

Abstract
Five di‐ and tetracyano‐substituted pyrene‐fused pyrazaacenes were synthesized and studied as potential electron acceptors in the solid state. Single crystals of all compounds were grown and the crystal packing studied by DFT calculations (transfer integrals and reorganization energies) to get insight into possible use for semiconducting charge transport.
Keywords: aggregation, crystal engineering, electron transfer, fused-ring systems, pyrazaacenes
Means to stream: Five di‐ and tetracyano‐substituted pyrene‐fused pyrazaacenes were synthesized and studied as potential electron acceptors in the solid state. Single crystals of all compounds were grown, and the crystal packing was studied by XRD and DFT calculations of transfer integrals and reorganization energies with a view to their possible use as n‐type semiconductors.

Introduction
Organic molecules with low lying LUMOs have many desirable properties, which make them an interesting class of materials for a wide range of applications, for example, as n‐type semiconductors in organic field‐effect transistors (OFETs),1 as electron acceptors in solar cells or for organic light‐emitting diodes.1a, 1e, 2 In the field of OFETs low LUMO levels favor electron injection and alignment with the work function of gold electrodes.3 Low‐lying LUMO levels further improve stability by reducing the decomposition of negatively charged species on exposure to oxygen or water and are therefore valuable for operating organic electronic devices under ambient conditions.1a, 4 However, only a few n‐type semiconductors with high electron mobility have been reported so far [e.g., aromatic diimides (μ e=1.30–8.60 cm2 V−1 s−1),5 tetraazapentacenes (μ e=11.0–13.3 cm2 V−1 s−1),6 cyano‐terminated quinoidal terthiophenes (μ e=3.00 cm2 V−1 s−1),7 and B‐N‐fused dibenzoazaacene (μ e=1.60 cm2 V−1 s−1).3a The design and synthesis of new electron‐transport materials therefore remains an attractive goal. The general strategy is to attach or include electron‐withdrawing groups or atoms (e.g., halogens, boron, cyano or acyl groups) to or in an aromatic backbone.1a, 7, 8 In this context, pyrene‐fused pyrazaacenes (PPAs) are especially promising due to their high stability and their already considerably low LUMO energies.3b, 9 By attaching cyano groups to PPAs, electron acceptors that fulfill certain criteria to be attractive for organic electronics should be readily available by condensation reactions. Surprisingly, to date only a few cyano‐substituted PPAs have been reported. To the best of our knowledge, the first such derivatives were mentioned in 1975 by Wöhrle et al.10 The synthesis as well as IR spectra and UV/Vis spectroscopic analysis of PQDC and H‐PPQTC were described. These compounds were studied for the formation of amino‐iminopyrazines as valuable precursors for phthalocyanines, which was realized by Nardi et al. for H‐PPQTC in 2014 on Cu surfaces.11 A tert‐butyl‐substituted derivative of PPQTC was synthesized by Liu and co‐workers in 2011.12 t Bu‐PPQTC and H‐PPQTC were thoroughly investigated by UV/Vis, photoluminescence, and cyclovoltammetry, which clearly identifies these compounds with LUMOs comparable to that of PCBM61 as potential electron acceptors for applications in organic electronics. In the same year Abdel‐Razik described the synthesis of the larger congener t Bu‐QPPTC as precursor for polymer networks based on phthalocyanine formation.13 Mateo‐Alonso obtained soluble derivatives of both core structures by attachment of triisopropylsilylethynyl substituents to the 2,7‐positions of the pyrene core.3b
Besides the molecular electronic structure, intermolecular orbital overlap plays a crucial role for charge transfer. Therefore, knowledge of the molecular arrangement of fused aromatic molecules in the solid state is of utmost importance to calculate transfer integrals for charge transfer to evaluate the potential of these structures for electronic applications in more detail.14
Despite the attractive low‐lying LUMOs of the few literature‐known cyano PPAs of Scheme 1, no X‐ray structures giving more detailed information about the above‐mentioned possible arrangement were described to date. The only exception is PQDC, for which a solid‐state structure was described recently.15 This contribution had a different focus, and therefore no discussion of transfer integrals was done. Because of the high potential of such cyano PPAs and the lack of detailed structural information in the solid state, we revisited this class of compounds to grow single crystals, study packing, and calculate transfer integrals.
Scheme 1.
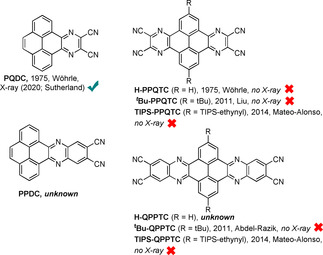
Overview of literature‐known cyano‐substituted PPAs.
Results and Discussion
Synthesis and characterization
Phenanthroquinoxalino dicarbonitrile (PQDC) and phenanthrophenazine dicarbonitrile (PPDC) was synthesized by acid‐catalyzed condensation of pyrenedione 1 16 with 2,3‐diaminomalonitrile 3 and 4,5‐diaminophthalonitrile 4, respectively (Scheme 2).3b Pyrazinophenanthroquinoxaline tetracarbonitrile derivatives H‐PPQTC and t Bu‐PPQTC, as well as quinoxalinophenanthrophenazine‐tetracarbonitrile derivative QPPTC were synthesized accordingly from pyrenetetraone 2 a.16, 17 After the reaction the crude products were isolated by filtration and washed with methanol. Subsequently, PQDC, H‐PPQTC, and t Bu‐PPQTC were heated to reflux in 30 % nitric acid and filtered while still hot.11
Scheme 2.
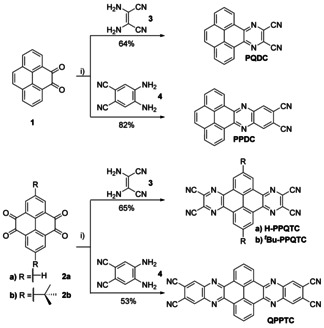
Synthesis of di‐ and tetracyanopyrazines PQDC, PPDC, H‐PPQTC, t Bu‐PPQTC, and QPPTC. Conditions: i) EtOH/AcOH (1:1), Ar, 80 °C, 11–16 h.
During this step a color change from dark brown to beige was observed. After filtration and washing with water and methanol the solids were extracted in a Soxhlet apparatus to obtain PQDC as a metallic golden powder and the two PPQTC derivatives as light‐yellow solids. The yields of the three compounds were 64–65 %. PPDC was purified by washing with THF and methanol and isolated as an orange powder in 82 % yield. Soxhlet extraction with THF gave QPPTC as yellow powder in 53 % yield. PPDC could be further purified by sublimation in a kugelrohrofen at 300 °C and (5–8)×10−3 mbar. PQDC and the two PPQTC derivatives could be sublimed under similar conditions, but showed additional signals in the 1H NMR spectra indicating decomposition (see Supporting Information).
All compounds were fully characterized by 1H and 13C NMR spectroscopy, IR spectroscopy, HRMS, and elemental analysis (for details, see Supporting Information).
Optoelectronic properties
All compounds were studied by UV/Vis and fluorescence spectroscopy in dichloromethane or ortho‐dichlorobenzene (oDCB) in the case of H‐PPQTC for solubility reasons (Figure 1). Absorption spectra of PQDC and H‐PPQTC have already been reported,10 but are included in the discussion as well. Pronounced differences for di‐ and tetrasubstituted compounds were observed, whereas similar spectra for di‐ and tetracyano compounds were obtained. PQDC shows absorption peaks at 447, 354, 313, 284, and 258 nm corresponding to n–π* and π–π* transitions.10, 18 Due to the elongation of the aromatic core, a bathochromic shift of the longest‐wavelength absorption peaks towards λ abs=485 nm is observed. Two peaks at 348 and 333 nm are much better resolved compared with PQDC. Both compounds have very similar emission spectra with a maximum at λ ex=533–534 nm and a blueshifted shoulder at 510 nm for PQDC, resulting in Stokes shifts of E Stokes=3609 cm−1 for PQDC and E Stokes=1891 cm−1 for PPDC, indicating a decrease of reorganization energy from the transition of the ground state to the first excited state.19 This might be explained by the larger aromatic system of PPDC, which is less influenced by structural change when excited. Photoluminescence quantum yields (PLQY) were 31 % for PQDC and 22 % for PPDC. Both H‐PPQTC and t Bu‐PPQTC have a weak absorption maximum at λ abs=426 nm and λ abs=437 nm and a broad absorption band at λ abs=330 nm and λ abs=345 nm, respectively. The π–π* transition bands (≈215 nm) are significantly reduced compared to those of the dicyano compounds PQDC and PPDC. H‐PPQTC emits at λ em=437 nm (PLQY of 37 %), resulting in a Stokes shift of E Stokes=1448 cm−1. For t Bu‐PPQTC the emission maximum is redshifted compared to H‐PPQTC by 24 nm to λ em=461 nm and shows an additional peak at 529 nm, which might be due to excimer formation. The corresponding Stokes shift (E Stokes=2231 cm−1) is larger than that of H‐PPQTC (1448 cm−1), and the PLQY (15 %) was lower than that of H‐PPQTC (37 %). Both values are in agreement with data reported in the literature.12 QPPTC shows distinct absorption maxima at λ abs=440, 412, 390, 335, and 309 nm. The emission maximum at λ em=449 nm with a small Stokes shift of E Stokes=455 cm−1 is indicative of the formation of a J‐aggregate.20 A second emission maximum is found at λ em=475 nm with a shoulder at approximately 526 nm. The PLQY was 8 %. The optical bandgaps estimated from the onset wavelength lie between 2.4 eV (PPDC) and 2.9 eV (H‐PPQTC).
Figure 1.
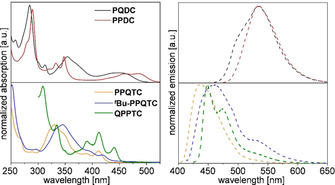
Absorption and emission spectra of PQDC, PPDC, H‐PPQTC, and t Bu‐PPQTC, measured in CH2Cl2 (1×10−6 mol L−1) at RT and of QPPTC measured in oDCB (1×10−6 mol L−1) at RT.
All compounds were studied by cyclovoltammetry in dichloromethane, except QPPTC, which was studied in oDCB (Figure 2). For PQDC only one quasireversible reduction potential at E red=−1.42 V was observed, whereas for the other three compounds two potentials were detected.
Figure 2.
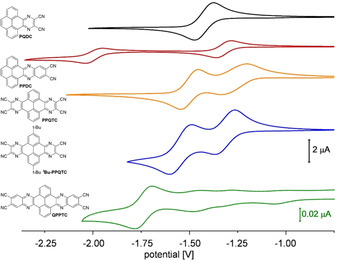
Cyclic voltammograms of PQDC, PPDC, H‐PPQTC, t Bu‐PPQTC (CH2Cl2, nBu4NPF6 (0.1 m), and QPPTC (oDCB, nBu4NPF6 (0.1 m), measured at room temperature with a Pt electrode, an Ag/Ag+ pseudo‐reference electrode, and Fc/Fc+ as internal reference (scanning speed: 50 mV s−1). Note, that the scale of the y axis of QPPTC is different from that of the other compounds because of the much lower signal intensity.
For PPDC, the first reduction potential was found at E red,1=−1.32 V and the second at E red,2=−1.99 V. Both H‐PPQTC (E red,1=−1.26 V; E red,2=−1.49 V) and t Bu‐PPQTC (E red,1=−1.31 V and E red,2=−1.54 V) showed similar reduction potentials, which is in agreement with prior reported values in the literature.12 For QPPTC the solubility is very low, even in oDCB, and therefore only a signal of low intensity could be found. QPPTC has one quasireversible reduction potential at E red=−1.74 V. From the first reduction potentials the electron affinities were estimated by the commonly used expression EA=−(E red,1+4.8) eV. They are 3.1 eV for QPPTC and 3.4–3.5 eV for the other four compounds.
DFT calculations (B3LYP/6‐311++G**) for all compounds were done to compare frontier molecular orbitals of these electron‐acceptor compounds (Figure 3). The HOMO of PQDC is found at E HOMO=−6.6 eV with orbital coefficients that are larger on the pyrene core than on the pyrazine units and the cyano substituents. This is similar for the HOMOs of all other compounds. The LUMO energy is calculated to be E LUMO=−3.3 eV, and here the orbital coefficients are mainly located at the pyrazine unit and the substituents. Again, this is similar for all other compounds of this series. PPDC has one more fused ring between the pyrene core and the pyrazine unit, and thus the gap between HOMO and LUMO is smaller with a slightly stabilized LUMO at E LUMO=−3.5 eV and a destabilized HOMO at E HOMO=−6.5 eV. In comparison with PQDC, for H‐PPQTC with two pyrazine units and four cyano substituents, both LUMO and HOMO are significantly stabilized (E LUMO=−3.9 eV and E HOMO=−7.4 eV). The frontier molecular orbital energies between PPDC and the larger QPPTC are comparable, with E LUMO=−3.9 eV and E HOMO=−7.3 eV. The energy levels of the two larger compounds H‐PPQTC and QPPTC are quite comparable. The introduction of two tert‐butyl groups at the pyrene 2,7‐positions (t Bu‐PPQTC) destabilizes the LUMO by about +0.2 eV to E LUMO=−3.7 eV. This is slightly lower than that of the corresponding triisopropylethynyl‐substituted tetracyano derivatives (E LUMO=−3.4 to −3.5 eV).3b The optoelectronic properties are summarized in Table 1.
Figure 3.
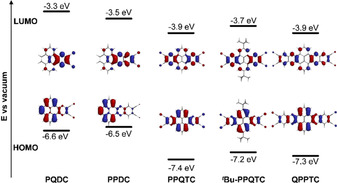
HOMO–LUMO diagrams of PQDC, PPDC, H‐PPQTC, t Bu‐PPQTC, and QPPTC (calculated using DFT‐B3LYP:6‐311++G**).
Table 1.
Optoelectronic and electrochemical properties of PQDC, PPDC, H‐PPQTC, t Bu‐PPQTC and QPPTC.
|
Compound |
λ abs [c] [nm] |
λ onset [nm] |
λ em [nm] (λ ex) [nm] |
E Stokes [cm−1] |
Φ [%] |
[eV] |
[V] |
EA [eV][f] |
[eV] |
[eV] |
[eV] |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
PQDC [a] |
447 |
492 |
510sh, 533 (445) |
3609 |
31 |
2.5 |
−1.42 |
−3.4 |
−6.6 |
−3.3 |
3.3 |
|||||
|
PPDC [a] |
485 |
514 |
534 (475) |
1891 |
22 |
2.4 |
−1.32, −1.99 |
−3.5 |
−6.5 |
−3.5 |
3.0 |
|||||
|
H‐PPQTC [a] |
411 |
426 |
437 (401) |
1448 |
37 |
2.9 |
−1.26, −1.49 |
−3.5 |
−7.4 |
−3.9 |
3.5 |
|||||
|
t Bu‐PPQTC [a] |
418 |
437 |
461, 529 (408) |
2231 |
15 |
2.8 |
−1.31, −1.54 |
−3.5 |
−7.2 |
−3.7 |
3.5 |
|||||
|
QPPTC [b] |
440,[h] 412 |
451,[h]427 |
449,[h] 475, 526sh[i] (430) |
455,[h] 3219 |
8.9 |
2.7,[h] 2.9 |
−1.74 |
−3.1 |
−7.3 |
−3.9 |
3.4 |
[a] Measured in CH2Cl2 at RT. [b] Measured in oDCB at RT. [c] absorption maximum at the longest wavelength. [d] Estimated from onset; E g(opt)= . [e] Cyclic voltammogram measured with a Pt electrode and nBu4NPF6 as electrolyte. Scan speed: 50 mV s−1; ferrocene/ferrocenium (Fc/Fc+) was used as internal reference. [f] Electron affinity estimated by EA=−(E red,1+4.8) eV. [g] Obtained from quantum‐chemical calculations at the DFT‐B3LYP/6‐311++G** level of theory. [h] J‐aggregate. [i] sh: shoulder.
Single‐crystal X‐ray structure analysis
Crystal packing plays a crucial role for charge‐transport in organic semiconductors. For all compounds except QPPTC, single crystals were obtained by vacuum sublimation with a kugelrohrofen. Crystals were also grown from solution for PQDC, H‐PPQTC, t Bu‐PPQTC, and QPPTC. In total, eight different modifications were obtained.
Crystals of PQDC were grown from solution, either by vapor diffusion of methanol into a THF solution or by slow evaporation of a CH2Cl2 solution, as well as by vacuum sublimation in a kugelrohrofen (200 °C, 5×10−3 mbar). In each case the same unit cell was obtained independent of the crystallization method, indicating that this packing motif is highly robust.
PQDC crystallized in the monoclinic space group P21/c with four molecules per unit cell and one molecule in the asymmetric unit (polymorph α). Recently, the same packing and unit cell have been reported by Sutherland et al.15 The cyano groups are slightly contorted out of planarity by approximately 2.5°. Adjacent molecules form π‐stacked dimers, in which these are rotated by 120° (Figure 4 a). The molecules assemble in parallel one‐dimensional π‐stacked columns with d A=3.38 Å along the crystallographic c axis (Figure 4 b). Adjacent columns interact either by twofold hydrogen bonding between the cyano groups and aromatic protons (d B=2.54–2.58 Å), or to protons of the K‐region (d C=2.60 Å, Figure 4 c). Similar twofold hydrogen‐bonding motifs are known for other aromatic 1,2‐dicyano compounds as well.21
Figure 4.
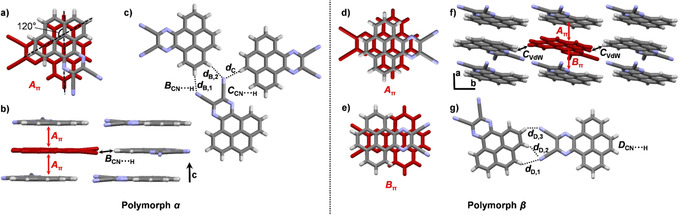
a–c) Single‐crystal X‐ray structure of PQDC (polymorph α) crystallized by sublimation in a kugelrohrofen (200 °C, 5×10−3 mbar), vapor diffusion of MeOH into a THF solution, or by evaporation of a CH2Cl2 solution. a) π‐Stacked dimer Aπ with d A=3.38 Å. b) Infinite face‐to‐face stacking pattern along the crystallographic c axis, indicated with red arrows. c) Hydrogen‐bonding motif in dimers B and C (d B,1=2.58 Å, d B,2=2.54 Å, d C=2.60 Å. d)–g) Polymorph β crystallized by slow evaporation of a CHCl3 solution. d), e) π‐Stacked dimers Aπ (d A=3.51 Å) and Bπ (d B=3.29 Å). f) Infinite face‐to‐face stacking pattern along the crystallographic a axis, indicated with red arrows. g) Hydrogen‐bonding motif in dimer D (d D,1=2.81 Å, d D,2=2.68 Å, d D,1=2.79 Å).
However, when PQDC was crystallized by slow evaporation of a chloroform solution two different crystals were found in the same batch. One was identical with the previously described α modification. The other one was a different polymorph in monoclinic space group P21/n, with four molecules in the unit cell and one molecule in the asymmetric unit (polymorph β). Here, the molecules form two different face‐to‐face π‐stacked dimers with antiparallel orientation of the two PQDC molecules and pronounced molecular overlap (Figure 4 d and e). Whereas dimer Aπ has the longest π–π distance (d A=3.51 Å) of all structures in this work, dimer Bπ has the shortest (d B=3.29 Å). PQDC forms one‐dimensional stacks of alternating dimers Aπ and Bπ along the crystallographic a axis (Figure 4 f). These stacks are arranged in layers within the ab plane and interact through van der Waals interactions (d ArH…HAr=2.43 Å). Adjacent layers are stabilized by triple hydrogen bonding of the cyano groups with aromatic protons (d CN…H=2.68–2.81 Å, Figure 4 g). By using the UNI force field method22 implemented in Mercury, intermolecular potentials between π‐stacked dimers for both polymorphs were calculated. In the α polymorph a potential of E π=−96 kJ mol−1 for dimer A was found; in the β polymorph the potentials are E π=−92 kJ mol−1 for dimer Aπ and E π=−112 kJ mol−1 for dimer Bπ. The total packing energies are similar for both modifications, that is, E tot=−182 kJ mol−1 for α and E tot=−187 kJ mol−1 for β, which explains the simultaneous competitive formation of both polymorphs from CHCl3. The robustness of the α polymorph under the other conditions seems to have kinetic rather than thermodynamic reasons.
Crystals of PPDC were grown by vacuum sublimation in a kugelrohrofen (250 °C, (3–6)×10−3 mbar). Like PQDC, PPDC also crystallized in monoclinic space group P21/c with four molecules in the unit cell and one molecule in the asymmetric unit. The molecules assemble in π‐stacked columns of two alternating dimers with d A=3.34 Å and d B=3.37 Å along the crystallographic b axis (Figure 5 a–c). In both dimers the molecules are oriented in an antiparallel fashion and have a pronounced molecular overlap of the aromatic backbone (Figure 5 a and b). Adjacent columns form a layered structure, which is stabilized by hydrogen bonding between cyano groups and protons of the K‐region. (d C=2.47 Å, Figure 5 c). Adjacent layers also form twofold hydrogen bonds (d D=2.50 Å, Figure 5 d). The parent structure of PPDC without cyano groups has been recently described:15 π‐stacked columns with a parallel orientation of the molecules. Thus, the antiparallel orientation observed in PPDC is related to the presence of the cyano groups and can be explained by the dipole moment induced by them favoring an alignment with close contacts of opposite poles.
Figure 5.
Single‐crystal X‐ray structure of PPDC (polymorph α) crystallized by sublimation in a kugelrohrofen (250 °C, (3–6)×10−3 mbar). a), b) π‐Stacked dimers Aπ (d A=3.34 Å) and Bπ (d B=3.37 Å). c) Packing; asymmetric unit colored in red. Infinite face‐to‐face stacking pattern along the crystallographic b axis, indicated with red arrows. d) Twofold hydrogen‐bonding motif in dimer D (d D,1=d D,2=2.50 Å).
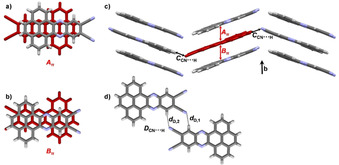
For H‐PPQTC two modifications could be obtained. The first one was grown by slow evaporation of a CH2Cl2 solution (solvate α). Here H‐PPQTC crystallized in triclinic space group P with four molecules in the unit cell and two molecules in the asymmetric unit. Two solvent molecules are enclathrated as well. Two different π‐stacked dimers can be found (Figure 6 a and b). In dimer Aπ adjacent molecules are twisted by approximately 90°, and in dimer Bπ the twist angle is 63°. The π–π distance is the same for both dimers (d A=d B=3.38 Å). Both dimers form independent π‐stacked columns along the crystallographic a axis (Figure 6 c), which interact through dipolar CN⋅⋅⋅CN interactions (d D1,2=3.49–3.55 Å) and hydrogen bonding (d D,3=2.75 Å) between adjacent cyano groups and aromatic protons (Figure 6 d).
Figure 6.
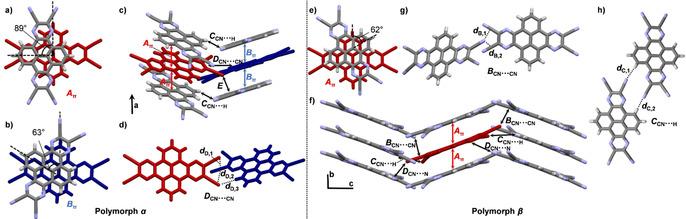
a–d) Single‐crystal X‐ray structure of H‐PPQTC (solvate α) crystallized by evaporation of a CH2Cl2 solution. a), b) π‐Stacked dimers Aπ (d A=3.38 Å) and Bπ (d B=3.38 Å). c) Packing; the colors correspond to molecules of the two different dimers shown in a) and b). Solvent molecules omitted for clarity. Infinite face‐to‐face stacking pattern along the crystallographic a axis, indicated with red and blue arrows. d) CN⋅⋅⋅CN short contacts and hydrogen‐bonding motif in dimer D (d D,1=3.49 Å, d D,2=3.55 Å, d D,3=2.75 Å). e–h) Polymorph β crystallized by sublimation in a kugelrohrofen (300 °C, 5×10−2 mbar). e) π‐Stacked dimer Aπ (d A=3.36 Å). f) Packing; asymmetric unit colored in red. Infinite face‐to‐face stacking pattern along the crystallographic b axis, indicated with red arrows. g) CN⋅⋅⋅CN dipolar interaction motif B (d B,1=3.58 Å, d B,2=3.68 Å). h) Twofold hydrogen‐bonding motif in dimer C (d C,1=2.65 Å, d C,2=2.69 Å).
Another polymorph of H‐PPQTC was obtained by vacuum sublimation in a kugelrohrofen (300 °C, 5×10−2 mbar) (polymorph β). This time it crystallized in the orthorhombic space group Pbca with eight molecules in the unit cell and one molecule in the asymmetric unit. Again, dimers with d A=3.36 Å and a twist of 62° (Figure 6 e) very similar to dimer Bπ found in the α modification are formed, which arrange in one‐dimensional π‐stacked columns along the crystallographic b axis (Figure 6 f). The columns are aligned in layers in the crystallographic bc plane and are stabilized by dipolar C⋅⋅⋅N interactions (d B=3.35–3.69 Å) between adjacent cyano groups (Figure 6 g) and by twofold hydrogen bonding (d C=2.65–2.69 Å, Figure 6 h). Short contacts between cyano groups and phenazine nitrogen atoms (d D=3.48 Å) are also found. Similar intermolecular potentials calculated by the UNI force field method exist for the π‐stacked dimers (−116 to −120 kJ mol−1) in both modifications, whereas the total packing energy is much higher for the solvate of the α polymorph with CH2Cl2 (E tot=−976 kJ mol−1) than for the structure of the sublimed β polymorph (E tot=−238 kJ mol−1), probably due to additional stabilization by interaction with the solvent molecules.
On slow evaporation of a solution in CH2Cl2, t Bu‐PPQTC crystallized in the monoclinic space group P21/n with four molecules in the unit cell and one molecule and one solvent molecule in the asymmetric unit (solvate α). The molecules form π‐stacked dimers (Figure 7 a) with d A=3.36 Å, which are arranged in one‐dimensional columns along the crystallographic a axis. Adjacent columns form sheets in the crystallographic ab plane (Figure 7 b), which are stabilized by dipolar CN⋅⋅⋅CN contacts (d B=3.26–3.66 Å) similar to those found in the previous structure (Figure 7 c). Adjacent sheets interact through van der Waals interactions of the tert‐butyl groups.
Figure 7.
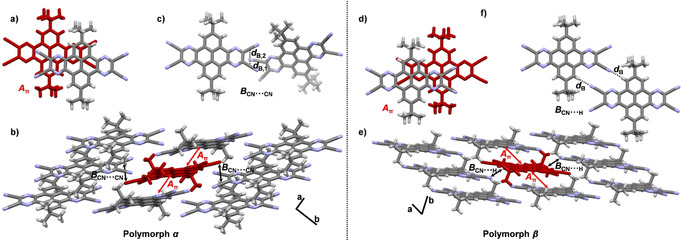
a–c) Single‐crystal X‐ray structure of t Bu‐PPQTC (solvate α) crystallized by evaporation of a CH2Cl2 solution. a) π‐Stacked dimer Aπ (d A=3.36 Å). b) Infinite face‐to‐face stacking pattern along the crystallographic a axis, indicated with red arrows. Solvent molecules omitted for clarity. c) CN⋅⋅⋅CN short‐contact motif in dimer B (d B,1=3.26 Å, d B,2=3.38 Å). d–f) Polymorph β crystallized by sublimation in a kugelrohrofen (250 °C, 5×10−3 mbar). d) π‐Stacked dimer Aπ (d A=3.37 Å). e) Infinite face‐to‐face stacking pattern along the crystallographic a axis, indicated with red arrows. f) Twofold hydrogen‐bonding motif in dimer B (d B=2.73 Å).
When sublimed in vacuo in a kugelrohrofen (250 °C, 5×10−3 mbar) t Bu‐PPQTC crystallized in triclinic space group P with one molecule per unit cell and half a molecule in the asymmetric unit (polymorph β). Similar to the α modification, π‐stacked dimers (Figure 7 d) with d A=3.37 Å form one‐dimensional columns along the crystallographic a axis, which are arranged in sheets in the ab plane (Figure 7 e). Within the sheets molecules interact through twofold hydrogen bonding (d B=2.73 Å) (Figure 7 f). Adjacent sheets are stabilized by van der Waals interactions. Similar to the previous two structures the intermolecular potentials between the π‐stacked dimers A are similar in both modifications (−140 to −143 kJ mol−1), whereas the total packing energy is higher for the solvate α (E tot=−314 kJ mol−1) than for the sublimed polymorph β (E tot=−277 kJ mol−1). A comparison between the crystal structures of H‐PPQTC and t Bu‐PPQTC reveals that the tert‐butyl groups influence the π stacking. Whereas H‐PPQTC π stacks with a twist of the π planes of either 60 or 90°, parallel π stacking with smaller overlap of the planes is found in the presence of tert‐butyl groups.
QPPTC was crystallized by slowly cooling a hot, saturated solution in benzonitrile. It crystallized in monoclinic space group P21 with two molecules of QPPTC and two solvent molecules in the asymmetric unit. One‐dimensional π‐stacked columns along the crystallographic c axis with d A=3.40 Å are formed (Figure 8 a), with pronounced molecular overlap (Figure 8 b). Adjacent columns are stabilized by a fourfold complementary combination of hydrogen bonding of the cyano nitrogen atom to the phenylene proton (d B=2.58–2.67 Å) and dipolar C⋅⋅⋅N interactions (d C=3.39–3.46 Å) (Figure 8 c). Enclathrated benzonitrile molecules are strongly contorted out of planarity (Figure 8 d), and this indicates that this fourfold interaction motif has a highly directing and stabilizing effect on the overall packing. Table 2 summarizes all crystallographic details.
Figure 8.
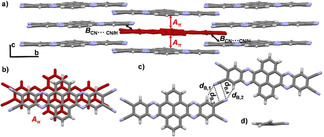
Single‐crystal X‐ray structure of QPPTC crystallized by cooling of a hot solution in benzonitrile. a) Infinite face‐to‐face stacking pattern along the crystallographic c axis, indicated with red arrows. Solvent molecules omitted for clarity. b) π‐Stacked dimer Aπ (d A=3.40 Å). c) Hydrogen‐bonding and dipolar C⋅⋅⋅N interactions in dimer B (d B,1=2.58 Å, d B,2=2.67 Å, d B,3=3.49 Å, d B,4=3.46 Å. d) Contortion of enclathrated benzonitrile.
Table 2.
Crystallographic parameters, distances, and short contacts of PQDC, PPDC, H‐PPQTC, t Bu‐PPQTC, and QPPTC.[a]
|
Compound |
Method |
Space group |
N asym [f] |
Z [g] |
d π–π [Å][h] |
Θ π–π [°][i] |
E π–π [kJ mol−1][j] |
E tot [kJ mol−1][j] |
Other short contacts [Å; °][a] |
|
|---|---|---|---|---|---|---|---|---|---|---|
|
PQDC |
THF/MeOH,[b] CH2Cl2, CHCl3,[c] sublimation,[d] α |
P21/c (monoclinic) |
1 |
4 |
3.38 |
120 |
−96 |
−182 |
2.54–2.60; 115–135 (CN⋅⋅⋅H), 3.30; 146 (CN⋅⋅⋅N) |
|
|
CHCl3,[c] β |
P21/n (monoclinic) |
1 |
4 |
3.51 (A), 3.29 (B) |
180, 180 |
−92 (A), −112 (B) |
−187 |
2.68–2.81; 107–160 (CN⋅⋅⋅H), 3.25; 105 (CN⋅⋅⋅N) |
||
|
PPDC |
sublimation[d] |
P21/c (monoclinic) |
1 |
4 |
3.34 (A), 3.37 (B) |
180, 180 |
−116 (A), −118 (B) |
−203 |
2.47–2.67; 110–136 (CN⋅⋅⋅H) |
|
|
CH2Cl2,[c] α |
P
(triclinic) |
4×0.5, 2×CH2Cl2 |
4 |
3.38 (A), 3.38 (B) |
89, 63 |
−116 (A), −120 (B) |
−976 |
3.42–3.82; 73–89 (CN⋅⋅⋅CN), 2.59–2.75; 128–132 (CN⋅⋅⋅H) |
||
|
H‐PPQTC |
sublimation,[d] β |
Pbca (orthorhombic) |
1 |
8 |
3.36 |
62 |
−118 |
−238 |
3.35–4.02; 81–113 (CN⋅⋅⋅CN), 2.49–2.69; 129–144 (CN⋅⋅⋅H), 3.12; 146 (CN⋅⋅⋅N) |
|
|
CH2Cl2,[c] α |
P21/n (monoclinic) |
2×0.5, 1×CH2Cl2 |
4 |
3.37 |
0 |
−140 |
−314 |
3.26–3.66; 72–85 (CN⋅⋅⋅CN), 2.67; 152 (CN⋅⋅⋅H) |
||
|
t Bu‐PPQTC |
sublimation,[d] β |
P
(triclinic) |
0.5 |
1 |
3.37 |
0 |
−143 |
−277 |
2.73; 135.9 (CN⋅⋅⋅H) |
|
|
QPPTC |
benzonitrile[e] |
P21 (monoclinic) |
2, 2×C6H5CN |
4 |
3.40 |
0 |
−178 |
−368 |
3.39–3.49, 88–94; 2.47–2.81; 127–142 (CN⋅⋅⋅H) |
[a] Values were determined for two adjacent molecules. For the determination procedure, see the Supporting Information. [b] Vapor diffusion. [c] Slow evaporation at RT. [d] Under vacuum in a kugelrohrofen. [e] Cooling a hot, saturated solution to RT. [f] Number of molecules in the asymmetric unit; refined molecules of solvation not counted. [g] Total number of molecules in the unit cell including refined molecules of solvation. [h] The π–π distances; the numbers in parentheses indicate the π‐stacking motifs shown in Figures 7–8; π distances were calculated as the mean distance of all atoms of the aromatic backbone (see the Supporting Information for details). [i] Torsion angle between two π‐stacked molecules as shown in Figures 7–8. [j] Intermolecular potentials between π‐stacked dimers and total packing energy calculated by the UNI force field method implemented in Mercury.22
The parent structure of QPPTC without cyano groups obtained by sublimation has already been described.23 Here, columnar π stacks with similar molecular overlap and π–π distance (3.42 Å) are found that only differ in the relative orientation of the columns.
Calculated charge‐transfer integrals
To evaluate the potential of the obtained structures for application as OFET materials, the electronic couplings between adjacent molecules of all obtained crystal structures were calculated. The electronic coupling (charge‐transfer integral) between molecules can be used to quantify the charge‐transport capability and depends on the relative orientation between the molecules. Different transfer integrals result in varying mobilities. For example, the charge‐transport mobility in an anthracene crystal differs in different directions with respect to the orientation of the molecules.24 Both HOMO transfer integrals t h responsible for hole transport and LUMO transfer integrals t e responsible for electron transport were calculated by the DFTB method (see the Supporting Information for details of the calculation).25 The results for π‐stacked dimers are listed in Table 3. Couplings of other molecular pairs with short contacts are negligibly small and are therefore not discussed herein (for details, see the Supporting Information). This also means that charge‐carrier transport is expected to be highly anisotropic and occurs predominantly along the π‐stacking axis, which is parallel to the shortest crystallographic axis in all structures, as indicated in Table 3. For most dimers in this work electron transport is favored over hole transport, as expected due to the electron‐deficient nature of the compounds and the low‐lying LUMOs.
Table 3.
Charge transfer integrals for hole (t h) and electron (t e) transport for all crystal structures of PQDC, PPDC, H‐PPQTC, t Bu‐PPQTC, and QPPTC and reorganization energies.
|
Compound |
Modification[a] |
t h [b] [meV] |
t e [b] [meV] |
Direction[c] |
λ h [d] [meV] |
λ e [d] [meV] |
|---|---|---|---|---|---|---|
|
PQDC |
α |
20 |
49 |
c |
160 |
256 |
|
β |
88 (A), 57 (B) |
113 (A), 252 (B) |
a |
|||
|
PPDC |
α |
13 (A) (B), 0 |
172 (A), 101 (B) |
b |
152 |
190 |
|
H‐PPQTC |
α |
37 (A), 26 (B) |
27 (A) (B), 46 |
a |
136 |
121 |
|
β |
12 |
36 |
b |
|||
|
t Bu‐PPQTC |
α |
53 |
76 |
a |
141 |
129 |
|
β |
50 |
69 |
a |
|||
|
QPPTC |
α |
4 |
108 |
c |
117 |
91 |
For the α polymorph of PQDC a moderate coupling for electron transport (t e=49 meV) and a lower coupling for hole transport (t h=20 meV) were calculated for π‐stacked molecules along the crystallographic c axis. For the β polymorph both electron and hole couplings are larger. Hole‐transfer integrals for the two alternating π‐stacked dimers along the a axis are t h,A=88 meV and t h,B=57 meV. For electron transport the transfer integrals are exceptionally large (t e,A=113 meV and t e,B=252 meV), which makes this polymorph highly promising for potential application in n‐type OFETs. However, due to similar packing energies of both α and β polymorphs, polymorphic mixtures are to be expected, which is a drawback for device fabrication. For PPDC, too, an exceptionally large transfer integral for electron transport for dimer Aπ (see Figure 5 a) was found (t e=172 meV). For dimer Bπ it is reduced to t e=101 meV, which is still a large value. Couplings in the same range have been previously reported for dimers of triptycene end‐capped quinoxalino‐phenanthrophenazines.9aa Both integrals are in the same range as for some perylene diimine derivatives (t e up to 141 meV).26 They are also similar to the transfer integral for hole transport of rubrene (t h=100 meV),27 which is known to have excellent charge‐carrier mobility in OFETs.28 Since both dimers alternate along the crystallographic b axis, the coupling in dimer Bπ most likely determines the overall charge transport. The large values make PPDC a promising candidate for potential application as an n‐type semiconductor in OFET devices.1 In contrast to the large transfer integrals for electron transport, couplings for hole transport are negligibly small (0–13 meV). For the α modification of H‐PPQTC moderate couplings for both hole and electron transport were calculated along the a axis. They vary between t h=37 meV and t e=27 meV for dimer Aπ and t h=26 meV and t e=46 meV for dimer Bπ (see Figure 6 a and b). Modification β has a similar integral for electron transport (t e=36 meV) and somewhat smaller integral for hole transport (t h=12 meV).
Both modifications of t Bu‐PPQTC show similar transfer integrals along the a axis. For hole transport they are t h=50 and 53 meV, whereas for electron transport fairly large couplings of t e=69 and 76 meV were obtained. These values are larger than those of H‐PPQTC due to more efficient orbital interaction, and make t Bu‐PPQTC promising for potential device applications, whereas H‐PPQTC seems to be unsuitable for efficient charge transfer. Thus, the addition of tert‐butyl groups seems to be beneficial for this purpose. The longest fused aromatic system, namely QPPTC, shows a large electron‐transfer integral along the crystallographic c axis (108 meV), whereas the hole coupling is negligible. This is contrary to the parent structure of QPPTC without cyano groups, for which hole‐transfer integrals (63 meV) are much larger than those for electron transfer (14 meV).23
Reorganization energies for hole and electron transport were calculated as well (DFT/B3LYP‐6‐31G*). They describe the strength of local electron–phonon coupling and strongly affect the charge‐transport properties.29 Small energies are desired for high carrier mobilities.14a, 30 The calculations revealed reorganization energies for hole transport between 117 and 160 meV, comparable to those of rubrene (159 meV)31 and unsubstituted QPP (118 meV)23 and slightly larger than that of pentacene (97 meV).32
For electron transport larger values were calculated for PQDC and PPDC (256 and 190 meV), whereas for the two PPQTC derivatives the energies were slightly smaller than for hole transport (121 and 129 meV). These values are rather low compared with electron‐transfer reorganization energies of other systems, such as perylene diimides (250–322 mV)26 QPPTC has the lowest reorganization energy (91 meV), which is only slightly higher than for its parent structure (80 meV).23 Since for QPPTC reorganization energy is lower than the transfer integral, this should lead to very efficient bandlike charge transport, which is promising for high charge transport mobilities.29b Table 3 summarizes transfer integrals and reorganization energies.
Conclusions
The synthesis and optoelectronic characterization of five di‐ and tetracyanopyrazines was presented. Due to the electron‐withdrawing effect of the cyano groups, all compounds have low‐lying LUMO levels of up to −3.9 eV, making them predestined for electron transport. All compounds could be crystallized by vacuum sublimation and/or from solution. Single‐crystal X‐ray diffraction revealed in all eight structures columnar face‐to‐face π‐stacking motifs with short π–π distances (≈3.29–3.51 Å; av 3.37 Å). Hydrogen‐bonding and dipolar CN⋅⋅⋅CN interactions are found in all structures and seem to have a strong impact on stabilizing the packing. PQDC crystallized in the same unit cell regardless of the crystallization conditions. Only in the case of chloroform a second polymorph was also formed. For two compounds (i.e., PPQTC and t Bu‐PPQTC) two different modifications were obtained, but the π‐stacking motifs were similar in both cases. Calculated charge‐transfer integrals for electron transport of 69–76 meV for t Bu‐PPQTC, 101–172 meV for PPDC, 108 meV for QPPTC and 113–252 meV for PQDC (β), together with low reorganization energies (91–256 meV) indicate that those four compounds are promising candidates for n‐type semiconductors. Their low molecular weight allows easy sublimation, which is beneficial both for purification and device fabrication. We are currently working on using these compounds in n‐type OFET devices.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to Deutsche Forschungsgemeinschaft supporting this project within the collaborative research center SFB1249 “N‐heteropolycyclic compounds as functional materials” (TP‐A04 and TP‐B02). Open access funding enabled and organized by Projekt DEAL.
L. Ueberricke, I. Ciubotaru, F. Ghalami, F. Mildner, F. Rominger, M. Elstner, M. Mastalerz, Chem. Eur. J. 2020, 26, 11634.
References
- 1.
- 1a. Zhan X., Facchetti A., Barlow S., Marks T. J., Ratner M. A., Wasielewski M. R., Marder S. R., Adv. Mater. 2011, 23, 268–284; [DOI] [PubMed] [Google Scholar]
- 1b. Newman C. R., Frisbie C. D., da Silva Filho D. A., Brédas J.-L., Ewbank P. C., Mann K. R., Chem. Mater. 2004, 16, 4436–4451; [Google Scholar]
- 1c. Zaumseil J., Sirringhaus H., Chem. Rev. 2007, 107, 1296–1323; [DOI] [PubMed] [Google Scholar]
- 1d. Wen Y., Liu Y., Adv. Mater. 2010, 22, 1331–1345; [DOI] [PubMed] [Google Scholar]
- 1e. Zhao X., Zhan X., Chem. Soc. Rev. 2011, 40, 3728–3743. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Xu R.-P., Li Y.-Q., Tang J.-X., J. Mater. Chem. C 2016, 4, 9116–9142; [Google Scholar]
- 2b. Salehi A., Fu X., Shin D.-H., So F., Adv. Funct. Mater. 2019, 29, 1808803. [Google Scholar]
- 3.
- 3a. Min Y., Dou C., Liu D., Dong H., Liu J., J. Am. Chem. Soc. 2019, 141, 17015–17021; [DOI] [PubMed] [Google Scholar]
- 3b. More S., Bhosale R., Mateo-Alonso A., Chem. Eur. J. 2014, 20, 10626–10631. [DOI] [PubMed] [Google Scholar]
- 4. de Leeuw D. M., Simenon M. M. J., Brown A. R., Einerhand R. E. F., Synth. Met. 1997, 87, 53–59. [Google Scholar]
- 5.
- 5a. He T., Stolte M., Würthner F., Adv. Mater. 2013, 25, 6951–6955; [DOI] [PubMed] [Google Scholar]
- 5b. Soeda J., Uemura T., Mizuno Y., Nakao A., Nakazawa Y., Facchetti A., Takeya J., Adv. Mater. 2011, 23, 3681–3685. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Xue G., Wu J., Fan C., Liu S., Huang Z., Liu Y., Shan B., Xin H. L., Miao Q., Chen H., Li H., Mater. Horiz. 2016, 3, 119–123; [Google Scholar]
- 6b. Xu X., Yao Y., Shan B., Gu X., Liu D., Liu J., Xu J., Zhao N., Hu W., Miao Q., Adv. Mater. 2016, 28, 5276–5283. [DOI] [PubMed] [Google Scholar]
- 7. Zhang C., Zang Y., Gann E., McNeill C. R., Zhu X., Di C.-a., Zhu D., J. Am. Chem. Soc. 2014, 136, 16176–16184. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Tang M. L., Oh J. H., Reichardt A. D., Bao Z., J. Am. Chem. Soc. 2009, 131, 3733–3740; [DOI] [PubMed] [Google Scholar]
- 8b. Molinari A. S., Alves H., Chen Z., Facchetti A., Morpurgo A. F., J. Am. Chem. Soc. 2009, 131, 2462–2463; [DOI] [PubMed] [Google Scholar]
- 8c. Delgado M. C. R., Pigg K. R., da Silva Filho D. A., Gruhn N. E., Sakamoto Y., Suzuki T., Osuna R. M., Casado J., Hernández V., Navarrete J. T. L., Martinelli N. G., Cornil J., Sánchez-Carrera R. S., Coropceanu V., Brédas J.-L., J. Am. Chem. Soc. 2009, 131, 1502–1512; [DOI] [PubMed] [Google Scholar]
- 8d. Yin J., Chaitanya K., Ju X.-H., J. Mater. Res. 2016, 31, 337–347. [Google Scholar]
- 9.
- 9a. Mateo-Alonso A., Chem. Soc. Rev. 2014, 43, 6311–6324; [DOI] [PubMed] [Google Scholar]
- 9b. Hu J., Zhang D., Jin S., Cheng S. Z. D., Harris F. W., Chem. Mater. 2004, 16, 4912–4915; [Google Scholar]
- 9c. Guo J., Xu Y., Jin S., Chen L., Kaji T., Honsho Y., Addicoat M. A., Kim J., Saeki A., Ihee H., Seki S., Irle S., Hiramoto M., Gao J., Jiang D., Nature Commun. 2013, 4, 2736; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Mateo-Alonso A., Kulisic N., Valenti G., Marcaccio M., Paolucci F., Prato M., Chem. Asian J. 2010, 5, 482–485; [DOI] [PubMed] [Google Scholar]
- 9e. Kulisic N., More S., Mateo-Alonso A., Chem. Commun. 2011, 47, 514–516; [DOI] [PubMed] [Google Scholar]
- 9f. More S., Bhosale R., Choudhary S., Mateo-Alonso A., Org. Lett. 2012, 14, 4170–4173; [DOI] [PubMed] [Google Scholar]
- 9g. García R., Melle-Franco M., Mateo-Alonso A., Chem. Commun. 2015, 51, 8037–8040; [DOI] [PubMed] [Google Scholar]
- 9h. Marco A. B., Cortizo-Lacalle D., Gozalvez C., Olano M., Atxabal A., Sun X., Melle-Franco M., Hueso L. E., Mateo-Alonso A., Chem. Commun. 2015, 51, 10754–10757; [DOI] [PubMed] [Google Scholar]
- 9i. Antonicelli G., Gozalvez C., Atxabal A., Melle-Franco M., Hueso L. E., Mateo-Alonso A., Org. Lett. 2016, 18, 4694–4697; [DOI] [PubMed] [Google Scholar]
- 9j. Cortizo-Lacalle D., Pertegás A., Melle-Franco M., Bolink H. J., Mateo-Alonso A., Org. Chem. Front. 2017, 4, 876–881; [Google Scholar]
- 9k. Mateo-Alonso A., Eur. J. Org. Chem. 2017, 7006–7011; [Google Scholar]
- 9l. Cortizo-Lacalle D., Gozalvez C., Melle-Franco M., Mateo-Alonso A., Nanoscale 2018, 10, 11297–11301; [DOI] [PubMed] [Google Scholar]
- 9m. Cortizo-Lacalle D., Mora-Fuentes J. P., Strutyński K., Saeki A., Melle-Franco M., Mateo-Alonso A., Angew. Chem. Int. Ed. 2018, 57, 703–708; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 711–716; [Google Scholar]
- 9n. Gozalvez C., Zafra J. L., Saeki A., Melle-Franco M., Casado J., Mateo-Alonso A., Chem. Sci. 2019, 10, 2743–2749; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9o. Mora-Fuentes J. P., Riaño A., Cortizo-Lacalle D., Saeki A., Melle-Franco M., Mateo-Alonso A., Angew. Chem. Int. Ed. 2019, 58, 552–556; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 562–566; [Google Scholar]
- 9p. Mora-Fuentes J. P., Papadopoulos I., Thiel D., Álvarez-Boto R., Cortizo-Lacalle D., Clark T., Melle-Franco M., Guldi D. M., Mateo-Alonso A., Angew. Chem. Int. Ed. 2020, 59, 1113–1117; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1129–1133; [Google Scholar]
- 9q. Gao B., Wang M., Cheng Y., Wang L., Jing X., Wang F., J. Am. Chem. Soc. 2008, 130, 8297–8306; [DOI] [PubMed] [Google Scholar]
- 9r. Sahoo P. K., Giri C., Haldar T. S., Puttreddy R., Rissanen K., Mal P., Eur. J. Org. Chem. 2016, 1283–1291; [Google Scholar]
- 9s. Li J., Chen S., Wang Z., Zhang Q., Chem. Rec. 2016, 16, 1518–1530; [DOI] [PubMed] [Google Scholar]
- 9t. Wang Z., Gu P., Liu G., Yao H., Wu Y., Li Y., Rakesh G., Zhu J., Fu H., Zhang Q., Chem. Commun. 2017, 53, 7772–7775; [DOI] [PubMed] [Google Scholar]
- 9u. Gu P.-Y., Wang Z., Liu G., Yao H., Wang Z., Li Y., Zhu J., Li S., Zhang Q., Chem. Mater. 2017, 29, 4172–4175; [Google Scholar]
- 9v. Zhang Z., Zhang Q., Mater. Chem. Front. 2020, 10.1039/C9QM00656G; [DOI] [Google Scholar]
- 9w. Hu B.-L., Zhang K., An C., Schollmeyer D., Pisula W., Baumgarten M., Angew. Chem. Int. Ed. 2018, 57, 12375–12379; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12555–12559; [Google Scholar]
- 9x. Hu B.-L., An C., Wagner M., Ivanova G., Ivanova A., Baumgarten M., J. Am. Chem. Soc. 2019, 141, 5130–5134; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9y. Kohl B., Rominger F., Mastalerz M., Org. Lett. 2014, 16, 704–707; [DOI] [PubMed] [Google Scholar]
- 9z. Kohl B., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2015, 54, 6051–6056; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6149–6154; [Google Scholar]
- 9aa. Ueberricke L., Holub D., Kranz J., Rominger F., Elstner M., Mastalerz M., Chem. Eur. J. 2019, 25, 11121–11134; [DOI] [PubMed] [Google Scholar]
- 9ab. Ueberricke L., Wieland S., Rominger F., Mastalerz M., Org. Mater. 2019, 01, 050–062; [Google Scholar]
- 9ac. Kohl B., Rominger F., Mastalerz M., Chem. Eur. J. 2015, 21, 17308–17313; [DOI] [PubMed] [Google Scholar]
- 9ad. Kohl B., Bohnwagner M. V., Rominger F., Wadepohl H., Dreuw A., Mastalerz M., Chem. Eur. J. 2016, 22, 646–655; [DOI] [PubMed] [Google Scholar]
- 9ae. Kohl B., Baumgärtner K., Rominger F., Mastalerz M., Eur. J. Org. Chem. 2019, 4891–4896. [Google Scholar]
- 10. Rothkopf H. W., Wöhrle D., Müller R., Koßmehl G., Chem. Ber. 1975, 108, 875–886. [Google Scholar]
- 11. Nardi E., Chen L., Clair S., Koudia M., Giovanelli L., Feng X., Müllen K., Abel M., J. Phys. Chem. C 2014, 118, 27549–27553. [Google Scholar]
- 12. Li Q., Li J., Ren H., Gao Z., Liu D., Synth. Commun. 2011, 41, 3325–3333. [Google Scholar]
- 13. Abdel-Razik H. H., Mahmoud K. H., J. Appl. Polym. Sci. 2012, 123, 1329–1339. [Google Scholar]
- 14.
- 14a. Coropceanu V., Cornil J., da Silva Filho D. A., Olivier Y., Silbey R., Brédas J.-L., Chem. Rev. 2007, 107, 926–952; [DOI] [PubMed] [Google Scholar]
- 14b. Reese C., Bao Z., J. Mater. Chem. 2006, 16, 329–333. [Google Scholar]
- 15. Hogan D. T., Gelfand B. S., Spasyuk D. M., Sutherland T. C., Mater. Chem. Front. 2020, 4, 268–276. [Google Scholar]
- 16. Hu J., Zhang D., Harris F. W., J. Org. Chem. 2005, 70, 707–708. [DOI] [PubMed] [Google Scholar]
- 17. Yamato T., Miyazawa A., Tashiro M., Chem. Ber. 1993, 126, 2505–2511. [Google Scholar]
- 18. Rieger R., Müllen K., J. Phys. Org. Chem. 2010, 23, 315–325. [Google Scholar]
- 19. Halder M., Datta S., Bolel P., Mahapatra N., Panja S., Vardhan H., Kayal S., Khatua D. K., Das I., Anal. Methods 2016, 8, 2805–2811. [Google Scholar]
- 20. Würthner F., Kaiser T. E., Saha-Möller C. R., Angew. Chem. Int. Ed. 2011, 50, 3376–3410; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3436–3473. [Google Scholar]
- 21.
- 21a. Janczak J., Kubiak R., Acta Crystallogr. Sect. C 1995, 51, 1399–1401; [Google Scholar]
- 21b. Li H., Fronczek F. R., Vicente M. G. H., J. Organomet. Chem. 2009, 694, 1607–1611; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Zorlu Y., Ün İ., Hirel C., Dumoulin F., Ahsen V., J. Chem. Crystallogr. 2013, 43, 636–645. [Google Scholar]
- 22.
- 22a. Gavezzotti A., Acc. Chem. Res. 1994, 27, 309–314; [Google Scholar]
- 22b. Gavezzotti A., Filippini G., J. Phys. Chem. 1994, 98, 4831–4837. [Google Scholar]
- 23. Wex B., El-Ballouli A. A. O., Vanvooren A., Zschieschang U., Klauk H., Krause J. A., Cornil J., Kaafarani B. R., J. Mol. Struct. 2015, 1093, 144–149. [Google Scholar]
- 24.
- 24a. Maruyama Y., Inokuchi H., Bull. Chem. Soc. Jpn. 1967, 40, 2073–2077; [Google Scholar]
- 24b. Kepler R. G., Phys. Rev. 1960, 119, 1226–1229. [Google Scholar]
- 25.
- 25a. Elstner M., Porezag D., Jungnickel G., Elsner J., Haugk M., Frauenheim T., Suhai S., Seifert G., Phys. Rev. B 1998, 58, 7260–7268; [Google Scholar]
- 25b. Kubas A., Hoffmann F., Heck A., Oberhofer H., Elstner M., Blumberger J., J. Chem. Phys. 2014, 140, 104105; [DOI] [PubMed] [Google Scholar]
- 25c. Kubas A., Gajdos F., Heck A., Oberhofer H., Elstner M., Blumberger J., Phys. Chem. Chem. Phys. 2015, 17, 14342–14354; [DOI] [PubMed] [Google Scholar]
- 25d. Heck A., Kranz J. J., Kubař T., Elstner M., J. Chem. Theory Comput. 2015, 11, 5068–5082; [DOI] [PubMed] [Google Scholar]
- 25e. Heck A., Kranz J. J., Elstner M., J. Chem. Theory Comput. 2016, 12, 3087–3096; [DOI] [PubMed] [Google Scholar]
- 25f. Kubař T., Elstner M., J. Royal Soc. Interface 2013, 10, 20130415; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25g. Kubař T., Elstner M., J. Phys. Chem. B 2010, 114, 11221–11240; [DOI] [PubMed] [Google Scholar]
- 25h. Lüdemann G., Solov'yov I. A., Kubař T., Elstner M., J. Am. Chem. Soc. 2015, 137, 1147–1156; [DOI] [PubMed] [Google Scholar]
- 25i. Lüdemann G., Woiczikowski P. B., Kubař T., Elstner M., Steinbrecher T. B., J. Phys. Chem. B 2013, 117, 10769–10778; [DOI] [PubMed] [Google Scholar]
- 25j. Holub D., Ma H., Krauß N., Lamparter T., Elstner M., Gillet N., Chem. Sci. 2018, 9, 1259–1272; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25k. Aradi B., Hourahine B., Frauenheim T., J. Phys. Chem. A 2007, 111, 5678–5684. [DOI] [PubMed] [Google Scholar]
- 26. Delgado M. C. R., Kim E.-G., d. S. Filho D. A., Bredas J.-L., J. Am. Chem. Soc. 2010, 132, 3375–3387. [DOI] [PubMed] [Google Scholar]
- 27. McGarry K. A., Xie W., Sutton C., Risko C., Wu Y., Young V. G., Brédas J.-L., Frisbie C. D., Douglas C. J., Chem. Mater. 2013, 25, 2254–2263. [Google Scholar]
- 28. Sundar V. C., Zaumseil J., Podzorov V., Menard E., Willett R. L., Someya T., Gershenson M. E., Rogers J. A., Science 2004, 303, 1644–1646. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Sakanoue K., Motoda M., Sugimoto M., Sakaki S., J. Phys. Chem. A 1999, 103, 5551–5556; [Google Scholar]
- 29b. Kim E.-G., Coropceanu V., Gruhn N. E., Sánchez-Carrera R. S., Snoeberger R., Matzger A. J., Brédas J.-L., J. Am. Chem. Soc. 2007, 129, 13072–13081. [DOI] [PubMed] [Google Scholar]
- 30. Brédas J.-L., Beljonne D., Coropceanu V., Cornil J., Chem. Rev. 2004, 104, 4971–5004. [DOI] [PubMed] [Google Scholar]
- 31. da Silva Filho D. A., Kim E.-G., Brédas J.-L., Adv. Mater. 2005, 17, 1072–1076. [Google Scholar]
- 32. Coropceanu V., Kwon O., Wex B., Kaafarani B. R., Gruhn N. E., Durivage J. C., Neckers D. C., Brédas J.-L., Chem. Eur. J. 2006, 12, 2073–2080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary