

Summary
Xinjiang has been the largest and highest yield cotton production region not only in China, but also in the world. Improvements in Upland cotton cultivars in Xinjiang have occurred via pedigree selection and/or crossing of elite alleles from the former Soviet Union and other cotton producing regions of China. But it is unclear how genomic constitutions from foundation parents have been selected and inherited. Here, we deep‐sequenced seven historic foundation parents, comprising four cultivars introduced from the former Soviet Union (108Ф, C1470, 611Б and KK1543) and three from United States and Africa (DPL15, STV2B and UGDM), and re‐sequenced sixty‐nine Xinjiang modern cultivars. Phylogenetic analysis of more than 2 million high‐quality single nucleotide polymorphisms allowed their classification two groups, suggesting that Xinjiang Upland cotton cultivars were not only spawned from 108Ф, C1470, 611Б and KK1543, but also had a close kinship with DPL15, STV2B and UGDM. Notably, identity‐by‐descent (IBD) tracking demonstrated that the former Soviet Union cultivars have made a huge contribution to modern cultivar improvement in Xinjiang. A total of 156 selective sweeps were identified. Among them, apoptosis‐antagonizing transcription factor gene (GhAATF1) and mitochondrial transcription termination factor family protein gene (GhmTERF1) were highly involved in the determination of lint percentage. Additionally, the auxin response factor gene (GhARF3) located in inherited IBD segments from 108Ф and 611Б was highly correlated with fibre quality. These results provide an insight into the genomics of artificial selection for improving cotton production and facilitate next‐generation precision breeding of cotton and other crops.
Keywords: Gossypium hirsutum, Xinjiang cotton improvement, identity by descent, resequencing
Introduction
By 2050, there will be approximately 9 billion people on the planet (Gerland et al., 2014). Plant breeding will play an important role in feeding this huge population and in dealing with challenges such as climate change, annual reductions and urbanization of arable land (Ritchie et al., 2018). Phenotype‐based selection by independent and local people 10–12 thousand years ago resulted in the dramatic phenotypic changes eventually seen in modern crops (Doebley et al., 2006; Meyer et al., 2012). As Mendelian and quantitative genetics theories were developed in the late nineteenth and early twentieth centuries, breeders could exploit family relationships and estimate breeding values accurately (Wallace et al., 2018). As early as the 1990s, molecular markers and genomic data were implemented in the selection of better lines and were used to complement phenotypic data in plant breeding (Tanksley et al., 1989). Genome‐wide association mapping has successfully identified quantitative trait loci (QTL) for relative traits, leading to genomic prediction approaches, which have become common practice in the comparison and selection of the best individuals for complex traits (Bevan et al., 2017; Heslot et al., 2015; Meuwissen et al., 2001). Nowadays, we can integrate genotype and phenotype data efficiently to identify causal genetic features that breeders can select and use to perform biological interventions (Ramstein et al., 2018; Wallace et al., 2018). Next‐generation sequencing (NGS) technologies have facilitated the wide availability of genome sequence assemblies, the resequencing of several hundred lines, the development of high‐density genetic maps, a range of marker genotyping platforms and the identification of markers associated with agronomic traits (Varshney et al., 2019).
Hybridizations between elite cultivars with desirable traits and intensive artificial selection pressures are essential for the development of new cultivars. These selection pressures have constricted genetic variation in local populations, so more desirable genes have accumulated in the most recently established cultivars. These cultivars exhibit phenotypes commensurate with important traits (Shinada et al., 2014), as exemplified in the development of Kitaake, Kyowa and Huanghuazhan in rice (Shinada et al., 2014; Zhou et al., 2016), B73, Mo17, etc. in maize (Lai et al., 2010; Smith et al., 2004; Wu et al., 2016), and Ekangmian 9 and CRI12 in cotton (Lu et al., 2018; Ma et al., 2018). Moreover, identity‐by‐descent (IBD) regions have been demonstrated to be powerful in relatedness evaluation and mapping of genetic loci associated with phenotypic variations in many studies (Browning and Browning, 2010; Browning and Thompson, 2012; Stevens et al., 2011; Westerlind et al., 2015) . The shift in genomic structure and the history of the genetic architecture of Huanghuazhan have been analysed, and the major effectual genomic regions were pinpointed to traceable genomic regions (Chen et al., 2017; Zhou et al., 2016). In addition, Fang et al. (2017a) used IBD detection to show that the genetic contributions of Deltapline 15 (DPL15), Stoneville 2B (STV2B) and Uganda cotton (UGDM) to seven widely grown cultivars in China were approximately 14.19%, 10.45% and 4.19%, respectively. This suggests that these three cultivars are very important in Chinese cotton breeding (Fang et al., 2017a, 2017b).
The Xinjiang Uygur autonomous region is the largest cotton‐growing region in the world. In Xinjiang, the cotton planting area spans more than 2.54 million hectares and produced 5.00 million tons of cotton lint in 2019, approximately accounting for 76.08% of the cotton planting area and 84.94% in China (http://www.stats.gov.cn) and ~19% in the world of cotton production. This suggests that Xinjiang is not only an essential cotton industry base in China, but also plays an irreplaceable role in the world’s cotton industry. The history of Xinjiang Upland cotton cultivars is complex. They have been developed by integrating elite alleles from the former Soviet Union, the United States and Uganda in Africa (Figures S1 and S2) (Abdullaev et al., 2013; Bowman et al., 2006; Huang, 1996; Tian et al., 2016). There is no genome‐wide account of the history of cotton breeding in Xinjiang, and the IBD segments from the formation of the accessions and their relevant functions remain largely uncharacterized. Thus, we deep‐sequenced seven historical and representative foundation parents, 108Ф, 611Б, C1470 and KK1543, which were introduced from the former Soviet Union and played a vital role in the early 1960s in Xinjiang, and three other landraces or cultivars—DPL15 and STV2B from the United States and UGDM from Uganda, which have significantly influenced modern cultivar improvement in Yangtze River and Yellow River cotton‐growing regions in China. Then, sixty‐nine Xinjiang modern varieties developed between 1969 and 2013 were re‐sequenced to allow an in‐depth analysis of genomic variation and selection segments in order to provide genome‐wide level insights into the development of modern cultivars from the given foundation parents, to shed light on the genetic mechanism of artificial selection during cotton breeding in Xinjiang, and to facilitate next‐generation precision breeding of cotton and other crops.
Results
Cultivar improvement over the cotton breeding process in Xinjiang
Seven foundation parents that were introduced to Xinjiang in the 1960s, and sixty‐nine modern cultivars bred between 1969 and 2013, were analysed to identify genomic signatures of breeding in Xinjiang. Nine fibre quality and yield‐related traits of these 76 accessions including seven founder landraces (DPL15, STV2B, UGDM, 108Ф, C1470, 611Б and KK1543) were measured in the northern (Shihezi) and the southern (Korla) regions in Xinjiang to investigate the yield and fibre quality improvements between these foundation parents and modern cultivars. These traits comprised yield traits such as boll weight (BW), boll number (BN), fruit branch number (FBN) and lint percentage (LP), and fibre qualities such as fibre elongation (FE), length (FL), micronaire (FM), strength (FS) and uniformity (FU; Figure S3). Their field performances in yield and fibre qualities were mostly consistent with their original recording (Huang, 1996; Tian et al., 2016). Line charts (Figure S4) revealed that all traits were simultaneously improved over the years as expected, with the exception of fibre fineness. Among these traits, LP was much higher in modern cultivars (more than 40%) than that in foundation parents (around 37%; Figure S4c), indicating that LP has been selected as a major target during cotton improvement in Xinjiang. BW was only slightly improved from about 5.31 to 5.77 g (Figure S4d) and fibre length was increased to 28.90 mm (Figure S4f), and strength to 31.20 cN/tex (Figure S4h), suggesting that lint yield and fibre quality had been improved gradually in Xinjiang.
Genomic variations and population structure
In order to explore genetic variations and genetic patterns that arose during cotton breeding, a total of 630 Gb of clean data with an average depth of 54.33‐fold were made available through whole‐genome resequencing of seven foundation parents. Of them, 99.29% of the reads were mapped to our newly assembled reference genome in Gossypium hirsutum acc. TM‐1 (Hu et al., 2019), and 98.31% of the genome was covered by their reads. We further obtained 1.19 Tb of clean data for the other 69 cultivars, which covered 97.53% of the TM‐1 genome, with an average 10.65‐fold depth and 94.74% mapping rate (Table S1).
We incorporated and filtered several single nucleotide polymorphisms (SNPs) for each sample. Ultimately, 2 395 681 SNPs met the rough filter standards (see Experimental procedures) and were used to investigate phylogenetic relationships. Of them, 26 SNPs were randomly selected for polymerase chain reaction (PCR)‐based sequencing in 10 accessions and we found that the accuracy was 94.12% (Table S11). Therefore, the quality of SNP calling was considered reliable for further analysis. Of 2 395 681 SNPs, 45 511 (1.90% of the total) were located within protein‐coding genes, 157 067 (6.56%) were located in upstream or downstream regions, and the remaining 2 080 375 (86.84%) SNPs were located in intergenic regions. In the coding regions, we annotated 28 654 (1.20% of the total, 62.96% of the CDs) nonsynonymous, 187 splicing, 174 stop‐loss and 605 stop‐gain SNPs that caused amino acid changes, elongated transcripts or caused premature stopping (Figure 1a).
Figure 1.
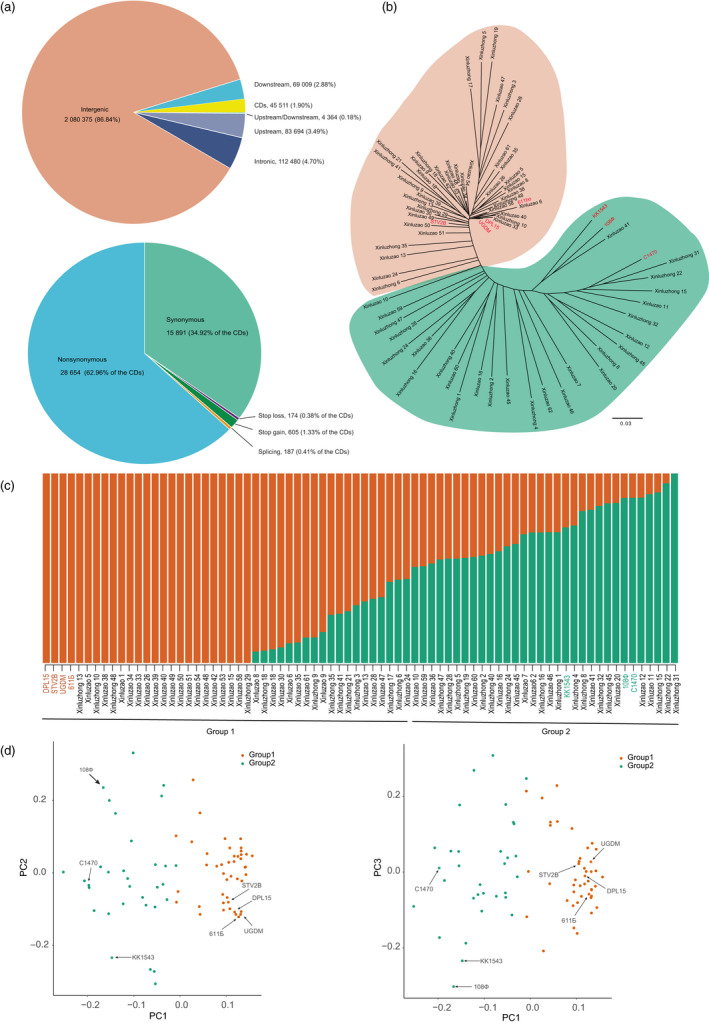
Single nucleotide polymorphisms (SNPs) annotation, phylogenetic tree, genetic structure and principal component analysis (PCA) of the 76 accessions. (a) Summary of SNP annotation. The upper pie chart shows the distribution of SNPs, while the lower pie chart shows the detailed distribution of SNPs in gene coding regions. (b) Phylogenetic analysis of 76 accessions. The neighbour‐joining tree was constructed using whole‐genome SNP data. The cotton samples were divided into clade 1 (orange) and clade 2 (green). (c) Population structure analysis of all accessions. The accessions were divided into 2 groups when K = 2. The y‐axis quantifies cluster membership, and the x‐axis represents the different accessions. (d) PCA plot of the first three components. The left plot shows PC1 and PC2, and the right shows PC1 and PC3. Group 1 and group 2 are orange and green, respectively.
Cross‐validation of the K (the number of populations modelled) test with figures ranging from 1 to 10 suggested that K = 2 was a sensible modelling choice (Figure S5). Combined with the neighbour‐joining (NJ) tree (Figure 1b), population structure (Figure 1c) and principal component analysis (Figure 1d, Table S2), these accessions could be divided into two groups: group 1 (n = 44) contained more Xinluzao cultivars (28, 40.58%) together with DPL15, STV2B, UGDM and 611Б, and group 2 (n = 32) contained slightly more Xinluzhong cultivars (16, 23.19%) than the Xinluzao (13, 18.84%), together with the former Soviet Union cultivars: 108Ф, C1470 and KK1543 (Figure 1b–d). These findings are consistent with the breeding history or cultivar pedigree in Xinjiang (Tian et al., 2016). 611Б is generally used in the northern part of Xinjiang to develop short season cultivars, whereas 108Ф and C1470 have been generally used in the southern part of the Xinjiang to develop middle season cotton cultivars. As an early maturation landrace, KK1543 has been used in both regions, which may be the reason that it is not as easy to completely classify these cultivars.
Genomic signatures of improvement in yield and fibre quality
To identify potential selective signatures during cotton cultivar improvement in Xinjiang, we scanned genomic regions using fixation index value (Fst) and nucleotide diversity (π) methods (Figure 2). The potential selective signals during the improvement were analysed using whole‐genome polymorphisms from the foundation parents to modern improved cultivars. The Fst values between the foundation parents and modern improved cultivars were 0.0271 and 0.0306 for the A sub‐genome and 0.0236 for the D sub‐genome, respectively. The nucleotide diversity levels of the foundation parents (πparents = 8.91E‐05) were a little higher than that of the modern varieties (πcultivars = 8.04E‐05), indicating that the modern improved cultivars were influenced by a modest artificial selection pressure during the cotton cultivar improvement in Xinjiang, which led to low genetic diversity.
Figure 2.
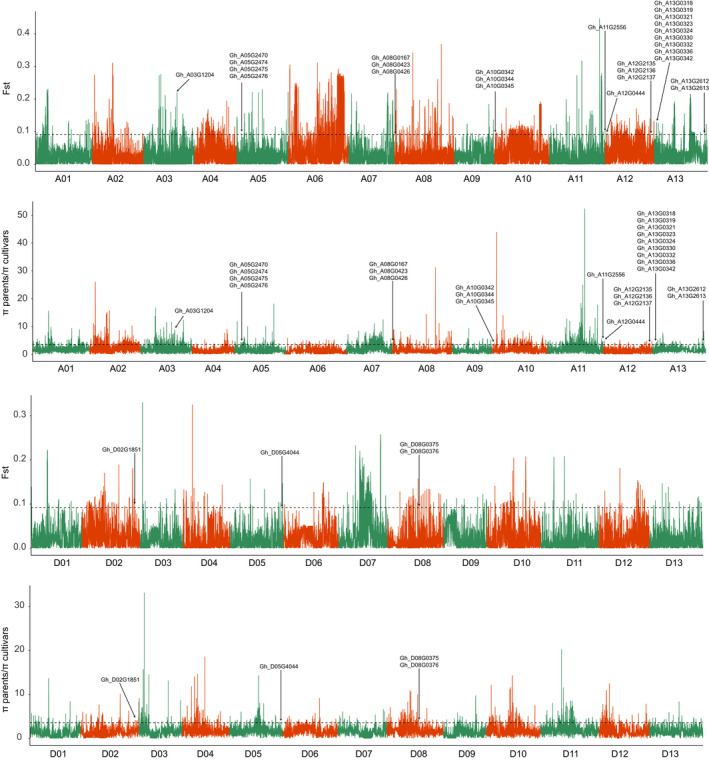
Genome‐wide screen of artificial selection sweeps. Whole‐genome analysis of the selective sweeps through the comparison of parents and cultivars. The genome‐wide thresholds of 3.5721 and 0.09157 were defined by the top 5% of the πparents/πcultivars and Fst values. The arrows indicate the sweeps that contained 31 genes with nonsynonymous SNPs.
We detected 156 improvement‐selective sweeps covering 23.86 Mb of the whole genome through comparisons of whole‐genome genetic diversities in the seven parents and 69 modern cultivars, with a top 5% genetic diversity cut‐off (top 5% Fst ≥ 0.09157, or πparents/πcultivars ≥ 3.5721; Table S3). Of them, 118 selective regions covering 16.32 Mb were located in the A sub‐genome and much higher than the 38 sweeps covering 7.54 Mb located in the D sub‐genome, further suggesting that the A sub‐genome is more important in modern cotton improvement in yield and fibre quality (Fang et al., 2017a). Interestingly, we found that 242 QTLs identified by linkage mapping in previous reports overlapped with these 123 improvement‐selective sweeps (Table S4). For instance, a selective sweep at A07:12.88–12.98Mb overlapped with a QTL hot spot region comprising five QTLs for BW, fibre strength and length, confirming that these regions have been artificially selected during cotton cultivar improvement in Xinjiang, China.
Identification of candidate improvement‐selective genes
Within the identified improvement‐selective sweeps, 318 candidate genes, 237 in the A sub‐genome and 81 in the D sub‐genome, respectively, were identified (Table S5), indicating that these genes have undergone artificial selection during the Upland cotton improvement in Xinjiang. We exploited the expression profiling data from RNA‐seq and functional annotation of the orthologues in Arabidopsis to determine whether the candidate selective genes are likely associated with lint yield, fibre quality or stress tolerance (Table S5, Figure S6). The expression patterns of 318 improvement‐selective genes were analysed in root, stem, leaf and fibre tissues from three stages of fibre development, and found that a number of genes may play vital roles in fibre development (Figure S6). For instance, the expression levels of Gh_A13G0335, which encodes a leucine‐rich repeat protein kinase, were higher during the fibre initiation and elongation stages than in the secondary‐wall‐synthesis stage, and the ATP binding of this protein has been reported to be related to fibre length and strength (Zhang et al., 2015). Gh_A08G0422, which encodes UDP‐glucose pyrophosphorylase 1, was found to be highly expressed during fibre development and may be involved in cell wall biosynthesis, a pathway that has been proved to be particularly important for fibre quality (Zhang et al., 2015). Gh_A05G1951 was expressed at high levels in 15, 20 and 25 DPA fibres, and Gh_A08G1220 was expressed dynamically in all tissues. Both genes encode C2H2 zinc finger proteins. Two NAC transcription factors, Gh_A05G1960 and Gh_A05G2474, were down‐regulated in all three fibre development stages. Together, these data suggest these genes may contribute to fibre quality improvement.
We also investigated the expression levels of improvement‐selective genes after stress treatments, comprising heat, cold, salt and drought, to study their involvement to stress responses (Figure S6). Gh_A02G0993, a protein kinase, was expressed at high levels 1 and 3 h after heat, 3 and 24 h after cold, 1 and 12 h after salt and 1, 3 and 6 h after drought stress, suggesting that it may be related to all stress tolerances. Gh_A05G1960, which encodes a NAC transcription factor, had significantly higher expression levels at each hour following heat treatment, at 12 and 24 h after cold, at 1 and 12 h after salt and 1, 3 and 6 h after drought stress. Moreover, Gh_A05G1954, which encodes a protein phosphatase 2C family protein, Gh_A08G0826, an indole‐3‐acetic acid‐induced ARG2, and Gh_A13G0346 bHLH, a transcription factor protein, were also found to be highly expressed at each hour post‐stress treatment. Taken together, these results suggest that the 318 candidate improvement‐selective genes play important roles not only in fibre quality improvement and increases in lint yield, but also in stress‐related responses. This indicates that they are worth researching further.
We then focused on 31 nonsynonymous genes (27 in the A sub‐genome and 4 in the D sub‐genome). With these genes, we integrated RNA‐seq data, carried out gene‐based association analysis and annotation of each gene to study their functions in lint yield and fibre quality (Figure 3; Figure S6). Interestingly, we identified two candidate improvement‐selective genes responsible for increasing LP and lint yield. One improvement‐selective sweep, ranging from 3.8 to 4.0 Mb on chr.A13, where Fst and πparents/πcultivars were significantly higher than the top 5% genetic diversity cut‐off values, contained two candidate genes, Gh_A13G0332 and Gh_A13G0336 (Figure 3a). Gh_A13G0332, an orthologous gene to AT5G61330 in Arabidopsis, encodes apoptosis‐antagonizing transcription factor (AATF) and was designated as GhAATF1. It has been reported that AATF genes play important roles in the regulation of gene transcription and cell proliferation (Haanpaa et al., 2009; Sharma, 2013). The GhAATF1 protein contains the AATF (PF13339) domain from amino acid 118 to 252 at the N‐terminal and the AATF (PF08164) domain from 331 to 406 amino acid in the C‐terminal. GhAATF1 contained a nonsynonymous SNP (A13:3842189: T/C) in the conserved domain at the 14bp position, resulting in an amino acid change from leucine to serine (Figure 3b). Haplotype analysis revealed that the accessions carrying the CC allele had significantly higher LP than those with the TT allele under two different environmental conditions (Figure 3c; P = 0.0061 in KRL and P = 0.04 in SHZ; Figure S7). The GhAATF1 gene was dominantly expressed during the early fibre development, especially in the 3, 5 and 10 DPA in ovules and fibres, indicating its important role in lint yield (Figure 3d). Another gene, Gh_A13G0336, which encodes a mitochondrial transcription termination factor family protein, designated as GhmTERF1, is orthologous to AT2G21710 in Arabidopsis. GhmTERF1 contains an mTERF domain (PF02536) from amino acid 293 to 600. The nonsynonymous SNP (A13:3968673: A/C) occurred at the 28bp position in the exon regions, resulting in an amino acid change from asparagine to histidine (Figure 3b). AtmTERF genes in Arabidopsis are related to seedling growth and embryo development (Babiychuk et al., 2011; Meskauskiene et al., 2009). The haplotypes (AA and CC) had positive phenotypic effects on the LP in two environments (P = 0.015 in KRL and P = 0.0039 in SHZ, respectively; Figure 3c, Figure S7). The expression of GhmTERF1 was higher at the early fibre development and elongation stages (Figure 3d). Therefore, GhAATF1 and GhmTERF1, with two nonsynonymous SNPs, are likely the most important genes involved in LP and lint yield increases in Xinjiang.
Figure 3.
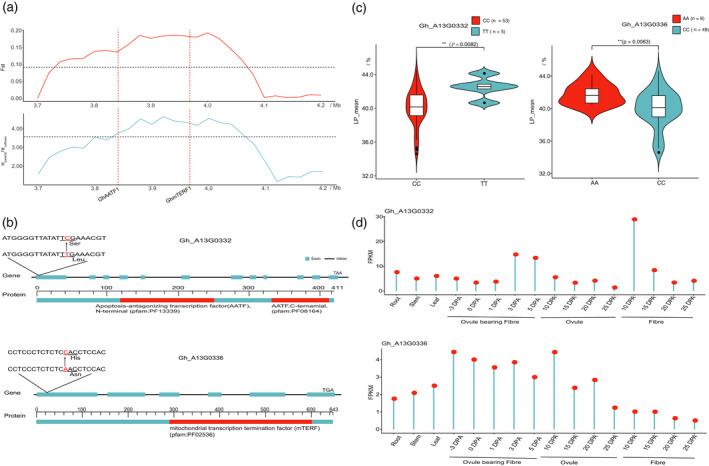
Identification of candidate genes related to trait development under artificial selection. (a) Selection signals from 3.7 to 4.2 Mb on chromosome A13. The upper line plot shows Fst in red, and the lower line plot shows πparents/πcultivars in blue. (b) Gene structure and polymorphisms of GhAATF1 and GhmTERF1. (c) Lint percentage (%) analyses of accessions with CC and TT genotypes of GhAATF1 (left). Lint percentage (%) analyses of accessions with AA and CC genotypes of GhmTERF1 (right). Centre line, median; box limits, upper and lower quartiles; and whiskers, 1.5x the interquartile range (**P < 0.01, two‐sided t‐test). (d) Transcriptomic patterns of GhAATF1 (upper) and GhmTERF1 (lower) in distinct tissues, based on the number of fragments per kilobase of the exon model per million mapped reads (FPKM) in a single experiment, including root, stem and leaf tissues during ovule and fibre development stages.
Variations and transmission of elite alleles to improve lint yield and fibre quality
To evaluate the contributions of various foundation parents to the current cultivars during historical cotton improvement in Xinjiang, we analysed the IBD segments between seven foundation parents and modern cultivars. Based on the numerous whole‐genome SNPs described above, a total of 10 385 IBD segments including 6122 segments located in the A sub‐genome, and 4263 located in the D sub‐genome, were identified (Figure 4, Figure S8, Table S6). Among them, 1623 (15.63%) IBD segments were inherited from C1470; 1528 (14.71%) from 108Ф; 1509 (14.53%) from KK1543; 1262 (12.15%) from DPL15; 1202 (11.57%) from STV2B; 1175 (11.31%) from UGDM; 1045 (10.06%) and from 611Б. In total, 5705 (54.93%) IBD segments were inherited from the former Soviet Union (C1470, 108Ф, KK1543 and 611Б), and 3639 (35.04%) were from America (DPL15 and STV2B) or Africa (UGDM). Therefore, in terms of quantity of IBD segments, it is obvious that modern cultivars in Xinjiang are closer to the introduced former Soviet Union varieties.
Figure 4.
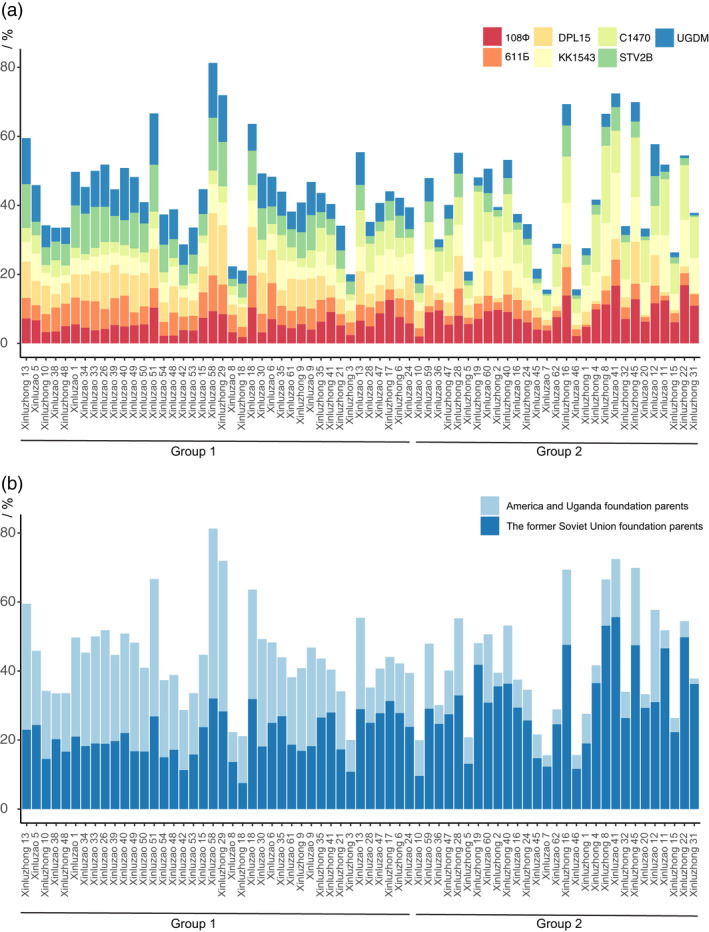
Distribution of modern cultivar identity‐by‐descents (IBDs) inherited from foundation parents. (a) Distribution of sixty‐nine modern cultivar IBDs inherited from seven foundation parents. The genetic constitutions of these sixty‐nine cultivars were identified from 108Ф, 611Б, DPL15, KK1543, C1470, STV2B and UGDM. Different colours represent different parents, and the corresponding colour of each parent is noted on the legend (top right). (b) The contribution of various genetic pools from different areas (the former Soviet Union and America or Africa) in modern cultivars.
Furthermore, of 10 385 IBD segments, 984 existed in more than two current cultivars (Table S7), suggesting they may be very important in improving lint yield and fibre qualities since they are selected and maintained in breeding. There were 1041 IBD regions (10.02%) that came from two or more foundation parents, suggesting their origin in American cotton and therefore the narrow kinship background of Xinjiang Upland cotton populations (Table S6, Table S7). For instance, apart from the IBD regions inherited from seven foundation parents in Xinluzao 1, two IBD segments, respectively, located on chr.A12, from 14 323 879 to 14 649 871 bp, and chr.D09, from 19 077 842 to 19 400 618, in Xinluzao 1 were inherited simultaneously from 108Ф and DPL15. In addition, one IBD segment inherited from 108Ф/611Б/KK1543 was located on chr.D01, from 42 914 274 to 42 992 363 in Xinluzao 1. Moreover, several IBD segments were inherited from more than two foundation parents. For example, we identified 108Ф/DPL15/STV2B, 108Ф/KK1543, 108Ф/KK1543/STV2B, 108Ф/STV2B, 108Ф/UGDM, 611Б/DPL15/STV2B, 611Б/KK1543, DPL15/STV2B, DPL15/UGDM, KK1543/UGDM, C1470/UGDM and STV2B/UGDM IBD segments in Xinluzao 1 (Table S6, Table S7), which may be because all seven foundation parents were inherited from American cotton landraces, such as King, Cook, Lone star, Delfos and Dixie Triumph. This suggests these IBD regions are very important for lint yield and fibre quality improvement in American cotton improvement, so they have been selected and almost fixed during cotton cultivar improvement (Figure S2).
By calculating the percentage of the length of each chromosome that comprises IBD fragments, the genetic constitutions of each modern cultivar derived from the seven foundation parents were variable (Figure 4a). The maximum percentage of IBD segments inherited from 108Ф was 16.87%, found in Xinluzhong 22, and the minimum 1.83%, in Xinluzhong 18; from C1470, the maximum was 22.40%, in Xinluzhong 8, and the minimum 1.05%, in Xinluzao 42; from KK1543, the maximum was 19.13%, in Xinluzao 41, and the minimum 0.98%, in Xinluzhong 18; and from 611Б, the maximum was 10.42%, in Xinluzao 6, and 0.63%, in Xinluzhong 1. The maximum percentages of IBD segments inherited from STV2B, DPL15 and UGDM, at 15.35%, 18.05% and 15.85%, respectively, were all located in Xinluzao 58, and the minimums, which were 0.46%, 0.41% and 0.69%, respectively, were all in Xinluzhong 31. The genetic contribution of the seven foundation parents to Xinluzao 58 totalled 81.28%: 18.05% from DPL15, 15.85% from UGDM, 15.35% from STV2B, 10.37% from 611Б, 9.34% from 108 Ф, 8.42% from KK1543 and 3.90% from C1470. The lowest contribution of the foundation parents was in Xinluzao 7, with only 15.57% in total derived from C1470 (4.63%), 108 Ф (3.71%), KK1543 (2.43%), DPL15 (1.59%), 611Б (1.51%), UGDM (1.20%) and STV2B (0.50%).
According to Xinjiang cultivar pedigree (Figure S2), Xinluzao 8 was selected as one of the parents from Xinluzao 1, which was derived from 611Б previously; Xinluzao 35 from KK1543; and Xinluzhong 28 from DPL 15 originally. Thus, we found that the highest percentages of IBD segments were in Xinluzao 8 (5.08%), which were inherited from 611Б, in Xinluzao 35 (8.74%), inherited from KK1543, and in Xinluzhong 28 (9.48%), inherited from DPL15 (Figure 4a). These were consistent with their pedigrees.
The average genetic constitutions of the modern cultivars in Xinjiang were calculated. The mean coverage percentages derived from C1470, 108Ф, KK1543, DPL15, UGDM, STV2B and 611Б were approximately 6.851%, 6.847%, 6.719%, 6.080%, 5.969%, 5.583% and 4.887%, respectively. Consequently, it is clear that foundation parents introduced from the former Soviet Union (total coverage percentage: 25.31%) played more important roles than those introduced directly from America and Africa (total coverage percentage: 17.63%) in modern Xinjiang cultivars. This is consistent with the breeding history in this region, where breeders introduced these former Soviet Union cotton varieties to improve cotton adaptation and yield in Xinjiang in the 1960s (Figure 4b). For instance, 55.62% of IBD regions of Xinluzao 41 were inherited from the former Soviet Union cultivars (19.13% from KK1543, 16.73% from 108Ф, 12.22% from C1470 and 7.54% from 611Б), while only 16.81% IBD segments were from America or Africa (6.87% from STV2B, 6.02% from DPL15 and 3.92% from UGDM). Similar results were shown in Xinluzhong 8, in which 53.14% of IBD segments were inherited from the former Soviet Union accessions (22.40% from C1470, 15.39% from KK1543, 11.27% from 108Ф and 4.07% from 611Б) and 13.37% came from American or African cultivars (5.47% from STV2B, 4.16% from DPL15 and 3.74% from UGDM). Naturally, according to the genetic structure (Figure 1), the average coverage of America and Uganda foundation parents in group 1 (22.88%) was much greater than that in group 2 (10.39%), while the average coverage of the former Soviet Union foundation parents in group 1 (21.14%) was less than that in group 2 (31.06%). This goes some way to explain why group 2 members were more closely related to the former Soviet Union accessions, while group 1 members were closer to DPL15, STV2B and UGDM.
Candidate genes to increase lint yield and improve fibre quality in IBD segments
The IBD segments involved in the artificial selection sweeps were analysed. A total of 192 unique IBD regions (143 in the A sub‐genome and 49 in the D sub‐genome), including 1174 genes were selected (Table S9, Table S10). Among these genes, 93 (61 in the A and 32 in the D sub‐genomes) contained nonsynonymous, stop‐gain or stop‐loss SNP mutations. When the annotation and expression profiles of these genes in different tissues and different stress treatment were integrated, we found that several genes may be involved in fibre development or tolerance response (Figure S9). For instance, Gh_A08G0167, which encodes a serine/threonine kinase protein, showed high expression levels at 3DPA and 5DPA ovule bearing fibres, at 15DPA fibres and at 15DPA ovules, while Gh_A10G0619, which encodes a DOF zinc finger protein, showed low expression levels in each fibre development stage, but high expression 1 and 12 h post‐heat treatment, and 12 and 24 h post‐cold treatment. Moreover, Gh_D01G1996, which encodes a bZIP transcription factor family protein, showed high expression levels at −3 and 1DPA ovule bearing fibres, at 10, 15 and 25DPA ovules, and at 20DPA fibres, suggesting that it functions in fibre initiation. This gene was also highly expressed each hour post‐heat treatment, 1 and 12 h post‐salt treatment, and 6 and 12 h post‐drought treatment, indicating that it is also involved in stress tolerance.
Most importantly, we identified a major gene, Gh_A10G0304, which encodes auxin response factor 3 (GhARF3) and is homologous to AT2G33860 in Arabidopsis. Gene‐based association or haplotype analysis and transcriptomic data revealed that this gene may be responsible for fibre quality improvement in terms of both fibre length and fibre strength. Gene‐based association showed that a nonsynonymous SNP from T to A occurred at 1487 bp in the CDS of GhARF3, which resulted in an amino acid change from isoleucine to asparagine (Figure 5a). The expression of GhARF3 was expressed at high level at −3 DPA and 0 DPA ovule bearing fibres and at 10 DPA and 15DPA ovules (Figure 5b). This was significantly associated with fibre length and strength in two environments (FL: P = 0.0035, FS: P = 0.033; Figure 5c, Figure S10). It has been reported that ARF genes can affect fibre cell initiation and fibre development (Sun et al., 2015; Xiao et al., 2018). GhARF3 was found to be located in the Xinluzao 13 IBD segment from 2 403 609 to 2 951 829 on chr.A10, which was inherited from 108Ф, and in the Xinluzao 35 IBD region from 1 198 185 to 2 888 834 on chr.A10, which was inherited from 611Б. Therefore, this gene was pedigree‐selected from the former Soviet Union foundation parents.
Figure 5.
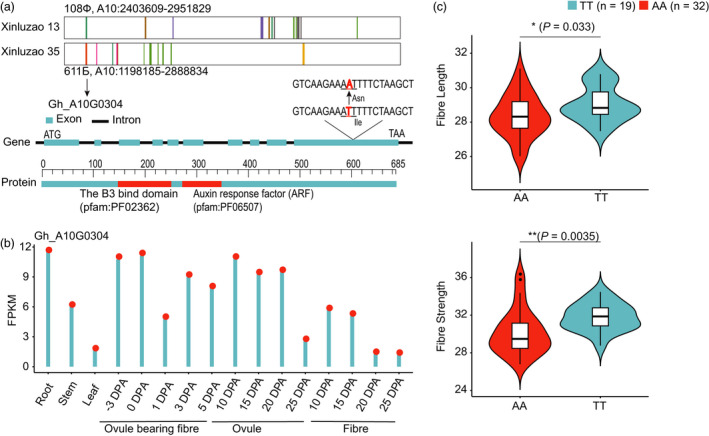
Identification of candidate gene GhARF3. (a) Identification of GhARF3 gene structure and location in identity‐by‐descent (IBD) regions. The IBD regions inherited from 108Ф and 611Б on chr.A10, a nonsynonymous SNP (T‐to‐A) resulted in a change from isoleucine to asparagine. (b) Transcriptomic patterns of GhARF3 in distinct tissues, including root, stem and leaf, in ovule and fibre development stages based on FPKM in a single experiment. (c) Fibre length (upper) and fibre strength (lower) analyses of accessions with AA and TT genotypes of GhARF3. Centre line, median; box limits, upper and lower quartiles; and whiskers, 1.5× the interquartile range (*P < 0.05, **P < 0.01, two‐sided t‐test).
Discussion
Genomic signatures of lint yield and fibre quality improvement in Upland cotton in Xinjiang
With the development of sequencing technology and molecular methods, we are embracing a new era of crop breeding. It is becoming easier to dissect the artificial selection processes and the constitution of cultivars, and these have been proven to be an effective strategy for driving genomic adaption and for revealing dynamic changes in traits to improve crop breeding. In this study, we re‐sequenced seven foundation parent cultivars and sixty‐nine modern cultivars in Xinjiang to analyse genomic variations and selection segments via whole‐genome resequencing technology. Based on a group of SNPs, a number of improvement‐selective sweeps involving a string of causal genes and IBD segments inherited from foundation parents have been identified, laying a foundation for germplasm resource analysis and breeding by design in the future. In recent years, an abundance studies have been performed to trace IBD segments and exploit key trait regions during the breeding process following their well‐defined genetic paths (Chen et al., 2017; Fang et al., 2017a, 2017b; Lai et al., 2010; Lu et al., 2018; Ma et al., 2018; Wu et al., 2016). Based on more than 2 million SNPs, the Xinjiang cotton population could be divided into two groups (Figure 1), preliminarily indicating that Upland cotton populations in Xinjiang were not only spawned from DPL15, STV2B and UGDM, which are the original germplasms used for modern Upland cotton breeding in Yangtze River and Yellow River cotton‐growing regions in China (Fang et al., 2017a, 2017b), but also have a close kinship with the former Soviet Union cotton landraces. For the first time, we report here that the genetic constitution of the Xinjiang cotton population is more similar to the former Soviet Union landraces by collecting data on the quality and coverage of IBD segments, thus providing evidence at the genomic level that Upland cotton breeding in Xinjiang originated from former Soviet Union cultivars (Figure 4, Figures S1 and S2, Table S6). The genetic constitution of modern cultivars in Xinjiang is more similar to C1470 (6.851%), 108Ф (6.847%), KK1543 (6.719%) and 611Б (4.887%) than DPL15 (6.080%), UGDM (5.969%) and STV2B (5.583%), suggesting that the background of modern Upland cotton cultivars in Xinjiang is intricate, and this is consistent with the breeding history in Xinjiang, which suggests that modern Upland cotton cultivars were mainly inherited from former Soviet Union accessions, but also from American and African cultivars (Figures S1 and S2). More elite alleles integrated from multiple American cotton landraces are likely responsible for the development of high lint yield cultivars in Xinjiang. Such a strong genetic background and the integration of elite alleles from distinct cotton landraces explain why cotton production in Xinjiang is significantly higher than that of other cotton planting areas or countries. This is likely one of the reasons that cotton production in China has recently moved to the inland region of northwest China, majorly in Xinjiang. In addition, it is foreseeable that cotton production in Xinjiang will be responsible for further improvements in lint yield and fibre quality.
Lint percentage plays an essential role in Xinjiang cotton production
Artificial selection signatures have offered a potentially powerful approach for identifying improvement‐related regions and candidate genes. The Fst between the foundation parents and modern cultivars was 0.0271, which is much lower than that between the G. hirsutum races and the cultivars (Fst = 0.10) (Fang et al., 2017a), and lower than that for maize (Fst = 0.14) (Jiao et al., 2012) and hexaploid wheat (Fst = 0.15) (Cavanagh et al., 2013). It is, however, slightly higher than that for sesame (Fst = 0.02) (Wei et al., 2015). These data, combined with the values for nucleotide diversity between foundation parents and modern cultivars, suggest that modern cultivars have undergone chronic artificial selection during Upland cotton improvement in Xinjiang.
Selective sweep analysis has been widely used to identify putative domestication or improvement events in crops (Lin et al., 2014; Wang et al., 2017). Only genomic regions from collateral cultivars bred can be reserved in offspring varieties. In total, 156 selective sweeps were identified in our populations (Table S3), 118 in the A sub‐genome and 38 in the D sub‐genome, further demonstrating that the A sub‐genome contributed more to cotton yield and fibre quality improvement than the D sub‐genome (Zhang et al., 2015).
An increase in yield has always been a fundamental breeding goal. By measuring nine traits of the accessions in two locations and comparing foundation parents with modern cultivars, the lint yield and fibre quality of Upland cotton in Xinjiang were found to be chronologically improved. Of them, LP was the fastest increased phenotype (Figure S4), which is consistent with the breeding practice in China. LP is a relative value obtained from the lint weight divided by seed cotton weight, which includes the seed weight and lint weight. So, theoretically, the higher the LP, the higher the lint yield. Therefore, the price of seed cotton harvested is always related to its LP in China. For this reason, much attention has always been paid to increases in LP in Chinese cotton breeding.
The candidate genes involved in artificial selection increased lint yield and improved fibre quality
We identified 318 candidate genes, including 31 genes with nonsynonymous SNPs that had undergone artificial selection, and 93 genes with nonsynonymous SNPs located in IBD segments. According to annotation and RNA‐seq expression profiling data, several candidate genes were associated with fibre development or stress‐related responses, including protein kinase family genes, transcription factors and UDP‐glucose‐related genes (Figures S6 and S9). These candidate genes were entered into a database and lay a solid foundation for the further exploration of the roles of these genes in fibre quality improvement or stress tolerance.
Lint percentage is a complex quantitative trait, related to both seed size and lint yield, and is regulated by multiple genes (Wang et al., 2017). We identified two candidate genes, GhAATF1 and GhmTERF1, that contribute to LP based on the selection sweeps (Figure 3). According to a previous study, AATF plays an important role in transcriptional regulation and interacts directly with nuclear hormone receptors to enhance their transactivation (Sharma, 2013). The mTERF family was known to blind mitochondrial DNA participating in transcription initiation, termination and modulation of DNA replication (Roberti et al., 2009; Robles et al., 2012). These genes have been shown to be related to growth and development in plants (Zhao et al., 2014). Thus, we infer that these two genes affect LP by regulating the transcription process in cotton. In addition, the nonsynonymous SNP in these two genes is significantly correlated with LP (Figure 3). Similarly, a nonsynonymous SNP in GhWAKL3 (Ma et al., 2018), two nonsynonymous SNPs in AIL6 (Fang et al., 2017a, 2017b) and two SNPs in Dof‐binding motif have all been associated with high lint yield (Wang et al., 2017).
Above all, fibre length and strength are the most important factors in determining fibre quality. Another candidate gene, GhARF3, which was inherited from 108Ф and 611Б, was found to be involved in fibre quality (Figure 5). Auxin signalling plays an essential role in regulating plant development; therefore, ARF genes have been characterized in Gossypium raimondii to elucidate their roles in fibre development (Sun et al., 2015). In addition, genome‐wide identification of the ARF gene family in G. hirsutum revealed that these genes could affect cotton fibre cell initiation (Xiao et al., 2018). Thus, it is believed that GHARF3 may be a key gene in cotton fibre development. Further work will be necessary to study all candidate gene functions in detail to confirm how these genes play roles in fibre initiation or stress‐related responses.
Experimental procedures
Sample preparation and DNA extraction
Seventy‐six accessions, comprising seven founder landraces (DPL15 and STV2B from the United States, UGDM from Uganda, and 108Ф, C1470, 611Б and KK1543 from the former Soviet Union), and sixty‐nine widely grown cultivars in Xinjiang, China (Table S1), were selected for Illumina sequencing. All samples were planted during the 2017 growing season at Shihezi (17SHZ, 85.94°E, 44.27°N) in North Xinjiang and Korla (17KRL, 86.06°E, 41.68°N) in South Xinjiang. All measurements were made according to the descriptors and data standards for cotton.
We collected young leaf tissues from each accession for genomic DNA extraction using a standard cetyltrimethylammonium bromide protocol (Paterson et al., 1993). Agarose gel electrophoresis was performed to verify the quality and purity of all DNA preparations.
Library construction and sequencing
Paired‐end sequencing libraries with insert sizes ranging from 300 to 500 bp were constructed according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). All libraries were sequenced on the Illumina HiSeq 2000 platform. For the 69 accessions from Xinjiang, a total of 1.2 terabases of genomic sequence clean data were generated with an average 10.65× genome coverage. We deep‐sequenced four of the former Soviet Union cultivars and generated 383Gb of clean data, also downloaded the other clean data from NCBI (DPL15: SRR5512448, STV2B: SRR5512449, UGDM: SRR5512442) and generated 247 Gb clean data.
Genotype calling and SNP identification
Clean paired‐end reads were aligned against the reference genome sequence (G. hirsutum acc.TM‐1) (Hu et al., 2019) using the BWA‐MEM algorithm from the BWA (Burrows–Wheeler Aligner, version v0.7.17) software platform (Li and Durbin, 2009). The sequence alignment files containing the overall mapping information created during the mapping process were counted, indexed and converted into binary BAM files using SAMtools software (version 1.6, settings: ‐bS) (Li et al., 2009). Potential PCR duplications were removed to reduce mismatches generated by PCR amplification using Picard tools (version 2.5.0; http://broadinstitute.github.io/picard). Then, the filtered BAM files were used to perform SNP calling for each sample. SNP detection was carried out using SAMtools software and BCFtools software (version 1.6) (Li et al., 2009). The following parameters were used for SNP calling: samtools mpileup –ug and bcftools call ‐vmO z.
To ensure the accuracy of the SNP variants, we performed a two‐step filter: (i) Genome Analysis Toolkit (GATK, version 3.8.0) (McKenna et al., 2010), with the following parameters: QualByDepth (QD) <2.0, FisherStrand (FS) >60.0, RMSMappingQuality (MQ) <40.0, MappingQualityRankSumTest (MQRankSum) <−12.5, ReadPosRankSumTest (ReadPosRankSum) <−8.0; and (ii) VCFtools software (version 1.6) (Danecek et al., 2011) with the minor allele frequency set to >0.05 and missing <0.2. Finally, we only analysed the SNPs that were located in the 26 pseudomolecules of the TM‐1 assembled genome, and the SNPs in the small scaffolds were removed. The SNPs were annotated using the GFF files (the annotation file of all coding regions of each gene) of the TM‐1 reference genome sequence (Hu et al., 2019) via ANNOVAR software (Wang et al., 2010).
SNP validation
We further randomly selected 26 SNPs and carried out PCR‐based sequencing in 10 randomly selected accessions with three replicates. We aligned all the PCR products against the TM‐1 genome using BLAST (Altschul et al., 1990), and the reads with mapping lengths >90% and identity >80% were used for SNP validation. Using the alignment results, the genotypes of 10 accessions for each SNP site were retrieved. Only the homozygous genotypes consistent across three replicates were used to calculate the accuracy, which was 94.12% (Tables S11 and S12).
Population structure analysis
After SNP identification and validation, we generated an SNP matrix of 76 accessions and obtained the simple matching coefficient of the whole‐genome SNPs as the genetic distance and then we exploited Phylip software (version 3.696) (Felsenstein, 1989) to generate the NJ tree. The Dendroscope (Huson et al., 2007) was used to display the phylogenetic tree. We performed population structure analysis using ADMIXTURE (version 1.3.0) (Zhou et al., 2011) and principal component analysis with GCTA software (version 1.26.0) (Yang et al., 2011).
Population genetics analysis
A sliding window method (100‐Kb sliding windows with a step of 20‐Kb) was used to calculate the genetic diversity (π) ratios (πparents/πcultivars) and genetic differentiation (Fst) between the foundation parents and Xinjiang modern cultivars. We empirically selected the genomic regions with simultaneous top 5% π ratios and top 5% Fst values as selective region signals across the genome. These were predicted to be candidate genes that underwent an artificial selection.
Identification of identity‐by‐descent segments
In order to detect the IBD segments of cultivars shared by inheritance from the foundation parents, we identified IBD regions using the algorithm from the BEAGLE (Browning and Browning, 2007) implementation of Refined IBD (Browning and Browning, 2013) with the following parameters: window = 1, length = 0.01, trim = 0.1, LOD = 3. The larger LOD values indicate greater evidence of IBDs. Due to the genome homology of different cultivars, some segments were difficult to identify among the seven foundation parents and were noted as overlapped, for example 108Ф/KK1543, 611Б/KK1543 and DPL15/STV2B (Table S6). However, when the genetic constitution of each of the modern cultivars inherited from the seven foundation parents was calculated, the overlapped IBD segments were added to each absolute value from the foundation parents, respectively.
RNA‐seq of gene expression levels
RNA‐seq data from distinct tissues and stresses have been reported in our previous TM‐1 genome sequencing research. In the present study, the raw transcriptomic data were downloaded from Sequence Read Archive (PRJNA490626) (Hu et al., 2019). We calculated the expression of each gene using the fragments per kilobase of exon model per million mapped reads (FPKM) with Cufflinks (version 2.1.1) (Trapnell et al., 2010). We firstly investigated the expression pattern of candidate genes in fibre from three stages of fibre development, the initiation stage (−3 and 0 DPA), cell‐elongation stage (1, 3, 5, 10 and 15 DPA) and secondary‐wall‐synthesis stage (20 and 25 DPA), as well as root, stem and leaf tissue. We also studied the expression level of candidate genes at 1, 3, 6, 12 and 24 h post‐treatment with heat (37 °C), cold (4 °C), salt and drought stress to analyse their responses to stress. When drawing the heat map, we performed Z‐score processing based on the FPKM values to make the data comparable.
Conflict of interest
The authors declare no competing financial interests.
Author’s contributions
T.Z conceptualized the research programme, designed the experiments and coordinated the project. T.Z., Y.H., Z.S., Q.T., A.S., L.F., X.L. and H.C. collected the 76 cotton samples and worked on the phenotyping. Y.H., Y.C., Y.Z., C.X. and W.S. extracted the high‐quality DNA. Z.H. and F.D. performed the genotyping and bioinformatics analyses. T.Z. and Z.H. analysed all the data and wrote the manuscript. All authors discussed the results and commented on the manuscript. All authors read and approved the final manuscript.
Supporting information
Figure S1 Summary of the route of Upland cotton introduction to Xinjiang.
Figure S2 Partial pedigree information for Xinjiang Upland cotton breeding.
Figure S3 Frequency distribution of phenotypic variation of 9 yield and fibre quality traits and correlation coefficients among the traits in 76 accessions.
Figure S4 Trait improvement over the breeding process in Xinjiang.
Figure S5 Population structure of all cotton accessions.
Figure S6 Heat map of transcriptomic patterns of 318 candidate genes in selection sweeps.
Figure S7 Box plot of GhAATF1 and GhmTERF1 related to lint percentages in two environments.
Figure S8 Identification of IBD segments inherited from foundation parents.
Figure S9 Heat map of transcriptomic patterns of 93 candidate nonsynonymous genes in IBD segments.
Figure S 10 Box plot of fibre length and strength in two environments.
Table S1 Summary information and sequencing data of the 76 Upland cotton accessions.
Table S2 Principal component analysis (PCA) results of the 76 Upland cotton accessions.
Table S3 156 Significant overlapping regions of top 5% artificial selection.
Table S 4 List of fibre quality and lint yield‐related QTLs overlapped with selected sweeps.
Table S5 Annotation information of 318 candidate genes (including 31 nonsynonymous genes) located in the significant selective sweeps.
Table S6 Summary of whole IBD segments in the modern varieties inherited from seven foundation parents.
Table S7 Overlapped IBD segments in more than two modern cultivars.
Table S8 IBD segments involved in significant artificial selection sweeps.
Table S9 1174 candidate genes (including 93 nonsynonymous genes) located in the significantly selected IBD segments.
Table S10 Annotation of 1174 candidate genes (including 93 nonsynonymous genes).
Table S11 SNP accuracy verified using PCR‐based sequencing.
Table S12 Primers for SNP accuracy validation.
Acknowledgements
This work was financially supported in part by grants from the National Natural Science Foundation of China (U1503284, 31701469), the Fundamental Research Funds for the Central Universities (2019XZZX004‐13), the earmarked fund for the China Agriculture Research System, Esquel Group, the Distinguished Discipline Support Program of Zhejiang University, and the Science Technology and Achievement Transformation Project of the Xinjiang Production and Construction Corps (2016AC02). We thank the National Medium‐term Gene Bank of Cotton in China for providing some of the cotton germplasm resource seeds.
Han, Z. , Hu, Y. , Tian, Q. , Cao, Y. , Si, A. , Si, Z. , Zang, Y. , Xu, C. , Shen, W. , Dai, F. , Liu, X. , Fang, L. , Chen, H. and Zhang, T. (2020) Genomic signatures and candidate genes of lint yield and fibre quality improvement in Upland cotton in Xinjiang. Plant Biotechnol. J., 10.1111/pbi.13356
Data availability statement
All the sequence data sets generated during the current study are available in the NCBI Sequence Read Archive (SRA) under accession PRJNA564187.
References
- Abdullaev, A. , Abdullaev, A.A. , Salakhutdinov, I. , Rizaeva, S. , Kuryazov, Z. , Ernazarova, D. and Abdurakhmonov, I. (2013) Cotton germplasm collection of Uzbekistan. Asian Australas. J. Plant Sci. Biotechnol. Glob. Sci. Books, 7, 1–15. [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Babiychuk, E. , Vandepoele, K. , Wissing, J. , Garcia‐Diaz, M. , De Rycke, R. , Akbari, H. , Joubes, J. et al. (2011) Plastid gene expression and plant development require a plastidic protein of the mitochondrial transcription termination factor family. Proc. Natl. Acad. Sci. USA, 108, 6674–6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan, M.W. , Uauy, C. , Wulff, B.B. , Zhou, J. , Krasileva, K. and Clark, M.D. (2017) Genomic innovation for crop improvement. Nature, 543, 346–354. [DOI] [PubMed] [Google Scholar]
- Bowman, D.T. , Gutierrez, O.A. , Percy, R.G. , Calhoun, D.S. and May, O.L. (2006) Pedigrees of upland and pima cotton cultivars released between 1970 and 2005. Bulletin, 11, 55–57. [Google Scholar]
- Browning, S.R. and Browning, B.L. (2007) Rapid and accurate haplotype phasing and missing‐data inference for whole‐genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 8, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning, S.R. and Browning, B.L. (2010) High‐resolution detection of identity by descent in unrelated individuals. Am. J. Hum. Genet. 86, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning, B.L. and Browning, S.R. (2013) Improving the accuracy and efficiency of identity‐by‐descent detection in population data. Genetics, 194, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning, S.R. and Thompson, E.A. (2012) Detecting rare variant associations by identity‐by‐descent mapping in case‐control studies. Genetics, 190, 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh, C.R. , Chao, S. , Wang, S. , Huang, B.E. , Stephen, S. , Kiani, S. , Forrest, K. et al. (2013) Genome‐wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars. Proc. Natl. Acad. Sci. USA, 110, 8057–8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Lin, Z. , Zhou, D. , Wang, C. , Li, H. , Yu, R. , Deng, H. et al. (2017) Genome‐wide study of an elite rice pedigree reveals a complex history of genetic architecture for breeding improvement. Sci. Rep. 7, 45685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. , Handsaker, R.E. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley, J.F. , Gaut, B.S. and Smith, B.D. (2006) The molecular genetics of crop domestication. Cell, 127, 1309–1321. [DOI] [PubMed] [Google Scholar]
- Fang, L. , Gong, H. , Hu, Y. , Liu, C. , Zhou, B. , Huang, T. , Wang, Y. et al. (2017a) Genomic insights into divergence and dual domestication of cultivated allotetraploid cottons. Genome Biol. 18, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, L. , Wang, Q. , Hu, Y. , Jia, Y. , Chen, J. , Liu, B. , Zhang, Z. et al. (2017b) Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat. Genet. 49, 1089. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. (1989) PHYLIP: phylogeny inference package (version 3.2). Cladistics, 5, 164–166. [Google Scholar]
- Gerland, P. , Raftery, A.E. , Sevcikova, H. , Li, N. , Gu, D. , Spoorenberg, T. , Alkema, L. et al. (2014) World population stabilization unlikely this century. Science, 346, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haanpaa, M. , Reiman, M. , Nikkil, J. , Erkko, H. , Pylkäs, K. and Winqvist, R. (2009) Mutation analysis of the AATF gene in breast cancer families. BMC Cancer, 9, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslot, N. , Jannink, J.L. and Sorrells, M.E. (2015) Perspectives for genomic selection applications and research in plants. Crop Sci. 55, 1–12. [Google Scholar]
- Hu, Y. , Chen, J. , Fang, L. , Zhang, Z. , Ma, W. , Niu, Y. , Ju, L. et al. (2019) Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748. [DOI] [PubMed] [Google Scholar]
- Huang, Z.K. (1996) Cotton Varieties and Their Genealogy in China. Beijing: China Agriculture Press. [Google Scholar]
- Huson, D.H. , Richter, D.C. , Rausch, C. , Dezulian, T. , Franz, M. and Rupp, R. (2007) Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics, 8, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, Y. , Zhao, H. , Ren, L. , Song, W. , Zeng, B. , Guo, J. , Wang, B. et al. (2012) Genome‐wide genetic changes during modern breeding of maize. Nat. Genet. 44, 812–815. [DOI] [PubMed] [Google Scholar]
- Lai, J. , Li, R. , Xu, X. , Jin, W. , Xu, M. , Zhao, H. , Xiang, Z. et al. (2010) Genome‐ wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 42, 1027–1030. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T. , Zhu, G. , Zhang, J. , Xu, X. , Yu, Q. , Zheng, Z. , Zheng, Z. et al. (2014) Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226. [DOI] [PubMed] [Google Scholar]
- Lu, X. , Fu, X. , Wang, D. , Wang, J. , Chen, X. , Hao, M. , Wang, J. et al. (2018) Resequencing of cv CRI‐12 family reveals haplotype block inheritance and recombination of agronomical important genes in artificial selection. Plant Biotechnol. J. 17, 945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, X. , Wang, Z. , Li, W. , Zhang, Y. , Zhou, X. , Liu, Y. , Ren, Z. et al. (2018) Resequencing core accessions of a pedigree identifies derivation of genomic segments and key agronomic trait loci during cotton improvement. Plant Biotechnol. J. 17, 762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meskauskiene, R. , Wursch, M. , Laloi, C. , Vidi, P.A. , Coll, N.S. , Kessler, F. , Baruah, A. et al. (2009) A mutation in the Arabidopsis mTERF‐related plastid protein SOLDAT10 activates retrograde signaling and suppresses (1) O(2)‐induced cell death. Plant J. 60, 399–410. [DOI] [PubMed] [Google Scholar]
- Meuwissen, T.H. , Hayes, B.J. and Goddard, M.E. (2001) Prediction of total genetic value using genome‐wide dense marker maps. Genetics, 157, 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, R.S. , DuVal, A.E. and Jensen, H.R. (2012) Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytol. 196, 29–48. [DOI] [PubMed] [Google Scholar]
- Paterson, A.H. , Brubaker, C.L. and Wendel, J.F. (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol. Biol. Rep. ‐ ISPMB (USA), 11, 122–127. [Google Scholar]
- Ramstein, G.P. , Jensen, S.E. and Buckler, E.S. (2018) Breaking the curse of dimensionality to identify causal variants in Breeding 4. Theor. Appl. Genet. 132, 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, H. , Reay, D.S. and Higgins, P. (2018) Beyond calories: a holistic assessment of the global food system. Front. Sustain. Food Systems, 2, 57. [Google Scholar]
- Roberti, M. , Polosa, P.L. , Bruni, F. , Manzari, C. , Deceglie, S. , Gadaleta, M.N. and Cantatore, P. (2009) The MTERF family proteins: mitochondrial transcription regulators and beyond. Biochim. Biophys. Acta. 1787, 303–311. [DOI] [PubMed] [Google Scholar]
- Robles, Pedro , José, L.M. and Víctor, Q. (2012) Unveiling plant mTERF functions. Mol. Plant, 5, 294–296. [DOI] [PubMed] [Google Scholar]
- Sharma, Monika . (2013) Apoptosis‐antagonizing transcription factor (AATF) gene silencing: role in induction of apoptosis and down‐regulation of estrogen receptor in breast cancer cells. Biotech. Lett. 35, 1561–1570. [DOI] [PubMed] [Google Scholar]
- Shinada, H. , Yamamoto, T. , Yamamoto, E. , Hori, K. , Yonemaru, J. , Matsuba, S. and Fujino, K. (2014) Historical changes in population structure during rice breeding programs in the northern limits of rice cultivation. Theor. Appl. Genet. 127, 995–1004. [DOI] [PubMed] [Google Scholar]
- Smith, J. , Stephen, C. , Donald, N.Duvick , Smith, Oscar S. , Mark, C. and Feng, L.Z. (2004) Changes in pedigree backgrounds of Pioneer brand maize hybrids widely grown from 1930 to 1999. Crop Sci. 44, 1935–1946. [Google Scholar]
- Stevens, E.L. , Heckenberg, G. , Roberson, E.D. , Baugher, J.D. , Downey, T.J. and Pevsner, J. (2011) Inference of relationships in population data using identity‐by‐descent and identity‐by‐state. PloS Genet. 7, e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, R. , Wang, K. , Guo, T. , Jones, D. , Cobb, J. , Zhang, B. and Wang, Q. (2015) Genome‐wide identification of auxin response factor (ARF) genes and its tissue‐specific prominent expression in Gossypium raimondii . Func. Integ. Genomics, 15, 481–493. [DOI] [PubMed] [Google Scholar]
- Tanksley, S.D. , Young, N.D. , Paterson, A.H. and Bonierbale, M.W. (1989) RFLP mapping in plant breeding: new tools for an old science. Biotechnology, 7, 257–264. [Google Scholar]
- Tian, X.M. , Li, X.Y. , Lv, X. , Li, B.C. , Chen, G.W. et al. (2016) Xinjiang Cotton Theory and Modern Cotton Technology. Beijing: Science Press. [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , Van Baren, M. , Saizberg, S.L. et al. (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney, R.K. , Pandey, K. , Bohra, A. , Singh, V.K. , Thudi, M. and Saxena, R.K. (2019) Toward the sequence‐based breeding in legumes in the post‐genome sequencing era. Theor. Appl. Genet. 132, 797–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, J.G. , Rodgers‐Melnick, E. and Buckler, E.S. (2018) On the road to Breeding 4.0: unraveling the good, the bad, and the boring of crop quantitative genomics. Ann. Review Genet. 52, 421–444. [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from next‐generation sequencing data. Nucl. Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Lin, M. , Lin, Z. , Wang, P. , Yang, Q. , Ye, Z. et al. (2017) Asymmetric sub‐genome selection and cis‐regulatory divergence during cotton domestication. Nat. Genet. 49, 579–587. [DOI] [PubMed] [Google Scholar]
- Wei, X. , Liu, K. , Zhang, Y. , Feng, Q. , Wang, L. , Zhao, Y. , Li, D. et al. (2015) Genetic discovery for oil production and quality in sesame. Nat. Commun. 6, 8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlind, H. , Imrell, K. , Ramanujam, R. , Myhr, K.M. , Celius, E.G. , Harbo, H.F. , Oturai, A. B. et al. (2015) Identity‐by‐descent mapping in a Scandinavian multiple sclerosis cohort. Eur. J. Hum. Genet. 23, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. , Li, Y. , Fu, J. , Li, X. , Li, C. , Zhang, D. , Shi, Y. et al. (2016) Exploring identity‐by‐descent segments and putative functions using different foundation parents in maize. PLoS ONE ONE, 1, e0168374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, G. , He, P. , Zhao, P. , Liu, H. , Zhang, L. , Pang, C. and Yu, J. (2018) Genome‐wide identification of the GhARF gene family reveals that GhARF2 and GhARF18 are involved in cotton fiber cell initiation. J. Exp. Bot. 69, 4323–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Lee, S. , Goddard, M. and Visscher, P. (2011) GCTA: a tool for genome‐wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Hu, Y. , Jiang, W. , Fang, L. , Guan, X. , Chen, J. , Zhang, J. et al. (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L.acc.TM‐1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531. [DOI] [PubMed] [Google Scholar]
- Zhou, H. , Alexander, D. and Lange, K. (2011) A quasi‐Newton acceleration for high‐dimensional optimization algorithms. Statist. Comput. 21, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. X. , Cai, M. , Zhang, X. , Li, Y. , Zhang, J. , Zhao, H. , Kong, F. et al. (2014) Genome-Wide Identification, Evolution and Expression Analysis of mTERF Gene Family in Maize. PLoS One 9, e94126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, D. , Chen, W. , Lin, Z. , Chen, H. , Wang, C. , Li, H. , Yu, R. et al. (2016) Pedigree‐based analysis of derivation of genome segments of an elite rice reveals key regions during its breeding. Plant Biotechnol. J. 14, 638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Summary of the route of Upland cotton introduction to Xinjiang.
Figure S2 Partial pedigree information for Xinjiang Upland cotton breeding.
Figure S3 Frequency distribution of phenotypic variation of 9 yield and fibre quality traits and correlation coefficients among the traits in 76 accessions.
Figure S4 Trait improvement over the breeding process in Xinjiang.
Figure S5 Population structure of all cotton accessions.
Figure S6 Heat map of transcriptomic patterns of 318 candidate genes in selection sweeps.
Figure S7 Box plot of GhAATF1 and GhmTERF1 related to lint percentages in two environments.
Figure S8 Identification of IBD segments inherited from foundation parents.
Figure S9 Heat map of transcriptomic patterns of 93 candidate nonsynonymous genes in IBD segments.
Figure S 10 Box plot of fibre length and strength in two environments.
Table S1 Summary information and sequencing data of the 76 Upland cotton accessions.
Table S2 Principal component analysis (PCA) results of the 76 Upland cotton accessions.
Table S3 156 Significant overlapping regions of top 5% artificial selection.
Table S 4 List of fibre quality and lint yield‐related QTLs overlapped with selected sweeps.
Table S5 Annotation information of 318 candidate genes (including 31 nonsynonymous genes) located in the significant selective sweeps.
Table S6 Summary of whole IBD segments in the modern varieties inherited from seven foundation parents.
Table S7 Overlapped IBD segments in more than two modern cultivars.
Table S8 IBD segments involved in significant artificial selection sweeps.
Table S9 1174 candidate genes (including 93 nonsynonymous genes) located in the significantly selected IBD segments.
Table S10 Annotation of 1174 candidate genes (including 93 nonsynonymous genes).
Table S11 SNP accuracy verified using PCR‐based sequencing.
Table S12 Primers for SNP accuracy validation.
Data Availability Statement
All the sequence data sets generated during the current study are available in the NCBI Sequence Read Archive (SRA) under accession PRJNA564187.