

Abstract
Intersystem crossing (ISC) of triplet photosensitizers is a vital process for fundamental photochemistry and photodynamic therapy (PDT). Herein, we report the co‐existence of efficient ISC and long triplet excited lifetime in a heavy atom‐free bodipy helicene molecule. Via theoretical computation and time‐resolved EPR spectroscopy, we confirmed that the ISC of the bodipy results from its twisted molecular structure and reduced symmetry. The twisted bodipy shows intense long wavelength absorption (ϵ=1.76×105 m −1 cm−1 at 630 nm), satisfactory triplet quantum yield (ΦT=52 %), and long‐lived triplet state (τ T=492 μs), leading to unprecedented performance as a triplet photosensitizer for PDT. Moreover, nanoparticles constructed with such helical bodipy show efficient PDT‐mediated antitumor immunity amplification with an ultra‐low dose (0.25 μg kg−1), which is several hundred times lower than that of the existing PDT reagents.
Keywords: bodipy, intersystem crossing, photodynamic therapy, photosensitizers, twisted π-systems
PDT with a twist: The intersystem crossing (ISC) mechanism of a heavy atom‐free helical bodipy derivative was elucidated. Due to the intense absorption at long wavelengths (ϵ=1.76×105 m −1 cm−1 at 630 nm), satisfactory triplet quantum yield (ΦT=52 %), and long‐lived triplet state (τ T=492 μs), a record low‐dose photodynamic therapy (PDT)‐augmented immunotherapy was achieved.
Introduction
PDT holds great promise and has attracted considerable interest as a less invasive tumor treatment option.1, 2, 3, 4, 5 When photosensitizers are photoexcited, they react with oxygen molecules in the microenvironment to produce singlet oxygen (1O2), thereby eliciting nearby tumor cell death. In this treatment, photosensitizing molecules that exhibit intense absorption of long‐wavelength light, long‐lived triplet states, and high singlet oxygen sensitization ability are essential for the potency of the therapy.
To date, only two porphyrin‐based PDT reagents have been approved by FDA for clinical cancer treatment.6 However, due to their low absorption for deep‐tissue‐penetrable long‐wavelength light and subsequent suboptimal singlet oxygen sensitization ability, high doses of these PDT reagents are required for operation. For example, PpIX has a low molar absorption efficiency (ϵ<5000 m −1 cm−1) at 635 nm. Hence, high concentrations (40 mg kg−1–200 mg kg−1) of 5‐aminolevulinic acid (ALA, the precursor of PpIX) are required for PDT treatment.7 Moreover, due to the low molar absorption coefficient (ϵ≈1170 m −1 cm−1) at 630 nm, Photofrin, the other FDA approved PDT reagent in clinical practice, also suffers from the same problem of requiring high doses for PDT treatment.1, 7 There are great concerns that the use of such high doses of photosensitizers can lead to prolonged undesired patient photosensitivity and certain side effects such as acid reflux, nausea, and flushing sensation.6, 8
In order to reduce the required drug dose, significant efforts have been made to develop non‐porphyrin PDT reagents that have strong absorption of long‐wavelength light and high singlet oxygen quantum yield. Yet, to date, the effective long‐wavelength‐light‐absorbing photosensitizers have to rely on heavy atoms to improve their ISC (the transition from the singlet to triplet state) and consequent singlet oxygen generation.9, 10 For instance, metal complexes (Pt, Ru, Ir)11, 12, 13, 14, 15 and brominated/iodinated chromophores16 are reported. Unfortunately, besides the potential toxicity of these heavy atoms, the dilemma here is that the improvement of the S1→Tn ISC is often at the cost of reducing the triplet state lifetime, as the undesired T1→S0 ISC (decay of the triplet excited state) can be enhanced by the heavy atom effect as well,9 which is especially significant for those long‐wavelength‐absorbing photosensitizers (Supporting Information, Table S2). Such a shortened triplet lifetime curtailed the reaction duration of oxygen and PDT reagents, thus being detrimental for singlet oxygen generation and concomitant PDT, especially for hypoxic environments such as those in tumors (Figure S9).4
To overcome this challenge, in this work, we sought to explore the possibility to develop metal/heavy atom‐free long‐wavelength‐absorbing triplet photosensitizers that have both satisfactory triplet state quantum yield and uncompromised long triplet excited state lifetime. Currently, heavy atom‐free photosensitizers are known to have much longer triplet lifetime17 but usually poor ISC.5 Efficient long‐wavelength heavy atom‐free triplet photosensitizers are rare (Table S3). In this study, we got inspiration from distorted conjugated systems, which display elevated ISC efficiency.18, 19, 20, 21, 22, 23, 24, 25, 26 In particular, the removal of the mirror‐plane symmetry of a planar π‐system is considered to allow for a stronger spin orbit coupling (SOC) between π–π* states with different spin manifolds (the singlet state and triplet state), hence facilitating ISC.27 The most notable exemplar molecule is the buckminsterfullerene (C60), which has a heavily curved π‐conjugation skeleton. This molecule was reported to show unity ISC efficiency but has only an extremely low molar extinction coefficient in the visible spectral range.21 Another exemplar are helicenes. Helicenes are helical compounds made of twisted ortho‐fused aromatic rings, which have been known to have an excellent ISC effect.27 Yet, most existing helicenes only absorb ultraviolet light, and it has been quite challenging to shift absorption towards the visible/near‐infrared region.28, 29, 30, 31
Herein, we report on the discovery of the co‐existence of satisfactory triplet state quantum yield and long triplet lifetime in a heavy atom‐free helicene molecule, based on a strong and robust visible‐light‐harvesting bodipy chromophore (helical‐BDP, Figure 1). In particular, such a heavy atom‐free molecule exhibits the co‐existence of satisfactory triplet quantum yield (ISC quantum yield ΦT=52 %) and the long‐lived triplet state (492 μs), as well as exceptionally intense absorption in the long‐wavelength range (molar absorption coefficient ϵ=1.76×105 m −1 cm−1 at 630 nm). The ISC mechanism of this heavy atom‐free molecule was elucidated by investigating the selective population of triplet excited state sublevels during ISC by time‐resolved electron paramagnetic resonance (TR‐EPR) spectroscopy and advanced theoretical computations. Moreover, by encapsulating such a triplet photosensitizer in a nanoparticle, the helical bodipy nanoparticles showed surprisingly highly efficient PDT‐augmented anti‐PD‐L1 antitumor immunity, with a record low dose (0.25 μg kg−1). This dose is hundred times lower than that of the PDT reagents used in the clinic and reported in the literature (at least 0.1 mg kg−1, Tables S1 and S4).
Figure 1.
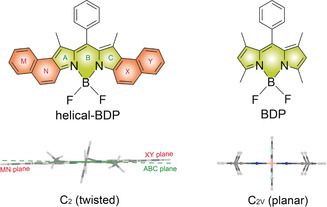
Molecular structures of helical‐BDP and BDP (top) and the side view of the RHF optimized ground structures (bottom).
Results and Discussion
Molecular Structures
Inspired by the ISC ability of helicene and the lack of long‐wavelength‐absorbing helicenes,31, 32 we were prompted to seek new helicene structures that are based on long‐wavelength‐light‐harvesting chromophores and explore their ISC properties. Bodipy is a popular fluorophore for its large molar absorption coefficient, bright fluorescence, and excellent stability. In this regard, we scrutinized few reported bodipy derivatives with twisted π‐conjugation planes (previously synthesized only as fluorophore candidates and the ISC properties were not studied).32, 33, 34, 35 Among these, through theoretical computations, we identified that a π‐conjugation framework of the naphthalene‐fused bodipy derivative (helical‐BDP, Figure 1), previously reported by Shen et al,35 is twisted in a way that is similar to that of helicene.36, 37, 38 As shown in Figure 1, there is a small but notable angle between the planes shown in green and orange. This geometry is distinctly different from the unsubstituted bodipy (BDP, Figure 1), and those substituted bodipy derivatives that have a strictly planar extended π‐conjugation framework,39 which are known to have negligible ISC.40
With the intuition that this helical‐twisted molecule (helical‐BDP, Figure 1) should inherit the strong ISC ability from helicene, we explored its ISC capability, triplet lifetime, and potential for PDT. The helical‐BDP was prepared based on reported methods.35 We observed the outstanding ISC of this twisted molecule by both nanosecond transient absorption (ns‐TA) spectroscopy and singlet oxygen photosensitizing experiments. The ISC mechanism for this twisted heavy atom‐free molecule was further revealed through TR‐EPR and theoretical computations. The parent bodipy (BDP, Figure 1) was used as a control compound throughout this study.
Photophysical Study: Absorption, Triplet Excited States, and ISC Kinetics
The absorption band of the helical‐BDP is centered at 630 nm, with the molar absorption coefficient (1.76×105 m −1 cm−1) almost twice that of BDP (Figure 2 a). Moreover, the vibrational progression of the helical‐BDP is more resolved than that of the BDP. Our results demonstrate that the twisted structure clearly altered the excited state properties of the bodipy chromophore.
Figure 2.
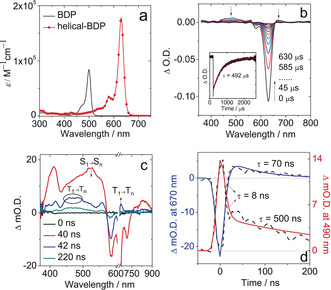
Steady‐state and transient absorption spectroscopies. a) Molar extinction coefficient of helical‐BDP in DCM. b) ns‐TA spectra of helical‐BDP (λ ex=628 nm, c=1.25×10−6 m). Inset: Decay trace of helical‐BDP at 610 nm, c=3.1×10−7 m. This was measured in deaerated DCM, recorded in the co‐linear measurement mode of the LP980 ns‐TA spectrometer, 20 °C. c) ns‐TA spectra of helical‐BDP in toluene (λ ex=640 nm) at 0 ns, 40 ns, 42 ns and 220 ns. The arrows indicate the evolution of various bands. d) The kinetic trace at 670 nm and 490 nm show the time constant for triplet state formation.
The triplet excited state of the helical‐BDP was investigated by ns‐TA spectroscopy (Figure 2 b). In particular, upon pulsed laser excitation at 628 nm, two negative bands centered at 625 nm and 579 nm were observed. These are the two ground state bleaching bands (GSB, see the steady state absorption of the compound, Figure 2 a). In addition, a weak excited state absorption (ESA) band in the range of 400–550 nm was observed. This is attributed to the absorption of the T1 triplet state (T1→Tn transition). Moreover, by monitoring the decay of the GSB signal at 610 nm, the triplet excited state was found to be long‐lived (hundreds of microseconds) in deaerated solution (inset of Figure 2 b). In contrast, in an aerated solution, the lifetime was greatly reduced to 224 ns (Figure S2). Such a significant reduction in its lifetime clearly confirmed the triplet state feature of the transient species.
Notably, the intrinsic triplet state lifetime (τ=492 μs, for details, see Supporting Information) is much longer than that of typical heavy atom‐containing triplet photosensitizers showing long‐wavelength absorption (Table S2, for instance, τ T=1.7 μs for the 2,6‐diiodo‐bisstyrylbodipy). It should be noted that heavy‐atom photosensitizers tend to have a significantly reduced triplet state lifetime (heavy‐atom effect), which is especially severe for chromophores that have long‐wavelength absorption with low triplet energy. For instance, with iodination, the triplet lifetime of 3,5‐distyrylbodipy was significantly reduced from 72 μs to 1.7 μs (Table S2). Such a shortened triplet lifetime is detrimental to applications, such as oxygen sensing,41 photocatalysis42 and PDT,4 as these applications are all based on intermolecular diffusion‐controlled triplet energy transfer or electron transfer, for which the efficiency is dependent on the triplet state lifetimes of the photosensitizer. Our study shows that the 1O2 sensitization ability (a basic process in PDT) of a triplet photosensitizer showing a short‐lived triplet state almost vanishes in hypoxic conditions; the singlet oxygen quantum yield drops from 45 % under normal conditions to 2 % under a hypoxic atmosphere. However, this did not occur for the long‐lived helical‐BDP (Φ Δ≈36 % in both normal atmosphere and hypoxic atmosphere, Figure S9). Hence, the long triplet state lifetime of the helical bodipy shows its unique advantage over the conventional method of using the heavy‐atom effect. Inspired by the excellent photophysical properties of helical‐BDP, we explored the feasibility of TTA upconversion by using this special heavy atom‐free photosensitizer. Outstanding overall red‐to‐yellow TTA upconversion brightness was obtained (η=ϵ Φ UC=1296 m −1 cm−1 at 250 mW cm−2 of 635 nm excitation) with helical‐BDP as an intense red‐light‐absorbing photosensitizer in conjunction with perylenebisimide (PBI) as the yellow emitter (Figures S5 and S6).
In order to study the kinetics of ISC, ns‐TA spectroscopy with higher time resolution was performed. As shown in Figure 2 c, the ESA at 525 nm is attributed to the S1→Sn absorption band. At a longer delay time (42 ns in Figure 2 c), the shape of the spectrum resembles that of Figure 2 b, clearly showing that the ISC process takes place. Hence, the ESA bands centered at 490 nm and 670 nm can be attributed to the T1→Tn absorption. The ISC time constant for helical‐BDP was determined to be 1/k ISC=8 ns (Figure 2 d). According to the population ratio of the sublevels of the triplet state measured by TR‐EPR spectra (Px/Py/Pz=1:0:0.2, see later section, Figure 4 c), the electron spin‐selective ISC rate constants (k ISC) to the triplet sublevels (Tx, Ty, Tz) of the T1 state are 0.10 ns−1, 0 ns−1, and 0.02 ns−1, respectively. The photophysical parameters of the compounds are listed in Table 1.
Figure 4.
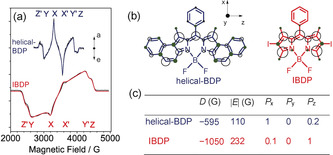
a) TR‐EPR spectra of helical‐BDP (λ ex=555 nm) and IBDP (λ ex=532 nm) in toluene/MeTHF (v/v, 3:1) frozen mixed solution, c=3.0×10−5 m, 80 K. The canonical orientations of each transition are indicated (the prime indicates the ms=0 → ms=+1 transitions). b) Spin density distribution of helical‐BDP and IBDP. Larger circles indicate larger (absolute) spin density value on the atom. The ZFS frame is also presented. c) Fitting parameters of the simulations in (a). P i is the relative population of the i‐th ZFS state, labeled as indicated in (a).
Table 1.
Photophysical parameters of the compounds.[a]
|
Compounds |
λ abs [b] |
ϵ [c] |
λ em [d] |
τ F [e] |
Φ F [f] |
τ T [g] |
Φ Δ [h] |
Φ T [i] |
|---|---|---|---|---|---|---|---|---|
|
helical‐BDP |
630 |
17.6 |
649 |
3.3 |
21 % |
492 |
36 % |
52 % |
|
BDP |
500 |
8.4 |
512 |
4.1 |
52 % |
–[j] |
–[j] |
–[j] |
[a] In dichloromethane (DCM), 25 °C. [b] Maximum absorption wavelength, in nm. [c] Molar absorption coefficient, 104 m −1 cm−1. [d] Maximum fluorescence emission wavelength, in nm. [e] Luminescence lifetimes, in nanoseconds (ns). [f] Fluorescence quantum yield with methyl blue as standard (Φ F=3 % in MeOH). [g] Intrinsic triplet excited state lifetime, in microseconds (μs), c=3.1×10−7 m, low concentration was used to reduce the self‐quenching effect. [h] Singlet oxygen quantum yield with methyl blue as standard (Φ Δ=57 % in DCM). [i] Triplet quantum yield determined by ns‐TA spectroscopy, by triplet‐triplet energy transfer method, with Methyl Blue as standard (Φ T=50 % in MeOH). [j] Not observed.
ISC Mechanism: Theoretical Computation and TR‐EPR Spectroscopy
The geometries of the compounds were examined with the intuition that they are pivotal to the ISC. Theoretical computations43, 44 show that the optimized ground‐state geometry of BDP has C 2v geometry (the π‐conjugation of the Bodipy chromophore is planar); while for the helical‐BDP, the C 2 geometry is most stable, followed closely by the C S geometry (both are non‐planar geometries, Table S7), whereas the C 2v geometry is a transition state.
In order to find out the relationship between the twisted structure and the enhanced ISC, the spin‐orbit coupling (SOC) matrix elements between S1 state and the sublevels (Tx, Ty, and Tz) of each triplet excited state were further determined by quantum chemical calculations (listed in Table 2). As the SOC in organic systems is considered to be a small correction to purely nonrelativistic electronic states,27 the magnitude of the calculated SOC values (between the singlet and triplet states) reflect the possibility of ISC in a certain degree. Since T1 is below the S1 state, and T2 is slightly above S1 by approximately 0.1 eV in both the helical‐BDP and BDP, both S1→T1 and S1→T2 are considered to be possible pathways of ISC (Table S7). Note that according to the CAS‐Cl calculations the states S1, T1 and T2 share the same symmetry.
Table 2.
Matrix elements (in units of cm−1) of the one‐electron Breit–Pauli Operator of the SOC between S1 and the lowest two triplet states.[a]
|
|
helical‐BDP |
BDP |
||
|---|---|---|---|---|
|
SOC states |
C 2V |
C 2 |
CS |
C 2V |
|
S1/T1 |
0 |
0.0977 x |
0.0024 z |
0 |
|
S1/T2 |
0 |
0.1689 x |
0.0781 z |
0 |
[a] Each value is labeled with the index (x, y, z) of the triplet sublevel. In both helical‐BDP and BDP, SOC is forbidden by symmetry in the structures with C 2v point group (see Supporting Information for details).
The selection rules for SOC between a singlet state S and a triplet state T require that the product of the irreducible representation of the spatial symmetries of these two states transform like one of the three rotations [see Supporting Information for details; Equation (1)]:
| (1) |
As a consequence, in molecules with C 2v geometry, SOC is forbidden between singlet and triplet states with the same spatial symmetry. Hence, SOC between S1/T1 or S1/T2 is forbidden in C 2v (Table 2 and Figure 3 b). This rationalizes the negligible ISC ability of the parent BDP.
Figure 3.
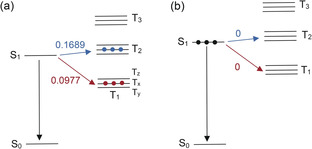
SOC matrix elements of the first excited singlet state with the nearest triplet states. a) SOC of helical‐BDP in C 2 geometry and b) SOC of BDP in C 2v geometry. The labelling of the substates are indicated (note that every excited triplet state consists of three non‐degenerate sublevels Tx, Ty and Tz). The larger the SOC value, the greater possibility of ISC.
With the molecular structure deviated from the C 2v geometry, these selection rules will be relaxed (Figure 3 a). Clearly, the magnitude of this coupling will depend on how much the twisted structures deviate from the idealized C 2v geometry. For the helical‐BDP, as the C 2‐geometry has the lowest energy, selective population of the Tx state is expected (see Figure 3 a and Table 2). Note that Tx, Ty, Tz are three non‐degenerate sublevels of the T1 triplet state.
The overpopulation of the Tx state was experimentally confirmed by TR‐EPR (Figure 4 c). The spin selectivity of the ISC reflects both in the electron spin polarization (ESP) of the three sublevels of the triplet embedded in a magnetic field and in the enhanced absorption/emission (a/e) character of the main features of the TR‐EPR transitions, as detailed in the Supporting Information. The TR‐EPR spectrum of the triplet state of helical‐BDP shows an (a, e, a, e, a, e) ESP pattern (Figure 4 a, blue line). A careful simulation reveals an overpopulation of the Tx state (Figure 4 c). The consistency between theoretical computation and experimental TR‐EPR validate that the enhanced ISC of helical‐BDP is due to the reduced symmetry in the twisted structure. It also enables us to deduce that the helical‐BDP possesses mainly a helical structure (C 2 geometry).
Moreover, the confinement of the triplet state wave functions of these compounds is examined, as it is crucial for rationalization of the ISC ability of the twisted structure.27 Experimentally, the zero‐field splitting (ZFS) parameter D‐value reflects the extension of the wave functions of the two unpaired electrons of the triplet state. This value can be easily obtained from the total width of the TR‐EPR spectra (see Supporting Information) of IBDP and helical‐BDP (Figure 4). We clearly noticed that the width of the TR‐EPR spectrum of IBDP is quite larger than that of the helical‐BDP (D=−595 G for helical‐BDP and D=−1050 G for IBDP, Figure 4 c). We deduce that the triplet wave function of helical‐BDP is far more delocalized than that of IBDP. This is supported by the calculation of the spin density distribution; Figure 4 b shows that the wave function spreads all over the π‐system of the helical‐BDP. Hence, we have a true helical π‐system. This is in agreement with the significant enhancement of ISC for the heavy atom‐free helical‐BDP; it is the delocalization of the electrons on the entire twisted molecular framework that increases the SOC for the ISC between the two π‐π* states, as twisting the π‐conjugation framework allows for non‐vanishing one‐center and two‐center integrals of all atomic angular momentum operators. On the contrary, if the triplet wave function was localized on the planar π‐conjugation framework as in the parent BDP, we would not expect any enhancement of SOC.
Record Low‐Dose PDT Augmented Checkpoint Blockade Immunotherapy
Inspired by the exceptionally strong long‐wavelength absorption (ϵ=1.76×105 m −1 cm−1 at 630 nm), satisfactory singlet oxygen quantum yield (ΦΔ=36 %) and the long‐lived triplet state (τT=492 μs), we explored the potential of helical‐BDP as a long‐wavelength‐absorbing heavy atom‐free PDT reagent to augment the checkpoint blockade immunotherapy.
Firstly, the PDT effect of helical‐BDP on tumor cells were examined in vitro. In order to encapsulate such hydrophobic photosensitizers to be used in aqueous solution, the helical‐BDP was wrapped in the octadecylamine substituted amphiphilic polymer (PSMA‐PEG‐OA) to generate such dye‐loaded nanoparticles (helical‐BDP‐NPs, Figure S8). These nanoparticles showed outstanding colloidal stability (Figure S18), high dye entrapment efficiency (76 %). Their singlet oxygen quantum yields (Φ Δ) were determined to be 21 %. The fluorescence spectrum of the helical‐BDP‐NPs is presented in Figure 5 a and the fluorescence quantum yield (Φ F) is measured to be 3.8 %. The helical‐BDP‐NPs exhibited high photocytotoxicity to the CT26 tumor cells (semi‐lethal concentration IC50=11.5 nm) under 656 nm light irradiation, showing remarkably improved performance (Figure 5 b,c) compared to the phthalocyanine photosensitizer IRDye 700DX, which is arguably one of the best photo‐immune therapy photosensitizers.45
Figure 5.
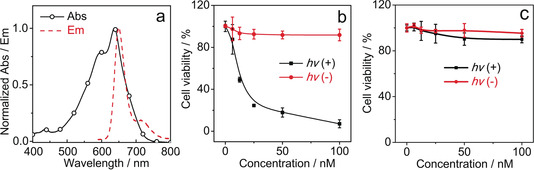
PDT in vitro. a) Normalized absorption and emission spectrum of helical‐BDP‐NPs. Cell viability of CT26 cells pre‐treated with increasing doses of b) helical‐BDP‐NPs and c) IRDye 700DX with and without light irradiation. Irradiation by 656 nm LED, light dose: 6 J cm−2, 20 °C.
In order to study the effect of PDT mediated checkpoint blockade immunotherapy, a bilateral model of CT26 tumors on BALB/c mice was established to artificially mimic metastasis. The tumor that is treated with light illumination is considered to be the “primary tumor”, and the tumor untreated at another side is considered to mimic the “metastatic tumor”. As shown in Figure 6 b, the mice were randomly divided into 5 groups: (1) hv only, (2) anti‐PD‐L1 only, (3) helical‐BDP‐NPs only; (4) helical‐BDP‐NPs + hv only, and (5) helical‐BDP‐NPs + anti‐PD‐L1 + hv. The therapeutic efficacies of different treatment groups were evaluated by measuring tumor volume.
Figure 6.
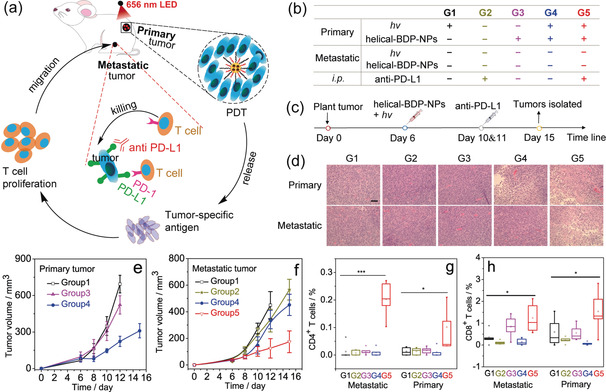
PDT augmented checkpoint blockade immunotherapy. a) Schematic indicating of the helical‐BDP‐NPs with anti‐PD‐L1 for cancer treatment. b) The experimental condition of each group (the mice were randomly divided into 5 groups). G=Group. The “+” represents the condition is satisfied and the “−” represents the condition is unsatisfied. c) Timeline of the treatment. d) Hematoxylin and eosin (H&E) staining of tumor tissue sections from different treatment groups at day 15. Scale bar=150 μm. e) Tumor volume of primary tumor. f) Tumor volume of artificial metastatic tumor. g) Analysis the immunity CD4+ T cells and h) CD8+ T cells of metastatic tumor and primary tumor. Data are expressed as means ± s.d. (n=5). *P<0.05, **P<0.01, and ***P<0.001.
The tumor volume growth rates of the primary tumor (treated with PDT) are presented in Figure 6 e. As compared to Group 3 (only the injection of helical‐BDP‐NPs) and Group 1 (only irradiation with light, hv), the primary tumor in Group 4 (helical‐BDP‐NPs + hv) shows significant inhibition of growth, indicating that helical‐BDP‐NPs is a potent PDT reagent for primary tumors.
The tumor volume growth rates of the metastatic tumor are presented in Figure 6 f. It is impressive that although the metastatic tumor was untreated with PDT, the combination of immune therapy with PDT treatment on primary tumors (Group 5=Group 2 + Group 4) presented significant metastatic tumor suppression (Figure 6 f) and obvious necrosis of metastatic tumor cells (H&E staining analysis, Figure 6 d). These experimental results demonstrated that the helical‐BDP‐NPs mediated PDT can significantly augment the checkpoint blockade immunotherapy efficacy and promote abscopal effects (Figure 6 a).
Specifically, anti‐PD‐L1 can inhibit the interaction between PD‐L1 (in tumor cells) and PD‐1 (in T‐cells), thus rescue the T cells (Figure 6 a).46 However, this kind of checkpoint blockade immunotherapy, being only effective for tumors with the presence of T‐cells, has low response rates (10–40 %).47 On the other hand, as shown in Figure 6 a, PDT can release tumor specific antigen through tumor cells apoptosis, inducing acute inflammation and increasing the infiltration of T‐cells.48, 49, 50, 51 Hence the PDT augmented the anti‐PD‐L1 checkpoint blockade immunotherapy, leading to an obviously inhibition to metastatic tumor. This is verified by the analysis of the antitumor immunity. The immune cell profiling in the spleen (Figure S16) shows that the cytotoxic CD8+ T cell and helper CD4+ T cell levels significantly increased in the treated group (Group 5) as compared to Group 1. Moreover, in Group 5, there is a significant increase of tumor‐infiltrating CD4+ T‐cells and CD8+ T‐cells in both primary and metastatic tumors (Figure 6 g,h). As a result, a significantly increased concentration of IFN‐γ in Group 5 was observed (Figure S21).52, 53 These results demonstrated that the combined effect of helical‐BDP‐NPs mediated PDT and anti‐PD‐L1 treatment significantly increased the infiltration of effector T cells to treat the metastasis.
More importantly, to the best of our knowledge, with this novel triplet photosensitizer helical‐BDP, PDT‐mediated antitumor immunity amplification was achieved with the lowest drug dose reported to date (0.25 μg kg−1) and an ultralow light dose (6 J cm−2), being thousands of times lower than other traditional/commercial PDT reagents that have been used in the literature (>1.4 mg kg−1, >18 J cm−2, Table S4). The highly efficient PDT effect can be attributed to the following reasons: 1) The exceptionally high molar extinction coefficient (ϵ=1.76×105 m −1 cm−1 at 630 nm) enables outstanding light harvesting to occur, 2) the super‐long triplet lifetime (τT=492 μs) is critical and favorable toward achieving efficient energy transfer to produce 1O2, especially in hypoxic tumors (Figure S9), and 3) the small size of the uniform spherical nanoparticles (31.5±5.2 nm, Figure S10) is beneficial for their accumulation and retention in the tumor. Collectively, super‐efficient PDT was achieved.
Conclusion
In summary, our results represent a new paradigm in the development of the next generation of triplet photosensitizers. For the first time, co‐existence of satisfactory ISC and long triplet lifetime was revealed in a heavy atom‐free bodipy helicene molecule. Advanced quantum chemical calculations and time‐resolved EPR spectroscopy indicate that torsion of the plane into a helix (C2‐symmetry) enhances the ISC, yielding the population of Tx substate. Moreover, the triplet state wave function (spin‐unpaired electrons) is found to be delocalized over the entire twisted π‐conjugate skeleton. We showed that such a twisted bodipy helicene molecule shows exceptionally intense long‐wavelength absorption (ϵ=1.76×105 m −1 cm−1 at 630 nm, twice that of the normal unsubstituted BDP), satisfactory triplet quantum yield (Φ T=52 %) and a long‐lived triplet state (τ T=492 μs), leading to unprecedented performance as a triplet photosensitizer. More importantly, by encapsulating such a heavy atom‐free triplet photosensitizer in a nanoparticle, a record low dose of the photosensitizers (0.25 μg kg−1) with an ultralow light dose (6 J cm−2) is achieved for effective PDT immunotherapy. This dose is hundred times lower than that of the existing PDT reagents. Taken together, this heavy atom‐free triplet photosensitizer shows long sought‐after advantages over conventionally used triplet photosensitizers. Since ISC of organic chromophores is fundamentally important in photochemistry as well as in many other important areas such as PDT, we believe that our study not only leads to the discovery of new heavy atom‐free photosensitizer molecular structural motifs with unique triplet properties, it also opens up a wide variety of opportunities in photonic/biophotonic fields.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
J.Z. thanks the NSFC (21673031, 21761142005, 21911530095 and 21421005), the State Key Laboratory of Fine Chemicals (ZYTS201901), the Fundamental Research Funds for the Central Universities (DUT19TD28), Dipartimento di Scienze Chimiche, Università degli Studi di Padova (Visiting Scientist) for support. B.D. thanks Dalian University of Technology for the Haitian Professorship support. L.H. and G.H. are supported by the startup funding of University of Massachusetts. Open access funding enabled and organized by Projekt DEAL.
Z. Wang, L. Huang, Y. Yan, A. M. El-Zohry, A. Toffoletti, J. Zhao, A. Barbon, B. Dick, O. F. Mohammed, G. Han, Angew. Chem. Int. Ed. 2020, 59, 16114.
Contributor Information
Prof. Jianzhang Zhao, Email: zhaojzh@dlut.edu.cn.
Dr. Antonio Barbon, Email: antonio.barbon@unipd.it.
Prof. Bernhard Dick, Email: Bernhard.Dick@chemie.uni-regensburg.de.
Prof. Omar F. Mohammed, Email: omar.abdelsaboor@kaust.edu.sa.
Prof. Gang Han, Email: Gang.Han@umassmed.edu.
References
- 1. Dolmans D. E., Fukumura D., Jain R. K., Nat. Rev. Cancer 2003, 3, 380–387. [DOI] [PubMed] [Google Scholar]
- 2. Lovell J. F., Liu T. W., Chen J., Zheng G., Chem. Rev. 2010, 110, 2839–2857. [DOI] [PubMed] [Google Scholar]
- 3. Detty M. R., Gibson S. L., Wagner S. J., J. Med. Chem. 2004, 47, 3897–3915. [DOI] [PubMed] [Google Scholar]
- 4. Lincoln R., Kohler L., Monro S., Yin H., Stephenson M., Zong R., Chouai A., Dorsey C., Hennigar R., Thummel R. P., J. Am. Chem. Soc. 2013, 135, 17161–17175. [DOI] [PubMed] [Google Scholar]
- 5. Li J., Pu K., Acc. Chem. Res. 2020, 53, 752–762. [DOI] [PubMed] [Google Scholar]
- 6. Van Straten D., Mashayekhi V., De Bruijn H. S., Oliveira S., Robinson D. J., Cancers 2017, 9, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dougherty T. J., Gomer C. J., Henderson B. W., Jori G., Kessel D., Korbelik M., Moan J., Peng Q., J. Natl. Cancer Inst. 1998, 90, 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sarezky D., Raquib A. R., Dunaief J. L., Kim B. J., Clin. Ophthalmol. 2016, 10, 1899–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.N. J. Turro, V. Ramamurthy, J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, CA, 2009.
- 10. Zhao J., Wu W., Sun J., Guo S., Chem. Soc. Rev. 2013, 42, 5323–5351. [DOI] [PubMed] [Google Scholar]
- 11. Campagna S., Puntoriero F., Nastasi F., Bergamini G., Balzani V., Top. Curr. Chem. 2007, 280, 117–214. [Google Scholar]
- 12. You Y., Nam W., Chem. Soc. Rev. 2012, 41, 7061–7084. [DOI] [PubMed] [Google Scholar]
- 13. Eryazici I., Moorefield C. N., Newkome G. R., Chem. Rev. 2008, 108, 1834–1895. [DOI] [PubMed] [Google Scholar]
- 14. Ji S., Wu W., Wu W., Guo H., Zhao J., Angew. Chem. Int. Ed. 2011, 50, 1626–1629; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1664–1667. [Google Scholar]
- 15. Chi Y., Chou P.-T., Chem. Soc. Rev. 2007, 36, 1421–1431. [DOI] [PubMed] [Google Scholar]
- 16. Gorman A., Killoran J., O'Shea C., Kenna T., Gallagher W. M., O'Shea D. F., J. Am. Chem. Soc. 2004, 126, 10619–10631. [DOI] [PubMed] [Google Scholar]
- 17. Hou Y., Liu Q., Zhao J., Chem. Commun. 2020, 56, 1721–1724. [DOI] [PubMed] [Google Scholar]
- 18. Nagarajan K., Mallia A. R., Muraleedharan K., Hariharan M., Chem. Sci. 2017, 8, 1776–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salla C. A., Farias G., Rouzières M., Dechambenoit P., Durola F., Bock H., de Souza B., Bechtold I. H., Angew. Chem. Int. Ed. 2019, 58, 6982–6986; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7056–7060. [Google Scholar]
- 20. Liu K., Lalancette R. A., Jäkle F., J. Am. Chem. Soc. 2017, 139, 18170–18173. [DOI] [PubMed] [Google Scholar]
- 21. Arbogast J. W., Darmanyan A. P., Foote C. S., Diederich F., Whetten R., Rubin Y., Alvarez M. M., Anz S. J., J. Phys. Chem. 1991, 95, 11–12. [Google Scholar]
- 22. Hu W., He T., Zhao H., Tao H., Chen R., Jin L., Li J., Fan Q., Huang W., Baev A., Angew. Chem. Int. Ed. 2019, 58, 11105–11111; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11222–11228. [Google Scholar]
- 23. Yu Z., Wu Y., Peng Q., Sun C., Chen J., Yao J., Fu H., Chem. Eur. J. 2016, 22, 4717–4722. [DOI] [PubMed] [Google Scholar]
- 24. Wu Y., Zhen Y., Ma Y., Zheng R., Wang Z., Fu H., J. Phys. Chem. Lett. 2010, 1, 2499–2502. [Google Scholar]
- 25. Menning S., Krämer M., Duckworth A., Rominger F., Beeby A., Dreuw A., Bunz U. H. F., J. Org. Chem. 2014, 79, 6571–6578. [DOI] [PubMed] [Google Scholar]
- 26. Nagarajan K., Mallia A. R., Reddy V. S., Hariharan M., J. Phys. Chem. C 2016, 120, 8443–8450. [Google Scholar]
- 27. Schmidt K., Brovelli S., Coropceanu V., Beljonne D., Cornil J., Bazzini C., Caronna T., Tubino R., Meinardi F., Shuai Z., J. Phys. Chem. A 2007, 111, 10490–10499. [DOI] [PubMed] [Google Scholar]
- 28. Kim S., Weissman S., J. Am. Chem. Soc. 1979, 101, 5863–5864. [Google Scholar]
- 29. Biet T., Martin K., Hankache J., Hellou N., Hauser A., Bürgi T., Vanthuyne N., Aharon T., Caricato M., Crassous J., Chem. Eur. J. 2017, 23, 437–446. [DOI] [PubMed] [Google Scholar]
- 30. Hsieh Y.-C., Wu C.-F., Chen Y.-T., Fang C.-T., Wang C.-S., Li C.-H., Chen L.-Y., Cheng M.-J., Chueh C.-C., Chou P.-T., J. Am. Chem. Soc. 2018, 140, 14357–14366. [DOI] [PubMed] [Google Scholar]
- 31. Delgado I. H., Pascal S., Wallabregue A., Duwald R., Besnard C., Guénée L., Nançoz C., Vauthey E., Tovar R. C., Lunkley J. L., Chem. Sci. 2016, 7, 4685–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ito H., Sakai H., Suzuki Y., Kawamata J., Hasobe T., Chem. Eur. J. 2020, 26, 316–325. [DOI] [PubMed] [Google Scholar]
- 33. Hayashi Y., Obata N., Tamaru M., Yamaguchi S., Matsuo Y., Saeki A., Seki S., Kureishi Y., Saito S., Yamaguchi S., Org. Lett. 2012, 14, 866–869. [DOI] [PubMed] [Google Scholar]
- 34. Wang J., Wu Q., Wang S., Yu C., Li J., Hao E., Wei Y., Mu X., Jiao L., Org. Lett. 2015, 17, 5360–5363. [DOI] [PubMed] [Google Scholar]
- 35. Zhou Z., Zhou J., Gai L., Yuan A., Shen Z., Chem. Commun. 2017, 53, 6621–6624. [DOI] [PubMed] [Google Scholar]
- 36. Gingras M., Chem. Soc. Rev. 2013, 42, 1051–1095. [DOI] [PubMed] [Google Scholar]
- 37. Zak J. K., Miyasaka M., Rajca S., Lapkowski M., Rajca A., J. Am. Chem. Soc. 2010, 132, 3246–3247. [DOI] [PubMed] [Google Scholar]
- 38. Wigglesworth T. J., Sud D., Norsten T. B., Lekhi V. S., Branda N. R., J. Am. Chem. Soc. 2005, 127, 7272–7273. [DOI] [PubMed] [Google Scholar]
- 39. Loudet A., Burgess K., Chem. Rev. 2007, 107, 4891–4932. [DOI] [PubMed] [Google Scholar]
- 40. Zhao J., Xu K., Yang W., Wang Z., Zhong F., Chem. Soc. Rev. 2015, 44, 8904–8939. [DOI] [PubMed] [Google Scholar]
- 41. Ji S., Wu W., Wu W., Song P., Han K., Wang Z., Liu S., Guo H., Zhao J., J. Mater. Chem. 2010, 20, 1953–1963. [Google Scholar]
- 42. Guo S., Chen K.-K., Dong R., Zhang Z.-M., Zhao J., Lu T.-B., ACS Catal. 2018, 8, 8659–8670. [Google Scholar]
- 43.A. A. Granovsky, Firefly version 8.0.0, http://classic.chem.msu.su/gran/firefly/index.html 2014.
- 44. Schmidt M. W., Baldridge K. K., Boatz J. A., Elbert S. T., Gordon M. S., Jensen J. H., Koseki S., Matsunaga N., Nguyen K. A., Su S., Windus T. L., Dupuis M., J. A. Montgomery, Jr. , J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar]
- 45. Mitsunaga M., Ogawa M., Kosaka N., Rosenblum L. T., Choyke P. L., Kobayashi H., Nat. Med. 2011, 17, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Swaika A., Hammond W. A., Joseph R. W., Mol. Immunol. 2015, 67, 4–17. [DOI] [PubMed] [Google Scholar]
- 47. Hu L., Cao Z., Ma L., Liu Z., Liao G., Wang J., Shen S., Li D., Yang X., Biomaterials 2019, 223, 119469. [DOI] [PubMed] [Google Scholar]
- 48. Castano A. P., Pawel M., Hamblin M. R., Nat. Rev. Cancer 2006, 6, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gollnick S. O., Evans S. S., Baumann H., Owczarczak B., Maier P., Vaughan L., Wang W. C., Unger E., Henderson B. W., Br. J. Cancer 2003, 88, 1772–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Galluzzi L., Kepp O., Kroemer G., EMBO J. 2012, 31, 1055–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Milla Sanabria L., Rodríguez M. E., Cogno I. S., Vittar N. B. R., Pansa M. F., Lamberti M. J., Rivarola V. A., Biochim. Biophys. Acta Rev. Cancer 2013, 1835, 36–45. [DOI] [PubMed] [Google Scholar]
- 52. Ni L., Lu J., Cancer Med. 2018, 7, 4509–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lan G., Ni K., Xu Z., Veroneau S. S., Song Y., Lin W., J. Am. Chem. Soc. 2018, 140, 5670–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary