

Abstract
In recent years, the non‐covalent interaction of halogen bonding (XB) has found increasing application in organocatalysis. However, reports of the activation of metal‐ligand bonds by XB have so far been limited to a few reactions with elemental iodine or bromine. Herein, we present the activation of metal‐halogen bonds by two classes of inert halogen bond donors and the use of the resulting activated complexes in homogenous gold catalysis. The only recently explored class of iodolium derivatives were shown to be effective activators in two test reactions and their activity could be modulated by blocking of the Lewis acidic sites. Bis(benzimidazolium)‐based halogen bonding activators provided even more rapid conversion, while the non‐iodinated reference compound showed little activity. The role of halogen bonding in the activation of metal‐halogen bonds was further investigated by NMR experiments and DFT calculations, which support the mode of activation occurring via halogen bonding.
Keywords: gold, halogen bonding, noncovalent interactions, organocatalysis, supramolecular chemistry
The activation of a gold‐chloride complex by halogen‐bonding‐based Lewis acids is described as the first case of metal‐ligand bond cleavage by inert halogen bond donors. Iodine(I) and iodine(III) derivatives provided excellent performance in two proof‐of‐principle reactions. Comparative experiments for both activators strongly suggest halogen bonding as the mode of activation.
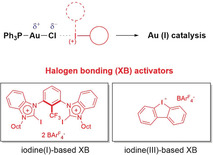
Halogen bonding (XB)1, 2 describes the non‐covalent interaction between electrophilic halogen substituents and Lewis bases.3 Its high directionality, tunability and complementarity to hydrogen bonding (HB) has led to frequent use4 of XB in the solid state, especially with numerous applications in crystal engineering.5, 6 Increasingly, the use of halogen bonding has also expanded into solution chemistry, with a multitude of reports on anion recognition7, 8 and the construction of supramolecular assemblies.9
In the last decade, halogen bonding has also been established in organocatalysis, with an ever increasing number of organic reactions reported which are promoted by XB donors.10, 11 In many of these cases, halide leaving groups were abstracted from organohalogen compounds in order to facilitate nucleophilic substitution reactions (Figure 1, left).
Figure 1.
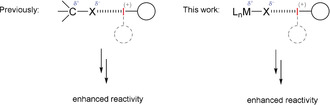
Activation of C‐X and M‐X bonds by halogen bonding.
At this time, multiple classes of halogen bonding catalysts have been established and applied to different types of organic reactions,11 including monodentate or multidentate neutral as well as cationic iodine(I)‐based XB donors, and also recently, iodine(III) derivatives.12 Lately, a first example has also been reported in which enantioselectivity in a Mukaiyama aldol reaction was induced by a chiral halogen bond donor devoid of any further coordinating groups.13
The application of halogen bonding in transition metal chemistry has yet mostly been limited to halogen bonding ligands which induce organization of the ligand sphere.14, 15, 16, 17, 18 Halogen bonds to halide19, 20, 21, 22 ligands as well as numerous other23 Lewis basic ligands in metal complexes have been observed for inorganic and organic halogen bond donors.24
Beyond coordination of these ligands by halogen bonding, only a handful25, 26, 27, 28 of instances exist, in which ligand exchange is observed. In these cases, elemental iodine or bromine were used as halide exchange reagents: even though intermediary halogen bonding was postulated, in the progress of the reaction the I−I or Br−Br bonds were cleaved. The transfer of the concept of C‐X activation to M‐X bonds using inert XB donors has, to the best of our knowledge, not yet been realized (Figure 1, right).
Herein, we present the first example of such, in two gold‐catalyzed reactions which are accelerated in the presence of two separate classes of halogen bond donors via Au−Cl bond activation. In this study, the scarcely explored class of halogen bonding catalysts12 based on an iodolium (cyclic diaryl iodonium) motif, 1 and 2, are utilized (Figure 2). Additionally, the tetrakis[3,5‐bis(trifluoromethyl)‐phenyl]borate (BArF 4 −) (4 a), triflate (4 b), and the newly synthesized hexafluorophosphate (4 c) salts of the previously tested, highly preorganized29 bis(benzimidazolium) halogen bond donor 4 30 were investigated.
Figure 2.
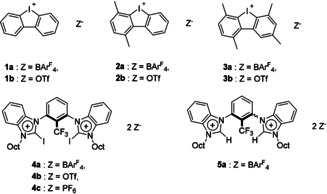
Overview of halogen bond donors and related reference compounds applied in this study.
As first test reaction, the gold(I)‐catalyzed cyclization of propargylic amide 6 to the oxazoline 7 was chosen (Scheme 1). This reaction has previously been used to benchmark catalyst activity and the effectiveness of added activators in homogenous gold catalysis.31, 32, 33, 34 The reaction was performed with 2 mol % of chloro(triphenylphosphine) gold and an equimolar amount of the halide abstracting agent in deuterated chloroform as solvent.
Scheme 1.
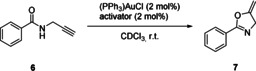
Au‐catalyzed cyclization of amide 6 to oxazoline 7.
Due to the subsequent slow aromatization of 7 to the corresponding oxazole34 (cf. Figure S1 in the Supporting Information), for accuracy, only the conversion of the starting material is considered, the extent of which could be monitored by 1H NMR spectroscopy. Without the presence of any halide‐abstracting agent, the reaction shows no background reactivity after more than 30 hours (Table 1, Entry 1). In contrast, addition of iodolium salt 1 a results in 60 % conversion after only 3 hours of reaction time (Table 1, Entry 2) and almost quantitative conversion is reached after 12 hours (Figure 3, yellow curve). Reference experiments showed no reaction when the activator was added with no gold complex present. Accordingly, doubly methylated halogen bond donor 2 a showed the expected diminished activity, with only 38 % conversion after 3 hours (Table 1, Entry 4; see also Figure 3, red curve).
Table 1.
Cyclization of 6 in the presence of activators and reference compounds.
|
Entry |
Activator[a] |
Conversion[b] of 6 [%] |
k rel [c] |
|---|---|---|---|
|
1 |
None |
≤5[d] |
‐ |
|
2 |
1 a [d] |
60 |
1100 |
|
3 |
1 b |
≤5 |
10 |
|
4 |
2 a [e] |
38 |
660 |
|
5 |
2 b |
≤5 |
3 |
|
6 |
3 a |
≤5 |
1 |
|
7 |
3 b |
≤5 |
‐ |
|
8 |
4 a |
92 |
3100 |
|
9 |
4 b |
≤5 |
18 |
|
10 |
4 c |
25 |
330 |
|
11 |
5 a |
≤5 |
26 |
|
12 |
NaBArF 4 |
95 |
3500 |
|
13 |
TMABArF 4 |
≤5 |
15 |
|
14 |
AgOTf |
5 |
100 |
|
15 |
AgBF4 |
14 |
180 |
|
16 |
AgPF6 |
44 |
740 |
|
17 |
I2 [f] |
≤5 |
‐ |
|
18 |
I2 [g] |
≤5 |
‐ |
[a] Activators added in 2 mol % unless indicated otherwise. [b] Conversion determined by 1H NMR after 3 hours of reaction time, unless indicated otherwise. An error margin of ≈5 % is assumed. All experiments were reproduced at least twice. [c] Relative initial rates after 70 minutes of reaction time, referenced to 3 a (and rounded to two valid digits). [d] 30 hours reaction time. [e] The propargylic amide 6 was added to a preformed solution of the activator and the gold complex. [f] Added in 1 mol %. [g] Added in 1 mol‰.
Figure 3.
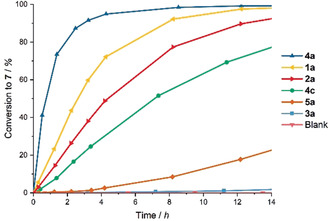
Kinetic plot for the reaction of amide 6 in the presence of various activators.
Gratifyingly, the doubly blocked iodolium salt 3 a showed only little activity, with less than 5 % conversion after 12 hours (Table 1, Entry 6). These results point to the mechanism of activation occurring via halogen bonding as the blockage of electrophilic axes results in markedly diminished activity. Initial rate accelerations for the iodolium salts revealed that 2 a results in an acceleration of k rel=660 relative to the fully blocked compound 3 a; conversely, the completely unblocked XB donor 1 a provides almost double the initial rate of 2 a (Table 1, Entries 2, 4, and 6). The analogous triflate salts 1–3 b all showed poor activity, with only slight conversion noticeable after 3 hours and comparatively poor rate accelerations (Table 1, Entries 3, 5, and 7).
This counterion‐rate dependence is likely due to the need for a weakly coordinating anion under these reaction conditions, as the addition of the strong halide abstracting agent silver triflate only results in 13 % conversion after 12 hours. In comparison, the analogous silver tetrafluoroborate and hexafluorophosphate salts provided markedly increased conversions in the same time (87 % and >95 %, see Supporting Information; compare also Table 1, Entries 14–16).
It is important to note, however, that rapid conversion was also reached by the addition of NaBArF 4, likely due to the strong driving force of NaCl formation; almost complete (95 %) conversion was obtained after only 3 hours of reaction time (Table 1, Entry 12). Therefore, possible activity of the BArF 4 anion itself was additionally tested by the addition of TMABArF 4, which resulted only in trace amounts of conversion, even after 12 hours of reaction time. This result, combined with the inactivity of 3 a, suggests that the observed activity in the cases of the iodolium compounds stems from the unblocked electrophilic axes on the iodine atom and thus, halogen bonding interactions.
With these promising results in hand, the highly preorganized bis(benzimidazolium)‐based halogen bond donor 4 a was tested and showed exceptional activity, with 92 % conversion after 3 hours and almost quantitative product formation after 8 hours of reaction time. The initial rate acceleration with 4 a was almost triple that of iodolium salt 1 a (Table 1, Entry 8; see also Figure 3, dark blue curve).
This observation agrees with previous results,12 in which iodolium salts were at times outperformed by a bidentate, dicationic XB donor; this highlights again the exceptional activity of this preorganized structural motif. The stability of 4 a during the reaction could be followed by 19F NMR: no visible decomposition was observed after several hours of reaction time (see Supporting Information). To exclude the effect of possible undetected decomposition of the XB donors, molecular iodine was tested in the reaction as a possible decomposition product. Addition of 1 mol‰ of I2 yielded no conversion and even 1 mol % of I2, which would result from complete decomposition of the halogen bond donor, showed no activity. Unsurprisingly, the triflate salt 4 b exhibited similar inactivity to the iodolium salts 1–2 b. (Table 1, Entry 9).
The hexafluorophosphate salt 4 c however, which could be synthesized by simple anion exchange from 4 b, showed markedly increased activity compared to the triflate salt, with 25 % conversion after 3 hours of reaction time (Figure 3). This compound, however, suffers from poor solubility in chloroform, which limits the direct comparison with the analogous 4 a and 4 b. To probe the nature of the activation with 4 a further, reference compound 5 a was synthesized; the latter is structurally analogous to 4 a, but incapable of halogen bonding (Figure 2). Expectedly, this salt showed drastically lower activity, with almost no conversion during the first 3 hours of reaction time (Table 1, Entry 11). It is interesting to note, however, that some activity is observed with longer reaction times; as much as 18 % conversion is achieved after 12 hours of reaction time (Figure 3, orange curve). The mechanism behind this activation needs to be investigated further but is unlikely to be related to the XB activation observed for compound 4 a, especially when considering the drastic differences in the initial rate accelerations (Table 1, cf. Entries 8 vs. 11).
As a whole, the presented results constitute, to the best of our knowledge, the first application of cationic XB donors as inert activators in transition metal catalysis and show the compatibility of these compounds with complex reaction systems.
We also tested the halogen bonding activators in the gold‐catalyzed cycloisomerization of 1,6‐enyne 8 (Scheme 2). This reaction forms a mixture of the cyclic products 9 a and 9 b, which for all tested activators were obtained in a ratio of approximately 85:15 in favor of the 5‐membered cyclic product 9 a (see Figure S5). Since this rearrangement is believed to proceed through cyclopropyl gold carbene species, its efficacy underlines that halogen bonding activation is beyond generating simple Lewis acidity.35, 36, 37 In this case, only negligible background reaction is observed in the absence of any halide abstracting agent, effecting less than 5 % yield after 15 hours of reaction time (Table 2, Entry 1 and Figure 4).
Scheme 2.
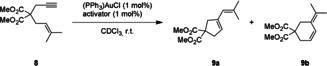
Au‐catalyzed cyclization of malonate ester 8.
Table 2.
Cyclization of 8 in the presence of 1 mol % of activators and reference compounds.
|
Entry |
Activator[a] |
Yield[b] of 9 a/b [%] |
k rel [c] |
|---|---|---|---|
|
1 |
None |
≤5[d] |
‐ |
|
2 |
1 a |
76 |
740 |
|
3 |
1 b |
≤5 |
14 |
|
4 |
2 a |
43 |
420 |
|
5 |
2 b |
≤5 |
10 |
|
6 |
3 a |
≤5 |
1 |
|
7 |
3 b |
≤5 |
10 |
|
8 |
4 a |
83 |
830 |
|
9 |
4 b |
≤5 |
3 |
|
10 |
5 a |
≤5 |
41 |
|
11 |
NaBArF 4 |
84 |
850 |
|
12 |
TMABArF 4 |
≤5 |
5 |
[a] Activators added in 1 mol %. [b] 1H NMR yield of 9 a/b after 40 minutes of reaction time, unless indicated otherwise. An error margin of ≈5 % is assumed. All experiments were reproduced at least twice. [c] Relative initial rates after 20 minutes of reaction time, referenced to 3 a (and rounded to two valid digits). [d] 15 hours reaction time.
Figure 4.
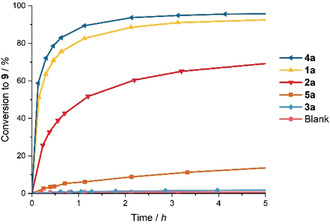
Kinetic plot for the reaction of malonate ester 8 in the presence of various activators.
Addition of 1 mol % of iodolium salt 1 a resulted in 76 % combined yield of products 9 a/b after only 40 minutes of reaction time (Table 2, Entry 2). The partially hindered XB donor 2 a again showed diminished activity, with only 43 % yield in comparable reaction time (Table 2, Entry 4). The inactivity of the tetramethylated salt 3 a was confirmed again; a negligible yield was observed after 5 hours of reaction time (Table 2, Entry 6). The relative rate acceleration of 1 a proved to be almost double that of 2 a, with both compounds being vastly more active than 3 a (Table 2, Entries 2, 4, and 6), which is in line with the results observed for the cyclization of 6.
The strong influence of the counterion was observable in this reaction as well, with triflate salt 1 b yielding under 5 % of product after 3 hours reaction time. The same trend was observed for the sterically hindered iodolium salts 2 b and 3 b: only slight activity was observed for 2 b (Table 2, Entries 3, 5, and 7).
Similarly to the previous reaction, NaBArF 4 proved to be highly active, providing 84 % yield after 40 minutes, while TMABArF 4 was shown to be almost completely inactive. This supports once again that the BArF 4 anion itself is inactive and that any observed activity is due to the corresponding cation (Table 2, Entries 11,12).
The bidentate, bis‐benzimidazolium‐based XB donor 4 a was found again to be the most active halogen bonding activator, with a performance comparable to NaBArF 4, providing a yield of 83 % after 40 minutes and an initial rate acceleration of 830 compared to 3 a (Table 2, Entry 8). Reference compound 5 a also showed poor activity in this reaction, with less than 5 % yield in the initial 40 minutes and only 11 % yield after 3 hours of reaction time (Table 2, Entry 10).
The parallel results obtained for two homogenous gold‐catalyzed reactions and two separate classes of XB donors thus strongly indicate that the mode of activation occurs via halogen bonding. Further insight into the activation of the gold chloride complex was sought by NMR experiments. 31P NMR spectroscopy could be utilized to track the phosphorus shift of the triphenylphosphine ligand on the gold complex used in the catalysis experiments. For the addition of AgOTf to (PPh3)AuCl, a shielding of the phosphine signal has been reported.38, 39 When NaBArF 4 is added however, a significant deshielding occurs, with a signal at 45.2 ppm being observed (Figure 5 b). This signal has commonly been attributed to an (Au(PPh3)2)+ species38, 39, 40 and indicates significant dissociation of the Au−Cl bond.41
Figure 5.
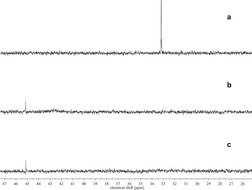
31P NMR shifts observed of (PPh3)AuCl (a) and its 1:1 mixtures with NaBArF 4 (b) and 4 a (c) in CDCl3.
When a 1:1 mixture of XB donor 4 a and (PPh3)AuCl was prepared, the same shift could be observed (Figure 5 c). This result demonstrates again the observed activation of the Au−Cl bond, which could be applied in the reactions demonstrated.
Finally, DFT calculations (M06‐2X42‐D343 def2‐TZVP(D)44 with and without intrinsic solvation using SMD1845 with parameters for chloroform) were performed to obtain a first molecular picture of the interaction of the strongest XB donor 4 a with the gold complex (Figure 6). In both cases, corresponding bidentate halogen bonded adducts were obtained as minima, the formation of which was predicted to be favorable by 27 kcal mol−1 (in the gas phase) or 5 kcal mol−1 (in chloroform). The Au−Cl bond length was elongated in both adducts compared to the isolated complex (2.42 Å vs. 2.32 Å in the gas phase and 2.41 Å vs. 2.37 Å in chloroform), in line with an activation of this bond by the XB donor. The further fate of the activated gold species will likely involve complex equilibria (leading to (Au(PPh3)2)+, see above), the modelling of which is outside the scope of this paper.
Figure 6.
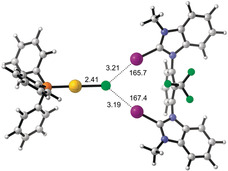
Adduct between halogen bond donor 4 a (with octyl groups replaced by methyl and counteranions omitted) and (PPh3)AuCl in chloroform according to DFT calculations (bond distances in Å, C‐I‐Cl angles in °). Graphics generated by CYLview.46.
In conclusion, two proof‐of‐principle reactions were presented, in which significant activation of a metal‐halogen bond was effected by halogen bond donors. Two classes of XB donors were shown to be efficient activators in homogenous gold catalysis, including one of the first applications of iodolium‐based XB donors. The activities of the tested activators agreed well with previously established trends. Different reference experiments were conducted for both classes of XB donors, which strongly suggest that halogen bonding is crucial to the activation observed. 31P NMR experiments strongly suggest activation of the Au−Cl bond, while computational results support the favorable interaction between the gold complex and halogen bond donors.
These initial results for gold‐catalyzed reactions show promise that the activation presented in this study can be expanded to more systems in transition metal catalysis. Halogen bond donors could present unique and desirable reactivity profiles for this type of activation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Julia Jurksch for performing orientating experiments and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (638337) for financial support. Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2033–390677874—RESOLV. Open access funding enabled and organized by Projekt DEAL.
J. Wolf, F. Huber, N. Erochok, F. Heinen, V. Guérin, C. Y. Legault, S. F. Kirsch, S. M. Huber, Angew. Chem. Int. Ed. 2020, 59, 16496.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.12107832.v1).
Contributor Information
Prof. Claude Y. Legault, Email: claude.legault@usherbrooke.ca.
Prof. Stefan F. Kirsch, Email: sfkirsch@uni-wuppertal.de.
Prof. Stefan M. Huber, Email: stefan.m.huber@rub.de.
References
- 1. Legon A. C., Phys. Chem. Chem. Phys. 2010, 12, 7736–7747. [DOI] [PubMed] [Google Scholar]
- 2.Recently, the term “HaB” has been proposed as an alternative abbreviation for halogen bonding: Scilabra P., Terraneo G., Resnati G., Acc. Chem. Res. 2019, 52, 1313–1324. [DOI] [PubMed] [Google Scholar]
- 3. Metrangolo P., Neukirch H., Pilati T., Resnati G., Acc. Chem. Res. 2005, 38, 386–395. [DOI] [PubMed] [Google Scholar]
- 4. Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G., Chem. Rev. 2016, 116, 2478–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mukherjee A., Tothadi S., Desiraju G. R., Acc. Chem. Res. 2014, 47, 2514–2524. [DOI] [PubMed] [Google Scholar]
- 6. Metrangolo P., Meyer F., Pilati T., Resnati G., Terraneo G., Angew. Chem. Int. Ed. 2008, 47, 6114–6127; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6206–6220. [Google Scholar]
- 7. Brown A., Beer P. D., Chem. Commun. 2016, 52, 8645–8658. [DOI] [PubMed] [Google Scholar]
- 8. Tepper R., Schubert U. S., Angew. Chem. Int. Ed. 2018, 57, 6004–6016; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6110–6123. [Google Scholar]
- 9. von der Heiden D., Vanderkooy A., Erdélyi M., Coord. Chem. Rev. 2020, 407, 213147. [Google Scholar]
- 10. Bulfield D., Huber S. M., Chem. Eur. J. 2016, 22, 14434–14450. [DOI] [PubMed] [Google Scholar]
- 11. Sutar R. L., Huber S. M., ACS Catal. 2019, 9, 9622–9639. [Google Scholar]
- 12. Heinen F., Engelage E., Dreger A., Weiss R., Huber S. M., Angew. Chem. Int. Ed. 2018, 57, 3830–3833; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3892–3896. [Google Scholar]
- 13. Sutar R. L., Engelage E., Stoll R., Huber S. M., Angew. Chem. Int. Ed. 2020, 59, 6806–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindsay V. N. G., Lin W., Charette A. B., J. Am. Chem. Soc. 2009, 131, 16383–16385. [DOI] [PubMed] [Google Scholar]
- 15. Lindsay V. N. G., Charette A. B., ACS Catal. 2012, 2, 1221–1225. [Google Scholar]
- 16. Shixaliyev N. Q., Gurbanov A. V., Maharramov A. M., Mahmudov K. T., Kopylovich M. N., Martins L. M. D. R. S., Muzalevskiy V. M., Nenajdenko V. G., Pombeiro A. J. L., New J. Chem. 2014, 38, 4807–4815. [Google Scholar]
- 17. Carreras L., Serrano-Torné M., van Leeuwen P. W. N. M., Vidal-Ferran A., Chem. Sci. 2018, 9, 3644–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carreras L., Benet-Buchholz J., Franconetti A., Frontera A., van Leeuwen P. W. N. M., Vidal-Ferran A., Chem. Commun. 2019, 55, 2380–2383. [DOI] [PubMed] [Google Scholar]
- 19. Zordan F., Brammer L., Sherwood P., J. Am. Chem. Soc. 2005, 127, 5979–5989. [DOI] [PubMed] [Google Scholar]
- 20. Mínguez Espallargas G., Brammer L., Sherwood P., Angew. Chem. Int. Ed. 2006, 45, 435–440; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 449–454. [Google Scholar]
- 21. Libri S., Jasim N. A., Perutz R. N., Brammer L., J. Am. Chem. Soc. 2008, 130, 7842–7844. [DOI] [PubMed] [Google Scholar]
- 22. Beweries T., Brammer L., Jasim N. A., McGrady J. E., Perutz R. N., Whitwood A. C., J. Am. Chem. Soc. 2011, 133, 14338–14348. [DOI] [PubMed] [Google Scholar]
- 23. Brammer L., Mínguez Espallargas G., Libri S., CrystEngComm 2008, 10, 1712. [Google Scholar]
- 24.For a related study, see: Scheiner S., CrystEngComm 2019, 21, 2875–2883. [Google Scholar]
- 25. Mosquera M. E. G., Gomez-Sal P., Diaz I., Aguirre L. M., Ienco A., Manca G., Mealli C., Inorg. Chem. 2016, 55, 283–291. [DOI] [PubMed] [Google Scholar]
- 26. Mosquera M. E. G., Egido I., Hortelano C., López-López M., Gómez-Sal P., Faraday Discuss. 2017, 203, 257–283. [DOI] [PubMed] [Google Scholar]
- 27. Bezzubov S. I., Kalle P., Bilyalova A. A., Tatarin S. V., Dolzhenko V. D., Chem. Eur. J. 2018, 24, 12779–12783. [DOI] [PubMed] [Google Scholar]
- 28. Bulatova M., Melekhova A. A., Novikov A. S., Ivanov D. M., Bokach N. A., Z. Kristallogr. - Cryst. Mater. 2018, 233, 371–377. [Google Scholar]
- 29. Voelkel M. H. H., Wonner P., Huber S. M., ChemistryOpen 2020, 9, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jungbauer S. H., Huber S. M., J. Am. Chem. Soc. 2015, 137, 12110–12120. [DOI] [PubMed] [Google Scholar]
- 31. Tšupova S., Rudolph M., Rominger F., Hashmi A. S. K., Adv. Synth. Catal. 2016, 358, 3999–4005. [Google Scholar]
- 32. Schießl J., Schulmeister J., Doppiu A., Wörner E., Rudolph M., Karch R., Hashmi A. S. K., Adv. Synth. Catal. 2018, 360, 2493–2502. [Google Scholar]
- 33. Wegener M., Huber F., Bolli C., Jenne C., Kirsch S. F., Chem. Eur. J. 2015, 21, 1328–1336. [DOI] [PubMed] [Google Scholar]
- 34. Hashmi A. S. K., Weyrauch J. P., Frey W., Bats J. W., Org. Lett. 2004, 6, 4391–4394. [DOI] [PubMed] [Google Scholar]
- 35. Jiménez-Núñez E., Echavarren A. M., Chem. Rev. 2008, 108, 3326–3350. [DOI] [PubMed] [Google Scholar]
- 36. Nieto-Oberhuber C., Muñoz M. P., Buñuel E., Nevado C., Cárdenas D. J., Echavarren A. M., Angew. Chem. Int. Ed. 2004, 43, 2402–2406; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2456–2460. [Google Scholar]
- 37. Inagaki F., Matsumoto C., Okada Y., Maruyama N., Mukai C., Angew. Chem. Int. Ed. 2015, 54, 818–822; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 832–836. [Google Scholar]
- 38. Zhdanko A., Ströbele M., Maier M. E., Chem. Eur. J. 2012, 18, 14732–14744. [DOI] [PubMed] [Google Scholar]
- 39. Kumar M., Jasinski J., Hammond G. B., Xu B., Chem. Eur. J. 2014, 20, 3113–3119. [DOI] [PubMed] [Google Scholar]
- 40. Brenzovich W. E., Benitez D., Lackner A. D., Shunatona H. P., Tkatchouk E., Goddard W. A., Toste F. D., Angew. Chem. Int. Ed. 2010, 49, 5519–5522; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5651–5654. [Google Scholar]
- 41.Previous studies on cationic gold species have invoked the presence of complex equilibria processes following the liberation of (PPh3)Au+, see for example ref. [33].
- 42. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 43. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132–133, 154104. [DOI] [PubMed] [Google Scholar]
- 44. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 45. Engelage E., Schulz N., Heinen F., Huber S. M., Truhlar D. G., Cramer C. J., Chem. Eur. J. 2018, 24, 15983–15987. [DOI] [PubMed] [Google Scholar]
- 46.CYLview, 1.0b; C. Y. Legault, Université de Sherbrooke 2009 (http://www.cylview.org).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary