and IMMP1L (inner mitochondrial membrane peptidase subunit 1) showing similar CFZ‐targeting between the two patients' cells. Overall, differential responses to CFZ were observed both between benign and tumor‐derived CR cells as well as between patients.
To more clearly define the effects of CFZ, IPA network analyses were performed on the gene‐sets exclusively regulated in tumor or normal CR cells, treated with CFZ, from both patients. The list of exclusively up‐ and downregulated genes by CFZ in both patients, was retrieved using Venny 2.1.0. 30 The numbers in the shaded regions represent differentially regulated genes shared in multiple comparisons. The description of these comparisons is provided in Table S5. We identified 23 common genes that were exclusively induced in the tumor CR cells (FDR 0.05; FC 1.5) while 58 shared genes were induced in the normal CR cells (Figure 5A). Among the significantly inhibited genes (FDR 0.05; FC 1.5), there were 45 genes inhibited that were shared in the tumor CR cells, with 56 shared genes inhibited in the normal CR cells (Figure 5B).
Figure 5.
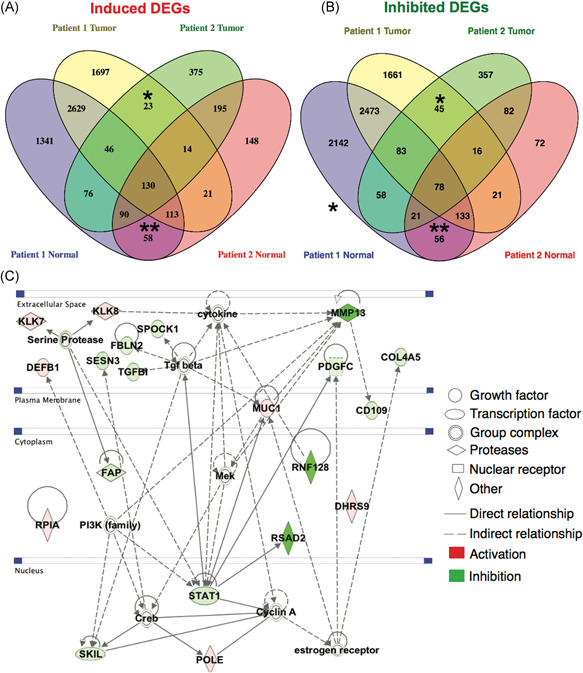
Genes exclusively regulated by CFZ in normal and tumor CR cells. A, Venn diagram of the sets of genes induced (FC 1.5; FDR 0.05) by CFZ in the CR cells from patients 1 and 2. *Induced genes shared between the tumor‐derived cells. **Induced genes shared between the normal cells. B, Venn diagram of the sets of inhibited genes (FC 1.5; FDR 0.05) by CFZ in the CR cells from patients 1 and 2. *Inhibited genes shared between the tumor‐derived cells. **Inhibited genes shared between the = normal cells (C) Ingenuity Pathway Network analyses of the genes that were regulated by CFZ exclusively in the tumor‐derived CR cells. CFZ, carfilzomib; CR, conditionally reprogrammed; FC, fold change; FDR, False Discovery Rate [Color figure can be viewed at wileyonlinelibrary.com]
The IPA network analyses identified genes involved in cell proliferation, including STAT1, TGFB, CD109, PDGFC), MMP13, and downstream targets of serine proteases (SESN3 and FAP) as being inhibited in both of the tumor‐derived CR cells (Figure 5C). KLK7 was induced by CFZ (Figure 5C) and KLK7 has been shown to be suppressed during PCa progression with its loss correlating with higher Gleason score. 33 No significant effects on these genes were observed in the benign CR cells (Figure S2).
4. DISCUSSION
Despite the potential for cost‐effectiveness, drug repurposing by computational and chemical screening approaches remains complex and can be time, labor, and resource intensive. 34 To address the aforementioned shortcomings, we used an integrated predictive network pharmacology platform to accurately and rapidly predict new drug‐target interactions and their new uses from the available transcriptomic data. 22 While other attempts at drug repurposing for CRPC have been attempted, we believe this is a first of its type study using treatment‐naive primary prostate cells in culture to identify and subsequently verify drug efficacy. 35 Our platform has successfully predicted the novel applications of celecoxib 36 and the antiparasitic, mebendazole 9 for use in cancer treatment. Similarly, the successes achieved with our CR approach to treat lethal diseases is evident by our earlier studies. 37 Our recent studies have further established that the CR culture conditions support long term genomic stability and can validate drug response in vivo and in vitro 18 and, therefore, may be vital resources for personalized drug identification.
In the present study, the transcriptomic data from normal and tumor‐derived primary prostate cells was used to identify the drug‐ligand interactions. The cells were then used to test the sensitivity to the predicted drugs, such as CFZ, and to begin to define the mechanisms of differential cell targeting. The increased sensitivity of the tumor CR cells to CFZ was associated with alterations in protease gene expression, however, the effects of CFZ differed between patients. For example, patient 1 tumor CR cells showed robust reductions in genes associated with the 26S proteasome and serine‐ and metalloproteases, while genes such as PSMD6 and PSMA3, which were induced in patient 2, were repressed in patient 1. We then chose to define targets that were co‐regulated between patients. Both PRSS23 and PSMG3 were induced in all CR cells, making these genes unlikely targets for differential drug sensitivity. Of the protease genes, only MMP13, which was repressed in the tumor‐derived CR cells, and IMMP1L, which was induced in the benign CR cells and below detection in the tumor‐derived CR cells, were differentially regulated. IMMP1L is a part of the mitochondrial inner membrane peptidase complex and is involved in generating mature proteins in the mitochondria. While little is known about its role in PCa, mitochondrial inner membrane permeabilization has been found to occur during BAX/BAK‐dependent induction of apoptotic cell death. 38 The induction of IMMP1L by CFZ may help protect the normal CR cells against apoptosis. Interestingly, MMP13 is induced in aggressive PCa 39 and MMP13 knockdown has been shown to impact proliferation in melanoma cells. 40 Our data suggest that MMP13 targeting by CFZ may have impacts on PCa cells beyond altering the extracellular matrix. In addition, both patients' tumor‐derived CR cells exhibited a reduction in various signaling pathway‐related genes, including STAT1, TGFβ, and PDGF. As CFZ failed to reduce the levels of these genes in the normal cells, their inhibition may contribute in part to the differential sensitivities observed.
Clinically, BTZ has been found to be partially effective in phase I trials in patients with metastatic CRPC, with 25% of the patients showing stable PSA and 4% of patients with more than 50% decline in PSA. 15 Modest responses were also noted in a phase II trial of CFZ in combination with other drugs in patients with metastatic CRPC, with a decline in PSA observed in 3.5% of patients. Similarly, while clinical 41 and epidemiological 42 , 43 studies have suggested that SFN rich diets may impact PCa progression, ‐ in clinical trial NCT01228084, only 1 out of 20 patients with recurrent PCa, treatmed with 200 μmoles/d of SFN rich extracts, showed more than a 50% decline in PSA in. 16 Interestingly our data with SFN established that the CR cells from patient 1 (Gleason 6), but not from patient 2 (Gleason 8) showed responses to SFN. Overall, these and other failed or inconclusive clinical trials underscore the need for biological testing and validation of potential/predicted drugs and drug combinations.
5. CONCLUSIONS
Chemoprevention of PCa progression has the capacity to greatly enhance the quality of life of men who have been treated for localized primary PCa. By combining the easily propagated, patient‐derived CR prostate cells with network pharmacology and subsequent in vitro modeling, our findings suggest that our integrated methodology provides broad applicability to rapidly identify and test approved drugs and provides a novel approach to perhaps prevent, or at least delay, biochemical recurrences in patients with primary PCa.
While we are not aware of any studies investigating their use in localized PCa, our data suggests that these drugs may be efficacious, either alone or in combination with standard of care interventions such a prostatectomy or radiation, in early stage and localized PCa. If biochemical recurrence could be delayed, or perhaps avoided, the need for ADT and the subsequent progression to CRPC may also be deferred, positively impacting both survivorship and quality of life. In addition, DrugGenEx‐Net also provides the ability for repeat applicability in clinical care. For example, should tumors recur, transcriptomic data from the patient's tumors can be used to rapidly identify drugs to further advance clinical care. Clinical trials are needed to investigate these intriguing possibilities.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supplementary Table S1 Complete list of the genes differentially expressed (P < .09, FC 1.5) in tumor vs normal CR cells for patient 1 and patient 2
Supplementary Table S2 Complete list of the predicted drug and target genes and pathways for patient 1
Supplementary Table S3 Complete list of the predicted drug and target genes and pathways for patient 2
Supplementary Table S4 Complete list of the genes differentially expressed (FDR < 0.05, FC 1.5) in tumor vs normal CR cells for patient 1 and patient 2 after treatment with carfilzomib
Supplementary Table S5. Categories of differentially regulated genes (FDR <0.05, FC 1.5) defined by venny 2.1.0
ACKNOWLEDGMENTS
The following Georgetown‐Lombardi Comprehensive Cancer Center core facilities were used throughout the study: the Genomics and Epigenomics Shared Resource, the Histology and Tissue Shared Resource, the Cell Culture Shared Resource and the Computational Chemistry Shared Resource The study was funded by DOD‐PC140268 (Albanese), DOD‐CA140882 (Dakshanamurthy), NIH‐P30 CA051008‐25 (Weiner), and NIH‐U01 CA194730‐04 (Kumar and Albanese). “Open access funding enabled and organized by Projekt DEAL.”
Naeem A, Dakshanamurthy S, Walthieu H, et al. Predicting new drug indications for prostate cancer: The integration of an in silico proteochemometric network pharmacology platform with patient‐derived primary prostate cells. The Prostate. 2020;80:1233–1243. 10.1002/pros.24050
Aisha Naeem and Sivanesan Dakshanamurthy contributed equally to the study.
REFERENCES
- 1. Mahal BA, Berman RA, Taplin ME, Huang FW. Prostate cancer‐specific mortality across Gleason scores in black vs nonblack men. JAMA. 2018;320(23):2479‐2481. [DOI] [PubMed] [Google Scholar]
- 2. Stangelberger A, Waldert M, Djavan B. Prostate cancer in elderly men. Rev Urol. 2008;10(2):111‐119. [PMC free article] [PubMed] [Google Scholar]
- 3. Antonarakis ES, Kibel AS, Yu EY, et al. Sequencing of sipuleucel‐T and androgen deprivation therapy in men with hormone‐sensitive biochemically recurrent prostate cancer: a phase II randomized trial. Clin Cancer Res. 2017;23(10):2451‐2459. [DOI] [PubMed] [Google Scholar]
- 4. Sydes MR, Spears MR, Mason MD, et al. Adding abiraterone or docetaxel to long‐term hormone therapy for prostate cancer: directly randomised data from the STAMPEDE multi‐arm, multi‐stage platform protocol. Ann Oncol. 2018;29(5):1235‐1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wallis CJD, Klaassen Z, Bhindi B, et al. Comparison of abiraterone acetate and docetaxel with androgen deprivation therapy in high‐risk and metastatic hormone‐naive prostate cancer: a systematic review and network meta‐analysis. Eur Urol. 2018;73(6):834‐844. [DOI] [PubMed] [Google Scholar]
- 6. Issa NT, Peters OJ, Byers SW, Dakshanamurthy S. RepurposeVS: a drug repurposing‐focused computational method for accurate drug‐target signature predictions. Comb Chem High Throughput Screen. 2015;18(8):784‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Issa NT, Kruger J, Wathieu H, Raja R, Byers SW, Dakshanamurthy S. DrugGenEx‐Net: a novel computational platform for systems pharmacology and gene expression‐based drug repurposing. BMC Bioinformatics. 2016;17(1):202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wathieu H, Issa NT, Fernandez AI, et al. Differential prioritization of therapies to subtypes of triple negative breast cancer using a systems medicine method. Oncotarget. 2017;8(54):92926‐92942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simbulan‐Rosenthal CM, Dakshanamurthy S, Gaur A, et al. The repurposed anthelmintic mebendazole in combination with trametinib suppresses refractory NRASQ61K melanoma. Oncotarget. 2017;8(8):12576‐12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ringer L, Sirajuddin P, Tricoli L, et al. The induction of the p53 tumor suppressor protein bridges the apoptotic and autophagic signaling pathways to regulate cell death in prostate cancer cells. Oncotarget. 2014;5(21):10678‐10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X, Ory V, Chapman S, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180(2):590‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pollock CB, McDonough S, Wang VS, et al. Strigolactone analogues induce apoptosis through activation of p38 and the stress response pathway in cancer cell lines and in conditionally reprogramed primary prostate cancer cells. Oncotarget. 2014;5(6):1683‐1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Timofeeva OA, Palechor‐Ceron N, Li G, et al. Conditionally reprogrammed normal and primary tumor prostate epithelial cells: a novel patient‐derived cell model for studies of human prostate cancer. Oncotarget. 2017;8(14):22741‐22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tricoli L, Naeem A, Parasido E, et al. Characterization of the effects of defined, multidimensional culture conditions on conditionally reprogrammed primary human prostate cells. Oncotarget. 2018;9(2):2193‐2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang H, Zonder JA, Dou QP. Clinical development of novel proteasome inhibitors for cancer treatment. Expert Opin Investig Drugs. 2009;18(7):957‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alumkal JJ, Slottke R, Schwartzman J, et al. A phase II study of sulforaphane‐rich broccoli sprout extracts in men with recurrent prostate cancer. Invest New Drugs. 2015;33(2):480‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moreau P, Mateos MV, Berenson JR, et al. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018;19(7):953‐964. [DOI] [PubMed] [Google Scholar]
- 18. Parasido E, Avetian GS, Naeem A, et al. The sustained induction of c‐MYC drives Nab‐paclitaxel resistance in primary pancreatic ductal carcinoma cells. Mol Cancer Res. 2019;17(9):1815‐1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palechor‐Ceron N, Suprynowicz FA, Upadhyay G, et al. Radiation induces diffusible feeder cell factor(s) that cooperate with ROCK inhibitor to conditionally reprogram and immortalize epithelial cells. Am J Pathol. 2013;183(6):1862‐1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Griffith M, Griffith OL, Coffman AC, et al. DGIdb: mining the druggable genome. Nat Methods. 2013;10(12):1209‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davis AP, Murphy CG, Saraceni‐Richards CA, Rosenstein MC, Wiegers TC, Mattingly CJ. Comparative Toxicogenomics Database: a knowledgebase and discovery tool for chemical‐gene‐disease networks. Nucleic Acids Res. 2009;37(Database issue):D786‐D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dakshanamurthy S, Issa NT, Assefnia S, et al. Predicting new indications for approved drugs using a proteochemometric method. J Med Chem. 2012;55(15):6832‐6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB‐‐a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37(Database issue):D623‐D628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang DW, Sherman BT, Tan Q, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(Web Server issue):W169‐W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447‐D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004;20(18):3705‐3706. [DOI] [PubMed] [Google Scholar]
- 27. Dudoit S, Gentleman RC, Quackenbush J. Open source software for the analysis of microarray data. Biotechniques. 2003;(suppl):45‐51. [PubMed] [Google Scholar]
- 28. Klipper‐Aurbach Y, Wasserman M, Braunspiegel‐Weintrob N, et al. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin‐dependent diabetes mellitus. Med Hypotheses. 1995;45(5):486‐490. [DOI] [PubMed] [Google Scholar]
- 29. Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO‐slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47(D1):D419‐D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oliveros JC. VENNY. An interactive tool for comparing lists with Venn Diagrams . http://bioinfogpcnbcsices/tools/venny/indexhtml; 2007.
- 31. Croglio MP, Haake JM, Ryan CP, et al. Analogs of the novel phytohormone, strigolactone, trigger apoptosis and synergize with PARP inhibitors by inducing DNA damage and inhibiting DNA repair. Oncotarget. 2016;7(12):13984‐14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hermanek P, Sobin LH International Union Against Cancer (UICC) . TNM Classification of Malignant Tumours. 4th ed. New York, NY: John Wiley & Sons; 1992. [Google Scholar]
- 33. Zhang CY, Zhu Y, Rui WB, Dai J, Shen ZJ. Expression of kallikrein‐related peptidase 7 is decreased in prostate cancer. Asian J Androl. 2015;17(1):106‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yella JK, Yaddanapudi S, Wang Y, Jegga AG. Changing trends in computational drug repositioning. Pharmaceuticals. 2018;11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim IW, Kim JH, Oh JM. Screening of drug repositioning candidates for castration resistant prostate cancer. Front Oncol. 2019;9:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Assefnia S, Dakshanamurthy S, Guidry Auvil JM, et al. Cadherin‐11 in poor prognosis malignancies and rheumatoid arthritis: common target, common therapies. Oncotarget. 2014;5(6):1458‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuan H, Myers S, Wang J, et al. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. N Engl J Med. 2012;367(13):1220‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riley JS, Quarato G, Cloix C, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018;37:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dudani JS, Ibrahim M, Kirkpatrick J, Warren AD, Bhatia SN. Classification of prostate cancer using a protease activity nanosensor library. Proc Natl Acad Sci U S A. 2018;115(36):8954‐8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meierjohann S, Hufnagel A, Wende E, et al. MMP13 mediates cell cycle progression in melanocytes and melanoma cells: in vitro studies of migration and proliferation. Mol Cancer. 2010;9:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Traka MH, Melchini A, Coode‐Bate J, et al. Transcriptional changes in prostate of men on active surveillance after a 12‐mo glucoraphanin‐rich broccoli intervention‐results from the Effect of Sulforaphane on prostate CAncer PrEvention (ESCAPE) randomized controlled trial. Am J Clin Nutr. 2019;109(4):1133‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richman EL, Carroll PR, Chan JM. Vegetable and fruit intake after diagnosis and risk of prostate cancer progression. Int J Cancer. 2012;131(1):201‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu B, Mao Q, Cao M, Xie L. Cruciferous vegetables intake and risk of prostate cancer: a meta‐analysis. Int J Urol. 2012;19(2):134‐141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 Complete list of the genes differentially expressed (P < .09, FC 1.5) in tumor vs normal CR cells for patient 1 and patient 2
Supplementary Table S2 Complete list of the predicted drug and target genes and pathways for patient 1
Supplementary Table S3 Complete list of the predicted drug and target genes and pathways for patient 2
Supplementary Table S4 Complete list of the genes differentially expressed (FDR < 0.05, FC 1.5) in tumor vs normal CR cells for patient 1 and patient 2 after treatment with carfilzomib
Supplementary Table S5. Categories of differentially regulated genes (FDR <0.05, FC 1.5) defined by venny 2.1.0