

Summary
Background
Vedolizumab, a gut‐selective α4β7 integrin antibody, is approved for moderately to severely active ulcerative colitis (UC) and Crohn's disease (CD).
Aim
To report the final results from the vedolizumab GEMINI long‐term safety (LTS) study.
Methods
The phase 3, open‐label GEMINI LTS study (initiated May 2009) enrolled patients with UC or CD from four prior clinical trials and vedolizumab‐naïve patients. Vedolizumab LTS was evaluated; efficacy and patient‐reported outcomes were exploratory endpoints.
Results
Enrolled patients (UC, n = 894; CD, n = 1349) received vedolizumab 300 mg IV every 4 weeks; median cumulative exposure was 42.4 months (range: 0.03‐112.2) for UC and 31.5 months (range: 0.03‐100.3) for CD. Over 8 years, adverse events (AEs) occurred in 93% (UC) and 96% (CD) of patients, with UC (36%) and CD (35%) exacerbations most frequent. Serious AEs were reported for 31% (UC) and 41% (CD) of patients. Vedolizumab discontinuation due to AEs occurred in 15% (UC) and 17% (CD) of patients. There were no new trends for infections, malignancies, infusion‐related reactions, or hepatic events, and no cases of progressive multifocal leukoencephalopathy. Of the ten deaths (UC, n = 4; CD, n = 6), two were considered drug‐related by local investigators (West Nile virus infection‐related encephalitis and hepatocellular carcinoma). Continuous vedolizumab maintained clinical response long‐term, with 33% (UC) and 28% (CD) of patients in clinical remission at 400 treatment weeks.
Conclusions
The safety profile of vedolizumab remains favourable with no unexpected or new safety concerns. These results further establish the safety of vedolizumab and support its long‐term use (NCT00790933/EudraCT 2008‐002784‐14).
1. INTRODUCTION
Ulcerative colitis (UC) and Crohn's disease (CD) are serious, chronic, idiopathic inflammatory bowel diseases (IBD) characterised by abdominal pain, faecal urgency and diarrhoea with blood and/or mucus. 1 , 2 In addition to negative effects on health‐related quality of life (HRQOL), these progressive diseases can lead to structural bowel damage, loss of function, disability and increase the potential for hospitalisation and surgery. Current treatment strategies typically include a combination of corticosteroids, immunosuppressors and biologic therapies to induce remission. Even with successful initial treatment, patients with UC or CD generally require long‐term maintenance therapy.
Vedolizumab is a gut‐selective monoclonal antibody directed against α4β7 integrin and approved for the treatment of moderately to severely active UC and CD. 3 , 4 Previous Phase 3, double‐blind, randomised, placebo‐controlled studies demonstrated that maintenance treatment with vedolizumab for up to 1 year was effective and well‐tolerated in patients with UC (GEMINI 1) or CD (GEMINI 2 and GEMINI 3). 5 , 6 , 7 The GEMINI long‐term safety (LTS) study continued assessment of patients treated in the GEMINI studies, in addition to enrolling vedolizumab‐naïve patients, with the primary goal of evaluating the LTS of vedolizumab in patients with UC or CD. 8 , 9 , 10 Interim analyses (based on 4 years of follow‐up) demonstrated that long‐term vedolizumab therapy was well‐tolerated and also provided clinical and HRQOL benefits. 8 , 9 In this final analysis that includes over 2,000 patients, some with over 9 years of follow‐up, we report the final safety outcomes, along with exploratory clinical and HRQOL outcomes.
2. METHODS
2.1. Study Design
GEMINI LTS (NCT00790933/EudraCT 2008‐002784‐14) was a Phase 3, single‐arm, open‐label, multinational study conducted in patients with moderately to severely active UC or CD (Figure 1). GEMINI LTS enrolled patients from a long‐term Phase 2 study (UC and CD; NCT00619489), three Phase 3 studies (GEMINI 1 [UC; NCT00783718/EudraCT 2008‐002782‐32]; GEMINI 2 [CD; NCT00783692/EudraCT 2008‐002783‐33]; GEMINI 3 [CD; NCT01224171/EudraCT 2009‐016488‐12]) and included a cohort of vedolizumab‐naïve (de novo) patients with UC or CD. Additional details regarding the GEMINI trials can be found in the Supporting Information and in previously published reports. 5 , 6 , 7 Data presented herein were collected from approximately 400 study sites in 39 countries between 22 May 2009 and 31 October 2017.
Figure 1.
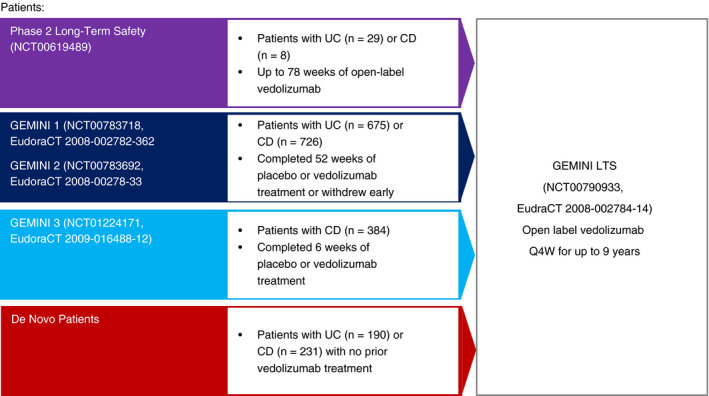
GEMINI LTS patient enrolment. Patients were eligible for enrolment in GEMINI LTS who had completed the Phase 2 long‐term safety study or one of the three Phase 3 GEMINI studies. In addition, a cohort of vedolizumab‐naïve patients were recruited directly into GEMINI LTS. CD, Crohn's disease; HBI, Harvey‐Bradshaw Index; IV, intravenous; LTS, long‐term safety; Q4W, every 4 weeks; UC, ulcerative colitis
2.2. Study Treatment
Patients entering the study from a prior vedolizumab study and de novo patients received vedolizumab 300 mg intravenously (IV) every 4 weeks (Q4W). Treatment duration varied by patient and continued if there was sustained clinical benefit or until loss to follow‐up for any reason, including vedolizumab approval and access to commercially available drug, or the presence of an expanded‐access programme at the local study site.
2.3. Study Assessments and Endpoints
The primary objective of GEMINI LTS was to evaluate the LTS profile of vedolizumab. Patients were evaluated per study protocol at visits at least every 4 weeks where vital signs, concomitant medications, adverse events (AEs) and serious AEs (SAEs; including signs and symptoms of progressive multifocal leukoencephalopathy [PML]) were recorded. A PML symptom checklist was administered before each dosing. AEs, captured in response to questioning from study personnel during study visits and spontaneously reported throughout the study, were coded according to the Medical Dictionary for Regulatory Activities (MedDRA). AEs of special interest were predefined as infections (including PML), malignancies, infusion‐related reactions, and hepatobiliary events. Infusion reactions were defined as any AE that occurred on the calendar day of or 1 calendar day after any vedolizumab infusion or any event defined by the investigator as infusion‐related.
Exploratory endpoints were related to HRQOL, clinical outcomes and IBD‐related procedures and hospitalisations. Patient‐reported HRQOL outcomes were assessed using validated tools including the Inflammatory Bowel Disease Questionnaire (IBDQ), the 36‐item Short‐Form Health Survey (SF‐36), and the European Quality of Life‐5 Dimension (EQ‐5D) Questionnaire. 11 , 12 , 13 A total IBDQ score was calculated by summing the scores from four subdomains (bowel symptoms, emotional function, social function and systemic function) for a final score ranging from 4 to 224 (higher scores indicated better QOL). An increase of ≥16 points in the total score represented a clinically meaningful improvement in patient QOL. For the total SF‐36 score, results from the physical component summary and the mental component summary were summed and converted to a percentage on the scale from 0 to 100 (higher scores indicated better QOL). EQ‐5D total score was calculated from individual scores from 5 domains—mobility, self‐care, usual activities, pain/discomfort and anxiety/depression—with scores of 1, 2 or 3 possible for each domain. The EQ‐5D visual analog scale (VAS) score was a self‐assigned rating of overall health with a score ranging from 0 as the worst to 100 as the best possible health. An increase of ≥7 points in EQ‐5D VAS score represented a clinically meaningful improvement in patient QOL. HRQOL outcomes were obtained at enrolment, Week 28, every 24 weeks for the first 4 years and yearly from Year 5 onward.
Clinical outcomes included changes in partial Mayo score in patients with UC or in Harvey‐Bradshaw Index (HBI) score in patients with CD. For patients with UC, clinical response was defined by a reduction in the partial Mayo score of ≥2 points and ≥25% from baseline (defined as the screening visit for de novo patients and as the last visit of the previous trial for rollover patients), with an accompanying decrease in the rectal bleeding subscore of ≥1 point or an absolute rectal bleeding subscore of ≤1 point; clinical remission was defined by a partial Mayo score of ≤2 with no individual subscore of >1. For patients with CD, clinical response was defined as ≥3‐point decrease from baseline in the HBI score, and clinical remission was defined by an HBI score ≤4. Major IBD‐related events such as hospitalisations, surgeries, or other procedures deemed related to UC or CD by the investigator were recorded.
2.4. Statistical Analyses
The safety population was defined as all patients who received any dose of vedolizumab in GEMINI LTS. The efficacy population consisted of all patients in the safety population who had ≥1 post baseline disease activity measurement.
All study endpoints were evaluated descriptively using point estimates and 95% confidence intervals (CIs). The incidence of AEs was calculated as exposure‐adjusted incidence rates, defined as the number of patients experiencing the event per 1000 person‐years (PYs) of exposure (defined as the first dose of vedolizumab in GEMINI LTS). Sub‐group analyses were performed according to prior tumour necrosis factor (TNF) antagonist failure status. TNF antagonist failure status was not collected in the long‐term Phase 2 study; hence, these patients were excluded from all assessments of prior TNF antagonist status. Changes from baseline in efficacy endpoints were assessed using the original baseline assessment before patients received their first dose of vedolizumab (ie from the start of the original study prior to enrolment in GEMINI LTS or, if de novo enrolment, from the start of GEMINI LTS). Efficacy outcomes were analysed post hoc using a last‐observation‐carried‐forward (LOCF) approach.
3. RESULTS
3.1. Patient Characteristics
A total of 2243 patients (894 UC, 1349 CD) enrolled in GEMINI LTS. Of these, 1822 participated in a prior vedolizumab randomised study; 421 were de novo patients (Figure 1). The safety population consisted of all 2243 patients; 2177 patients were included in the efficacy population. Overall, 766 of 2243 (34.2%) patients completed GEMINI LTS (36.9% UC, 32.3% CD). The most common reasons for treatment discontinuation were lack of efficacy (26.6% UC, 28.3% CD), AEs (14.4% UC, 16.5% CD), and withdrawal by the patient (16.9% UC, 14.8% CD). Additional reasons for withdrawal were not captured but likely included the availability of an expanded‐access programme at a local study site or the availability of commercial vedolizumab.
The mean age of patients with UC at enrolment in GEMINI LTS was 41.2 years; roughly one third were younger than 35 years and 4.7% were ≥65 years of age. At enrolment, the mean partial Mayo score of patients with UC was 6.0 and the mean disease duration was 8.1 years. For patients with CD, the mean age at enrolment was 37.8 years and almost half (47.8%) were younger than 35; only 2.5% were ≥65 years of age. The mean HBI score at enrolment was 10.9 and the mean duration of CD was 10.1 years. All patients with UC or CD had received ≥1 prior conventional therapy and approximately half of patients with UC and two‐thirds of patients with CD had previously failed therapy with a TNF antagonist. At enrolment, 36.9% and 40.4% of patients with UC and CD, respectively, were taking concomitant corticosteroids (Table 1).
Table 1.
Baseline demographics and clinical characteristics a
| Parameter | Ulcerative colitis (N = 894) | Crohn's disease (N = 1349) |
|---|---|---|
| Age, b y, mean (SD) | 41.2 (13.6) | 37.8 (12.7) |
| ≤35 years of age, n (%) | 333 (37.2) | 645 (47.8) |
| ≥65 years of age, n (%) | 42 (4.7) | 34 (2.5) |
| Male sex, n (%) | 522 (58.4) | 606 (44.9) |
| Race, n (%) | ||
| White | 762 (85.2) | 1219 (90.4) |
| Black | 14 (1.6) | 28 (2.1) |
| Asian | 105 (11.7) | 82 (6.1) |
| Other/not reported | 13 (1.5) | 20 (1.5) |
| Disease duration, years, mean (SD) | 8.1 (7.0) | 10.1 (8.3) |
| Partial Mayo score, mean (SD) | 6.0 (1.5) | NA |
| HBI score, mean (SD) | NA | 10.9 (3.4) |
| CDAI score, mean (SD) | NA | 314.0 (63.2) |
| History of extraintestinal disease manifestations, n (%) c | 299 (34.6) | 1124 (83.8) |
| Concomitant medications corticosteroids at enrolment, n (%) | 330 (36.9) | 545 (40.4) |
| Prior therapy, n (%) | ||
| Corticosteroids | 868 (97.1) | 1295 (96.0) |
| Immunomodulators | 666 (74.5) | 1158 (85.8) |
| TNF antagonist therapies | 415 (46.4) | 898 (66.6) |
| Prior TNF antagonist failure, d n (%) | 380 (46) | 848 (65) |
| Vedolizumab exposure, months, median (range) e | 42.4 (0.03‐112.2) | 31.5 (0.03‐100.3) |
Abbreviation: CDAI, Crohn's disease activity index; HBI, Harvey‐Bradshaw Index; NA, not available; SD, standard deviation; TNF, tumour necrosis factor.
Baseline for rollover patients was defined as the last assessment before the first dose of vedolizumab in the previous studies. Baseline for de novo patients was defined as the last assessment before the first dose of vedolizumab in GEMINI LTS.
Age was defined as (1 + first dose date in GEMINI LTS—birth date)/365.25.
History of extraintestinal disease manifestations were not collected for patients in the Phase 2 long‐term safety study.
Calculation includes only patients for whom TNF antagonist failure status was known.
Includes exposure to vedolizumab in previous clinical studies, without incorporating data up to 16 weeks after last dose.
Inclusive of previous trials, 1350 of the 2243 patients (60.2%) completed ≥24 infusions (2 years of vedolizumab exposure), with similar proportions of patients with UC or CD (63.8% and 57.8%, respectively). More than half (51.9%) completed ≥36 infusions (3 years of exposure) and 24.8% completed ≥72 infusions (6 years of exposure). Five patients with UC received ≥116 infusions (≥9 years of exposure). The median cumulative vedolizumab exposure was 42.4 months in patients with UC and 31.5 months in patients with CD, with maximum exposure times of 9.3 and 8.4 years, respectively (Table 1).
3.2. Adverse Events
Inclusive of preceding trials (for patients who rolled over from a prior study) and GEMINI LTS, almost all patients experienced at least 1 AE (92.7% UC, 96.0% CD), the majority of which were mild or moderate in severity. The overall incidence rate of AEs was 1220.5 and 1799.2/1000 PYs for patients with UC and CD, respectively. The most frequent AEs were disease exacerbations (35.9% UC, 35.3% CD), nasopharyngitis (28.2% UC, 25.4% CD) and arthralgia (17.3% UC, 24.4% CD). Fewer than half of AEs (39.7% UC, 46.2% CD) were considered by the treating physician to be vedolizumab‐related (Table 2). SAEs were less frequent (31.0% UC and 40.6% CD) with an incidence of 90.9 and 146.5/1000 PYs for patients with UC and CD, respectively. The most common SAEs were disease exacerbation (13.3% UC, 16.6% CD) and abdominal pain (1.0% UC, 3.0% CD). SAEs were deemed treatment related in 4.1% and 5.9% of patients UC and CD, respectively.
Table 2.
Safety overview
| Parameter | Ulcerative colitis (N = 894) | Crohn's disease (N = 1349) | ||
|---|---|---|---|---|
| n (%) | Incidence/1000 person‐years a | n (%) | Incidence/1000 person‐years a | |
| Any AE | 829 (92.7) | 1220.5 | 1295 (96.0) | 1799.2 |
| Disease exacerbation | 321 (35.9) | 105.2 | 476 (35.3) | 121.4 |
| Nasopharyngitis | 252 (28.2) | 93.9 | 342 (25.4) | 94.1 |
| Arthralgia | 155 (17.3) | 51.6 | 329 (24.4) | 90.2 |
| Abdominal pain | 111 (12.4) | 34.4 | 309 (22.9) | 80.0 |
| Headache | 164 (18.3) | 55.5 | 290 (21.5) | 76.4 |
| Upper respiratory tract infection | 166 (18.6) | 55.7 | 213 (15.8) | 53.2 |
| Nausea | 105 (11.7) | 32.3 | 231 (17.1) | 57.7 |
| Pyrexia | 80 (8.9) | 24.3 | 201 (14.9) | 48.6 |
| Severity of AE | ||||
| Mild | 163 (18.2) | NA | 223 (16.5) | NA |
| Moderate | 451 (50.4) | NA | 656 (48.6) | NA |
| Severe | 215 (24.0) | NA | 415 (30.8) | NA |
| Treatment‐related AEs | 355 (39.7) | NA | 623 (46.2) | NA |
| Treatment withdrawn due to AE | 137 (15.3) | NA | 229 (17.0) | NA |
| SAEs | 277 (31.0) | 90.9 | 548 (40.6) | 146.5 |
| Disease exacerbation | 119 (13.3) | 34.8 | 224 (16.6) | 50.3 |
| Abdominal pain | 9 (1.0) | 2.6 | 41 (3.0) | 9.0 |
| Anal abscess | 0 | 0 | 33 (2.4) | 7.3 |
| Small intestinal obstruction | 4 (<1) | 1.1 | 25 (1.9) | 5.5 |
| Treatment‐related SAEs | 37 (4.1) | NA | 79 (5.9) | NA |
| AESIs | ||||
| Total infections | 591 (66.1) | 388.9 | 937 (69.5) | 492.1 |
| Serious infections | 61 (6.8) | 18.0 | 146 (10.8) | 33.6 |
| Malignancies | 58 (6.5) | 17.2 | 92 (6.8) | 20.8 |
| Infusion reactions | 36 (4.0) | NA | 67 (5.0) | NA |
| Hepatic events | 29 (3.2) | 8.4 | 63 (4.7) | 14.1 |
| PML | 0 | 0 | 0 | 0 |
| Deaths | 4 (<1) b | NA | 6 (<1) c | NA |
| Treatment‐related death | 1 (<1) d | NA | 1 (<1) e | NA |
Abbreviations: AE, adverse event; AESIs, adverse events of special interest; NA, not available; PML, progressive multifocal leukoencephalopathy; SAE, serious adverse event.
Time‐adjusted incidence rate per 1000 patient‐years = (number of patients experiencing an AE of interest/total person time in years) × 1000.
Respiratory failure, acute stroke, West Nile virus encephalitis, pulmonary embolism.
Traumatic intracranial haemorrhage, hepatocellular carcinoma, suicide, pneumonia, septicaemia, leiomyosarcoma.
One patient who received long‐term vedolizumab therapy subsequently died because of West Nile virus encephalitis. However, based on the mechanism of action of vedolizumab and available information, there is no known association between this event and vedolizumab therapy. There are also no other safety signals linking vedolizumab to the West Nile virus. Therefore, although this event was recorded by the principal investigator as treatment‐related, there is no evidence to support this association.
Death of 1 patient with Crohn's disease (hepatocellular carcinoma) was considered treatment‐related by the treating physicians.
Of the 76 patients (3.4%) who were ≥65 years old, 93.0% reported an AE, comparable to 94.8% in patients <65 years old. However, patients ≥65 versus <65 years old had numerically lower rates of the most common AEs such as arthralgia (19.7% vs 21.8%), nasopharyngitis (15.5% vs 26.4%) and headache (15.5% vs 20.2%).
Patients with UC who had failed prior TNF antagonist therapy reported slightly more AEs than patients who had not (95.5% vs 90.7%). Numerically more patients with UC and prior TNF antagonist failure had treatment‐related AEs and SAEs than patients with no prior TNF antagonist failure (51.3% vs 31.5% and 35.5% vs 28.2%). Patients with CD also reported slightly more AEs with prior TNF antagonist failure versus without (97.5% vs 93.5%), more treatment‐related AEs (51.3% vs 36.9%), and more SAEs (42.7% vs 36.9%) (Table S1).
At enrolment, 299 patients with UC (34.6%) and 1124 patients with CD (83.8%) had a history of prior extraintestinal manifestations (EIM; Table 1). More patients with prior TNF antagonist failure had arthralgia/arthritis versus those without (36.3% vs 22.3% UC, 70.3% vs 60.0% CD). The most common EIM was arthralgia/arthritis (28.4% UC, 66.5% CD). More patients with UC who had previously failed prior TNF antagonist therapy reported a history of EIMs compared with those who had not failed TNF antagonist therapy (41.8% vs 28.9%). Similarly, more patients with CD and prior TNF antagonist failure had a prior history of EIMs than those with no prior failure (87.0% vs 78.3%).
Inclusive of prior vedolizumab trials and GEMINI LTS, treatment‐emergent EIMs occurred in 172 patients with UC (19.2%; 70.6/1000 PYs) and 443 patients with CD (32.8%; 157.5/1000 PYs). Arthralgia was the most common event, reported by 13.4% of patients with UC (47.0/1000 PYs) and 19.9% of patients with CD (84.1/1000 PYs), followed by arthritis (1.9% UC, 3.5% CD), anal fissure (1.7% UC, 3.0% CD) and anal fistula (<1% UC, 7.0% CD). Among patients with treatment‐emergent EIMs of arthralgia or arthritis, 39.5% of those with UC and 74.8% of those with CD had a history of arthralgia or arthritis. Additional treatment‐emergent EIMs are reported in Table 3.
Table 3.
Incidence of treatment‐emergent extraintestinal manifestations in patients with UC or CD
| Ulcerative colitis (N = 894) | Crohn's disease (N = 1349) | |||
|---|---|---|---|---|
| n (%) | Incidence/1000 person‐years (95% CI) a | n (%) | Incidence/1000 person‐years (95% CI) a | |
| Any extraintestinal manifestation | 172 (19.2) | 70.6 (59.3, 81.9) | 443 (32.8) | 157.5 (140.6, 174.5) |
| Arthralgia | 120 (13.4) | 47.0 (38.2, 55.8) | 269 (19.9) | 84.1 (73.2, 94.9) |
| Arthritis | 17 (1.9) | 6.0 (3.1, 8.8) | 47 (3.5) | 12.4 (8.9, 16.0) |
| Anal fissure | 15 (1.7) | 5.3 (2.6, 7.9) | 40 (3.0) | 10.5 (7.2, 13.8) |
| Anal fistula | 2 (<1) | 0.7 (0, 1.7) | 95 (7.0) | 25.5 (20.2, 30.8) |
| Aphthous stomatitis | 14 (1.6) | 4.9 (2.3, 7.5) | 54 (4.0) | 14.3 (10.4, 18.2) |
| Erythema nodosum | 6 (<1) | 2.1 (0.4, 3.8) | 30 (2.2) | 7.8 (5.0, 10.7) |
| Iritis/uveitis b | 12 (1.3) | 4.2 (1.8, 6.6) | 13 (1.0) | 3.4 (1.5, 5.2) |
| Pyoderma gangrenosum | 3 (<1) | 1.0 (0, 2.2) | 3 (<1) | 0.8 (0, 1.6) |
Abbreviation: CI, confidence interval.
Time‐adjusted incidence rate per 1000 person‐years = (number of patients experiencing an adverse event of interest/total person time in years) × 1000.
Includes iris and uveal tract infections, irritations and inflammations.
Vedolizumab was discontinued because of an AE by 137 patients with UC (15.3%) and 229 patients with CD (17.0%) (Table 2); UC or CD exacerbations (8.8% and 8.0%, respectively) were the most frequent reasons for discontinuation. All other AEs that led to discontinuation were reported in <1% of patients. There was a total of 10 deaths (4 UC, 6 CD). Two deaths were considered related to treatment by local investigators (Table 2), one patient who died of West Nile virus infection‐related encephalitis and a second who died of hepatocellular carcinoma. Narratives for these 10 patient deaths can be found in the Supporting Information.
3.3. Infections
Infections were reported in 591 patients with UC (66.1%; 388.9/1000 PYs) and 937 patients with CD (69.5%; 492.1/1000 PYs). Serious infections occurred in 6.8% of patients with UC (18.0/1000 PYs) and 10.8% of patients with CD (33.6/1000 PYs; Table 2). The most common serious infections were anal abscess (0 UC, 33 CD), pneumonia (9 UC, 12 CD), gastroenteritis (5 UC, 14 CD) and appendicitis (9 UC, 8 CD). In patients with UC and CD, 17.9% and 21.6% of infections, respectively, were considered treatment‐related by the investigator.
Among gastrointestinal infections of special interest, pathogen‐unspecified abdominal or gastrointestinal infections were experienced by 92 patients with UC (10.3%; 34.9/1000 PYs) and 141 patients with CD (10.5%; 39.6/1000 PYs; Table 4). Opportunistic infections were reported in 61 patients (31 UC, 30 CD), the most common of which were clostridia infections. Most opportunistic infections were mild (22 of 61) or moderate in severity (35 of 61). One or more clostridial infections were reported in 26 patients with UC (2.9%) and 21 patients with CD (1.6%). Of clostridial events, 12 were severe, while the remainder were moderate (23 events) or mild (15 events); 4 patients (2 UC, 2 CD) discontinued the study because of clostridial infections. Fourteen patients with UC and 14 with CD developed Clostridium difficile infections (Table S2); 11 patients (4 UC, 7 CD) were hospitalised.
Table 4.
Incidence of gastrointestinal infections of special interest in patients with UC or CD
| Parameter | Ulcerative colitis (N = 894) | Crohn's disease (N = 1349) | ||
|---|---|---|---|---|
| n (%) | Incidence/1000 person‐years (95% CI) a | n (%) | Incidence/1000 person‐years (95% CI) a | |
| Gastrointestinal infections of special interest | 122 (13.6) | 46.9 (38.3, 55.6) | 162 (12.0) | 46.1 (38.8, 53.4) |
| Abdominal and gastrointestinal infections (pathogen unspecified) b | 92 (10.3) | 34.9 (27.6, 42.3) | 141 (10.5) | 39.6 (32.9, 46.3) |
| Clostridium infections c | 26 (2.9) | 9.1 (5.6, 12.6) | 21 (1.6) | 5.4 (3.1, 7.8) |
| Campylobacter infections d | 9 (1.0) | 3.1 (1.1, 5.2) | 8 (<1) | 2.1 (0.6, 3.5) |
| Shigella infections e | 0 | 0 | 1 (<1) | 0.3 (0, 0.8) |
| Yersinia infections f | 1 (<1) | 0.3 (0, 1.0) | 0 | 0 |
| Bacterial infections (bacteria unspecified) g | 0 | 0 | 1 (<1) | 0.3 (0, 0.8) |
Abbreviation: CI, confidence interval; MedDRA, Medical Dictionary for Regulatory Activities.
Time‐adjusted incidence rate per 1000 person‐years = (number of patients experiencing an adverse event of interest/total person time in years) × 1000.
MedDRA preferred terms: gastroenteritis, diarrhoea infectious, gastrointestinal infection, enteritis infectious, enterocolitis infectious and gastric infection.
MedDRA preferred terms: Clostridium difficile colitis, Clostridial infection and Clostridium colitis.
MedDRA preferred terms: Campylobacter gastroenteritis, Campylobacter intestinal infection and Campylobacter infection.
MedDRA preferred term gastroenteritis shigella.
MedDRA preferred term gastroenteritis yersinia.
Bacterial infections represent MedDRA preferred term gastroenteritis bacterial.
Of the 2243 patients, 294 (13.1%) were enrolled from countries with high rates of tuberculosis (TB) infection (>20 cases/100 000 people) (Table S3). Over the course of the study, 4 patients (1 UC, 3 CD), all living in countries with higher endemic rates of TB, were diagnosed with primary pulmonary TB; 3 of the 4 patients discontinued the study because of this event. Three of the cases of primary pulmonary TB were considered SAEs. One additional patient developed TB from an undetected latent infection and discontinued the study. No cases of gastrointestinal TB were reported.
Nine patients (5 UC, 4 CD) reported salmonella infections and 7 patients (5 UC, 2 CD) experienced giardiasis; no patient discontinued due to these infections. Meningitis, cryptosporidiosis and alveolar osteitis were each reported in one patient but did not lead to study discontinuation. Finally, there were no cases of herpes encephalitis, disseminated herpes zoster or PML.
3.4. Neoplasms
Benign or malignant neoplasms occurred in 58 patients with UC (6.5%; 17.2/1000 PYs) and 92 patients with CD (6.8%; 20.8/1000 PYs; Table 2). The most common benign neoplastic events were skin papillomas (<1% UC, 1.2% CD), and melanocytic naevus (<1% in UC and CD) (Table S4).
The most common malignant neoplasm in patients with UC or CD was basal cell carcinoma with <1% in UC and CD. The next most common neoplasms were lung neoplasm in <1% of patients with UC and CD and colon carcinomas in <1% of patients with UC and CD (Table S4). One patient developed a malignant hepatic neoplasm, which was considered by the investigator to be related to vedolizumab. No trend was observed between the development of malignancy and age, sex, type of malignancy or duration of vedolizumab exposure.
3.5. Infusion‐Related Reactions
Investigator‐defined infusion‐related reactions were reported in 36 patients with UC (4.0%) and 67 patients with CD (5.0%) (Table 2). Two patients discontinued treatment because of infusion reactions. The most common infusion‐related reactions were nausea (3 UC, 11 CD), dizziness (3 UC, 8 CD) and headache (4 UC, 7 CD). Each infusion‐related reaction event (cumulative over the patients’ previous study and GEMINI LTS) occurred in <1% of patients overall.
3.6. Other AEs
Overall, hepatobiliary events were reported for 29 patients with UC (3.2%) and 63 patients with CD (4.7%) (Table 2). All events occurred in <1% of patients except for cholelithiasis, which occurred in 25 patients (1.1%). Three patients (2 UC, 1 CD) discontinued the study because of a hepatobiliary disorder.
Thromboembolic events were reported for 5 patients with UC (<1%) and 20 patients with CD (1.5%). Three events (deep vein thrombosis, thrombophlebitis, and venous thrombosis in a limb in 1 patient each) were considered related to study drug.
3.7. Efficacy Outcomes
Clinical response rates and clinical remission rates were likewise maintained long‐term. At 400 weeks of treatment, 217 of 658 (33.0%) patients with UC and 197 of 700 (28.1%) patients with CD were in clinical remission and 230 of 657 (35.0%) and 232 of 700 (33.1%), respectively, had a clinical response (Figures 2 and 3). Clinical response and clinical remission rates trended lower in both patients with UC and CD who had failed TNF antagonist therapy relative to patients who had not (Figures S1 and S2).
Figure 2.
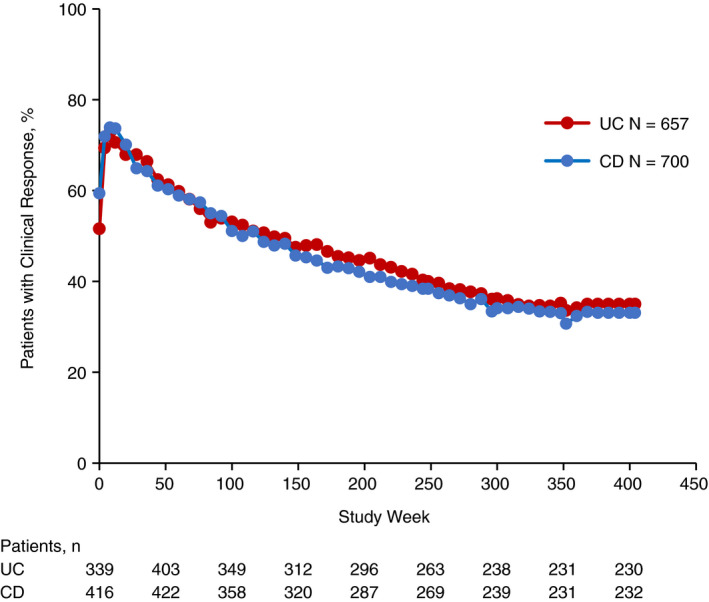
Clinical response over time in patients with UC or CD. Clinical response was assessed at baseline, weeks 4, 8, 12 and every 8 weeks thereafter. For patients with UC, clinical response was defined as a decrease in the partial Mayo score of ≥2 points and ≥25% from baseline, with an accompanying decrease in rectal bleeding subscore of ≥1 point from baseline or absolute rectal bleeding subscore of ≤1 point. For patients with CD, clinical response was defined as a ≥3‐point decrease from baseline in the HBI score. Baseline was defined as the last assessment prior to the first dose of study drug administration in GEMINI 1 for patients with UC and GEMINI 2 for patients with CD. For patients who discontinued the study, the final assessed value of clinical response was carried forward to the end of the study. CD, Crohn's disease; HBI, Harvey‐Bradshaw Index; UC, ulcerative colitis
Figure 3.
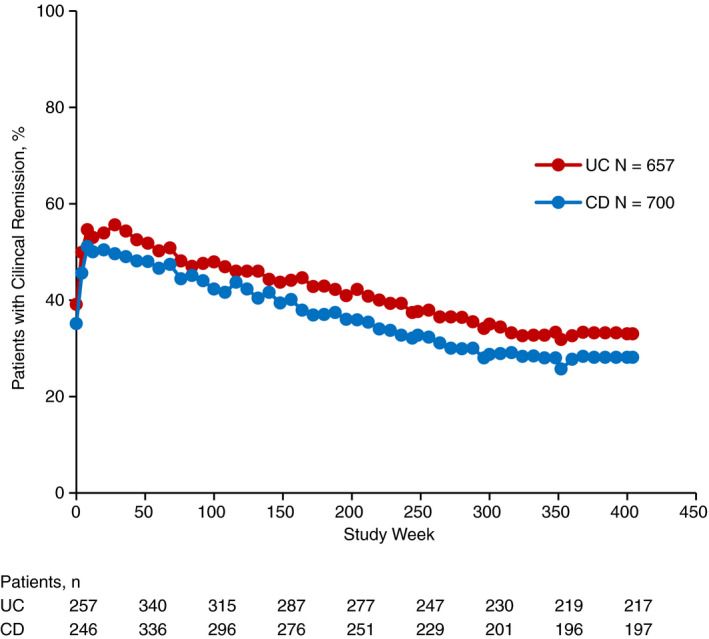
Clinical remission over time in patients with UC or CD. Clinical remission was assessed at baseline, Weeks 4, 8, 12 and every 8 weeks thereafter. For patients with UC, clinical remission was defined as a partial Mayo score of ≤2 with no individual subscore >1. For patients with CD, clinical remission was defined as an HBI score ≤4. For patients who discontinued the study, the final assessed value of clinical response was carried forward to the end of the study. CD, Crohn's disease; HBI, Harvey‐Bradshaw Index; UC, ulcerative colitis
HRQOL estimates, based upon IBDQ, SF‐36, EQ‐5D and EQ‐5Q VAS scores remained stable in patients with UC and CD over the duration of GEMINI LTS (Figure 4A‐D). HRQOL was comparable between patients with UC or CD who had failed prior TNF antagonist therapy versus those who had not (Figures S3 and S4).
Figure 4.
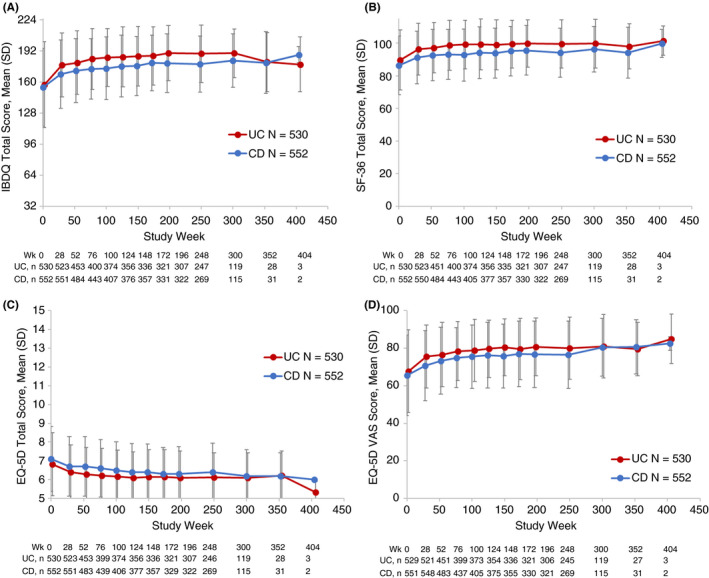
Health‐related quality of life (HRQOL) by indication and assessment tool in patients with UC or CD. HRQOL was assessed at baseline and week 26 and then every 24 weeks for the first 4 years, and yearly from Year 5 onward. Assessments tools included A. IBDQ total score, B. SF‐36 total score, C. EQ‐5D total score, and D. EQ‐5D VAS
Finally, 142 patients with UC (16.8%) experienced ≥1 UC‐related hospitalisation, colectomy or UC‐related procedure and 365 patients with CD (28.1%) experienced ≥1 CD‐related hospitalisation, bowel resection or CD‐related procedure. These events were more frequent among patients who had previously failed TNF antagonist therapy versus those who had not (23% vs 12% UC, 30% vs 24% CD). Of all hospitalisations, the most common reasons for admission of patients with UC were UC (34.4%), abdominal/gastrointestinal infection (4.5%) and abdominal/gastrointestinal pain (3.0%). Among patients with CD, the most common reasons for hospitalisation included CD (29.1%), abdominal/gastrointestinal infection (7.4%), and abdominal/gastrointestinal pain (6.2%) (Table S5). A total of 8 patients (<1%) with UC and 33 (2.4%) with CD developed postoperative complications. The most common postoperative event was intestinal anastomosis complication (1 UC, 8 CD). Additional complications are summarised in Table S6.
4. DISCUSSION
GEMINI LTS is the largest and longest clinical study yet performed to evaluate the risks and benefits of long‐term vedolizumab therapy in patients with moderately to severely active UC or CD. Previous interim analyses reported interim safety summaries, while this final report presents in‐depth safety over a median cumulative vedolizumab exposure period of 42.4 months (range: 0.03‐112.2 months) for patients with UC and 31.5 months (range: 0.03‐100.3 months) for patients with CD. 8 , 9 Study patients, all of whom had previously responded to vedolizumab induction therapy, had in total 7999 PYs (3451 PYs [UC] and 4548 PYs [CD]) of vedolizumab exposure. The large number of patients (N = 2243) and long cumulative duration of vedolizumab treatment provides valuable information to clinicians regarding the expected incidence of AEs with long‐term vedolizumab treatment in responding patients.
In this final data analysis, we were able to closely evaluate the potential for systemic infections with the gut‐selective vedolizumab and contrast that with previous reports of biologics with a broader mechanism of action. An analysis utilising the HealthCore Integrated Research Database (HIRD) reported that the incidence rates of a range of infections including C difficile(HR 1.39), TB (HR, 2.43), rectal or peritoneal abscesses (HR 3.12) were higher in patients currently taking TNF antagonist therapies compared with TNF antagonist naïve patients. 14 In GEMINI LTS, vedolizumab treatment was associated with relatively low rates of serious infections (18.0 and 33.6/1000 PYs in UC and CD, respectively). In a pivotal trial of adalimumab, rates of serious infection were roughly double with 35 and 67/1000 PYs, respectively. 15 In VARSITY, a double‐blind, head‐to‐head study of patients treated with vedolizumab or adalimumab, vedolizumab was associated with lower rates of overall and serious infections than adalimumab (23.4 vs 34.6/100 PYs and 1.6 vs 2.2/100 PYs, respectively). 16
While these data suggest a decreased risk of systemic infections with vedolizumab, an increased risk of gastrointestinal infections is plausible given the gut‐selective mechanism of action and evidence for reduced immune response in the gastrointestinal tract. 17 In this study, gastrointestinal infections were infrequent during long‐term vedolizumab exposure (34.9 and 39.6/1000 PYs for UC and CD, respectively), and the rate of C difficile (4.9 and 3.6/1000 PYs for UC and CD, respectively) was consistent with recent reports from other clinical studies including the HIRD, which reported rates of 3.14/1000 PY for patients with moderate to severe IBD. 14 The potential for increased risk of gastrointestinal infections continues to be evaluated.
GEMINI LTS identified no cases of PML with 7999 PYs of vedolizumab exposure. There have also been no confirmed cases of PML reported in vedolizumab clinical trials (>6000 patients treated with vedolizumab). In over 470 000 PYs of postmarketing exposure, only a single confirmed case of PML has been reported. This patient had multiple contributory factors for developing PML, specifically human immunodeficiency virus (HIV) infection with a CD4 count of 300 cells/mm3 and prolonged prior and concomitant immunosuppression use. The case was adjudicated by an Independent PML Adjudication Committee which concluded that the most likely cause of PML in this patient was their HIV diagnosis and prolonged immunosuppression. A PML risk estimation model explored the expected number of vedolizumab‐associated PML cases up to 2034. 18 The model showed that assuming the risk is the same as in the general population, the first PML case would have most likely occurred by 2018 (55% probability). Assuming the risk is the same as in rheumatoid arthritis (an inflammatory condition), the first PML case would have most likely occurred by 2018 (≥90% probability). The occurrence of this case is consistent with the predictions of the model.
Some treatments that suppress the immune system, including treatments for IBD, increase the risk of cancer. 19 Previous reports indicate that rates of lymphoma, skin cancers, head and neck cancers and cervical cancer may be increased with immunosuppressive regimens while the rates of other tumours including lung and breast cancers are less affected. 20 In the HIRD analysis, the incidence of solid tumours overall was similar between TNF antagonist exposed and TNF antagonist naïve patients, 8.21 and 9.95/1000 PYs (HR 0.80), but rates of colon cancer (1.45 vs 1.08/1000 PYs [HR 1.25]), rectal cancer (1.74 vs 0.64/1000 PYs [HR 2.81]) and cervical dysplasia (8.91 vs 4.07/1000 PYs [HR 1.93]) were increased in patients on TNF antagonist therapy. 14 A meta‐analysis of malignancies following TNF antagonist therapy reported similar rates for all‐site malignancies ranging from 3.88 to 9.34/1000 PYs. 21 The rate of all‐site malignancies in GEMINI LTS patients was also in line with these reports in patients with UC or CD (9.8 and 8.3/1000 PY, respectively) suggesting that vedolizumab, like TNF antagonist therapies, does not impact the overall rate of malignancies. However, in GEMINI LTS, vedolizumab was associated with lower rates of colon and rectal cancers (1.0 and 0.3/1000 PYs for UC and 0.3 and 0.5/1000 PYs for CD).
EIM such as arthralgia and arthritis are common in IBD. In this final analysis of GEMINI LTS, arthralgia was the most common treatment‐emergent EIM (13.4% of patients with UC [47.0/1000 PYs] and 19.9% of patients with CD [84.1/1000 PYs]). Although this analysis includes longer cumulative exposure to vedolizumab, the rate of arthralgia is similar to a previous post‐hoc analysis of the GEMINI trials (9% UC, 28% CD). 22 Likewise, the pivotal GEMINI studies reported that arthralgia was the most common treatment‐emergent AE in the first 52 weeks of vedolizumab treatment (9% UC, 13.5% CD). 8 , 9 The GEMINI LTS interim analyses with approximately 4 years of follow‐up was slightly higher (14% UC, 20% CD) 8 , 9 but consistent with this final analysis (17% UC, 24% CD).
Long‐term vedolizumab therapy maintained or improved HRQOL in patients with moderately to severely active UC or CD. The range of validated assessment tools used to demonstrate improvements in HRQOL in GEMINI LTS provides further evidence of the consistent positive benefits provided by long‐term vedolizumab therapy in terms of both disease‐specific and global measures of patient well‐being. 11 , 12 , 13 In addition, GEMINI LTS demonstrated that continuous vedolizumab treatment was efficacious over the long‐term (in some patients for as long as 9 years) in patients with UC or CD. The previous interim analyses also reported that patients with UC or CD experienced clinical improvements with continuous vedolizumab treatment. 8 , 9
Study limitations include potential bias that may be associated with non‐blinded, non‐randomised, uncontrolled studies. In addition, per the study design, only patients up to 65 years of age were initially enrolled, skewing the population younger than what is observed in clinical practice. The frequency of maintenance dosing in GEMINI LTS (Q4W) was determined prior to regulatory approval and is thus more frequent than the approved Q8W dosing used in clinical practice in the US. A further consideration is that endoscopy was not prospectively conducted in GEMINI LTS, precluding evaluation of mucosal healing and changes with long‐term vedolizumab treatment. Finally, GEMINI LTS efficacy analyses were limited by the loss of patients over time primarily due to vedolizumab approval or expanded‐access programme availability at the local study site, with 378/2243 patients receiving vedolizumab for ≥6 years. The rate of patient dropout (1477/2243) should be interpreted with caution, as the study period was extended well beyond the original duration through the addition of study protocol amendments to enable continued patient access to vedolizumab in countries where commercial or extended access to vedolizumab was not otherwise available. Consequently, to approximate patient numbers, LOCF analyses were performed to assess clinical efficacy. The data show a steady rate of both clinical remission and response.
In conclusion, the final analysis of GEMINI LTS comprehensively demonstrates that vedolizumab therapy has a safety and tolerability profile suitable for long‐term treatment of patients with moderately to severely active UC or CD. This is a key consideration in the management of IBDs given that they are chronic conditions that typically require lifelong therapy.
AUTHORSHIP
Guarantor of the article: Edward V. Loftus.
Author contributions: All authors had access to the study data; contributed to manuscript drafting, critical review and revision. All authors approved the final manuscript for submission.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
This study was sponsored by Takeda. The authors thank the patients who participated in this study, their caregivers, and the study investigators and members of the GEMINI LTS study team. The authors thank Harisha Kadali for support with additional analyses of the safety database, and to Liz Gooch, PhD, of ProEd Communications, Inc for medical writing support funded by Takeda.
Declaration of personal interests: Edward V. Loftus: Financial support for research: AbbVie, Takeda, Janssen, UCB, Amgen, Pfizer, Genentech, Celgene, Receptos, Gilead, Bristol‐Myers Squibb, and Robarts Clinical Trials; Consultancy: AbbVie, Takeda, Janssen, UCB, Amgen, Pfizer, Eli Lilly, Celltrion Healthcare, Allergan, Bristol‐Myers Squibb, Gilead, Genentech, Celgene, and Boehringer Ingelheim. Brian G. Feagan: Consultancy: Abbott/AbbVie, ActoGeniX, Akros, Albireo Pharma, Amgen, Astra Zeneca, Avaxia Biologics Inc, Avir Pharma, Axcan, Baxter Healthcare Corp., Biogen Idec, Boehringer‐Ingelheim, Bristol‐Myers Squibb, Calypso Biotech, Celgene, Elan/Biogen, enGene, Ferring Pharma, Roche/Genentech, gICare Pharma, Gilead, Given Imaging Inc, GSK, Ironwood Pharma, Janssen Biotech (Centocor), JnJ/Janssen, Kyowa Hakko Kirin Co Ltd., Lexicon, Lilly, Lycera BioTech, Merck, Mesoblast Pharma, Millennium, Nektar, Nestles, Novartis, Novo Nordisk, Pfizer, Prometheus Therapeutics and Diagnostics, Protagonist, Receptos, Salix Pharma, Serono, Shire, Sigmoid Pharma, Synergy Pharma Inc, Takeda, Teva Pharma, TiGenix, Tillotts, UCB Pharma, Vertex Pharma, VHsquared Ltd., Warner Chilcott, Wyeth, Zealand, and Zyngenia; Grant/research support: Abbott/AbbVie, Amgen, Astra Zeneca, Bristol‐Myers Squibb (BMS), Janssen Biotech (Centocor), JnJ/Janssen, Roche/Genentech, Millennium, Pfizer, Receptos, Santarus, Sanofi, Tillotts, and UCB Pharma; Membership (board of directors): Robarts Clinical Trials Inc. Remo Panaccione: Consultancy: AbbVie, Amgen, Allergan, AstraZeneca, Baxter, BMS, Celgene, Cubist, Eisai, Eli Lilly, Ferring, Genentech, Gilead, Janssen, Merck, Robarts, Salix, Samsung, Schering‐Plough, Shire, Centocor, Elan, Glaxo‐Smith Kline, UCB, Pfizer, and Takeda; Speaker's Bureau: AbbVie, Abbott, Aptalis, AstraZeneca, Ferring, Janssen, Merck, Prometheus, Shire, Takeda; Research/Educational Support from: AbbVie, Abbott, Ferring, Janssen Pharmaceuticals, and Takeda. Jean‐Frédéric Colombel: Consultancy/advisory board membership: AbbVie, Amgen, Boehringer‐Ingelheim, Celgene Corporation, Celltrion, Enterome, Ferring, Genentech, Janssen Pharmaceuticals, Medimmune, Merck & Co., Pfizer, Protagonist, Second Genome, Seres, Takeda, and Theradiag; Speaker: AbbVie, Ferring, Takeda, and Shire; Research support: AbbVie, Genentech, and Takeda; Stock options: Intestinal Biotech Development, and Genfit. William J. Sandborn: Research grants from Atlantic Healthcare Limited, Amgen, Genentech, Gilead Sciences, AbbVie, Janssen, Takeda, Lilly, Celgene/Receptos, Pfizer, Prometheus Laboratories (now Prometheus Biosciences); consulting fees from AbbVie, Allergan, Amgen, Arena Pharmaceuticals, Avexegen Therapeutics, BeiGene, Boehringer Ingelheim, Celgene, Celltrion, Conatus, Cosmo, Escalier Biosciences, Ferring, Forbion, Genentech, Gilead Sciences, Gossamer Bio, Incyte, Janssen, Kyowa Kirin Pharmaceutical Research, Landos Biopharma, Lilly, Oppilan Pharma, Otsuka, Pfizer, Progenity, Prometheus Biosciences (merger of Precision IBD and Prometheus Laboratories), Reistone, Ritter Pharmaceuticals, Robarts Clinical Trials (owned by Health Academic Research Trust, HART), Series Therapeutics, Shire, Sienna Biopharmaceuticals, Sigmoid Biotechnologies, Sterna Biologicals, Sublimity Therapeutics, Takeda, Theravance Biopharma, Tigenix, Tillotts Pharma, UCB Pharma, Ventyx Biosciences, Vimalan Biosciences, Vivelix Pharmaceuticals; and stock or stock options from BeiGene, Escalier Biosciences, Gossamer Bio, Oppilan Pharma, Prometheus Biosciences (merger of Precision IBD and Prometheus Laboratories), Progenity, Ritter Pharmaceuticals, Ventyx Biosciences, Vimalan Biosciences. Spouse: Opthotech ‐ consultant, stock options; Progenity ‐ consultant, stock; Oppilan Pharma ‐ employee, stock options; Escalier Biosciences ‐ employee, stock options; Prometheus Biosciences (merger of Precision IBD and Prometheus Laboratories) ‐ employee, stock options; Ventyx Biosciences – employee, stock options; Vimalan Biosciences – employee, stock options. Bruce E. Sands: Consultancy: 4D Pharma, AbbVie, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, Boehringer Ingelheim, Capella Bioscience, Celgene, Celltrion Healthcare, Eli Lilly and Company, EnGene, F. Hoffmann‐La Roche, Ferring, Gilead Sciences, Ironwood Pharmaceuticals, Janssen, Lyndra, MedImmune, Oppilan Pharma, Otsuka America Pharmaceutical, Palatin Technologies, Pfizer, Progenity, Prometheus Laboratories, Protagonist Therapeutics Rheos Medicines, Seres Therapeutics, Shire, Sienna Biopharmaceuticals, Synergy Pharmaceuticals, Takeda, Target PharmaSolutions, Theravance Biopharma, TiGenix, UCB, Valeant Pharmaceuticals North America, and Vivelix Pharmaceuticals; Research support: Takeda, Janssen, Theravance Biopharma, and Pfizer. Silvio Danese: Lecture fee(s): AbbVie, Ferring, Hospira, Johnson and Johnson, Merck, MSD, Takeda, Mundipharma, Pfizer Inc, Tigenix, UCB Pharma, Vifor, Biogen, Celgene, Allergan, Celltrion, Sandoz, and Boehringer Ingelheim; Consultancy: AbbVie, Ferring, Hospira, Johnson and Johnson, Merck, MSD, Takeda, Mundipharma, Pfizer Inc, Tigenix, UCB Pharma, Vifor, Biogen, Celgene, Allergan, Celltrion, Sandoz, and Boehringer Ingelheim. Geert D'Haens: Financial support for research: AbbVie, Buhlmann Laboratories, Ferring, DrFALK Pharma, Johnson and Johnson, GlaxoSmithKline, Medtronics, Millennium/Takeda, MSD, Photopill, Prometheus laboratories, Robarts Clinical Trials, and Setpoint; Lecture fee(s): AbbVie, Biogen, Ferring, DrFALK Pharma, Johnson and Johnson, Giuliani, Millennium/Takeda, MSD, Norgine, Otsuka, Shire, Tillotts, UCB, and Vifor; Consultancy: AbbVie, Ablynx, Actogenix, Amakem, Amgen, AM Pharma, AstraZeneca, Biogen, BMS, Boehringer Ingelheim, Cosmo, Elan, Ferring, Celgene/Receptos, Celltrion, Johnson and Johnson, Engene, Galapagos, Gilead, GlaxoSmithKline, Immunic Therapeutics, Lycera, Medimetrics, Medtronics, Millennium/Takeda, Mitsubishi Pharma, MSD, Mundipharma, Novo Nordisk, Otsuka, Hospira/Pfizer, PDL, Prometheus Laboratories, Protagonist, Salix, Samsung Bioepis, Sandoz, Setpoint, Shire, TEVA, Tigenix, Tillotts, Topivert, UCB, Versant, and Vifor; Directorship(s): Robarts Clinical Trials. David T. Rubin: Consultancy: AbbVie, Abgenomics, Allergan Inc, Amgen, Celgene Corporation, Forward Pharma, Genentech/Roche, Janssen Pharmaceuticals, Merck & Co Inc, Miraca Life Sciences, Mitsubishi Tanabe Pharma Development America Inc, Napo Pharmaceuticals, Pfizer, Salix Pharmaceuticals, Samsung Bioepis, Sandoz Pharmaceuticals, Shire, Takeda, and Target PharmaSolutions Inc; Grant support: AbbVie, Genentech/Roche, Janssen Pharmaceuticals, Prometheus Laboratories, Shire, Takeda, and UCB Pharma. Ira Shafran: Lecture fee(s): Takeda; Grant support: Takeda. Andrejus Parfionovas: Employee of Takeda. Raquel Rogers: Employee of Takeda. Richard A. Lirio: Employee of Takeda. Séverine Vermeire: Financial support for research: MSD, AbbVie, Takeda, Pfizer, and Johnson & Johnson; Lecture fee(s): MSD, AbbVie, Takeda, Ferring, Centocor, Hospira, Pfizer, J&J, Tillotts Pharma and Genentech/Roche; Consultancy: MSD, AbbVie, Takeda, Ferring, Centocor, Hospira, Pfizer, J&J, Genentech/Roche, Celgene, Mundipharma, Celltrion, SecondGenome, Prometheus, Gilead, Galapagos, Robarts Clinical Trials, Amgen, GlaxoSmithKline, Shire, Arena and ProDigest.
Appendix 1. The authors’ complete affiliation
Edward V. Loftus Jr, Mayo Clinic College of Medicine, Rochester, MN, USA; Brian G. Feagan, Western University, London, ON, Canada; Remo Panaccione, University of Calgary, Inflammatory Bowel Disease Unit, Calgary, Alberta, Canada; Jean‐Frédéric Colombel, Icahn School of Medicine at Mount Sinai Hospital, New York, NY, USA; William J. Sandborn, University of California San Diego, La Jolla, USA; Silvio Danese, Humanitas Clinical and Research Center – IRCCS; Humanitas University, Department of Biomedical Sciences, Milan, Italy; Geert D’Haens, Amsterdam University Medical Centers, Amsterdam, Netherlands; David T. Rubin, University of Chicago Medicine Inflammatory Bowel Disease Center, Chicago, IL, USA; Ira Shafran, Shafran Gastroenterology Research Center, Winter Park, FL, USA; Andrejus Parfionovas, Raquel Rogers, and Richard A. Lirio, Takeda Development Center Americas Inc, Cambridge, MA, USA; Séverine Vermeire, University Hospitals Leuven, Leuven, Belgium
Loftus EV Jr, Feagan BG, Panaccione R, et al. Long‐term safety of vedolizumab for inflammatory bowel disease. Aliment Pharmacol Ther. 2020;52:1353–1365. 10.1111/apt.16060
The Handling Editor for this article was Professor Jonathan Rhodes, and it was accepted for publication after full peer‐review.
The complete list of authors’ affiliation list are listed in Appendix 1.
Funding information
This study was sponsored by Takeda. Medical writing support was provided by Liz Gooch, PhD, of ProEd Communications, Inc, and was funded by Takeda.
REFERENCES
- 1. Torres J, Mehandru S, Colombel J‐F, Peyrin‐Biroulet L. Crohn's disease. Lancet. 2017;389:1741‐1755. [DOI] [PubMed] [Google Scholar]
- 2. Ungaro R, Mehandru S, Allen PB, Peyrin‐Biroulet L, Colombel J‐F. Ulcerative colitis. Lancet. 2017;389:1756‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. ENTYVIO (vedolizumab) prescribing information. Takeda Pharma/S. Revised 04/01/2019.
- 4. Vedolizumab [summary of product characteristics]. Taastrup, Denmark: Takeda Pharma A/S; Revised 04/01/2019. [Google Scholar]
- 5. Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369:699‐710. [DOI] [PubMed] [Google Scholar]
- 6. Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. 2013;369:711‐721. [DOI] [PubMed] [Google Scholar]
- 7. Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn's disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147:618‐627.e3. [DOI] [PubMed] [Google Scholar]
- 8. Loftus EV Jr, Colombel JF, Feagan BG, et al. Long‐term efficacy of vedolizumab for ulcerative colitis. J Crohns Colitis. 2017;11:400‐411. [DOI] [PubMed] [Google Scholar]
- 9. Vermeire S, Loftus EV Jr, Colombel JF, et al. Long‐term efficacy of vedolizumab for Crohn's disease. J Crohns Colitis. 2017;11:412‐424. [DOI] [PubMed] [Google Scholar]
- 10. Colombel J‐F, Sands BE, Rutgeerts P, et al. The safety of vedolizumab for ulcerative colitis and Crohn's disease. Gut. 2017;66:839‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. König H‐H, Ulshöfer A, Gregor M, et al. Validation of the EuroQol questionnaire in patients with inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2002;14:1205‐1215. [DOI] [PubMed] [Google Scholar]
- 12. McColl E, Han SW, Barton JR, Welfare MR. A comparison of the discriminatory power of the Inflammatory Bowel Disease Questionnaire and the SF‐36 in people with ulcerative colitis. Qual Life Res. 2004;13:805‐811. [DOI] [PubMed] [Google Scholar]
- 13. Pallis AG, Mouzas IA, Vlachonikolis IG. The inflammatory bowel disease questionnaire: a review of its national validation studies. Inflamm Bowel Dis. 2004;10:261‐269. [DOI] [PubMed] [Google Scholar]
- 14. McAuliffe ME, Lanes S, Leach T, et al. Occurrence of adverse events among patients with inflammatory bowel disease in the HealthCore Integrated Research Database. Curr Med Res Opin. 2015;31:1655‐1664. [DOI] [PubMed] [Google Scholar]
- 15. Colombel J‐F, Sandborn WJ, Reinisch W, et al. Long‐term safety of adalimumab in clinical trials in adult patients with Crohn's disease or ulcerative colitis. Aliment Pharmacol Ther. 2018;47:219‐228. [DOI] [PubMed] [Google Scholar]
- 16. Sands BE, Peyrin‐Biroulet L, Loftus EV, et al. Vedolizumab versus adalimumab for moderate‐to‐severe ulcerative colitis. N Engl J Med. 2019;381:1215‐1226. [DOI] [PubMed] [Google Scholar]
- 17. Wyant T, Leach T, Sankoh S, et al. Vedolizumab affects antibody responses to immunisation selectively in the gastrointestinal tract: randomised controlled trial results. Gut. 2015;64:77‐83. [DOI] [PubMed] [Google Scholar]
- 18. Card T, Xu J, Liang H, Bhayat F. What is the risk of progressive multifocal leukoencephalopathy in patients with ulcerative colitis or crohn's disease treated with vedolizumab? Inflamm Bowel Dis. 2018;24:953‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Magro F, Peyrin‐Biroulet L, Sokol H, et al. Extra‐intestinal malignancies in inflammatory bowel disease: results of the 3rd ECCO Pathogenesis Scientific Workshop (III). J Crohns Colitis. 2014;8:31‐44. [DOI] [PubMed] [Google Scholar]
- 20. Gallagher MP, Kelly PJ, Jardine M, et al. Long‐term cancer risk of immunosuppressive regimens after kidney transplantation. J Am Soc Nephrol. 2010;21:852‐858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mariette X, Matucci‐Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta‐analysis. Ann Rheum Dis. 2011;70:1895‐1904. [DOI] [PubMed] [Google Scholar]
- 22. Feagan BG, Sandborn WJ, Colombel J‐F, et al. Incidence of arthritis/arthralgia in inflammatory bowel disease with long‐term vedolizumab treatment: post hoc analyses of the GEMINI trials. J Crohns Colitis. 2019;13:50‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material