

Abstract
Key points
AMP‐activated protein kinase (AMPK) is considered a major regulator of skeletal muscle metabolism during exercise.
However, we previously showed that, although AMPK activity increases by 8–10‐fold during ∼120 min of exercise at ∼65% in untrained individuals, there is no increase in these individuals after only 10 days of exercise training (longitudinal study).
In a cross‐sectional study, we show that there is also a lack of activation of skeletal muscle AMPK during 120 min of cycling exercise at 65% in endurance‐trained individuals.
These findings indicate that AMPK is not an important regulator of exercise metabolism during 120 min of exercise at 65% in endurance trained men.
It is important that more energy is directed towards examining other potential regulators of exercise metabolism.
Abstract
AMP‐activated protein kinase (AMPK) is considered a major regulator of skeletal muscle metabolism during exercise. Indeed, AMPK is activated during exercise and activation of AMPK by 5‐aminoimidazole‐4‐carboxyamide‐ribonucleoside (AICAR) increases skeletal muscle glucose uptake and fat oxidation. However, we have previously shown that, although AMPK activity increases by 8–10‐fold during ∼120 min of exercise at ∼65% in untrained individuals, there is no increase in these individuals after only 10 days of exercise training (longitudinal study). In a cross‐sectional study, we examined whether there is also a lack of activation of skeletal muscle AMPK during 120 min of cycling exercise at 65% in endurance‐trained individuals. Eleven untrained (UT; = 37.9 ± 5.6 ml.kg−1 min−1) and seven endurance trained (ET; = 61.8 ± 2.2 ml.kg−1 min−1) males completed 120 min of cycling exercise at 66 ± 4% (UT: 100 ± 21 W; ET: 190 ± 15 W). Muscle biopsies were obtained at rest and following 30 and 120 min of exercise. Muscle glycogen was significantly (P < 0.05) higher before exercise in ET and decreased similarly during exercise in the ET and UT individuals. Exercise significantly increased calculated skeletal muscle free AMP content and more so in the UT individuals. Exercise significantly (P < 0.05) increased skeletal muscle AMPK α2 activity (4‐fold), AMPK αThr172 phosphorylation (2‐fold) and ACCβ Ser222 phosphorylation (2‐fold) in the UT individuals but not in the ET individuals. These findings indicate that AMPK is not an important regulator of exercise metabolism during 120 min of exercise at 65% in endurance trained men.
Keywords: AMPK activity, endurance, exercise, metabolism, trained, training, signalling
Key points
AMP‐activated protein kinase (AMPK) is considered a major regulator of skeletal muscle metabolism during exercise.
However, we previously showed that, although AMPK activity increases by 8–10‐fold during ∼120 min of exercise at ∼65% in untrained individuals, there is no increase in these individuals after only 10 days of exercise training (longitudinal study).
In a cross‐sectional study, we show that there is also a lack of activation of skeletal muscle AMPK during 120 min of cycling exercise at 65% in endurance‐trained individuals.
These findings indicate that AMPK is not an important regulator of exercise metabolism during 120 min of exercise at 65% in endurance trained men.
It is important that more energy is directed towards examining other potential regulators of exercise metabolism.
Introduction
The signalling events that regulate skeletal muscle exercise metabolism have not been fully elucidated (Richter & Hargreaves, 2013). There is evidence for feed forward (calcium activated CaMK) (Wright et al. 2004; Wright et al. 2005; Jensen et al. 2007; Witczak et al. 2010) and feedback (AMP‐activated protein kinase; AMPK) (Hayashi et al. 1998; Mu et al. 2001; Lee‐Young et al. 2009; Abbott et al. 2011) regulation being involved, as well as nitric oxide (Balon & Nadler, 1997; Roberts et al. 1997; Bradley et al. 1999; Inyard et al. 2007; Ross et al. 2007; Merry et al. 2010a ) and reactive oxygen species production (Toyoda et al. 2004; Sandstrom et al. 2006; Merry et al. 2010a ), which are increased during contraction (Roberts et al. 1999; Reid & Durham, 2002; Linden et al. 2011). There is also some evidence that cytosketetal forces during contraction may signal glucose uptake via Rac1 (Sylow et al. 2013; Sylow et al. 2015).
Skeletal muscle AMPK activity increases during exercise in rodents (Winder & Hardie, 1996; Lee‐Young et al. 2009) and humans (Chen et al. 2000; Fujii et al. 2000; Wojtaszewski et al. 2000; Musi et al. 2001; Mortensen et al. 2013) and, given that activation of AMPK by 5‐aminoimidazole‐4‐carboxyamide‐ribonucleoside (AICAR) increases skeletal muscle glucose uptake and fat oxidation (Merrill et al. 1997; Hayashi et al. 2000; Jorgensen et al. 2004), it has been assumed that activation of AMPK during exercise increase fat and glucose metabolism in humans. However, a number of studies have shown dissociations between activation of AMPK and glucose uptake during muscle contraction in rodents (Jorgensen et al. 2004; Fujii et al. 2005; Kjobsted et al. 2017) and during exercise in humans (Wojtaszewski et al. 2000; McConell et al. 2005; Mortensen et al. 2013). In addition, several studies have shown that fat oxidation increases normally during contraction (Jeppesen et al. 2011) and during exercise (Jeppesen et al. 2013) in AMPK dominant negative/kinase dead mice.
Further evidence that questions a role for AMPK in glucose and fat metabolism during exercise is that substantial increases in glucose uptake and fat oxidation are seen during low intensity exercise (40–45% ) in humans even though AMPK signalling is not increased at such intensities (Wojtaszewski et al. 2002; Chen et al. 2003). In addition, in a longitudinal study, we showed in previously untrained men that 10 days of exercise training abolishes the ∼10‐fold increase in AMPK α2 activity during exercise at 65% despite fat oxidation being higher and glucose disposal, although attenuated, still being substantially increased during exercise (McConell et al. 2005). We found no relationship between AMPK activation (and AMPK αThr172 phosphorylation and ACCβ phosphorylation) and muscle glycogen use, glucose uptake and fat oxidation during exercise (McConell et al. 2005). There is no doubt that AMPK activation during exercise is important for post‐exercise adaptations (Winder et al. 2000; McGee et al. 2003), although these findings imply that AMPK activation during moderate intensity exercise is not necessary for normal increases in glucose uptake and fat oxidation during exercise.
It can be argued that there is less activation of AMPK after short‐term exercise training because there is less of an energy deficit in skeletal muscle during exercise. Indeed, we found this to be the case in humans in our short‐term training study reporting no activation of AMPK during exercise at 65% of pretraining (McConell et al. 2005). The exercise after training was conducted at the pre‐exercise training workload; as such, the exercise was at a little lower relative workload after training (McConell et al. 2005). However, it has subsequently been shown in humans that, after 12 weeks of exercise training, there is less activation of skeletal muscle AMPK when the post training exercise was conducted at the same relative intensity of 65% as the pre‐training exercise (Mortensen et al. 2013).
Another way of examining the effect of exercise training on AMPK activation is to conduct a cross‐sectional study comparing endurance trained with untrained individuals exercising at the same relative intensity. Nielsen et al. (2003) found similar increases in AMPK α2 activity during 20 min of exercise at 80% in trained compared with untrained individuals. This was surprising because they and others have found less of a skeletal muscle energy imbalance and therefore less of an increase in skeletal muscle AMP and ADP at 80% in trained compared with untrained men (Baldwin et al. 1999; Nielsen et al. 2003). It is not known whether skeletal muscle AMPK activity would increase during prolonged moderate intensity exercise in well trained individuals. This is important to determine because, if AMPK does not increase during such exercise, this would imply that AMPK is not important for the regulation of exercise metabolsim during such exercise.
Therefore, the present study aimed to examine whether long‐term endurance exercise trained individuals have an increase in skeletal muscle AMPK during 120 min of exercise at 65% . Based on our findings of no activation at this workload in previously untrained individuals after 10 days of exercise training (McConell et al. 2005), we hypothesized that skeletal muscle AMPK would not be activated during 120 min of exercise at 65% in endurance trained men.
Methods
Ethical approval
The present study was approved by the Human Research Ethics Committee of the University of Melbourne (Study number 040090) and conducted in accordance with the Declaration of Helsinki, except for registration in a database.
Subjects
Seven endurance‐trained cyclists and triathletes (26 ± 2 years; 72 ± 4 kg; = 4.4 ± 0.35 L min−1, mean ± SD) and eleven healthy but otherwise untrained (23 ± 3 years; 69 ± 9 kg; = 2.6 ± 0.4 L min−1, mean ± SD) non‐smoker males participated in the study (Table 1). Trained participants cycled on average 300 ± 100 km week–1, whereas untrained participants undertook no regular exercise.
Table 1.
Subject characteristics
| Parameter | Untrained | Trained |
|---|---|---|
| Age (years) | 23 ± 3 | 26 ± 2 |
| Weight (kg) | 69 ± 9 | 72 ± 4 |
| Height (m) | 1.75 ± 0.35 | 1.76 ± 0.39 |
| BMI (kg m–2) | 22 ± 2.6 | 23 ± 0.76 |
| (L min−1) | 2.60 ± 0.4* | 4.44 ± 0.35 |
| (ml kg−1 min−1) | 37.9 ± 5.6* | 61.8 ± 2.2 |
Values are the mean ± SD, n = 11 untrained and 7 exercise trained participants, BMI, body mass index, , oxygen consumption. *Significantly different to corresponding trained value (P < 0.05).
Experimental design
Participants were required to attend the laboratory on three separate occasions. The first visit involved a peak pulmonary oxygen consumption test during cycling (), followed 2–3 days later by a 30 min familiarisation ride at a workload calculated from the test to be ∼65% to confirm the power output for the experimental trials. Approximately 1 week later, participants returned to the laboratory for an exercise trial, which involved cycling for 120 min at ∼ 65% (untrained; 100 ± 21 W; trained 190 ± 15) (Table 2).
Dietary and exercise controls
All participants were asked to refrain from any formal exercise for 48 h prior to the experimental trial to minimise any acute exercise training effects and to avoid drinking alcohol or consumption of caffeine for 24 h prior. To ensure the energy intake was controlled between groups, participants were supplied with a diet to consume over the 24 h prior to each experimental trial containing ∼199 kJ kg–1 consisting of ∼65% of carbohydrates, ∼15% proteins and ∼ 20% fats. Participants were instructed to adhere to the diet but to consume water ad libitum and to finish the food by 10pm the evening prior to the experimental trial to enable attending the laboratory in a fasted state.
Exercise trials
On the morning of the exercise trial, a 22 gauge Teflon catheter (Optiva; Ethicon Endo‐Surgery, Cincinnati, OH, USA) was inserted into an antecubital forearm vein for blood sampling. The exercise protocol consisted of cycling for 120 min at 65% of . Blood was sampled 10 min prior to the commencement of exercise and then every 30 min during exercise for the measurement of plasma glucose, lactate, insulin, glycerol and free fatty acids. Expired air was collected into Douglas bags every 30 min during exercise and heart rate (Polar Favor, Oulu, Finland) was recorded every 30 min during exercise. and the respiratory exchange ratio were calculated from the expired air samples. Participants received 8 ml kg−1 body weight of water at the start of exercise, followed by a further 2 ml kg−1 body weight every 15 min of exercise and were fan cooled throughout the trial. At rest and after 30 and 120 min of exercise, muscle was obtained from the vastus lateralis under local anaesthesia, using the percutaneous needle biopsy technique, with suction. Muscle samples were rapidly (8–12 s from stopping exercise) frozen and stored in liquid N2 for later analysis of AMPK α1 and α2 activity, AMPK αThr172 phosphorylation and ACCβ Ser222 phosphorylation and muscle metabolites.
Analytical techniques
Blood
Plasma glucose, lactate (Lowry OH, 1972) and glycerol (Chernick, 1969) were determined using an enzymatic fluorometric procedure, plasma non‐esterified fatty acids (NEFA) by an enzymatic colorimetric method (NEFA‐C test, Wako, Osaka, Japan) and plasma insulin using a human insulin‐specific radioimmunoassay kit (Linco Research, St Charles, MO, USA).
Muscle metabolites
A portion of each muscle sample (∼20 mg) was freeze‐dried and subsequently crushed to a powder and any visible connective tissue was removed. The extraction of muscle glycogen commenced by incubating the sample in HCl before being neutralized with NaOH and subsequently analysed for glucosyl units using an enzymatic flurometric method (Passonneau & Lauderdale, 1974). The metabolites (ATP, CrP, Cr and lactate) were extracted firstly with precooled perchloric acid/EDTA before the addition of precooled KHCO3 to the supernatant. The metabolites were analysed in triplicate using an enzymatic flurometric method as reported by Harris et al. (1974). PCr, Cr and ATP were normalised to the participant's highest total creatine (Cr + CrP). The concentration of ADP (ADPfree) and AMP (AMPfree) was calculated based on the near equilibrium nature of the CK and adenylate kinase reactions, respectively. ADPfree was calculated from the measured ATP, Cr, PCr levels and the estimated H+ concentration, which was calculated from a formula based on the muscle lactate content for dry muscle (Mannion et al. 1993). The observed equilibrium constant (K obs) of 1.66 × 109 was used for creatine kinase (Lawson & Veech, 1979). An estimation of AMPfree was calculated from the measured ATP and estimated ADPfree, using a K obs of 1.05 for adenylate kinase (Lawson & Veech, 1979). Estimated ADPfree and AMPfree were expressed as μmol per kilogram of dry muscle mass (μmol kg−1 dry muscle).
Immunoblotting
Frozen skeletal muscle was homogenised in ice cold lysis buffer on ice [10 μl mg–1 tissue; 50 mm Tris‐HCl, pH 7.5, containing 1 mm EDTA, 1 mm EGTA, 10% v/v glycerol, 1% v/v Triton X‐100, 50 mm NaF, 5 mm Na4P2O7, 1 mm dithiothreitol, 1 mm phenylmethylsulphonyl fluoride, 1 μl ml−1 trypsin inhibitor and 5 μl ml−1 protease inhibitor cocktail (P8340; Sigma, St Louis, MO, USA)], incubated on ice for 20 min and centrifuged at 16 000 g for 20 min at 4°C. The protein concentration of samples was determined using the bicinchoninic acid protein assay (Pierce, Rockford, IL, USA) with BSA as the standard. All primary antibodies were diluted to a final concentration of 1:1000. Phosphospecific antibodies for AMPK αThr172 and ACCβ Ser221 were purchased from Upstate Biotechnology (Lake Placid, NY, USA; catalogue no. 07–626 and 05–673, respectively). Polyclonal rabbit antibody specific for total AMPK α protein was purchased from Cell Signalling Technology (Beverley, MA, USA; catalogue no. 2532). ACCβ was detected using IRDyeTM 800‐labelled streptavidin (Rockland, Gilbertsville, PA, USA; catalogue no. S000‐45).
Skeletal muscle lysates (80 μg) were heated in SDS sample buffer and subjected to SDS‐PAGE. Binding of purified proteins was detected by immunoblotting following an overnight incubation with primary antibody. Membranes were incubated in Odyssey anti‐rabbit IRDye™ 800‐ or anti‐mouse IRDye™ 700‐ labelled secondary antibody (Rockland, Gilbertsville, PA, USA), washed in PBS Tween 20 and were scanned for infra‐red fluorescence using an Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA). When both total protein and protein phosphorylation were measured, membranes were probed first for total protein, stripped of antibodies (2% SDS in 25 mm glycine, pH 2.0) and re‐probed with the anti‐phospho antibody. Phosphorylation was expressed relative to the total protein of the specific protein of interest.
AMPK activity
Skeletal muscle lysates (50 μg) were combined with 15 μl of protein A sepharose beads (Pierce), bound to either AMPK α1 (raised to the non‐conserved region of the AMPK α1 isoform, amino acid sequence 373–390 of rat AMPK α1) or AMPK α2 (amino acid sequences 351–366 and 490–516 of rat AMPKα2) polyclonal antibodies (a gift from Professor Bruce Kemp, St Vincent's Institute of Medical Research, Fitzroy, VIC, Australia) and incubated for 2 h at 4 °C. Immunocomplexes were washed in lysis buffer containing 0.5 m NaCl and resuspended in 25 μl of 0.05 m Tris buffer (pH 7.5). To commence the assay, 25 μl of reaction mixture containing (final concentrations) 50 mm Tris/HCl, pH 7.5, 0.1 mm EGTA, 0.1% (by vol) 2‐mercaptoethanol, 10 mm magnesium acetate, 0.1 mm [32P]ATP (∼200 cpm/pmol; PerkinElmer Life and Analytical Sciences, Boston, MA, USA), 30 μm AMARA peptide (Upstate Biotechnology) and 200 μm AMP was added to each sample at 30°C for 20 min with agitation. Then, 40 μl of each sample was transferred onto P81 chromatography paper and washed 3 × 10 min in 75 mm H3PO4, once in 100% ethanol, and air dried. P81 paper was then placed in organic scintillation fluid (Opti‐Fluor O; PerkinElmer) and radioactivity was counted on a β counter (PerkinElmer). AMARA peptide has the same AMPK phosphorylation site as ACCβ; therefore, AMPK activities were calculated as units of γ‐[32P]‐ATP incorporated into the AMARA peptide [ACCα (73‐87)A77] min−1 mg−1 total protein subjected to immunoprecipitation.
Statistical analysis
Data are expressed as the mean ± SD. Untrained and exercise trained group values were compared using a two‐factor (training × time) repeated measures ANOVA and, if there was a significant interaction, the ANOVA was followed by a post hoc comparison using a least significant difference test. P < 0.05 was considered statistically significant.
Results
Subjects
There was no significant difference in age, weight, height or body mass index between the untrained and exercise trained groups (Table 1). However, as expected, was significantly higher in the endurance trained group (P < 0.05) (Table 1) and the trained group cycled on average at almost twice the workload of the untrained group (P < 0.05) (Table 2). Importantly, the relative exercise intensity was the same between groups, with no significant difference for (as a percentage of ), heart rate or rating of perceived exhaustion (Table 2).
Table 2.
Physiological responses during 120 min of steady‐state exercise at ∼ 65%
| Parameter | Untrained | Trained |
|---|---|---|
| % of (ml.kg−1 min−1) | 66 ± 4.5 | 65 ± 1.6 |
| Workload (watts) | 100 ± 21 * | 190 ± 15 |
| RER | 0.91 ± 0.56 | 0.91 ± 0.38 |
| RPE | 12.4 ± 1.2 | 11.0 ± 1.5 |
| Heart rate (beats min–1) | 147 ± 9 | 143 ± 7 |
Data are the mean ± SD, n = 11 untrained and 7 exercise trained participants. , oxygen consumption, RER, respiratory exchange ratio, RPE, rating of perceived exhaustion.
Significantly different to corresponding trained value (P < 0.05).
Muscle metabolites
Measured muscle lactate and Cr and estimated ADPfree, AMPfree and the AMPfree:ATP ratio all increased progressively with exercise in both groups (Table 3). The exercise induced increase in muscle lactate, Cr and AMPfree was attenuated in the exercise trained compared to the untrained group (P < 0.05) (Table 3). Skeletal muscle AMPfree increased (P < 0.05) 17‐fold in the untrained group and significantly (P < 0.05) less (5‐fold) in the endurance trained group following 120 min of exercise and the muscle lactate concentration increased by 5.9‐fold in the untrained and 3.1‐fold in the well trained individuals (P < 0.05) (Table 3). PCr and the PCr:(PCr+Cr) ratio decreased progressively in both groups, and this decrease was greater in untrained subjects (∼26% decrease in untrained, ∼44% decrease in endurance trained groups, P < 0.05) (Table 3). There was no difference in measured ATP levels during exercise or between groups (P > 0.05) (Table 3).
Table 3.
Measured and calculated muscle metabolites at rest (0 min), and after 30 min and 120 min of steady‐state exercise at ∼ 65% of
| Metabolite | 0 min | 30 min | 120 min | ||
|---|---|---|---|---|---|
| Lactate | Untrained | § | 4.6 ± 1.2 | 11.9 ± 3.4 | 27.5 ± 11.6* |
| (mmol kg−1 dm) | Trained | §‡ | 3.8 ± 2.1 | 8.9 ± 3.6 | 11.6 ± 4.9 |
| PCr | Untrained | § | 70.1 ± 15.5 | 46.1 ± 12.8 | 42.9 ± 11.5 |
| (mmol kg−1 dm) | Trained | §‡ | 78.9 ± 13.7 | 61.5 ± 17.0 | 56.6 ± 13.8 |
| Cr | Untrained | § | 38.5 ± 7.0 | 61.4 ± 18.3 | 69.7 ± 17.6 |
| (mmol kg−1 dm) | Trained | §‡ | 39.1 ± 6.1 | 55.1 ± 11.1 | 58.8 ± 15.9 |
| PCr:(PCr + Cr) | Untrained | § | 0.64 ± 0.07 | 0.44 ± 0.12 | 0.37 ± 0.13 |
| Trained | §‡ | 0.66 ± 0.07 | 0.52 ± 0.12 | 0.50 ± 0.12 | |
| ATP | Untrained | 24.3 ± 2.2 | 24.1 ± 2.5 | 24.3 ± 3.5 | |
| (mmol kg−1 dm) | Trained | 24.4 ± 4.1 | 26.5 ± 6.2 | 24.1 ± 3.0 | |
| ADPfree | Untrained | § | 131 ± 36 | 338 ± 239 | 409 ± 199 |
| (μmol kg−1 dm) | Trained | § | 109 ± 20 | 228 ± 66 | 248 ± 117 |
| AMPfree | Untrained | § | 0.8 ± 0.5 | 2.3 ± 0.8 | 13.5 ± 4.1* |
| (μmol kg−1 dm) | Trained | §‡ | 0.6 ± 0.3 | 2.4 ± 1.5 | 3.2 ± 2.6 |
| AMPfree:ATP | Untrained | § | 0.04 ± 0.02 | 0.08 ± 0.03 | 0.56 ± 0.17 |
| Trained | §‡ | 0.03 ± 0.02 | 0.10 ± 0.07 | 0.13 ± 0.09 |
Values are the mean ± SD, n = 11 untrained and 7 exercise trained participants. PCr, creatine phosphate; Cr, creatine; ADPfree, free adenosine diphosphate; AMPfree, free adenosine monophosphate, dm; dry muscle. *Significantly different to corresponding trained value. §Main effect for time. ‡Main effect for untrained compared to trained (P < 0.05).
Resting muscle glycogen levels were 93% higher (P < 0.05) in the exercise trained group, and remained higher than the untrained values at 30 min (130%, P < 0.05) and 120 min of exercise (440%, P < 0.05) (Fig. 1). However, net glycogen utilization during exercise was similar between the two groups.
Figure 1. Muscle glycogen measurements in muscle samples.
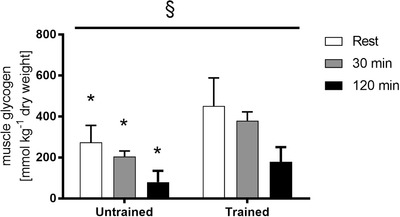
Muscle glycogen measured from muscle samples obtained before exercise (rest), after 30 min and immediately following 120 min of steady‐state exercise at ∼65% in untrained and exercise trained participants. Data are the mean ± SD (n = 11 untrained, 7 trained. *Significantly different to corresponding trained value (P < 0.05). §Main effect for time (P < 0.05).
Plasma glucose lactate, insulin, glycerol and NEFA
Plasma glucose concentration remained at a similar level during exercise in untrained and endurance trained subjects (P > 0.05) (Fig. 2A ). Plasma lactate was similar at rest in the two groups; however, it was elevated by ∼110% in the untrained group during exercise, which was significantly higher than the trained group (P < 0.05) (Fig. 2B ). Plasma insulin decreased progressively throughout exercise in both groups; however, fasting plasma insulin was 40% higher in the untrained compared to the trained group at rest, and remained ∼25% higher than the trained values throughout 120 min of exercise (P < 0.05) (Fig. 2C ). Both plasma glycerol and NEFA increased progressively throughout exercise in both groups; however, NEFA levels were attenuated in the exercise trained group at 90 and 120 min (P < 0.05) (Fig. 3).
Figure 2. Plasma glucose, lactate and insulin concentrations.
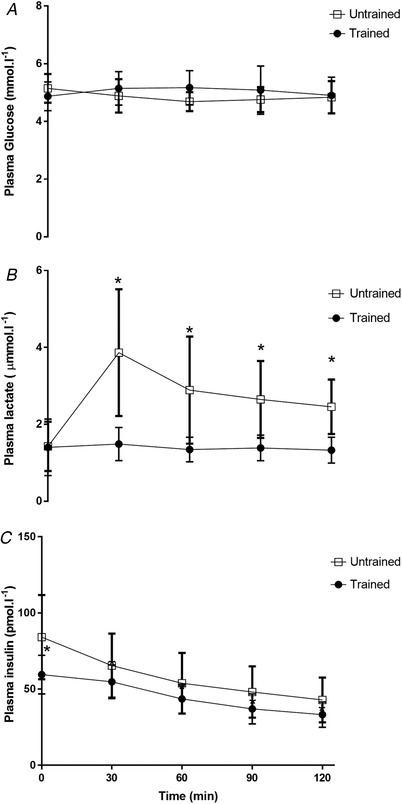
Plasma glucose (A), plasma lactate (B) and plasma insulin (C) concentrations at rest and during 120 min of steady‐state exercise at ∼65% in untrained and exercise trained participants. Data are the mean ± SD, n = 11 untrained, 7 trained. *Significantly different from corresponding trained value (P < 0.05).
Figure 3. Plasma glycerol and nonesterified fatty acid concentrations.
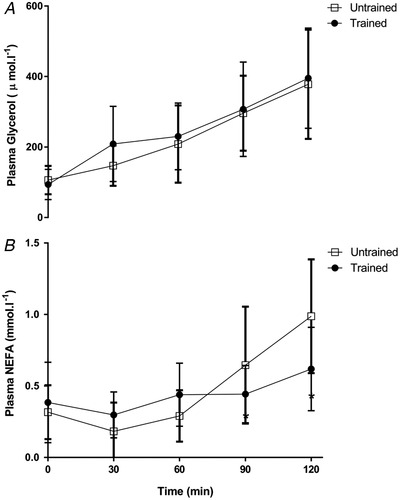
Plasma glycerol (A) and plasma nonesterified fatty acid (NEFA) (B) concentrations at rest and during 120 min of steady‐state exercise at ∼65% in untrained and exercise trained participants. Data are the mean ± SD, n = 11 untrained, 7 trained.
AMPK signalling
Basal AMPK α1 activity was 90% higher (P < 0.05) in the exercise trained group, whereas there was no difference in basal AMPK α2 activity between the two groups (Fig. 4). AMPK α1 activity increased by 220% and AMPK α2 activity increased by 370% during exercise in the untrained group at 120 min (P < 0.05). However, neither AMPK α1, nor AMPK α2 activity increased during exercise in the endurance trained group (P > 0.05) (Fig. 4). AMPK αThr172 (115%, P < 0.05) phosphorylation and ACCβ Ser222 phosphorylation (100%, P < 0.05) increased with exercise in the untrained group after 120 min of exercise (Fig. 5). However, there was no significant increase in AMPK αThr172 phosphorylation or ACCβ Ser222 phosphorylation during exercise in the trained group (P > 0.05) (Fig. 5).
Figure 4. Skeletal muscle AMPK α1 and α2 activity.
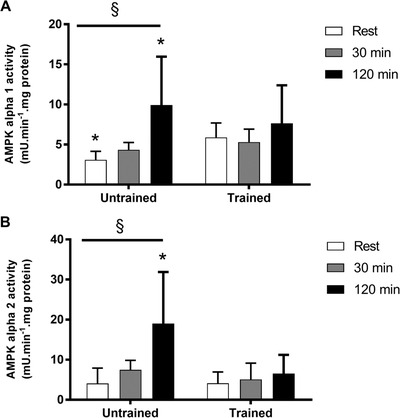
AMPK α1 (A) and α2 activity (B). Muscle samples were obtained before exercise (rest), after 30 min and immediately following 120 min of steady‐state exercise at ∼65% in untrained and exercise trained participants. Data are the mean ± SD, n = 11 untrained, 7 trained. *Significantly different from corresponding trained value (P < 0.05). §Main effect for time (P < 0.05).
Figure 5. Skeletal muscle AMPK αThr172 and ACC‐βSer221 phosphorylation.
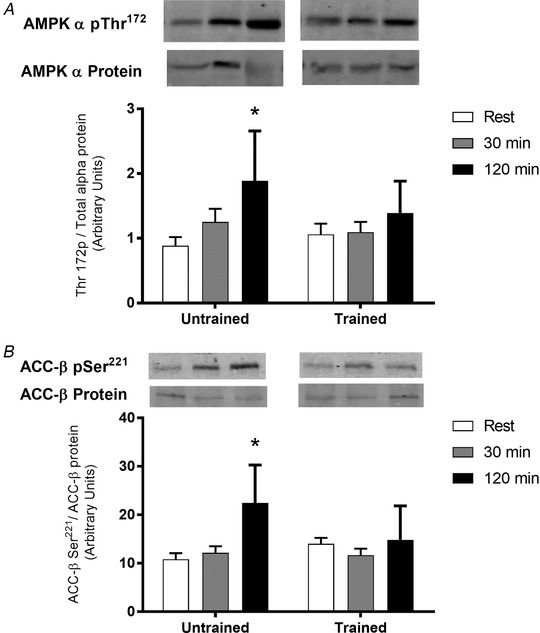
Representative immunoblot of AMPK αThr172 phosphorylation (A) measured using a phosphospecific antibody for AMPK αThr172, normalized to total AMPK α protein; and ACC‐βSer221 phosphorylation (B) measured using a phosphospecific antibody specific to ACC‐βSer221, normalized to total ACC‐β. Muscle samples were obtained before exercise (rest), after 30 min and immediately following 120 min of steady‐state exercise at ∼65% in untrained and exercise trained participants. Samples of pAMPK or AMPK for each participant were run on the same western blot. The representative western blots for pAMPK or AMPK shown are from the same membrane but are rearranged for clarity. Data are the mean ± SD, n = 11 untrained, 7 trained. *Significantly different from corresponding trained value (P < 0.05). §Main effect for time (P < 0.05).
Discussion
The results of the present study show that AMPK activity is not increased during prolonged steady‐state, moderate intensity exercise in endurance trained individuals. Indeed, AMPK α1 and α2 activity was significantly elevated following 120 min of exercise in the untrained group (220% and 370%, respectively), whereas no increase in AMPK activity was observed during exercise in the trained participants (Fig. 4). Given that there is a substantial amount of glucose and fat oxidised during 120 min of exercise at 65% in endurance trained individuals (Romijn et al. 1993; van Loon et al. 2001), these results indicate that AMPK activation is not important for exercise metabolism under these circumstances.
Almost every study investigating AMPK states in their Introduction that AMPK regulates glucose uptake and fat oxidation during exercise, despite much evidence to the contrary. Indeed, our current findings are consistent with our previous results indicating that, after 10 days of exercise training, there was no increase from rest in skeletal muscle AMPK activity, AMPK αThr172 phosphorylation or ACCβ phosphorylation during 120 min of exercise at 65% of pre‐training despite substantial glucose disposal and higher fat oxidation during exercise after (compared to before) exercise training (McConell et al. 2005). Other studies similarly report in humans that skeletal muscle AMPK activation is barely increased during exercise at 65% after 12 weeks of exercise training (Mortensen et al. 2013). Although not universally the case (Mu et al. 2001; Sakamoto et al. 2005; Lee‐Young et al. 2009), many rodent studies have found that there are normal increases in glucose uptake and fat oxidation during contraction or during exercise in AMPK kinase dead or AMPK knockout mice (Jorgensen et al. 2004; Fujii et al. 2005; Merry et al. 2010b ; Jeppesen et al. 2011; Jeppesen et al. 2013; Kjobsted et al. 2017).
Unlike the present study, it was reported previously that skeletal muscle AMPK activity increases similarly in untrained and well trained people cycling for 20 min at 80% (Nielsen et al. 2003). In addition, Clark et al. (2004) found AMPK activity increased similarly during repeated bouts of exercise at 85% after exercise training despite evidence of lower energy deficit after the exercise training. In the present study, exercise was performed at ∼65% of (untrained 66%; trained 65%) (Table 2); some may argue that this workload is quite easy for trained individuals and so no increase in AMPK activation would be expected. However, because there are 2–3‐fold increases from rest in whole‐body glucose uptake and fat oxidation during moderate intensity exercise at ∼65% in endurance trained individuals (Romijn et al. 1993; van Loon et al. 2001), factors that are important to mediating exercise metabolism should be enhanced at these workloads. Therefore, activation of skeletal muscle AMPK is probably not necessary for increases in glucose uptake and fat oxidation during moderate intensity exercise in humans. These findings are also supported by other human studies that found dissociations between AMPK activation and glucose uptake and fat oxidation during prolonged exercise at 40–45% (Chen et al. 2000; Wojtaszewski et al. 2002).
AMPK activity is increased by upstream kinase phosphorylation and allosterically by increases in AMP (Hardie, 2004; Sakamoto et al. 2005). In addition, ADP increases AMPK activity by preventing dephosphorylation of AMPK (Xiao et al. 2011). Exercise increases skeletal muscle free ADP and AMP (McConell et al. 1999) and exercise training attenuates the normal increases in ADP and AMP during exercise compared to untrained muscle (Gollnick & Hermansen, 1973). Indeed, in the present study, we report an attenuated increase in calculated AMP and ADP during exercise in the well trained compared to the untrained participants (Table 3) and thus would have expected a similar attenuated response in AMPK activity. In the present study, despite the five‐fold increase in AMPfree during exercise in the trained individuals AMPK activity was not increased from rest. Future studies should investigate whether there is a reduction in AMPK sensitivity to AMP and ADP during moderate intensity exercise after exercise training. This could be carried out in highly trained endurance subjects by using exercise intensity to titrate the free AMP levels to compare the threshold for AMPK activation during exercise in endurance trained vs. untrained individuals.
It is possible that AMPK activation during exercise in the trained individuals was restrained by the higher muscle glycogen since muscle glycogen content has also been implicated as a potential regulator of AMPK activation during exercise. Indeed, the β subunit contains a glycogen binding domain that associates with glycogen in cell free assays (Hudson et al. 2003; Polekhina et al. 2003). High muscle glycogen has been shown to inhibit contraction induced AMPK activation without influencing glucose uptake in rats (Derave et al. 2000). However, there is evidence to suggest that AMPK does not associate with glycogen in vivo (Viollet et al. 2003; Parker et al. 2007). Indeed our laboratory has shown that muscle glycogen is not responsible for abolished AMPK activity during exercise in humans. Following short‐term exercise training, there is no increase in AMPK activity during exercise commenced with normal or high glycogen levels (McConell et al. 2005). In the present study, muscle glycogen content was 93% higher in the trained subjects at rest, and remained higher than untrained values throughout the exercise trial (Fig. 1). However, given the findings of our short‐term exercise training study (McConell et al. 2005), as well as other evidence of a lack of importance of AMPK for in vivo muscle glycogen content (Viollet et al. 2003), the higher muscle glycogen content in the endurance trained individuals was probably not responsible for the abolished increase in AMPK activity during exercise.
It is not clear why activation of AMPK αThr172 phosphorylation and AMPK activity did not occur during exercise in the exercise trained individuals; however, there is some evidence to suggest that training is associated with changes in the regulation of upstream AMPKK(s). For example, endurance training in rats decreases basal LKB1 activity (Hurst et al. 2005) and decreases AMPK activity during exercise (Durante et al. 2002). It is also not known what effect exercise training has on PP2C activity, and it may be that increases in PP2C activity following training are preventing AMPK αThr172 phosphorylation and increases in AMPK activity during exercise. PP2C has previously been shown to inhibit the activity of both AMPK α1 and α2 isoforms (Davies et al. 1995); however, further studies are required to determine whether changes in PP2C activity occur following exercise training.
In summary, the present study found that, unlike untrained individuals, endurance trained men have no increase in skeletal muscle AMPK activity, AMPK phosphorylation or ACC phosphorylation during 120 min of cycling exercise at ∼65% . This finding is consistent with results following short‐term exercise training where no increase in AMPK activation during moderate intensity exercise is also observed (McConell et al. 2005). Given that skeletal muscle AMPK is not activated but fat oxidation and glucose uptake are substantial during moderate intensity exercise after exercise training, these results indicate that AMPK does not regulate metabolism under these circumstances.
Additional information
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by a grant from the National Health and Medical Research Council (NHMRC) of Australia (GKM: 237002).
Author contributions
GKM, KCL and GDW designed the study. GKM, KCL conducted the exercise tests. GKM, KCL, GDW and KLP conducted the experiments. KCL and GDW conducted the analysis. GKM and KCL drafted the manuscript. All authors revised the manuscript critically.
Supporting information
Statistical Summary Document
Acknowledgements
We thank the participants for taking part in this study and acknowledge the technical assistance of Mrs Judith Gooley, Assistant Professor Sean McGee, Dr Rob Lee‐Young and Professor Benedict Canny. This work was supported by a grant from the National Health and Medical Research Council (NHMRC) of Australia (GKM: 237002). GKM was a Danish Diabetes Academy Visiting Professor during the writing of this manuscript (supported by a research grant from the Danish Diabetes Academy, which is funded by the Novo Nordisk Foundation Grant NNF17SA0031406).
Biography
Glenn K. McConell is a Professor of Exercise Metabolism at Victoria University. He has 30 years of experience in exercise metabolism research and is internationally recognized with regard to the regulation of skeletal muscle glucose uptake in normal physiology and in diabetes. His research focuses on glucose metabolism and exercise, including examining the factors regulating skeletal muscle glucose uptake during exercise and the increase in insulin sensitivity after exercise. He has also been examining whether exercise early in life can prevent the insulin resistance associated with being born small for gestational age or having an obese father, and has he begun examining whether exercise can overcome the insulin resistance caused by shift work.

Edited by: Michael Hogan & Troy Hornberger
Linked articles: This article is highlighted in a Journal Club article by Frangos et al. To read this article, visit https://doi.org/10.1113/JP280447.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Abbott MJ, Bogachus LD & Turcotte LP (2011). AMPKalpha2 deficiency uncovers time dependency in the regulation of contraction‐induced palmitate and glucose uptake in mouse muscle. J Appl Physiol (1985) 111, 125–134. [DOI] [PubMed] [Google Scholar]
- Baldwin J, Snow RJ, Carey MF & Febbraio MA (1999). Muscle IMP accumulation during fatiguing submaximal exercise in endurance trained and untrained men. Am J Physiol 277, R295–R300. [DOI] [PubMed] [Google Scholar]
- Balon TW & Nadler JL (1997). Evidence that nitric oxide increases glucose transport in skeletal muscle. J Appl Physiol 82, 359–363. [DOI] [PubMed] [Google Scholar]
- Bradley SJ, Kingwell BA & McConell GK (1999). Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans [published erratum appears in Diabetes 1999 Dec;48(12):2480]. Diabetes 48, 1815–1821. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE & McConell GK (2003). Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes 52, 2205–2212. [DOI] [PubMed] [Google Scholar]
- Chen ZP, McConell GK, Michell BJ, Snow RJ, Canny BJ & Kemp BE (2000). AMPK signaling in contracting human skeletal muscle: acetyl‐CoA carboxylase and NO synthase phosphorylation. Am J Physiol Endocrinol Metab 279, E1202–E1206. [DOI] [PubMed] [Google Scholar]
- Chernick S (1969). Determination of Glycerol in Acyl Glycerols. Academic Press, New York, NY. [Google Scholar]
- Clark SA, Chen ZP, Murphy KT, Aughey RJ, McKenna MJ, Kemp BE & Hawley JA (2004). Intensified exercise training does not alter AMPK signaling in human skeletal muscle. Am J Physiol Endocrinol Metab 286, E737–E743. [DOI] [PubMed] [Google Scholar]
- Davies SP, Helps NR, Cohen PT & Hardie DG (1995). 5'‐AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP‐activated protein kinase. Studies using bacterially expressed human protein phosphatase‐2C alpha and native bovine protein phosphatase‐2AC. FEBS Lett 377, 421–425. [DOI] [PubMed] [Google Scholar]
- Derave W, Ai H, Ihlemann J, Witters LA, Kristiansen S, Richter EA & Ploug T (2000). Dissociation of AMP‐activated protein kinase activation and glucose transport in contracting slow‐twitch muscle. Diabetes 49, 1281–1287. [DOI] [PubMed] [Google Scholar]
- Durante PE, Mustard KJ, Park SH, Winder WW & Hardie DG (2002). Effects of endurance training on activity and expression of AMP‐activated protein kinase isoforms in rat muscles. Am J Physiol Endocrinol Metab 283, E178–E186. [DOI] [PubMed] [Google Scholar]
- Fujii N, Hayashi T, Hirshman MF, Smith JT, Habinowski SA, Kaijser L, Mu J, Ljungqvist O, Birnbaum MJ, Witters LA, Thorell A & Goodyear LJ (2000). Exercise induces isoform‐specific increase in 5'AMP‐activated protein kinase activity in human skeletal muscle. Biochem Biophys Res Commun 273, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM & Goodyear LJ (2005). AMP‐activated protein kinase alpha2 activity is not essential for contraction‐ and hyperosmolarity‐induced glucose transport in skeletal muscle. J Biol Chem 280, 39033–39041. [DOI] [PubMed] [Google Scholar]
- Gollnick PD & Hermansen L (1973). Biochemical adaptations to exercise: anaerobic metabolism. Exerc Sport Sci Rev 1, 1–43. [PubMed] [Google Scholar]
- Hardie DG (2004). The AMP‐activated protein kinase pathway–new players upstream and downstream. J Cell Sci 117, 5479–5487. [DOI] [PubMed] [Google Scholar]
- Harris RC, Hultman E & Nordesjo LO (1974). Glycogen, glycolytic intermediates and high‐energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scand J Clin Lab Invest 33, 109–120. [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA & Goodyear LJ (2000). Metabolic stress and altered glucose transport: activation of AMP‐activated protein kinase as a unifying coupling mechanism. Diabetes 49, 527–531. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Kurth EJ, Winder WW & Goodyear LJ (1998). Evidence for 5' AMP‐activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 47, 1369–1373. [DOI] [PubMed] [Google Scholar]
- Hudson ER, Pan DA, James J, Lucocq JM, Hawley SA, Green KA, Baba O, Terashima T & Hardie DG (2003). A novel domain in AMP‐activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol 13, 861–866. [DOI] [PubMed] [Google Scholar]
- Hurst D, Taylor EB, Cline TD, Greenwood LJ, Compton CL, Lamb JD & Winder WW (2005). AMP‐activated protein kinase kinase activity and phosphorylation of AMP‐activated protein kinase in contracting muscle of sedentary and endurance‐trained rats. Am J Physiol Endocrinol Metab 289, E710–E715. [DOI] [PubMed] [Google Scholar]
- Inyard AC, Clerk LH, Vincent MA & Barrett EJ (2007). Contraction stimulates nitric oxide independent microvascular recruitment and increases muscle insulin uptake. Diabetes 56, 2194–2200. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Jorgensen SB, Brandt N, Schjerling P, Wojtaszewski JF & Richter EA (2007). Possible CaMKK‐dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab 292, E1308–E1317. [DOI] [PubMed] [Google Scholar]
- Jeppesen J, Albers PH, Rose AJ, Birk JB, Schjerling P, Dzamko N, Steinberg GR & Kiens B (2011). Contraction‐induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J Lipid Res 52, 699–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen J, Maarbjerg SJ, Jordy AB, Fritzen AM, Pehmoller C, Sylow L, Serup AK, Jessen N, Thorsen K, Prats C, Qvortrup K, Dyck JR, Hunter RW, Sakamoto K, Thomson DM, Schjerling P, Wojtaszewski JF, Richter EA & Kiens B (2013). LKB1 regulates lipid oxidation during exercise independently of AMPK. Diabetes 62, 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA & Wojtaszewski JF (2004). Knockout of the alpha2 but not alpha1 5'‐AMP‐activated protein kinase isoform abolishes 5‐aminoimidazole‐4‐carboxamide‐1‐beta‐4‐ribofuranosidebut not contraction‐induced glucose uptake in skeletal muscle. J Biol Chem 279, 1070–1079. [DOI] [PubMed] [Google Scholar]
- Kjobsted R, Munk‐Hansen N, Birk JB, Foretz M, Viollet B, Bjornholm M, Zierath JR, Treebak JT & Wojtaszewski JF (2017). Enhanced Muscle Insulin Sensitivity After Contraction/Exercise Is Mediated by AMPK. Diabetes 66, 598–612. [DOI] [PubMed] [Google Scholar]
- Lawson JW & Veech RL (1979). Effects of pH and free Mg2+ on the Keq of the creatine kinase reaction and other phosphate hydrolyses and phosphate transfer reactions. J Biol Chem 254, 6528–6537. [PubMed] [Google Scholar]
- Lee‐Young RS, Griffee SR, Lynes SE, Bracy DP, Ayala JE, McGuinness OP & Wasserman DH (2009). Skeletal muscle AMP‐activated protein kinase is essential for the metabolic response to exercise in vivo. J Biol Chem 284, 23925–23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden KC, Wadley GD, Garnham AP & McConell GK (2011). Effect of l‐arginine infusion on glucose disposal during exercise in humans. Med Sci Sports Exerc 43, 1626–1634. [DOI] [PubMed] [Google Scholar]
- Lowry OH (1972). Academic Press, New York. [Google Scholar]
- Mannion AF, Jakeman PM & Willan PL (1993). Determination of human skeletal muscle buffer value by homogenate technique: methods of measurement. J Appl Physiol (1985) 75, 1412–1418. [DOI] [PubMed] [Google Scholar]
- McConell G, Snow RJ, Proietto J & Hargreaves M (1999). Muscle metabolism during prolonged exercise in humans: influence of carbohydrate availability. J Appl Physiol 87, 1083–1086. [DOI] [PubMed] [Google Scholar]
- McConell GK, Lee‐Young RS, Chen ZP, Stepto NK, Huynh NN, Stephens TJ, Canny BJ & Kemp BE (2005). Short‐term exercise training in humans reduces AMPK signalling during prolonged exercise independent of muscle glycogen. J Physiol 568, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee SL, Howlett KF, Starkie RL, Cameron‐Smith D, Kemp BE & Hargreaves M (2003). Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes 52, 926–928. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG & Winder WW (1997). AICA riboside increases AMP‐activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 273, E1107–E1112. [DOI] [PubMed] [Google Scholar]
- Merry TL, Lynch GS & McConell GK (2010a). Downstream mechanisms of nitric oxide‐mediated skeletal muscle glucose uptake during contraction. Am J Physiol Regul Integr Comp Physiol 299, R1656–R1665. [DOI] [PubMed] [Google Scholar]
- Merry TL, Steinberg GR, Lynch GS & McConell GK (2010b). Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am J Physiol Endocrinol Metab 298, E577–E585. [DOI] [PubMed] [Google Scholar]
- Mortensen B, Hingst JR, Frederiksen N, Hansen RW, Christiansen CS, Iversen N, Friedrichsen M, Birk JB, Pilegaard H, Hellsten Y, Vaag A & Wojtaszewski JF (2013). Effect of birth weight and 12 weeks of exercise training on exercise‐induced AMPK signaling in human skeletal muscle. Am J Physiol Endocrinol Metab 304, E1379–E1390. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr. , Valladares O, Bucan M & Birnbaum MJ (2001). A role for AMP‐activated protein kinase in contraction‐ and hypoxia‐ regulated glucose transport in skeletal muscle. Mol Cell 7, 1085–1094. [DOI] [PubMed] [Google Scholar]
- Musi N, Fujii N, Hirshman MF, Ekberg I, Froberg S, Ljungqvist O, Thorell A & Goodyear LJ (2001). AMP‐activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 50, 921–927. [DOI] [PubMed] [Google Scholar]
- Nielsen JN, Mustard KJ, Graham DA, Yu H, MacDonald CS, Pilegaard H, Goodyear LJ, Hardie DG, Richter EA & Wojtaszewski JF (2003). 5'‐AMP‐activated protein kinase activity and subunit expression in exercise‐trained human skeletal muscle. J Appl Physiol (1985) 94, 631–641. [DOI] [PubMed] [Google Scholar]
- Parker GJ, Koay A, Gilbert‐Wilson R, Waddington LJ & Stapleton D (2007). AMP‐activated protein kinase does not associate with glycogen alpha‐particles from rat liver. Biochem Biophys Res Commun 362, 811–815. [DOI] [PubMed] [Google Scholar]
- Passonneau JV & Lauderdale VR (1974). A comparison of three methods of glycogen measurement in tissues. Anal Biochem 60, 405–412. [DOI] [PubMed] [Google Scholar]
- Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC, Jennings IG, Campbell DJ, Witters LA, Parker MW, Kemp BE & Stapleton D (2003). AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol 13, 867–871. [DOI] [PubMed] [Google Scholar]
- Reid MB & Durham WJ (2002). Generation of reactive oxygen and nitrogen species in contracting skeletal muscle: potential impact on aging. Ann N Y Acad Sci 959, 108–116. [DOI] [PubMed] [Google Scholar]
- Richter EA & Hargreaves M (2013). Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev 93, 993–1017. [DOI] [PubMed] [Google Scholar]
- Roberts CK, Barnard RJ, Jasman A & Balon TW (1999). Acute exercise increases nitric oxide synthase activity in skeletal muscle. Am J Physiol 277, E390–E394. [DOI] [PubMed] [Google Scholar]
- Roberts CK, Barnard RJ, Scheck SH & Balon TW (1997). Exercise‐stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am J Physiol Endocrinol Metab 273, E220–E225. [DOI] [PubMed] [Google Scholar]
- Romijn JA, Coyle EF, Sidossis LS, Gastaldelli A, Horowitz JF, Endert E & Wolfe RR (1993). Regulation of endogenous fat and carbohydrate metabolism in relation to exercise intensity and duration. Am J Physiol Endocrinol Metab 265, E380–E391. [DOI] [PubMed] [Google Scholar]
- Ross RM, Wadley GD, Clark MG, Rattigan S & McConell GK (2007). Local NOS inhibition reduces skeletal muscle glucose uptake but not capillary blood flow during in situ muscle contraction in rats. Diabetes 56, 2885–2892. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A & Alessi DR (2005). Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J 24, 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstrom ME, Zhang SJ, Bruton J, Silva JP, Reid MB, Westerblad H & Katz A (2006). Role of reactive oxygen species in contraction‐mediated glucose transport in mouse skeletal muscle. J Physiol 575, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Jensen TE, Kleinert M, Mouatt JR, Maarbjerg SJ, Jeppesen J, Prats C, Chiu TT, Boguslavsky S, Klip A, Schjerling P & Richter EA (2013). Rac1 is a novel regulator of contraction‐stimulated glucose uptake in skeletal muscle. Diabetes 62, 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Moller LL, Kleinert M, Richter EA & Jensen TE (2015). Stretch‐stimulated glucose transport in skeletal muscle is regulated by Rac1. J Physiol 593, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Inoue G, Otaka A, Sato K, Fushiki T & Nakao K (2004). Possible involvement of the alpha1 isoform of 5'AMP‐activated protein kinase in oxidative stress‐stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab 287, E166–E173. [DOI] [PubMed] [Google Scholar]
- van Loon LJ, Greenhaff PL, Constantin‐Teodosiu D, Saris WH & Wagenmakers AJ (2001). The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol 536, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Flamez D, Mu J, Wojtaszewski JF, Schuit FC, Birnbaum M, Richter E, Burcelin R & Vaulont S (2003). Physiological role of AMP‐activated protein kinase (AMPK): insights from knockout mouse models. Biochem Soc Trans 31, 216–219. [DOI] [PubMed] [Google Scholar]
- Winder WW & Hardie DG (1996). Inactivation of acetyl‐CoA carboxylase and activation of AMP‐activated protein kinase in muscle during exercise. Am J Physiol 270, E299–E304. [DOI] [PubMed] [Google Scholar]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M & Holloszy JO (2000). Activation of AMP‐activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol 88, 2219–2226. [DOI] [PubMed] [Google Scholar]
- Witczak CA, Jessen N, Warro DM, Toyoda T, Fujii N, Anderson ME, Hirshman MF & Goodyear LJ (2010). CaMKII regulates contraction‐ but not insulin‐induced glucose uptake in mouse skeletal muscle. Am J Physiol Endocrinol Metab 298, E1150–E1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JF, Mourtzakis M, Hillig T, Saltin B & Pilegaard H (2002). Dissociation of AMPK activity and ACCbeta phosphorylation in human muscle during prolonged exercise. Biochem Biophys Res Commun 298, 309–316. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA & Kiens B (2000). Isoform‐specific and exercise intensity‐dependent activation of 5'‐AMP‐activated protein kinase in human skeletal muscle. J Physiol 528, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DC, Geiger PC, Holloszy JO & Han DH (2005). Contraction‐ and hypoxia‐stimulated glucose transport is mediated by a Ca2+‐dependent mechanism in slow‐twitch rat soleus muscle. Am J Physiol Endocrinol Metab 288, E1062–E1066. [DOI] [PubMed] [Google Scholar]
- Wright DC, Hucker KA, Holloszy JO & Han DH (2004). Ca2+ and AMPK both mediate stimulation of glucose transport by muscle contractions. Diabetes 53, 330–335. [DOI] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D & Gamblin SJ (2011). Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Summary Document
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.