

Abstract
Diverse iridium peptide bioconjugates and the corresponding iridium/gold bimetallic complexes have been synthesized starting from a cyclometallated carboxylic acid substituted IrIII complex [Ir(ppy)2(Phen‐5‐COO)] by solid phase peptide synthesis (SPPS). The selected peptide sequences were an enkephalin derivative Tyr‐Gly‐Gly‐Phe‐Leu together with the propargyl‐substituted species Tyr‐Gly‐Pgl‐Phe‐Leu to allow gold coordination (Pgl: propyrgyl‐glycine, HC≡C‐Gly), and a specific short peptide, Ala‐Cys‐Ala‐Phen, containing a cysteine residue. Introduction of the gold center has been achieved via a click reaction with the alkynyl group leading to an organometallic Au−C(triazole) species, or by direct coordination to the sulfur atom of the cysteine. The photophysical properties of these species revealed predominantly an emission originating from the Ir complex, using mixed metal‐to‐ligand and ligand‐to‐ligand charge transfer excited states of triplet multiplicity. The formation of the peptide bioconjugates caused a systematic redshift of the emission profiles. Lysosomal accumulation was observed for all the complexes, in contrast to the expected mitochondrial accumulation triggered by the gold complexes. Only the cysteine‐containing Ir/Au bioconjugate displayed cytotoxic activity. The absence of activity may be related to the lack of endosomal/lysosomal escape for the cationic peptide conjugates. Interestingly, the different coordination sphere of the gold atom may play a crucial role, as the Au−S(cysteine) bond may be more readily cleaved in a biological environment than the Au−C(triazole) bond, and thus the Au fragment could be released from or trapped in the lysosomes, respectively. This work represents a starting point in the development of bimetallic peptide bioconjugates as theranostics and in the knowledge of factors that contribute to anti‐proliferative activity.
Keywords: bimetallic compounds, cell imaging, gold, iridium, peptide bioconjugates, theranostic agents
Iridium peptide bioconjugates and the corresponding iridium/gold bimetallic species were synthesized. Lysosomal accumulation was observed for all the complexes and cytotoxic activity only observed for the bimetallic Au−S(cysteine) bonded bioconjugate, showing the crucial role of the gold coordination sphere in the design of bimetallic peptide bioconjugates as theranostic agents.

Introduction
In recent years heterometallic species have been identified as emerging tools for theranosis.1 Typically, such compounds are formed by two distinct metallic fragments connected through a linker, where each of the fragments is specifically designed for either cell imaging or therapeutic applications. Examples of this type of compounds were pioneered and developed by Gimeno and Fernández‐Moreira,2 Casini,3 Patra,4 or Gornitzka,5 among others. The examples from those groups basically rely on ReI, RuII and EuIII complexes as luminescent probes, combined with the well‐established bioactivity of AuI (Auranofin), RuII (NAMI‐A) and PtII (Cisplatin) analogues. Luminescent transition metal complexes have some advantages for life science applications over the more commonly used organic dyes. Their superior physicochemical properties (higher photostability, larger Stokes shifts and long‐lived excited states, often combined with high quantum efficiencies) make them optimal candidates for cellular imaging techniques. In particular, ReI, RuII, and IrIII derivatives were investigated for this purpose. Orthometallated IrIII complexes of the type [Ir(N C)2(N N)]+, where N C represents a phenyl pyridine (ppy) and N N a bisimine ligand are highly luminescent species with demonstrated superior capability for cellular imaging.6 However, one of the drawbacks of such cyclometallated iridium species is their poor aqueous solubility, which could be overcome with the introduction of biological vectors. To this end, peptides,7 polyether chains (PEG)8 or cell penetrating peptides (CPP)9 have been used to maximize solubility and cell permeability of organometallic IrIII complexes. Of these, peptides are particularly versatile. Metal–peptide conjugates in general are gaining importance in biochemical and pharmaceutical studies⋅10 At the moment, a significant number of peptides conjugated with bioactive compounds have been explored as chemotherapeutics or radiolabels.11
Extending this approach to heterobimetallic compounds with potential for theranostic applications we surmised that peptides would make ideal linkers between both, the emissive and bioactive metallic fragments. In this way the new bimetallic molecules will be gaining not only water solubility but also advantageous biological targeting/transport capacity as well as the possibility of triggering a biological response. Building on prior experiences in our groups, we decided to combine an emissive IrIII fragment with bioactive AuI derivatives around suitable peptide backbones. A few AuI–peptide species were reported in the literature by the Metzler‐Nolte and Gimeno groups with excellent antitumor activities, see Figure 1.12 Over and beyond those, and to the best of our knowledge, there is no mention of Au/Ir heterometallic theranostic agents.
Figure 1.

Gold(I) peptide derivatives A, B, C and D.12
Results and Discussion
Leu‐enkephalin (Leu‐Enk) is a pentapeptide (Tyr‐Gly‐Gly‐Phe‐Leu) that binds to the opiate receptors in the central nervous system. It has potential as biotherapeutic agent but its application is dramatically hindered by rapid metabolism/degradation and low bioavailability after administration because the pentapeptide itself is unable to cross the blood–brain barrier (BBB).13 We have reported the first organometallic derivatives of enkephalin,14 some of which showed enhanced permeation of the BBB due to the attachment of lipophilic metal complexes.15 More recent studies suggest that the combination of opioid derivatives with anticancer agents could generate a positive synergy effect onto the overall anticancer activity.12c, 16 In addition to that, it is known that some carcinomas such as colorectal and pulmonary17 cancer contain high levels of opioid peptides and their corresponding membrane‐bound opioid receptors. In these cases, cancer cell recognition and cellular uptake of drugs will be facilitated when the actual anticancer drugs are combined with opioid peptides.18
In general, small peptides are best prepared via stepwise amino acid incorporation by solid phase peptide synthesis (SPPS).19 One of the main challenges in the synthesis of metal bioconjugates is the chemoselective insertion of the metal, keeping in mind that some metal complexes may not be compatible with the conditions of SPPS, and more specifically cleavage from the resin as the last synthesis step. The chemoselectivity issue is obviously aggravated when two different metal complexes are to be inserted, both in a regio‐ and chemoselective manner. In this work, the Ir fragments were linked to the peptide via amide bonds using a carboxylic acid‐functionalized diimine ligand, and we chose alkynyl groups as handles for insertion of the Au fragment since these provide a means for mild, biorthogonal functionalization of the peptides during as well as after SPPS. Alkynyl gold(I) complexes have recently shown promise as metallodrug candidates with potential application as anticancer agents.20 In this work, we introduced the unnatural amino acid propargyl glycine (Pgl) as the alkyne handle for Au conjugation.
The carboxylate‐functionalized Ir complex [Ir(ppy)2(Phen‐COO)] (1) (ppy=2‐(2‐pyridinyl‐κN)phenyl‐κC) was prepared following the reaction sequence depicted in Scheme 1. The carboxylate‐functionalized phenanthroline ligand, phen‐5‐CO2Na, was synthesized by a modified literature procedure and reacted with the dimeric IrIII complex [Ir(ppy)2Cl]2 to yield 1 in 92 % yield. No attempt was made to separate the possible stereoisomers, as is also seen and described in the solid state structure of 1 (below).
Scheme 1.
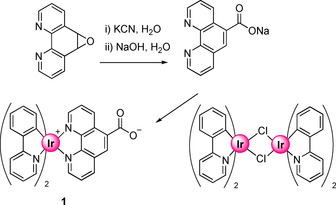
Synthetic procedure for the preparation of complex 1.
Suitable crystals of 1 for X‐ray diffraction were obtained by slow diffusion of diethyl ether into a dichloromethane solution of 1. The molecule crystallizes in the space group P21/n. The asymmetric unit is formed by one molecule of 1 and two molecules of dichloromethane from the crystallization solvent. Four molecules of 1, specifically two of each enantiomer, are present in the unit cell. As expected for these type of iridium complexes, the phenylpyridine ligands are coordinated to the metal center in a way that both pyridyl nitrogen atoms are trans to each other.21 Consequently, the phenyl carbon centers are trans to the phenanthroline derivative thereby completing octahedral coordination sphere around the IrIII metal center. Moreover, their trans influence renders longer Ir−N bond lengths for phenanthroline ligand in comparison with that seen for the Ir−N bond lengths in the phenylpyridine (Figure 2). Distortion from the ideal octahedral disposition is mainly imposed by the phenanthroline bite angle, which forces an N(4)‐Ir‐N(3) angle of 78.1(5)° instead of the ideal 90°. The molecular structure together with selected bond distances and angles are presented in Figure 2 and Table S1 (Supporting Information).
Figure 2.

POV‐ray view of one of the molecules present in the asymmetric unit of complex 1. Most relevant bond distances (Å) and angles (°): Ir(1)−C(11) 2.008(14), Ir(1)−C(22) 2.008(12), Ir(1)−N(1) 2.048(11), Ir(1)−N(2) 2.033(11), Ir(1)−N(3) 2.143(11), Ir(1)−N(4) 2.155(11); N(1)‐Ir(1)‐N(2) 173.5(4), N(4)‐Ir(1)‐N(3) 78.1(5).
We first established that Ir complex 1 was indeed a suitable building block for peptide synthesis. The pentapeptide Leu‐Enk with the primary sequence Tyr‐Gly‐Gly‐Phe‐Leu was synthesized by solid phase peptide synthesis (SPPS), specifically the standard Fmoc‐SPPS method.22 This synthetic method provides a peptide sequence after the stepwise addition and coupling of N‐protected amino acids to an insoluble polymer, in this specific case to a Wang resin. Each new amino acid will face a synthetic cycle consisting of: i) cleavage of the N‐protecting group of the amino acid anchored on the resin (deprotection step), ii) activation and coupling of the second N‐protected amino acid, and iii) cleavage of the full peptide from the resin when the peptide sequence is complete.
Thus, in this specific case, the resin already loaded with the first amino acid (Fmoc‐Leu) was subjected a Fmoc deprotection using 20 % of piperidine in DMF. Thereafter, Fmoc‐Phe was added together with the activators, TBTU (2‐(1H‐benzotriazole‐1‐yl)‐1,1,3,3‐tetramethylaminium tetrafluoroborate) and HOBT (hydroxybenzotriazole), as well as di‐isopropyl‐ethylamine (DIPEA) as base in DMF affording the successful coupling reaction to the N‐terminal leucine. This process was repeated with two subsequent glycines and the N‐terminal tyrosine. Eventually, Ir complex 1 was coupled to the N‐terminal amino group after Fmoc deprotection as if it were one additional amino acid. After cleavage from the resin by treatment with 95 % of TFA (trifluoroacetic acid), 2.5 % TIPS (triisopropylsilane) and 2.5 % H2O conjugate 2 was obtained successfully, see Scheme 2. Purification by semipreparative HPLC led to the desired compound in 7 % overall yield starting from the Fmoc‐Phe‐loaded Wang resin. MALDI mass spectrometry clearly showed the cation molecular peak at m/z=1262.4, which agrees completely with the calculated value for [M‐TFA] (C66H61IrN9O8).
Scheme 2.

Representation of solid phase peptide synthesis (SPPS) of compound 2.
After the success of the synthesis of the iridium peptide bioconjugate, it is necessary to introduce a functionalization in one of the amino acids to allow coordination of a gold center. For this we have chosen alkynyl groups since these provide a means for mild, biorthogonal functionalization of the peptides during as well as after SPPS. Consequently, we introduced the unnatural amino acid propargyl glycine (Pgl) as the alkyne handle for Au conjugation.
For the synthesis of compound 3, the Fmoc‐SPPS method was used as described for complex 2, with the introduction of Pgl as the third amino acid, replacing a glycine residue. As the last step, the luminescent IrIII complex 1 was coupled to the N‐terminal tyrosine. Cleavage of the bioconjugated organometallic IrIII species from the resin afforded conjugate 3 (Figure 3). The resulting solid was purified by column chromatography using a mixture of CH2Cl2:MeOH (10:2) as eluent. Mass spectrometry (MALDI‐MS) confirmed the expected structure. A peak for the molecular ion [M‐TFA] at m/z=1300.4267 was observed that perfectly matches the calculated mass for (C66H61IrN9O8).
Figure 3.

Chemical structure of compound 3 obtained by SPPS.
Thereafter, different methods were explored to coordinate the triphenylphosphine gold(I) fragment to the alkynyl group. In an effort to obtain a Au‐alkynyl species of the type Ph3PAu‐C≡C‐R directly, bases such as K2CO3, DIPEA or KOH in different solvents (dichloromethane, dimethylformamide and methanol) with [AuClPPh3] were tried, but did not afford the desired compound. A similar result was obtained when [Au(acac)PPh3] was used in either methanol or dichloromethane. Eventually, the use of [AuN3PPh3] as the gold(I) source allowed a (3+2) cycloaddition reaction (“click” reaction) to take place affording a Au‐substituted triazole (Scheme 3).12b, 23 The reaction mechanism is not entirely known23 but a mechanism similar to that originally proposed by Sharpless and Fokin for the classical Cu‐assisted azide‐alkyne cycloaddition reaction (Cu‐AAC) is assumed.24 All spectroscopic data confirm the successful formation of the bimetallic peptide, with the Au bound to the Pgl side chain. Compound 4 was purified by HPLC, The 31P{1H} NMR spectrum showed a single signal at 43.4 ppm in DMSO characteristic for the P‐Au‐C(triazole) unit (shifted from 31 ppm for the starting material Au azide). Moreover, a peak at mass m/z=1801.7648 observed by MALDI‐MS that matches the mass of the molecular ion (C84H76AuIrN12O8P)+ was also observed. These results are in agreement with data reported by Metzler‐Nolte and co‐workers for similar reactions.12b
Scheme 3.

Synthetic procedure of complex 4.
In order to explore alternative links of the Au fragment to the peptide side chain a peptide sequence containing a cysteine residue within the structure was synthesized and its coordination to the Ph3PAu fragment explored. Specifically, a short peptide with the Ala‐Cys‐Ala‐Phe sequence was chosen, where the N‐terminal alanine would be conjugated to the iridium fragment as for 4, and the Au fragment would be linked to the cysteine sulfur atom. Handling cysteine residues has an added difficulty because of the potential oxidation of the sulfur atom, therefore, a thiol protected cysteine residue, S‐Trityl‐cysteine, was used for the stepwise solid‐phase peptide synthesis (SPPS). In a similar procedure as described before the desired Ir peptide bioconjugate still bonded to the resin was prepared. Scheme 4 shows the specific peptide sequence containing the protected cysteine, and the possible approaches for binding the gold center to the peptide. Two pathways are conceivable to yield the desired heterobimetallic complex 5. In the first place, thiol deprotection followed by coordination of the gold fragment and finally cleavage from the Wang resin as the final step might yield conjugate 5. Alternatively, cleavage of the iridium peptide from the resin could be the first step, with concomitant removal of the thiol‐protecting trityl group, followed by coordination of the gold fragment to the cysteine sulfur atom. Attempts to obtain compound 5 by the first pathway however only led to decomposition of the product, possibly because cleavage conditions were too harsh for the S‐AuPPh3 fragment, as have been previously observed by us in gold thiolate amino acid systems if mild conditions were not used.12f In contrast, when the second synthetic pathway was used, compound 5 was cleanly obtained after purification by HPLC. MALDI mass spectrometry was used once more to corroborate the successful synthesis and the molecular peak was observed at 1575.6 m/z, which agrees with the calculated value for C71H62AuIrN8O6PS+ (1575.4).
Scheme 4.

Depiction of the synthetic pathways to obtain compound 5. Reaction conditions: i) DCM/TFA/TIS (90/5/5); ii) TFA/TIS/H2O (95/2.5/2.5); iii) [AuClPPh3], DIPEA, DCM followed by TFA/DCM/Phenol (50/40/10); iv) [AuClPPh3], DIPEA, DCM.
Absorption and emission spectroscopy
UV/Vis absorption spectra of complexes 1–5 were recorded in DMSO solution at 298 K and the most relevant data are summarized in Table 1. The absorption spectra for all the compounds presented a similar pattern with a highly intense absorption band around 265 nm and a set of weaker bands in the region between 370–500 nm (see Figure S1). The higher energy bands around 265 nm with extinction coefficients in the order of 104 dm3 mol−1 cm−1 can be attributed to a spin allowed ligand centered (1LC) transition, π→π* transition, mainly within the ppy and phen ligands.25 Then, at lower energy (λ>370 nm) charge transfer transitions are observed. The nature of these transitions is not well defined, and assumed to correspond to a mixture of ligand‐to‐ligand‐charge transfer (LLCT) transitions from the ppy to phen derivative, and to a metal‐to‐ligand‐charge transfer (MLCT) transition from iridium to the diimine ligand.26 This type of transition has been previously described as metal‐ligand‐to‐ligand charge transfer (MLLCT) transition, specifically as a dπIr‐C→π*N N transition.27 In this particular case, it can be suggested that bands from 375 to 415 nm, whose ϵ is in the range of (1–10)×103 m −1 cm−1, originate from spin‐allowed 1MLLCT transitions. Alternatively, lowest energy lying bands (>415 nm) with small molar absorption coefficient (ϵ <103 m −1 cm−1) are ascribed to spin‐forbidden 3MLLCT transitions.26 Despite the fact that in this specific case those bands are not clearly observed due to the small ϵ, they can be surmised, Figure S1. Emission and excitation spectra were also recorded for complexes 1–5 in DMSO solution, see Table 1 and Figure 4. Upon irradiation all complexes showed a broad emission around 582 nm for complex 1 and around 615 nm for the bioconjugated species, respectively. This redshift can be attributed to the electron‐withdrawing amide substituent that stabilises the π* orbitals of the diimine. However, the presence of the gold fragment does not seem to affect the emissive behaviour of the complexes. In all cases this emission is associated to 3MLCT (dπIr‐C→π*N N) transition and a 3LLCT πppy→π*N N by comparison with similar iridium structures.28 In addition, the quantum yields for emission were also determined in aerated DMSO at room temperature by an absolute method. For all complexes, quantum yields ranging from 13 to 21 % were observed. Interestingly, the bimetallic Ir/Au complexes had slightly higher values around 20 %.
Table 1.
Absorption, emission, excitation and quantum yields of complexes 1–5 measured in DMSO at 298 K.
|
|
UV/Vis (×103 ϵ [dm3 mol−1 cm−1]) |
λem (λexc) [nm] |
Ø |
|---|---|---|---|
|
1 |
266 (48.3), 374 (6.3), 415 (3.2) |
582 (443) |
17 |
|
2 |
267 (56.2), 375 (6.9), 417 (3.3) |
616 (433) |
13 |
|
3 |
269 (38.6), 378 (5.5), 416 (2.9) |
617 (426) |
13 |
|
4 |
266 (26.6), 381 (3.4), 416 (1.8) |
613 (375) |
21 |
|
5 |
267 (16.9), 382 (2.4), 416 (1.2) |
613 (385) |
19 |
Figure 4.

Emission spectra of complexes 1–5 recorded in DMSO and RT.
Cytotoxic activity
The antiproliferative activity of 1, 2, 4, and 5 was determined by the MTT assay in the lung cancer cell line A549. All complexes were incubated for 24 hours with the tumor cell line up to a maximal concentration of 50 μm. Neither the bioconjugated iridium compound 2, nor the parent complex 1 showed cytotoxicity up to the studied concentration, see Table 2. Surprisingly, the bimetallic bioconjugated compound 4 did not show cytotoxic activity, which is quite unexpected for a gold derivative. On the other hand, compound 5 displayed an antiproliferative effect with IC50=12±1.8 μm, see Table 2. This result demonstrates the importance of the direct gold coordination sphere for cytotoxicity. Here, the S‐Au‐P unit shows markedly higher cytotoxicity than the Au‐C‐triazol species with a C‐Au‐P unit. A plausible explanation could be related with the strength of the bond between Au−S and Au−C, being higher in the latter and preventing the metal center to react so easily with their biological targets. A comparison with the cytotoxicity of the reference Cisplatin complex in the same conditions 114.2±9.1 μm,29 although measured in water, revealed a much higher activity for the bioconjugate complex 5. It is worth mentioning that all bioconjugated complexes 2–5 had better solubility in the biological media than the parent 1 complex, demonstrating the expected positive effect of introducing the peptide.
Table 2.
IC50 values on A549 cells of 1–2 and 4–5 measured by the MTT assay after 24 h.
|
|
|
IC50 (A549‐24 h [μM])[a] |
|---|---|---|
|
Cisplatin |
|
114.2±9.1
|
|
1 |
|
>50 |
|
2 |
|
>50 |
|
4 |
|
>50 |
|
5 |
|
12.3±1.83 |
[a] Analyzed concentrations ranged from 1.25 to 50 μm, data are average +/‐SD of 3 independent measurements each one for quadruplicate. (Auranofin IC50=7.59 μM in A549, 24 h)31
Moreover, as many gold complexes have been reported to induce oxidative stress,30 the possible generation of reactive oxygen species (ROS) was analyzed for complex 5 by flow cytometry. Indeed, Figure 5 clearly shows an increment on the generation of ROS for those cells treated with complex 5.
Figure 5.

Flow cytometry analysis of ROS production induced by 5.
Cell imaging
The metal‐peptide bioconjugated complexes prepared here have the appropriate luminescent properties, with strong emission around 615 nm, to elucidate their cellular distribution by fluorescence cell microscopy. Specifically, A549 cells were incubated with the complexes at a concentration of 25 μm and colocalization was investigated with MitoTracker and LysoTracker, to detect mitochondrial or lysosomal localization, respectively. After 24 h of incubation, cells were irradiated at 473 nm to reveal the localization of the complexes, see Figures 6, Figure 7 and S2. Both MitoTracker and LysoTracker can be visualized after irradiation at 588 nm. Also, superimposition images of metal complexes and cellular localization dyes are shown in Figure 6 and Figure 7. In Figure 6, the colocalization for the monometallic Ir conjugate 2 and the bimetallic Ir/Au bioconjugate 4 is investigated. Localization of the complexes was seen in specific spots inside the cytoplasmic area of the cells, close to the nucleus but without penetrating the nuclear membrane. As can be clearly seen from the superimposition images, no localization of the bioconjugates occurs in mitochondria. Also for higher concentrations, a precipitate was found outside the cells, indicating that the complexes were not completely soluble (Figure 6).
Figure 6.

Fluorescence microscopy images from colocalization experiments of complexes 2 and 4 (green) with MitoTracker incubated with A549 cells. (A and A’) superimposition images. (B and B’) superimposition images including the phase contrast.
Figure 7.

Fluorescence microscopy images from colocalization experiments of complexes 4 and 5 (green) with LysoTracker incubated with A549 cells. (A and A’) superimposition images. (B and B’) superimposition images including the phase contrast.
This result is surprising, not only because some cyclometallated iridium complexes were shown to specifically target mitochondria,32 but also because mitochondria are a well‐known biological target for gold compounds.33 However, the addition of a peptide surely affects the interaction of the cyclometallated complex with the cell from the very beginning, possibly directing their internalization mechanism already, which might be eventually reflected on their biodistribution.
Interestingly, a more distinct localization of the conjugates was detected in lysosomes, through good overlay with the LysoTracker dye with Pearson coefficients ranging from 0.8–0.9. Figure 7 show the images for the bimetallic species 4 and 5 and S2 for the monometallic complex 2. Such localization has already been reported for some neutral34 and cationic35 cyclometallated IrIII complexes, all of them containing protonatable basic substituents.36 Lysosomes are known to be more acidic organelles compared with cytoplasm or other subcellular compartments (pH 5.0–5.5 vs. 7.4 respectively). Thus, the presence of protonatable basic substituents, as in this case N‐amide group, could be facilitating the lysosomal localization. Velders and co‐workers have described a series of cyclometallated IrIII derivatives containing three lysine residues that localized in the lysosomes.37 Furthermore, other cyclometallated IrIII derivatives containing cationic peptides were also described to accumulate in lysosomes,38 which agrees with the pattern observed in this case.
Interestingly, the lack of cytotoxic activity of these monometallic or bimetallic peptide bioconjugates may be related to the inability of the cationic complexes for a lysosomal escape. This situation appears somewhat different for the neutral bimetallic bioconjugate with a cysteine moiety, which is the only one with cytotoxic activity. Localization of this derivate is also observed in the lysosomes but a slow escape of the whole compounds may be possible or/and the gold phosphine fragment is slowly released from the peptide and translocates to mitochondria, which naturally would not be detectable by fluorescence microscopy. The higher strength of the Au−C bond in comparison with that of Au−S could be preventing such release from the lysosomes and eventually inhibiting the cytotoxicity of compound 4.
Conclusions
Solid phase peptide synthesis (SSPS) was used to prepare small peptides which were further linked to either a luminescent iridium(III) fragment or both, luminescent iridium(III) and bioactive gold(I) fragments. The iridium fragment was functionalized with a carboxylic acid group on a phenanthroline ligand, and bioconjugated to the N‐terminal amino group of three different peptides in the last step of an SPPS Scheme. For the Au fragment, two different biorthogonal conjugation strategies were explored. For one, the gold fragment was attached via a cycloaddition reaction of a gold azido derivative to the triple bond of propargyl‐glycine, affording a C‐coordinated Au‐triazole derivative 4. Alternatively, the Au fragment could be successfully bound to the sulfur atom of a cysteine residue 5. The excellent luminescence properties of the Ir fragment translated into the bioconjugates as well, which presents red shifted emissions and quantum yields around 20 %. All compounds could be readily tracked inside A549 lung cancer cells. Lysosomal localization after 24 h was confirmed by co‐staining with LysoTracker Red, which is consistent with an uptake mechanism through lysosomes, and the fact that this is still observable even 24 h after incubation is indicative for a very slow lysosomal escape for these conjugates. Mitochondrial activity, for example, through inhibition of mitochondrial thioredoxin reductase, is believed to be the mechanism of action of Au anticancer drug candidates.31 Surprisingly, no mitochondrial localization was observed, despite the fact that this has been observed for large lipophilic cations (LLCs),39 such as the Ir fragment, as well as for small molecule Au anticancer drug candidates. Interestingly, the lack of significant mitochondrial localization might correlate with the low anti‐proliferative activity of our metal‐peptide conjugates: Only the heterobimetallic complex 5 presented significant activity towards the A549 lung cancer cells, all other compounds were inactive up to a concentration of 50 μm. While a complete elucidation of the exact course of action would require significantly more experiments, this lack of activity may well have to do with lack of endosomal/ lysosomal escape for the (positively charged) peptide conjugates. Also, the strength of the Au–ligand bond may play a crucial role in that it is more readily cleaved in a biological environment for 5, having an Au‐S bond. The Au−C(triazole) bond in 4 is presumably more stable, and thus the Au fragment will be more reliably remain linked to the peptide, which in turn is trapped in lysosomes. This work therefore provides crucial insight into the factors that contribute to anti‐proliferative activity of generally toxic metal fragments in bioconjugates. While chemical tools and reactions exist to link metal fragments to selected sites in peptides almost at will, the exact choice of linker and the resulting coordination environment crucially contribute to the overall activity of the conjugate.
Experimental Section
General measurements and analysis instrumentation: Mass spectra were recorded on a Bruker Esquire 3000 PLUS, with the electrospray (ESI) technique and on a Bruker Microflex (MALDI‐TOF). 1H, 13C{1H} and 31P{1H} including 2D experiments, were recorded at room temperature on a Bruker Avance 400 spectrometer (1H 400.0 MHz, 13C 100.6 MHz, 31P 162.0 MHz) or on a Bruker Avance II 300 spectrometer (1H 300.0 MHz, 13C 75.5 MHz, 31P 121.5 MHz) with chemical shifts (δ, ppm) reported relative to the solvent peaks of the deuterated solvent. All J values are given in Hz. IR spectra were recorded in neat samples on a PerkinElmer Spectrum 100FT‐IR spectrometer. Room temperature steady‐state emission and excitation spectra were recorded with a Jobin–Yvon‐Horiba Fluorolog FL3‐11 spectrometer. UV‐vis spectra were recorded with 1 cm quartz cells on an Evolution 600 spectrophotometer. Quantum yields were measured in using an absolute method provided by Hamamatsu Photonics Quantaurus‐QY C11347‐11. Specifically, each sample was measured using the excitation scanning mode in aerated DMSO solution at rt, after recording a reference sample (neat aerated DMSO at rt). Excitation of the samples was made from 350 to 470 nm in 10 nm intervals. The quantum yield value given is the one obtained at maxima intensity excitation, that is, 1 (440 nm), 2 (430 nm), 3 (380 nm), 4 (390 nm), 5 (440 nm) The experiment was repeated three times to ensure reproducibility.
Semi‐preparative HPLC purifications and analysis were performed on a Knauer HPLC system using either one of the following columns: Macherey–Nagel VP 125/10 Nucleodur 100‐5 C18ec for purification and Macherey–Nagel EC 125/4 Nucleodur C18 Pyramid 5 μm or Agilent ZORBAX Eclipse XDB‐C18 4.6×150 mm for analysis.
Crystal structure determination: Crystals were mounted in inert oil glass fibres and transferred to the cold gas stream of an SMART APEX diffractometer equipped with a low‐temperature attachment. Data were collected using monochromated Mo Kα radiation (λ=0.71073 Å). Scan type ω. Absorption corrections based on multiple scans were applied with the program SADABS.40 The structures were solved by direct methods and refined on F 2 using the program SHELXL‐2016.41 All non‐hydrogen atoms were refined anisotropically. In most of the cases, hydrogen atoms were included in calculated positions and refined using a riding model; some of the hydrogen atoms have been located in the diffraction map. Refinements were carried out by full‐matrix least‐squares on F 2 for all data.
Deposition Number 1980164 contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Antiproliferative studies: MTT assay. Exponentially growing cells (A549) were seeded at a density of approximately 104 cells per well in 96‐well flat‐bottomed microplates and allowed to attach for 24 h prior to addition of compounds. The complexes were dissolved in DMSO and added to cells in concentrations ranging from 50 to 0.5 μm in quadruplicate. Cells were incubated with our compounds for 24 h at 37 °C. Ten microliters of MTT (5 mg mL−1) were added to each well and plates were incubated for 2 h at 37 °C. Finally, the growth media was eliminated and DMSO (100 μl per well) was added to dissolve the formazan precipitates. The optical density was measured at 550 nm using a 96‐well multiscanner autoreader (ELISA). The IC50 was calculated by nonlinear regression analysis.
ROS determination by flow cytometry: Exponentially growing cells (A549) were seeded at a density of approximately 5×105 cells per well in 6‐well flat‐bottomed plates and allowed to attach for 24 h prior to addition of the compound. Complex 5 was dissolved in DMSO and added to cells up to concentrations of 9.8 and 7.4 μm in duplicate. Cells were incubated with the complex for 48 h at 37 °C. The medium was removed and they were trypsinized. Thereafter the CellROX®Green Flow cytometry assay kit (Molecular Probes, C10493) was used to evaluate the generation of ROS in a Gallios Flow Cytometer (Beckman Couter).
Cell fluorescence microscopy study: European Collection of Cell Cultures were maintained in Hepes modified minimum essential medium (DMEM) supplemented with 5 % fetal bovine serum, penicillin, and streptomycin. A549 cells were detached from the plastic flask using trypsin‐EDTA solution and suspended in an excess volume of growth medium. 300 μl of a homogeneous cell suspension was then distributed into μ‐slide 8 well ibiTreat they were allowed to attach for 24 h prior to addition of compounds. Then, 200 μl of culture medium was removed and 100 μl of a solution of the corresponding complexes were added to the cells up to a final concentration of 19 or 25 μm. The complexes were incubated with the cells for 24 h at 37 °C. Thereafter, 50 μl of MitoTracker Red or LysoTracker red‐DND‐99 was added up to a final concentration of 10 nm. They were incubated with the cells for 15 min (MitoTracker) or 30 min (LysoTracker) at room temperature. Eventually the medium was replaced with fresh medium without phenol red. Preparations were viewed using an Olympus FV10‐i Oil type compact confocal laser microscope using a ×10 or ×60 objective, with excitation wavelength at 473 and 559 nm.
Materials and procedures: The starting materials [AuCl(tht)],42 [Au(acac)PPh3],43 [AuClPPh3],44 and [AuN3PPh3],23 were prepared according to published procedures. IrCl3⋅n H2O was purchased from Strem Chemicals and used as received. Fmoc‐protected amino acids and resins for SPPS were purchased from Novabiochem. All other reagents were commercially available and were used without further purification. Solvents were dried with a SPS solvent purification system. 1H, 13C{1H} NMR spectra, HR‐MALDI spectra and HPLC Chromatograms can be consulted in the ESI.
Synthesis of complex 1: [Ir(μ‐Cl)(ppy)2]2 (1 equiv, 536.5 mg, 0.499 mmol), sodium 1,10 phenanthroline‐5‐carboxylate (2.28 equiv, 281 mg, 1.14 mmol) and sodium carbonate (19.1 equiv, 780 mg, 9.51 mmol) were suspended in a 1:1 mixture of DCM and methanol (54 mL) and heated at 55 °C until consumption of the limiting reagent (ca 2 h). The solvent was then evaporated and the crude redissolved in DCM, filtrated over celite and washed with water (2×15 mL). The organic phase was dried with anhydrous magnesium sulphate and evaporated (233.2 mg, 92 %). 1 H NMR (300 MHz, DMSO): δ 9.87 (dd, J=8.6, 1.4 Hz, 1 H, H13 or H22), 8.87 (dd, J=8.3, 1.4 Hz, 1 H, H13 or H22), 8.53 (s, 1 H, H17), 8.25 (d, J=7.9 Hz, 2 H, H2 + H2’), 8.12 (dd, J=5.0, 1.4 Hz, 2 H, H15 + H20), 8.00–7.91 (m, 4 H, H8 + H8’ or H11 + H11’, + H14 + H21), 7.90–7.83 (m, tap, J=7.9 Hz, 2 H, H3 + H3’), 7.45 (tap, J=6.0 Hz, 2 H, H9 + H9’ or H10 + H10’), 7.11–6.84 (m, 6 H, H4 + H4’ + H5 + H5’ + H9 + H9’ or H10 + H10’), 6.30 (d, J=7.5 Hz, 2 H, H8 + H8’ or H11 + H11’). 13 C NMR (75 MHz, DMSO): δ 166.88 (s, C6 or C6’), 166.86 (s, C6 or C6’), 166.55 (s, C25), 150.59 (s, C12 or C12’ or C7 or C7’), 150.29 (s, C12 or C12’ or C7 or C7’), 149.71 (s, C15 or C20), 149.58 (s, C15 or C20), 148.99 (s, C9 or C9’ or C10 or C10’), 148.95 (s, C9 or C9’ or C10 or C10’), 146.23 (s, C23 or C24), 145.66 (s, C23 or C24), 144.07 (s, C12 or C12’ or C7 or C7’), 144.05 (s, C12 or C12’ or C7 or C7’), 141.63 (s, C17), 139.53 (s, C13 or C22), 138.77 (s, C13 or C22), 138.61 (s, C3 or C3’), 138.58 (s, C3 or C3’), 131.30 (s, C8 or C8’ or C11 or C11’), 131.27 (s, C8 or C8’ or C11 or C11’), 130.71 (s, C16 or C19), 130.66 (s, C16 or C19), 130.16 (s, 2C, C9 + C9’ or C10 + C10’), 126.74 (s, C14 or C21), 126.59 (s, C14 or C21), 125.99 (s, C17), 125.03 (s, C8 or C8’ or C11 or C11’), 123.87 (s, C4 or C4’), 123.84 (s, C4 or C4’), 122.24 (s, 2C, C5 + C5’), 119.93 (s, 2C, C2 + C2’). IR (cm−1): 3035 υ(CAr‐H), 1595, 1475 υ(COO−). HRMS (m/z): 725.1490 [M+H+], C35H24IrN4O2 725.1525.
Synthesis of complex 2: Solid‐phase peptide synthesis was performed manually in 5 mL plastic syringes with a porous disc as filter. The peptide sequence (Tyr(tBu)‐Gly‐Gly‐Phe) was assembled on Fmoc‐protected Leucine loaded Rink amide resin (Novabiochem, 362 mg, loading 0.69 mmol g−1) following general procedure. Then 211.2 mg (0.06 mmol) of the peptide‐loaded resin were weighed and reacted with 1 (94 mg, 0.129 mmol), TBTU (41.7 mg, 0.129 mmol) and DIPEA (45.3 μL, 0.260 mmol) in DMF as an additional peptide coupling reaction for 4 h. The resin was then washed twice with fresh DMF and the coupling step was repeated with fresh reagents for 8 additional hours for 2 additional hours, when finally, Kaiser Test confirmed that no free primary amines were present on the resin. Cleavage was performed with TFA/TIS/H2O (95/2.5/2.5) for 3 h, then precipitation in a mixture 1:1 of ice‐cold diethyl ether/hexane yielded the crude product, which was purified by semi preparative HPLC. Fractions containing the pure product were pooled, frozen in liquid nitrogen and lyophilized. Yellow solid (11.21 mg, 7 %). HR‐MALDI (m/z): 1262.4278 [M‐TFA], C63H59IrN9O8 1262.4111.
Synthesis of complex 3: Solid‐phase peptide synthesis was performed manually in 5 mL plastic syringes with a porous disc as filter. The peptide sequence (Tyr(tBu)‐Gly‐Pgl‐Phe) was assembled on Fmoc‐protected Leucine loaded Rink amide resin (Novabiochem, 390.62 mg, loading 0.69 mmol g−1) following general procedure for SPPS. Then 242.6 mg (0.205 mmol) of the peptide‐loaded resin were weighed and reacted with 39 (148.38 mg, 0.205 mmol), TBTU (65.83 mg, 0.205 mmol), HOBT (27.7 mg, 0.205 mmol) and DIPEA (71.4 μL, 0.41 mmol) in DMF as an additional peptide coupling reaction overnight. The resin was then washed twice with fresh DMF, and the coupling step was repeated with fresh reagents for 8 additional hours, when finally a Kaiser Test confirmed that no free primary amines were present on the resin. Cleavage was performed with TFA/TIS/H2O (95/2.5/2.5) for 4 h 30, then precipitation in a mixture 1:1 of ice‐cold diethyl ether/hexane yielded the crude product, which was purified by alumina column chromatography using CH2Cl2/MeOH (10:2) as eluent. Yellow solid (21.65 mg, 11 %). HR‐MALDI (m/z): 1300.4267 [M‐TFA], C66H61IrN9O8 1300.4267.
Synthesis of complex 4. 3: (13.66 mg, 0.01 mmol) was dissolved in DMF (2 mL) and DIPEA (1.7 μL, 0.01 mmol) followed by [AuN3PPh3] (24.2 mg, 0.048 mmol) were added. The solution was stirred for 2 days, then the solvent was evaporated and the resulting solid was washed twice with DCM. The crude product was was purified by semi preparative HPLC. Fractions containing the pure product were pooled, frozen in liquid nitrogen and lyophilized. Yellow solid (3.81 mg, 20 %). MALDI (m/z): 1801.8 [M‐TFA], C84H76AuIrN12O8P 1801.8.
Synthesis of complex 5: The peptide sequence (Ala‐Cys(Trt)‐Ala) was assembled on Fmoc‐protected Phenylalanine loaded Rink amide resin (Novabiochem, 308 mg, loading 0.65 mmol g−1) following general procedure for SPPS. Then 190 mg (0.09 mmol) of the peptide‐loaded resin were weighed and reacted with 1 (124 mg, 0.17 mmol), TBTU (55.2 mg, 0.017 mmol), HOBT (23.2, 0.017 mmol) and DIPEA (60 μL, 0.34 mmol) in DMF as an additional peptide coupling reaction overnight. The resin was then washed twice with fresh DMF and 1 TBTU and DIPEA were added again in DMF and the syringe was placed on a shaker overnight, when finally, Kaiser Test confirmed that no free primary amines were present on the resin. Cleavage was performed with 3 mL of TFA/TIS/H2O (95/2.5/2.5) for 3 h in a two‐necked round bottom flask under argon atmosphere. The resulting solution containing iridium bioconjugate was transferred to a weighed flask under argon via cannula. The solvent was evaporated, and the weight of the crude was calculated in order to add an equimolar quantity of AuClPPh3 (20.4 mg, 0.041 mmol), DCM (10 mL) and K2CO3 in excess. After 1 night at r.t., the mixture was filtered over celite, concentrated and precipitated with ether. The crude product was then purified by semi preparative HPLC. Fractions containing the pure product were pooled, frozen in liquid nitrogen and lyophilized. Yellow solid (6 mg, 2 %).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Authors thank the Ministerio de Ciencia Innovación (projects CTQ2016‐75816‐C2‐1‐P, PID2019‐104379RB‐C21, RTI2018‐097836‐J‐I00, RYC2018‐025872‐I and RED2018‐102471‐T) and Gobierno de Aragón‐Fondo Social Europeo (E07 20R) for financial support. A. L. thanks the Gobierno de Aragón for a predoctoral fellowship as well as Universidad de Zaragoza, Fundación Bancaria Ibercaja and Fundación Cai for a doctoral stay fellowship (CB 7/17).
A. Luengo, I. Marzo, M. Reback, I. M. Daubit, V. Fernández-Moreira, N. Metzler-Nolte, M. C. Gimeno, Chem. Eur. J. 2020, 26, 12158.
Contributor Information
Dr. Vanesa Fernández‐Moreira, Email: vanesa@unizar.es.
Prof. Dr. Nils Metzler‐Nolte, Email: nils.metzler-nolte@rub.de.
Prof. Dr. M. Concepción Gimeno, Email: gimeno@unizar.es.
References
- 1. Fernández-Moreira V., Gimeno M. C., Chem. Eur. J. 2018, 24, 3345–3353. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Luengo A., Fernández-Moreira V., Marzo I., Gimeno M. C., Inorg. Chem. 2017, 56, 15159–15170; [DOI] [PubMed] [Google Scholar]
- 2b. Luengo A., Fernández-Moreira V., Marzo I., Gimeno M. C., Organometallics 2018, 37, 3993–4001; [Google Scholar]
- 2c. Fernández-Moreira V., Marzo I., Gimeno M. C., Chem. Sci. 2014, 5, 4434–4446. [Google Scholar]
- 3. Wenzel M., de Almeida A., Bigaeva E., Kavanagh P., Picquet M., Le Gendre P., Bodio E., Casini A., Inorg. Chem. 2016, 55, 2544–2557. [DOI] [PubMed] [Google Scholar]
- 4. Chandra A., Singh K., Singh S., Sivakumar S., Patra A. K., Dalton Trans. 2016, 45, 494–497. [DOI] [PubMed] [Google Scholar]
- 5. Boselli L., Carraz M., Mazhres S., Paloque L., González G., Benoit-Vical F., Valentin A., Hemmert C., Gornitzka H., Organometallics 2015, 34, 1046–1055. [Google Scholar]
- 6.
- 6a. Caporale C., Bader C. A., Sorvina A., MaGee K. D. M., Skelton B. W., Gillam T. A., Wright P. J., Raiteri P., Stagni S., Morrison J. L., Plush S. E., Brooks D. A., Massi M., Chem. Eur. J. 2017, 23, 15666–15679; [DOI] [PubMed] [Google Scholar]
- 6b. Yu H., Xiao Y., Jin L., J. Am. Chem. Soc. 2012, 134, 17486–17489; [DOI] [PubMed] [Google Scholar]
- 6c. Moromizato S., Hisamatsu Y., Suzuki T., Matsuo Y., Abe R., Aoki S., Inorg. Chem. 2012, 51, 12697–12706. [DOI] [PubMed] [Google Scholar]
- 7. Wang X., Jia J., Huang Z., Zhou M., Fei H., Chem. Eur. J. 2011, 17, 8028–8032. [DOI] [PubMed] [Google Scholar]
- 8. Li S., Liu H., Zhang K., Lo K. K., Chem. Eur. J. 2010, 16, 8329–8339. [DOI] [PubMed] [Google Scholar]
- 9. Dolan C., Moriarty R. D., Lestini E., Devocelle M., Forster R. J., Keyes T. E., J. Inorg. Biochem. 2013, 119, 65–74. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Albericio F., Kruger H. G., Future Med. Chem. 2012, 4, 1527–1531; [DOI] [PubMed] [Google Scholar]
- 10b. Vlieghe P., Lisowski V., Martinez J., Khrestchatisky M., Drug Discovery Today 2010, 15, 40–56; [DOI] [PubMed] [Google Scholar]
- 10c. Albada H. B., Metzler-Nolte N., Chem. Rev. 2016, 116, 11797—11839. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Zwanziger D., Beck-Sickinger A. G., Curr. Pharm. Des. 2008, 14, 2385–2400; [DOI] [PubMed] [Google Scholar]
- 11b. Metzler-Nolte N. in Bioorganometallics, (Eds.: G. Jaouen) Wiley-VCH, Weinheim, 2006, pp. 125–179. [Google Scholar]
- 12.
- 12a. Caddy J., Hoffmanns U., Metzler-Nolte N., Z. Naturfosch. Sect. B 2007, 62, 460–466; [Google Scholar]
- 12b. Lemke J., Pinto A., Niehoff P., Vasylyeva V., Metzler-Nolte N., Dalton Trans. 2009, 7063–7070; [DOI] [PubMed] [Google Scholar]
- 12c. Köster S. D., Alborzinia H., Can S., Kitanovic I., Wölfl S., Rubbiani R., Ott I., Riesterer P., Prokop A., Merz K., Metzler-Nolte N., Chem. Sci. 2012, 3, 2062–2072; [Google Scholar]
- 12d. Gutiérrez A., Gimeno M. C., Marzo I., Metzler-Nolte N., Eur. J. Inorg. Chem. 2014, 2512–2519; [Google Scholar]
- 12e. Gutiérrez A., Marzo I., Cativiela C., Laguna A., Gimeno M. C., Chem. Eur. J. 2015, 21, 11088–11095; [DOI] [PubMed] [Google Scholar]
- 12f. Gutiérrez A., Gracia-Fleta L., Marzo I., Cativiela C., Laguna A., Gimeno M. C., Dalton Trans. 2014, 43, 17054–17066. [DOI] [PubMed] [Google Scholar]
- 13. Gopalakrishnan G., Lepetre S., Maksimenko A., Mura S., Desmaële D., Couvreur P., Cancer Adv. Healthcare Mater. 2015, 4, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Staveren D. R., Metzler-Nolte N., Chem. Commun. 2002, 1406–1407. [DOI] [PubMed] [Google Scholar]
- 15. Pinto A., Hoffmanns U., Ott M., Fricker G., Metzler-Nolte N., ChemBioChem 2009, 10, 1852–1860. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Chougule M., Patel A. R., Sachdeva P., Jackson T., Singh M., Lung Cancer 2011, 71, 271–282; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Meier S. M., Novak M., Kandioller W., Jakupec M. A., Arion V. B., Metzler-Nolte N., Keppler B. K., Hartinger C. G., Chem. Eur. J. 2013, 19, 9297–9307. [DOI] [PubMed] [Google Scholar]
- 17. Roth K. A., Barchas J. D., Cancer 1986, 57, 769–773. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Rigaudy P., Charcosset J.-Y., Garbay-Jaureguiberry C., Jacquemin-Sablon A., Roques B. P., Cancer Res. 1989, 49, 1836–1842; [PubMed] [Google Scholar]
- 18b. Strack M., Bedini A., Yip K. T., Lombardi S., Siegmund D., Stoll R., Spampinato S. M., Metzler-Nolte N., Chem. Eur. J. 2016, 22, 14605—14610. [DOI] [PubMed] [Google Scholar]
- 19. Palomo J. M., RSC Adv. 2014, 4, 32658–32672. [Google Scholar]
- 20. Cerrada E., Fernández-Moreira V., Gimeno M. C., Adv. Organom. Chem., Vol. 71, Ed.: P. J. Pérez, Elsevier, 2019, pp. 227–258. [Google Scholar]
- 21. Jiang W., Hong C., Wei H., Wu Z., Bian Z., Huang C., Inorg. Chim. Acta 2017, 459, 124–130. [Google Scholar]
- 22. Kirin S. I., Noor F., Metzler-Nolte N., Mier W., J. Chem. Educ. 2007, 84, 108–110. [Google Scholar]
- 23. Partyka D. V., J. B. Updegraff III , Zeller M., Hunter A. D., Gray T. G., Organometallics 2007, 26, 183–186. [Google Scholar]
- 24. Himo F., Lovell T., Hilgraf R., Rostovtsev V. V., Noodleman L., Sharpless K. B., Fokin V. V., J. Am. Chem. Soc. 2005, 127, 210–216. [DOI] [PubMed] [Google Scholar]
- 25. Langdon-Jones E. E., Ward B. D., Pope S. J. A., J. Organomet. Chem. 2018, 861, 234–243. [Google Scholar]
- 26. Bevernaegie R., Marcélis L., Laramée-Milette B., De Winter J., Robeyns K., Gerbaux P., Hanan G. S., Elias B., Inorg. Chem. 2018, 57, 1356–1367. [DOI] [PubMed] [Google Scholar]
- 27. Tamayo A. B., Garon S., Sajoto T., Djurovich P. I., Tsyba I. M., Bau R., Thompson M. E., Inorg. Chem. 2005, 44, 8723–8732. [DOI] [PubMed] [Google Scholar]
- 28. Lo K. K.-W., Chan B. T.-N., Liu H.-W., Zhang K. Y., Li S. P.-Y., Tang T. S.-M., Chem. Commun. 2013, 49, 4271–4273. [DOI] [PubMed] [Google Scholar]
- 29. Frik M., Fernández-Gallardo J., Gonzalo O., Mangas-Sanjuan V., González-Alvarez M., Serrano del Valle A., Hu C., González-Alvarez I., Bermejo M., Marzo I., Contel M., J. Med. Chem. 2015, 58, 5825–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mármol I., Virumbrales-Muñoz M., Quero J., Sánchez-de-Diego C., Fernández L., Ochoa I., Cerrada E., Yoldi M. J. R., J. Inorg. Biochem. 2017, 176, 123–133. [DOI] [PubMed] [Google Scholar]
- 31. Hatem E., Azzi S., El Banna N., He T., Héneman-Masurel A., Vernis L., Baïlle D., Masson V., Dingli F., Loew D., Azzarone B., Eid P., Baldacci G., Er-Huang M., J. Natl. Cancer Inst. 2019, 111, 597–608. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Wang B., Liang Y., Dong H., Tan T., Zhan B., Cheng J., Lo K. K.-W., Lam Y. W., Cheng S. H., ChemBioChem 2012, 13, 2729–2737; [DOI] [PubMed] [Google Scholar]
- 32b. Law W. H.-T., Lee L. C.-C., Louie M.-W., Liu H.-W., Ang T. W.-H., Lo K. K.-W., Inorg. Chem. 2013, 52, 13029–13041; [DOI] [PubMed] [Google Scholar]
- 32c. Tso K. K.-S., Leung K.-K., Liu H.-W., Lo K. K.-W., Chem. Commun. 2016, 52, 4557–4560. [DOI] [PubMed] [Google Scholar]
- 33.
- 33a. Bindoli A., Rigobello M. P., Scutari G., Gabbiani C., Casini A., Messori L., Coord. Chem. Rev. 2009, 253, 1692–1707; [Google Scholar]
- 33b. Barnard P. J., Berners-Price S. J., Coord. Chem. Rev. 2007, 251, 1889–1902; [Google Scholar]
- 33c. Pia Rigobello M., Messori L., Marcon G., Cinellu M. A., Bragadin M., Folda A., Scutari G., Bindoli A., J. Inorg. Biochem. 2004, 98, 1634–1641; [DOI] [PubMed] [Google Scholar]
- 33d. McKeage M. J., Maharaj L., Berners-Price S. J., Coord. Chem. Rev. 2002, 232, 127–135. [Google Scholar]
- 34. Qiu K., Huang H., Liu B., Liu Y., Huang Z., Chen Y., Ji L., Chao H., ACS Appl. Mater. Interfaces 2016, 8, 12702–12710. [DOI] [PubMed] [Google Scholar]
- 35. He L., Li Y., Tan C.-P., Ye R.-R., Chen M.-H., Cao J.-J., Ji L.-N., Mao Z.-W., Chem. Sci. 2015, 6, 5409–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Caporale C., Massi M., Coord. Chem. Rev. 2018, 363, 71–91; [Google Scholar]
- 36b. Qiu K., Zhu H., Rees T. W., Ji L., Zhang Q., Chao H., Coord. Chem. Rev. 2019, 398, 113010; [Google Scholar]
- 36c. Yang Y., Guo L., Ge X., Tian Z., Gong Y., Zheng H., Du Q., Zheng X., Liu Z., Dyes Pigm. 2019, 161, 119–129. [Google Scholar]
- 37. Steunenberg P., Ruggi A., Van Den Berg N. S., Buckle T., Kuil J., Van Leeuwen F. W. B., Velders A. H., Inorg. Chem. 2012, 51, 2105–2114. [DOI] [PubMed] [Google Scholar]
- 38. Yokoi K., Hisamatsu Y., Naito K., Aoki S., Eur. J. Inorg. Chem. 2017, 5295–5309. [Google Scholar]
- 39.
- 39a. Murphy M. P., Biochim. Biophy. Acta 2008, 1777, 1028–1031; [DOI] [PubMed] [Google Scholar]
- 39b. Finichiu P. G., James A. M., Larsen L., Smith R. A. J., Murphy M. P., J. Bioenerg. Biomembr. 2013, 45, 165–173; [DOI] [PubMed] [Google Scholar]
- 39c. Modica-Napolitano J. S., Aprille J. R., Adv. Drug Delivery Rev. 2001, 49, 63–70. [DOI] [PubMed] [Google Scholar]
- 40.G. M. Sheldrick, SADABS, Program for adsorption correction, University of Göttingen, Göttingen, Germany, 1996.
- 41. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Usón R., Laguna A., Laguna M., Briggs D. A., Murray H. H., J. P. Fackler, Jr. , Inorg. Synth., ed. H. D. Kaesz, 1989, 85–90; [Google Scholar]
- 42b. Brauer G., Handbuch der Präparativen Anorganischen Chemie, Ferdinand Enke Verlag, Stuttgart, 1978, p. 1014. [Google Scholar]
- 43. Gibson D., Johnson B. F. G., Lewis J., J. Chem. Soc. A 1970, 367–369. [Google Scholar]
- 44. Batchelor L. K., Paunescu E., Soudani M., Scopelliti R., Dyson P. J., Inorg. Chem. 2017, 56, 9617–9633. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary