

Abstract
Metal–organic framework (MOF)‐derived Co‐N‐C catalysts with isolated single cobalt atoms have been synthesized and compared with cobalt nanoparticles for formic acid dehydrogenation. The atomically dispersed Co‐N‐C catalyst achieves superior activity, better acid resistance, and improved long‐term stability compared with nanoparticles synthesized by a similar route. High‐angle annular dark‐field–scanning transmission electron microscopy, X‐ray photoelectron spectroscopy, electron paramagnetic resonance, and X‐ray absorption fine structure characterizations reveal the formation of CoIINx centers as active sites. The optimal low‐cost catalyst is a promising candidate for liquid H2 generation.
Keywords: cobalt, dehydrogenation, energy, nanoparticles, single-atom catalysts
The small print: Co‐N‐C single‐atom catalysts were derived from metal– organic frameworks and compared with related nanoparticles in the selective dehydrogenation of formic acid. The highly dispersed single CoIINx sites demonstrated improved reactivity and resistance to acid, constituting the current state‐of‐the‐art in low‐cost earth‐abundant catalysts for this transformation.

Supported metal single‐atom catalysts (SACs) have stimulated strong interest because they offer improved atom utilization efficiency and potentially higher catalytic turnover frequencies (TOFs) compared to established heterogeneous materials.1 Because of their unique properties, recently SACs have been studied for pollutant removal (CO oxidation), the water–gas shift (WGS) reaction, value‐added production of chemicals (for example, selective hydrogenation), transformation of biomass platform molecules (oxygenation of alcohols), methanol steam reforming, photo‐ and even electro‐catalytic reactions (the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and carbon dioxide reduction reaction (CO2RR)), and so on.2
Although the characteristics of quantum size effects, tunable electronic structure, and strong metal–support interaction make SACs advantageous compared to small metal clusters and nanoparticles (NPs), they often exhibit similar or lower activity than NPs.3 As an example, the activity of Pd/Fe3O4 in styrene hydrogenation exhibits an initially sharp rise with increasing metal size, but then gradually reduces.4 A similar reactivity trend has been observed for specific Co NPs, which demonstrated excellent activity and selectivity for the hydrogenation of nitroarenes towards anilines, while related catalyst with atomically dispersed CoNx species led to azo products.5
Notably, the high surface free energy of metals on the SACs surface favors aggregation to larger NPs—especially with harsh reaction conditions.6 To understand such behavior, and with the objective to rationally design optimal heterogeneous catalysts, comparison of the catalytic performance of SACs and NPs for the same reaction under similar conditions is required.3, 7
Based on our continuing interest in the development of base metal catalysts, herein we report a detailed comparison of the activity and stability of Co‐based SACs with related NPs for hydrogen generation from formic acid (FA; Scheme 1).
Scheme 1.
Concept of CO2/HCO2H cycle with consequent generation of H2 for use in a proton‐exchange membrane (PEM) (top) and potential advantages of Co‐N‐C (SACs) application (bottom).
In general, liquid organic hydrogen carriers8 are considered as promising candidates for reversible hydrogen storage and release. Among them FA,9 with a hydrogen capacity of 53 g L−1 or 4.4 wt %, has attracted considerable attention.10 In spite of the extremely high TOF and selectivity gained with some existing catalysts, several challenges still exist.11 For example, most of the known catalysts for FA dehydrogenation are based on noble‐metal‐based metallic or bimetallic NPs.12 Based on environmental and economic considerations, the use of non‐noble metal catalysts is appealing despite their lower activity.13 To date, both SACs and NPs have proven to be effective catalysts for FA dehydrogenation but there is a lack of direct comparison under the same or similar experimental conditions. For instance, in 2017 a CoNx catalyst demonstrated good activity for FA dehydrogenation, but both single‐atom and sub‐nano sites were found on the catalyst, so the real active species was unclear.14
Initially, we compared the performance of potential Co catalysts with atomically dispersed metal atoms and related NPs. The supported materials were prepared by pyrolysis of zeolitic imidazolate frameworks (ZIFs).15 As shown in Figure 1 A, Co‐ZIF and ZnCo‐ZIF were formed by sodalite coordination of Co2+ and Co2+/Zn2+ nodes with 2‐methylimidazole using methanol as a solvent at room temperature. With the introduction of Zn2+, Co2+ ions in the ZIF could be spatially isolated and bonded by linkers. During the pyrolysis processes under inert conditions (Ar), the metal ions were reduced by carbonized organic linkers. Because of Zn evaporation, metal ions were dispersed at the atomic level on N‐doped porous carbon.15b Without the help of Zn2+, larger Co NPs on N‐doped carbon were formed.
Figure 1.
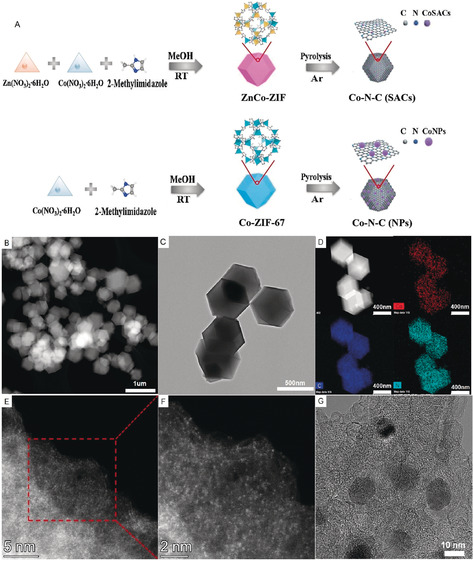
A) The synthesis route of SACs and NPs. B,C) HRTEM images of SACs. D) The EDS mapping of SACs shows the homogeneous distribution of Co and N on Carbon. E,F) Magnified AC‐HAADF‐STEM images of SACs reveals the atomic dispersed Co sites. G) HRTEM images of NPs.
Transmission electron microscopy (TEM) measurements of the material pyrolyzed at 1000 °C reveal that Co‐N‐C(SACs) retain the initial dodecahedral shape of ZIF (ca. 300 nm; Figures 1 B,C). The elemental mapping images of SACs by energy‐dispersive X‐ray spectroscopy (EDS) indicate that the distributions of Co, N, and C are homogenously dispersed over the entire architectures (Figure 1 D). Atomically dispersed Co centers on carbon were observed by sub‐angstrom resolution aberration‐corrected high‐angle annular dark‐field scanning transmission electron microscopy (AC‐HAADF‐STEM) analysis (Figure 1 E). These active sites were clearly identified by several separated bright dots under a high magnification mode (Figure 1 F). This result matches well with powder X‐ray diffraction (XRD) curves (Supporting Information, Figure S5), which present only a broad shoulder peak assigned to the (002) plane of the graphitic carbon.16 In the case of Co‐N‐C(NPs) pyrolyzed at 800 °C, an ellipsoid morphology and NPs with a diameter of approximately 10 nm on the carbon support were found (Figure 1 G). According to the XRD results, several peaks that are characteristic of metallic Co (01‐077‐7451) were observed. This indicates the formation of face‐centered cubic Co crystals (Figure S5).
To further investigate the geometry and electronic structure of Co‐N‐C materials, electron paramagnetic resonance (EPR) measurements were performed (Figure 2 A). Co‐N‐C(NPs) presented a broad peak with a g value of 2.87 arising from the strong ferromagnetic properties of Co NPs. Notably, Co‐N‐C(SACs) present no EPR signal associated with ferromagnetic Co clusters and no signal associated with single Co2+ sites, even at 100 K, because of their short relaxation time. Only a carbon radical signal at g=2.005 from the support was observed. However, when Ph3P was added as a ligand to SACs to probe the presence of isolated Co2+ sites, a new signal at g=2.279 assigned to Co2+ single sites17 coordinated to Ph3P ligand appeared. To obtain further surface information about the catalysts, Co 2p (Figure 2 B) and N 1s (Figure 2 C) X‐ray photoelectron spectroscopy (XPS) investigations were performed. According to the deconvoluted Co 2p1/2 curves, three sub‐peaks were assigned to surface Co3+, Co2+, and Co at around 782, 780.7, and 779.8 eV, respectively.18 Only Co2+ species were found on the SACs surface, while some Co3+ and Co were observed on the NP catalyst. Apparently, the formation of Co2+−N species is favorable for the SACs, which coincides well with prior literature.5b, 18, 19
Figure 2.
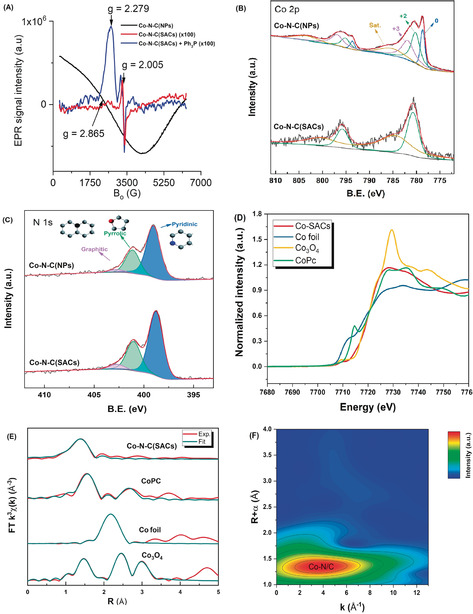
A) EPR spectra. B,C) XPS Co 2p (B), and N 1s (C) spectra. D) XANES, and E,F) EXAFS spectra of Co‐N‐C(SACs).
The N 1s spectrum revealed that three types of N species coexist: graphitic N (403 eV), pyrrolic N (401.2 eV), and pyridinic N (399 eV).13e, 20 Compared with NPs, a binding energy shift of pyridinic N (purple dashed line) could be found for SACs and no changes on the position of graphitic N and pyrrolic N. Combined with the normalized surface Co and N concentration based on the integration of these subpeaks (Supporting Information, Table S5), we believe that pyridinic N serves as the major anchor points for atomically dispersed Co sites in the case of SACs. Co valence states of SACs were also identified in the X‐ray absorption near‐edge structure (XANES) spectra (Figure 2 D). The absorption edge energy at 7728.3 eV in the XANES spectrum of SACs is similar to that of the Co phthalocyanine (CoPc) reference, suggesting that Co2+ is the dominating Co species. Finally, extended X‐ray absorption fine structure (EXAFS) analysis was performed to further verify the coordination environment of Co sites on SACs (Figures 2 E,F). As shown in Figure 2 E (R space), atomically dispersed Co on Co‐N‐C(SACs) exhibits a prominent peak at approximately 1.38 Å arising from Co−N coordination, the Co−Co peak (ca. 2.17 Å) evident in Co foil at higher R is absent. Wavelet transform (WT) was adopted to further investigate Co K‐edge EXAFS oscillations (Figure 2 F). Only one area with an intensity maximum at 3.8 Å−1 attributed to Co−N coordination was observed, confirming that Co single sites were the main species on the Co‐N‐C(SACs).
Subsequently, the two characterized samples of Co‐N‐C(SACs) and Co‐N‐C(NPs) were tested for the benchmark dehydrogenation reaction of FA under different conditions (Table 1). When H2O was used as solvent, NPs presented a better gas evolution rate than SACs within the first 10 minutes, but the activity decreased with time (Figure S2). This increased gas formation is likely a result of the increased Co content of the NPs. Notably, the gas production in Table 1 is not normalized on metal content. Meanwhile, a solution color change was observed for NPs but not for SACs (Figures S8 and S9). This is explained by the stronger Co−N interaction in the SAC material. Moreover, when applying Co‐N‐C(NPs) the pH and presence of additives did not change the gas formation rate and stability significantly. In contrast, when the solvents 2‐methyltetrahydrofuran (MeTHF) and propylene carbonate (PC) were used no significant color change was observed in the dehydrogenation reaction (Figure S10). In both cases, the gas production rates of SACs were significantly better, while the hydrogen selectivities were different. On the other hand, PC offered good hydrogen selectivity. Compared with NPs, SACs were around 150 % more productive in PC at 98 °C (Table 1). Moreover, SACs also demonstrated activity at the lower temperature of 75 °C. Neither NPs nor the previously described Co(1)/phen(7)/C are able to operate at these conditions. Applying PC not only resulted in the best catalytic performance but also improved stability of active sites for both kinds of Co‐N‐C materials.
Table 1.
Comparing Co‐NPs and Co‐SACs for FA dehydrogenation.[a]

|
Entry |
Catalyst |
Solvent [mL] |
T set [°C] |
Gas production rate[b] [mL g−1 h−1] |
|---|---|---|---|---|
|
1 |
Co‐N‐C(SACs) |
H2O |
100 (88) |
167.9 |
|
2 |
Co‐N‐C(NPs) |
H2O |
100 (88) |
195.8 |
|
3 |
Co‐N‐C(SACs) |
MeTHF |
85 (75) |
181.7 |
|
4 |
Co‐N‐C(NPs) |
MeTHF |
85 (75) |
79.2 |
|
5 |
Co‐N‐C(SACs) |
PC |
110 (98) |
319.2 |
|
6 |
Co‐N‐C(NPs) |
PC |
110 (98) |
229.2 |
|
7 |
Co‐N‐C(SACs) |
PC |
85 (75) |
68.8 |
|
8 |
Co‐N‐C(NPs) |
PC |
85 (75) |
0 |
|
9[c] |
Co(1)/phen(7)/C |
PC |
85 (75) |
0 |
|
10 |
– |
PC |
110 (98) |
0 |
|
11 |
Zn‐ZIF‐8 |
PC |
110 (98) |
1.6 |
|
12 |
Co‐ZIF‐67 |
PC |
110 (98) |
16.7 |
|
13 |
ZnCo‐ZIF |
PC |
110 (98) |
20.8 |
|
14 |
Co(NO3)2⋅6 H2O |
PC |
110 (98) |
29 |
[a] Reaction conditions: FA (10 mmol), catalyst (30 mg) or Co(NO3)2⋅4 H2O (5 mg) in specified solvents and reaction time of 4 h. The set temperature was around 10–12 °C higher than the actual reaction temperature shown in parentheses. [b] Total gas production of H2 and CO2 measured by manual burette; the rate is based on gram of catalyst per hour. [c] A nano‐Co catalyst prepared using phenanthroline was chosen. Key: propylene carbonate (PC), 2‐methyltetrahydrofuran (MeTHF). Experiments were performed at least twice (except entries 11–14) with reproducibility differences of 1.3 to 10.8 %.
Based on the results of atomic absorption spectroscopy (AAS) measurements (Table S6), used SACs present reduced Co (10.8 wt %) and N loss (0.6 wt %) compared to that of NPs (Co: 38.1 wt % and N: 41.5 wt %). This implies that CoIINx on SACs also have a stronger acid resistance than metallic Co on NPs. Considering that the pyrolysis temperature might have a significant effect on the coordination number of single Co sites, a series of catalysts with different pyrolysis temperatures was prepared and investigated for FA dehydrogenation (Supporting Information). The normalized gas rates (based on gram of Co per hour) of Co‐N‐C materials with different pyrolysis temperatures are provided in Figure 3. It was found that the rise of pyrolysis temperature had a negative impact on the reactivity of Co‐N‐C(NPs). During the pyrolysis process, metal–imidazolate–metal linkages would be cleaved to release CN fragments and trigger agglomeration of Co NPs upon weakening of metal−N bonds. Conversely, Co‐N‐C(SACs)‐1000 possessed the best gas rate of 23.6 L gCo −1 h−1 among the three Co‐N‐C(SACs) catalysts. To the best of our knowledge, this is also the best performance for an earth‐abundant‐based heterogeneous catalyst for dehydrogenation of FA.14 This activity is also comparable to most heterogeneous noble‐metal‐based catalysts (Table S1).
Figure 3.
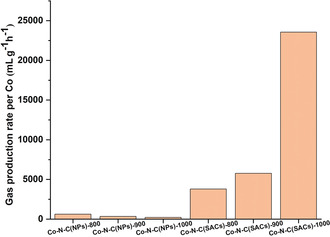
Catalysts with different pyrolysis temperature and their normalized catalytic gas production rates per Co in FA dehydrogenation. Reaction conditions: FA (10 mmol, 377 μL), catalyst (30 mg), PC (6 mL), T set=110 °C, T actual=98 °C, 4 h. Pyrolysis temperatures of catalysts shown behind. Experiments were performed at least twice.
The optimal activity of the Co‐N‐C(SACs)‐1000 is explained by the prevalence of CoIIN2 sites in this material. On the one hand, although Zn cannot be completely removed during the pyrolysis step, a significant Zn effect on activity can be neglected according to the low activity of Zn‐ZIF‐8 (Table 1, entry 11) and no indication of Co−Zn interaction was indicated by the EXAFS results (Figures 2 E, F). According to Wu et al., atomically dispersed Co catalysts with Co−N4, Co−N3, and Co−N2 sites can be obtained at different pyrolysis temperatures.15a Thus, changing Co−N coordination from Co−N4 and Co−N3 to Co−N2 might enhance the surface energy, thereby improving the reactivity of atomically dispersed Co sites. Therefore, CoIIN2 sites on Co‐N‐C(SACs) are considered the best catalytic centers for FA dehydrogenation.
EPR measurements were carried out to explore the stability of active sites on the optimal catalyst Co‐N‐C(SACs)‐1000. The EPR spectrum of the used Co‐N‐C(SACs)‐1000 catalyst is similar to that in Figure 2 A, which showed no signal corresponding to ferromagnetic Co NPs, suggesting that the active sites are stable against aggregation during the reaction course. Addition of Ph3P to the used catalyst to probe the presence of isolated Co2+ sites presented a spectrum similar to that in Figure 2 A, thereby supporting our hypothesis. A further proof for the increased stability of SACs compared to NPs is shown in Figure S3. Here, a linear increase of gas production was observed for Co‐N‐C(SACs), whereas saturation curves were observed for Co‐N‐C(NPs). These results highlight the better stability of CoNx moieties on SACs compared to metallic Co on NPs. Finally, long‐term experiments with a large substrate‐to‐catalyst ratio were carried out to further demonstrate the stability of these novel SACs. Even after 120 hours of reaction, the Co‐N‐C(SACs) showed only a slight deactivation and more than double gas production volume compared to Co‐N‐C(NPs) (Figure S11). EPR measurements showed that CoNx sites on Co‐N‐C(SACs) remained after this time since no ferromagnetic signal was observed (Figure S12). The AAS results demonstrated a decreased Co and N loss of SACs compared to NPs (Table S6). Nevertheless, a large excess of HCOOH at high temperature inevitably causes loss of atomically dispersed CoNx moieties, although they have a better stability than that of metallic NPs.
In summary, a detailed comparison of supported Co single‐atom and NP catalysts has been performed. Several SACs and NPs were prepared and characterized by HAADF‐STEM, EPR, XAFS, and XPS. Their catalytic performance was compared for FA dehydrogenation whereby atomically dispersed Co catalysts exhibited better efficiency, selectivity, and stability than NP catalysts. Further investigations demonstrated that the highly dispersed single CoIINx sites with high reactivity and acidic resistance enabled improved catalytic activities and stabilities. We believe the presented findings will be of use for the development of other improved non‐noble metal‐based catalysts, too.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support by the China Scholarship Council (201806025040), the National Natural Science Foundation of China (21906004), the BMBF, and the Germany and European Research Council (EU project 670986‐NoNaCat). We thank Alexander Léval, Jacob Schneidewind, Maximilian Marx, Dr. Rui Sang, and Anja Kammer for scientific discussions and valuable suggestions. We thank Dr. Nils Rockstroh, Reinhard Eckelt, Dr. Stephan Bartling, and Kathleen Schubert for their technical and analytical support (all LIKAT). We thank Dr. Valérie Briois for preliminary XAS measurements at ROCK beamline‐SOLEIL (supported by the French National Research Agency as part of the “Investissements d'Avenir” program (reference: ANR10‐EQPX45)). QEERI Core lab (Dr. Said Mansour and Janarthanan Ponraj) are greatly acknowledged for TEM analysis.
X. Li, A.-E. Surkus, J. Rabeah, M. Anwar, S. Dastigir, H. Junge, A. Brückner, M. Beller, Angew. Chem. Int. Ed. 2020, 59, 15849.
References
- 1.
- 1a. Zhang H. B., Liu G. G., Shi L., Ye J. H., Adv. Energy Mater. 2018, 8, 1701343; [Google Scholar]
- 1b. Mitchell S., Vorobyeva E., Perez-Ramirez J., Angew. Chem. Int. Ed. 2018, 57, 15316–15329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15538–15552. [Google Scholar]
- 2.
- 2a. Fu Q., Saltsburg H., Flytzani-Stephanopoulos M., Science 2003, 301, 935–938; [DOI] [PubMed] [Google Scholar]
- 2b. Gu J., Hsu C. S., Bai L., Chen H. M., Hu X., Science 2019, 364, 1091–1094; [DOI] [PubMed] [Google Scholar]
- 2c. Guo X., Fang G., Li G., Ma H., Fan H., Yu L., Ma C., Wu X., Deng D., Wei M., Tan D., Si R., Zhang S., Li J., Sun L., Tang Z., Pan X., Bao X., Science 2014, 344, 616–619; [DOI] [PubMed] [Google Scholar]
- 2d. Jones J., Xiong H., DeLaRiva A. T., Peterson E. J., Pham H., Challa S. R., Qi G., Oh S., Wiebenga M. H., Pereira Hernández X. I., Wang Y., Datye A. K., Science 2016, 353, 150–154; [DOI] [PubMed] [Google Scholar]
- 2e. Liu P., Zhao Y., Qin R., Mo S., Chen G., Gu L., Chevrier D. M., Zhang P., Guo Q., Zang D., Wu B., Fu G., Zheng N., Science 2016, 352, 797–800; [DOI] [PubMed] [Google Scholar]
- 2f. Yao S., Zhang X., Zhou W., Gao R., Xu W., Ye Y., Lin L., Wen X., Liu P., Chen B., Crumlin E., Guo J., Zuo Z., Li W., Xie J., Lu L., Kiely C. J., Gu L., Shi C., Rodriguez J. A., Ma D., Science 2017, 357, 389–393; [DOI] [PubMed] [Google Scholar]
- 2g. Yang L., Cheng D. J., Zeng X. F., Wan X., Shui J. L., Xiang Z. H., Cao D. P., Proc. Natl. Acad. Sci. USA 2018, 115, 6626–6631; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h. Zhang H. B., Wei J., Dong J. C., Liu G. G., Shi L., An P. F., Zhao G. X., Kong J. T., Wang X. J., Meng X. G., Zhang J., Ye J. H., Angew. Chem. Int. Ed. 2016, 55, 14310–14314; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14522–14526. [Google Scholar]
- 3. Liu L. C., Corma A., Chem. Rev. 2018, 118, 4981–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rossell M. D., Caparrós F. J., Angurell I., Muller G., Llorca J., Seco M., Rossell O., Catal. Sci. Technol. 2016, 6, 4081–4085. [Google Scholar]
- 5.
- 5a. Jagadeesh R. V., Murugesan K., Alshammari A. S., Neumann H., Pohl M.-M., Radnik J., Beller M., Science 2017, 358, 326–332; [DOI] [PubMed] [Google Scholar]
- 5b. Liu W. G., Zhang L. L., Yan W. S., Liu X. Y., Yang X. F., Miao S., Wang W. T., Wang A. Q., Zhang T., Chem. Sci. 2016, 7, 5758–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Yang X. F., Wang A. Q., Qiao B. T., Li J., Liu J. Y., Zhang T., Acc. Chem. Res. 2013, 46, 1740–1748; [DOI] [PubMed] [Google Scholar]
- 6b. Su X., Yang X.-F., Huang Y., Liu B., Zhang T., Acc. Chem. Res. 2019, 52, 656–664. [DOI] [PubMed] [Google Scholar]
- 7. Ding K., Gulec A., Johnson A. M., Schweitzer N. M., Stucky G. D., Marks L. D., Stair P. C., Science 2015, 350, 189–192. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Okada Y., Sasaki E., Watanabe E., Hyodo S., Nishijima H., Int. J. Hydrogen Energy 2006, 31, 1348–1356; [Google Scholar]
- 8b. Preuster P., Papp C., Wasserscheid P., Acc. Chem. Res. 2017, 50, 74–85. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Li Z., Xu Q., Acc. Chem. Res. 2017, 50, 1449–1458; [DOI] [PubMed] [Google Scholar]
- 9b. Mellmann D., Sponholz P., Junge H., Beller M., Chem. Soc. Rev. 2016, 45, 3954–3988; [DOI] [PubMed] [Google Scholar]
- 9c. Dalebrook A. F., Gan W., Grasemann M., Moret S., Laurenczy G., Chem. Commun. 2013, 49, 8735–8751; [DOI] [PubMed] [Google Scholar]
- 9d. Grasemann M., Laurenczy G., Energy Environ. Sci. 2012, 5, 8171–8181; [Google Scholar]
- 9e. Johnson T. C., Morris D. J., Wills M., Chem. Soc. Rev. 2010, 39, 81–88; [DOI] [PubMed] [Google Scholar]
- 9f. Wang W. H., Himeda Y., Muckerman J. T., Fujita E., Adv. Inorg. Chem. 2014, 66, 189–222. [Google Scholar]
- 10. Williams R., Crandall R. S., Bloom A., Appl. Phys. Lett. 1978, 33, 381–383. [Google Scholar]
- 11.
- 11a. Fukuzumi S., Yamada Y., Suenobu T., Ohkubo K., Kotani H., Energy Environ. Sci. 2011, 4, 2754–2766; [Google Scholar]
- 11b. Boddien A., Mellmann D., Gärtner F., Jackstell R., Junge H., Dyson P. J., Laurenczy G., Ludwig R., Beller M., Science 2011, 333, 1733–1736; [DOI] [PubMed] [Google Scholar]
- 11c. Fellay C., Dyson P. J., Laurenczy G., Angew. Chem. Int. Ed. 2008, 47, 3966–3968; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4030–4032; [Google Scholar]
- 11d. Fellay C., Yan N., Dyson P. J., Laurenczy G., Chem. Eur. J. 2009, 15, 3752–3760; [DOI] [PubMed] [Google Scholar]
- 11e. Gan W., Dyson P. J., Laurenczy G., ChemCatChem 2013, 5, 3124–3130; [Google Scholar]
- 11f. Montandon-Clerc M., Dalebrook A. F., Laurenczy G., J. Catal. 2016, 343, 62–67; [Google Scholar]
- 11g. Loges B., Boddien A., Junge H., Beller M., Angew. Chem. Int. Ed. 2008, 47, 3962–3965; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4026–4029; [Google Scholar]
- 11h. Boddien A., Loges B., Gärtner F., Torborg C., Fumino K., Junge H., Ludwig R., Beller M., J. Am. Chem. Soc. 2010, 132, 8924–8934. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Tedsree K., Li T., Jones S., Chan C. W. A., Yu K. M. K., Bagot P. A. J., Marquis E. A., Smith G. D. W., Tsang S. C. E., Nat. Nanotechnol. 2011, 6, 302–307; [DOI] [PubMed] [Google Scholar]
- 12b. Li S. J., Zhou Y. T., Kang X., Liu D. X., Gu L., Zhang Q. H., Yan J. M., Jiang Q., Adv. Mater. 2019, 31, 1806781; [DOI] [PubMed] [Google Scholar]
- 12c. Zhu C. Z., Fu S. F., Shi Q. R., Du D., Lin Y. H., Angew. Chem. Int. Ed. 2017, 56, 13944–13960; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14132–14148; [Google Scholar]
- 12d. Wang Q., Tsumori N., Kitta M., Xu Q., ACS Catal. 2018, 8, 12041–12045; [Google Scholar]
- 12e. Onishi N., Iguchi M., Yang X., Kanega R., Kawanami H., Xu Q., Himeda Y., Adv. Energy Mater. 2019, 9, 1801275. [Google Scholar]
- 13.
- 13a. Liu Q., Yang X., Huang Y., Xu S., Su X., Pan X., Xu J., Wang A., Liang C., Wang X., Zhang T., Energy Environ. Sci. 2015, 8, 3204–3207; [Google Scholar]
- 13b. Yi N., Saltsburg H., Flytzani-Stephanopoulos M., ChemSusChem 2013, 6, 816–819; [DOI] [PubMed] [Google Scholar]
- 13c. Weng Z., Jiang J., Wu Y., Wu Z., Guo X., Materna K. L., Liu W., Batista V. S., Brudvig G. W., Wang H., J. Am. Chem. Soc. 2016, 138, 8076–8079; [DOI] [PubMed] [Google Scholar]
- 13d. Zhu Q. L., Tsumori N., Xu Q., J. Am. Chem. Soc. 2015, 137, 11743–11748; [DOI] [PubMed] [Google Scholar]
- 13e. Bulushev D. A., Zacharska M., Shlyakhova E. V., Chuvilin A. L., Guo Y., Beloshapkin S., Okotrub A. V., Bulusheva L. G., ACS Catal. 2016, 6, 681–691; [Google Scholar]
- 13f. Bulushev D. A., Zacharska M., Lisitsyn A. S., Podyacheva O. Y., Hage F. S., Ramasse Q. M., Bangert U., Bulusheva L. G., ACS Catal. 2016, 6, 3442–3451; [Google Scholar]
- 13g. Zacharska M., Bulusheva L. G., Lisitsyn A. S., Beloshapkin S., Guo Y., Chuvilin A. L., Shlyakhova E. V., Podyacheva O. Y., Leahy J. J., Okotrub A. V., Bulushev D. A., ChemSusChem 2017, 10, 720–730. [DOI] [PubMed] [Google Scholar]
- 14. Tang C., Surkus A. E., Chen F., Pohl M. M., Agostini G., Schneider M., Junge H., Beller M., Angew. Chem. Int. Ed. 2017, 56, 16616–16620; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16843–16847. [Google Scholar]
- 15.
- 15a. Wang X., Chen Z., Zhao X., Yao T., Chen W., You R., Zhao C., Wu G., Wang J., Huang W., Yang J., Hong X., Wei S., Wu Y., Li Y., Angew. Chem. Int. Ed. 2018, 57, 1944–1948; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1962–1966; [Google Scholar]
- 15b. Yin P. Q., Yao T., Wu Y., Zheng L. R., Lin Y., Liu W., Ju H. X., Zhu J. F., Hong X., Deng Z. X., Zhou G., Wei S. Q., Li Y. D., Angew. Chem. Int. Ed. 2016, 55, 10800–10805; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10958–10963. [Google Scholar]
- 16. Zhao Y., Liu Z., Chu W., Song L., Zhang Z., Yu D., Tian Y., Xie S., Sun L., Adv. Mater. 2008, 20, 1777–1781. [Google Scholar]
- 17.
- 17a. Baumgarten M., Winscom C. J., Lubitz W., Appl. Magn. Reson. 2001, 20, 35–70; [Google Scholar]
- 17b. Ikeda-Saito M., Yamamoto H., Yonetani T., J. Biol. Chem. 1977, 252, 8639–8644. [PubMed] [Google Scholar]
- 18. Biesinger M. C., Payne B. P., Grosvenor A. P., Lau L. W. M., Gerson A. R., Smart R. S. C., Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar]
- 19.
- 19a. Sun T., Xu L., Li S., Chai W., Huang Y., Yan Y., Chen J., Appl. Catal. B 2016, 193, 1–8; [Google Scholar]
- 19b. Zhang L., Wang A., Wang W., Huang Y., Liu X., Miao S., Liu J., Zhang T., ACS Catal. 2015, 5, 6563–6572. [Google Scholar]
- 20. Arrigo R., Schuster M. E., Xie Z., Yi Y., Wowsnick G., Sun L. L., Hermann K. E., Friedrich M., Kast P., Hävecker M., Knop-Gericke A., Schlögl R., ACS Catal. 2015, 5, 2740–2753. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary