

Abstract
We investigated the effects of cannabidiol (CBD; 21‐day maintenance dose) on the pharmacokinetics (PK) of clobazam (CLB) and monitored the safety of CBD (or placebo) plus CLB in 20 patients with uncontrolled epilepsy on stable doses of CLB. Blood samples collected until 24 hours postdose were evaluated by liquid chromatography tandem mass spectrometry. PK parameters of CLB and major metabolite N‐desmethylclobazam (N‐CLB), valproic acid, stiripentol, levetiracetam, topiramate, plant‐derived highly purified CBD (Epidiolex in the United States; 100 mg/mL oral solution) and its major metabolites were derived using noncompartmental analysis. There was no evidence of a drug‐drug interaction (DDI) between CBD and CLB: geometric mean ratio (GMR) of day 33:day 1 CLB was 1.0 (90%CI, 0.8‐1.2) for Cmax and 1.1 (90%CI, 0.9‐1.2) for AUCtau. There was a significant DDI between CBD and N‐CLB: the GMR of day 33:day 1 N‐CLB was 2.2 (90%CI, 1.4‐3.5) for Cmax and 2.6 (90%CI, 2.0‐3.6) for AUCtau. Placebo had no effect on CLB or N‐CLB; CBD had no effect on levetiracetam. Data were insufficient regarding DDIs with other antiepileptic drugs. The safety profile of CBD (20 mg/kg/day) with CLB was acceptable; all but 1 adverse events (AEs) were mild or moderate. One serious AE (seizure cluster) led to CBD discontinuation. One patient withdrew after intolerable AEs. Although there was no evidence of a CBD and CLB DDI, there was a significant DDI between CBD and N‐CLB. The safety profile of GW Pharmaceuticals’ CBD formulation with CLB was consistent with other GW‐sponsored trials.
Keywords: cannabinoid, cannabidiol, clobazam, drug, interaction, epilepsy
Highly purified cannabidiol (CBD) oral solution is approved for the treatment of seizures associated with Lennox‐Gastaut syndrome or Dravet syndrome (DS) in patients ≥2 years of age 1 , 2 , 3 , 4 , 5 , 6 and is available in the United States as Epidiolex and in Europe as Epidyolex, where it is used in conjunction with clobazam (CLB). Data from nonclinical studies and the scientific literature support at least 3 targets for the mechanism of anticonvulsant action of CBD: modulation of intracellular Ca2+ by antagonism of G‐protein‐coupled receptor 55 and desensitization of transient receptor potential vanilloid type 1 channels, and inhibition of adenosine reuptake via inhibition of the equilibrative nucleoside transporter 1. 7 , 8 , 9 Importantly, CBD lacks detectable euphoric effects associated with a propensity for abuse because of limited or no interaction with the cannabinoid receptors, CB1 and CB2. 10
CBD is extensively metabolized in the liver by cytochrome P450 (CYP) enzymes, mainly CYP2C19 and, to a lesser extent, CYP3A4. 11 , 12 These CYP enzymes are induced by several antiepileptic drugs (AEDs) (eg, carbamazepine, topiramate, and phenytoin) and inhibited by others (eg, valproic acid [VPA]). CBD may also inhibit the CYP2C family of isozymes. 12 , 13 In vivo data show that coadministration of CBD increases plasma concentrations of CYP2C19 substrates and may increase the risk of adverse reactions with these substrates. 4
CLB is also extensively metabolized in the liver, mainly by CYP3A, with minor contributions from CYP2B6 and CYP2C19, resulting in formation of its active metabolite, N‐desmethylclobazam (N‐CLB). N‐CLB is then further metabolized by CYP2C19. 14 , 15 Based on animal and in vitro receptor‐binding data, the relative potency of N‐CLB is estimated to be between one‐fifth to equally as potent as CLB, and moderate‐to‐strong inhibitors of CYP2C19 may result in increased exposure to N‐CLB. 16 Indeed, several uncontrolled studies found that levels of N‐CLB are increased by CBD. 17 , 18 , 19 As such, a formal assessment of any drug‐drug interaction (DDI) between CBD and CLB (and N‐CLB) in patients with epilepsy is important and of clinical interest.
Methods
Trial Design
All relevant trial‐related documents, including the protocol, were reviewed by the Essex Research Ethics Committee (UK) and the Unidad de Soporte al CEIC ethics committee (Spain), and approval for the trial was granted on March 20, 2015. All patients provided written informed consent for participation in the trial, which was performed in full conformity with the current Declaration of Helsinki, 20 the International Council for Harmonisation guidelines for Good Clinical Practice, 21 and all other applicable regulations. The trial was performed between January 20, 2016, and July 21, 2016, at 8 sites (The Barberry, Birmingham, UK; Royal Sussex County Hospital, Brighton, UK; Epilepsy Unit, Western Infirmary, Glasgow, UK; Department of Neurology, Leeds General Infirmary, Leeds, UK; Department of Neurology, Salford Royal NHS Foundation Trust, Salford, UK; Department of Neurology, Hospital Clinic, Barcelona, Spain; Hospital Universitario La Paz, Department of Neurology, Madrid, Spain; Hospital Universitari Vall d'Hebron, Barcelona, Spain). The trial was performed considering the European Medicines Agency 22 recommendations for clinical investigation of medicinal products in the treatment of epileptic disorders and the Food and Drug Administration guidance for the clinical evaluation of AEDs in adults and children. 23
The trial enrolled adult patients with poorly controlled epilepsy already on a stable dose of CLB. Patients were randomized 4:1 to receive a pharmaceutical formulation of highly purified CBD derived from Cannabis sativa L. plant in an oral solution (100 mg/mL Epidiolex in the United States; GW Research Ltd., Cambridge, UK) or placebo from days 2 to 33. On day 1, patients did not receive CBD, but they were already receiving stable doses of CLB. Patients titrated their CBD or placebo dose for 10 days (days 2 to 11) to 20 mg/kg/day CBD (or placebo equivalent volume) split into 2 equal doses (twice daily), taken immediately after the patients’ CLB dose. The titration period was followed by a 21‐day maintenance dose period (days 12 to 32). This maintenance period duration was chosen to provide a reliable steady‐state assessment. The final morning dose of CBD and final PK visit were on day 33. Patients then tapered their CBD or placebo dose or were invited into an open‐label extension (OLE) study.
Inclusion and Exclusion Criteria
The inclusion criteria specified that the trial population consisted of male and female patients with epilepsy aged 18‐65 years, taking stable CLB (≤20 mg/day for 4 weeks prior to screening), and no more than 2 other AEDs.
Female subjects of childbearing potential were nonpregnant and nonlactating at screening. Male and female subjects agreed to use highly effective contraception for the duration of the trial and for 3 months thereafter. Patients had experienced at least 1 seizure of any type within the 2 months prior to randomization. Patients had no other clinically significant illness in the 4 weeks prior to screening or any significantly impaired hepatic function.
Trial Assessments
Materials
Reference and internal standards for CLB and N‐CLB bioanalysis were supplied by Cerilliant (Round Rock, Texas). Reference and internal standards for CBD, 6‐hydroxy‐cannabidiol (6‐OH‐CBD), 7‐hydroxy‐cannabidiol (7‐OH‐CBD), and 7‐carboxy‐cannabidiol (7‐COOH‐CBD) bioanalysis were supplied by GW Research Ltd., Cerilliant, or BDG Synthesis (Wellington, New Zealand). As Δ9‐tetrahydrocannabinol (THC) is present as a trace impurity in the CBD formulation measured in this trial, plasma concentrations of THC, 11‐hydroxy‐Δ9‐tetrahydrocannabinol (11‐OH‐THC), and 11‐nor‐9‐carboxy‐Δ9‐tetrahydrocannabinol (11‐COOH‐THC), and reference and internal standards for each analyte were supplied by GW Research Ltd., Cerilliant, or BDG Synthesis.
Plasma Sample Preparations
CBD and metabolite samples were extracted from plasma by protein precipitation with isopropyl alcohol and acetonitrile. THC and metabolite samples were extracted by liquid‐liquid extraction. CLB and N‐CLB samples were extracted by protein precipitation.
Bioanalysis and Pharmacokinetic Assessment
Using validated high‐performance liquid chromatography (HPLC) with tandem mass spectrometry methods, plasma concentrations of CBD and its major metabolites, THC and its metabolites were determined at LGC (Fordham, UK). Plasma concentrations of clobazam and N‐CLB were determined at Covance Laboratories Ltd (Harrogate, UK). Details of the analytical procedures used are provided in Table 1. Internal standards were d3‐CBD, d3‐THC d9‐11‐COOH‐THC, d3‐11‐OH‐THC (all supplied by Cerilliant), d5‐7‐COOH‐CBD, and d5‐7‐OH‐CBD (both supplied by BDG Synthesis), and d5‐CLB and d5‐N‐CLB (both supplied by Alsachim, Illkirch‐Graffenstaden, France). There were no significant interfering peaks observed at the retention times for any of the analytes, indicating adequate selectivity of the methods. The precision (coefficient of variation [%CV]) and accuracy (relative error [RE%]/mean % different [Bias%]) of the HPLC method were acceptable for all analytes (≤15% [20% at the lower limit of quantification (LLOQ)]).
Table 1.
Bioanalytical Methods Used
| Laboratory | LGC Fordham | Covance Harrogate | ||
|---|---|---|---|---|
| Assay Validation | CBD and Metabolite Assays | THC and Metabolite Assays | CLB and N‐CLB Assays | |
| Range | Low range | High range | Single range | Single range |
| Sample volume (μL) | 200 | 200 | 100 | 25 |
| Extraction technique | Protein precipitation | Protein precipitation | Liquid‐liquid extraction | Protein precipitation |
| Mobile phase |
A — 0.1% ammonia in methanol B — 5 mM ammonium formate (pH 9) |
A — 0.1% ammonia in isopropanol; methanol (20:80) B — 5 mM ammonium formate (pH 9) |
A — water:formic acid (100:0.05) B — methanol:formic acid (100:0.05) |
|
| CBD | THC | CLB | ||
|---|---|---|---|---|
| MS mode | APCI positive | APCI positive | ESI positive | ESI positive |
| MS/MS transition | 315→193 | 315→193 | 315→193 | 301→259 |
| LLOQ (ng/mL) | 2 | 90 | 0.125 | 1 |
| ULOQ (ng/mL) | 2000 | 10 000 | 62.5 | 400 |
| Curve parameters | Quadratic (1/x) | Quadratic (1/x) | Linear (1/x2) | Linear (1/x2) |
| 7‐OH‐CBD | 11‐OH‐THC | N‐CLB | ||
|---|---|---|---|---|
| MS mode | APCI negative | APCI negative | ESI positive | ESI positive |
| MS/MS transition | 329→268 | 329→268 | 331→193 | 287→245 |
| LLOQ (ng/mL) | 0.25 | 11.25 | 0.25 | 1 |
| ULOQ (ng/mL) | 250 | 1250 | 125 | 400 |
| Curve parameters | Linear (1/x2) | Linear (1/x2) | Linear (1/x2) | Linear (1/x2) |
| 7‐COOH‐CBD | 11‐COOH‐THC | |||
|---|---|---|---|---|
| MS mode | APCI negative | APCI negative | ESI negative | |
| MS/MS transition | 343→179 | 343→179 | 343→245 | |
| LLOQ (ng/mL) | 0.25 | 180 | 0.25 | |
| ULOQ (ng/mL) | 250 | 20000 | 125 | |
| Curve parameters | Linear (1/x2) | Quadratic (1/x) | Linear (1/x2) | |
7‐COOH‐CBD, 7‐carboxy‐cannabidiol; 7‐OH‐CBD, 7‐hydroxy‐cannabidiol; 11‐OH‐THC, 11‐hydroxy‐tetrahydrocannabinol; APCI, atmospheric pressure chemical ionization; CBD, cannabidiol; CLB, clobazam; ESI, electrospray ionization; LLOQ, lower limit of quantification; MS, mass spectrometry; N‐CLB, N‐desmethylclobazam; THC, tetrahydrocannabinol; ULOQ, upper limit of quantification.
The recovery of CBD, 6‐OH‐CBD, 7‐OH‐CBD, and 7‐COOH‐CBD from human plasma was between 92% and 100% at the 3 concentrations tested (low, medium, and high), and was considered acceptable. The recovery of THC, 11‐OH‐THC, and 11‐COOH‐THC from human plasma was between 56.3% and 124% at the 3 concentrations tested (low, medium, and high). Although the recovery for THC and its metabolites varied between quality control levels, this did not affect the linearity of the assay and therefore was not considered to impact the validity of the data. The recovery was 97.4% for CLB and 100.7% for N‐CLB; these were considered adequate to obtain precise and accurate quantification of the analytes.
Blood samples were taken from patients via an indwelling intravenous catheter or direct venipuncture. Resultant plasma was stored in a freezer at −80°C. Blood samples for pharmacokinetic (PK) analysis were taken at the following times: predose and then 0.25, 0.5, 1, 1.5, 2, 4, 6, 12, and 24 hours postdose.
PK parameters were determined by noncompartmental analysis. PK parameters evaluated included maximum measured plasma concentration (Cmax), area under the plasma concentration‑time curve over a dosing interval, where tau is the dosing interval (AUCtau), and time to maximum plasma concentration (tmax).
Safety Assessments
The safety and tolerability of CBD, including abuse liability and withdrawal symptoms, were evaluated by recording the incidence and severity of adverse events (AEs) throughout the trial, review of clinical laboratory tests, vital signs, 12‐lead electrocardiogram, physical examinations, seizure diary, and Columbia‐Suicide Severity Rating Scale (C‐SSRS). If there were symptoms of toxicity suspected to be from a DDI, the principal investigator was permitted to adjust the dose of CBD/placebo, CLB, or other AEDs after discussion with the medical monitor.
Statistical Analysis
The primary objective of the trial was to determine whether CBD affected the PK profile of CLB and its major metabolite, N‐CLB, in patients with epilepsy. Secondary objectives were to evaluate the safety and tolerability of CBD in patients with epilepsy taking CLB. Descriptive statistics of patient demographics and safety outcomes were based on the safety analysis set (all patients who received any CBD).
All PK analyses were performed on the PK analysis set, which included all patients who received CBD and provided sufficient data to derive PK parameters at PK visits on days 1 and 33. Noncompartmental methods were used to estimate PK parameters for all analytes with sufficient data above the LLOQ from the concentration‐time profiles. SAS statistical software version 9.2 or higher was used for descriptive statistics of the PK parameters, which included Cmax, AUCtau, and tmax.
To assess whether the presence of CBD altered the PK profile of CLB (or N‐CLB), a standard 90% confidence interval (CI) approach for the between‐time point ratios of geometric means of Cmax, AUCtau, and AUC0‐∞ was carried out on a logarithmic scale using a linear mixed‐effects model. The no‐effect boundary was set between 0.5 and 2.0, and if the 90%CI for the ratio of the geometric means of a PK variable fell within the interval (0.5‐2.0), a lack of meaningful effect was declared. The lower boundary aligns with the convention for classifying an investigational drug as a moderate or strong inducer (≥50% decrease in AUC), and the upper boundary aligns with the convention for classifying an investigational drug as a moderate or strong inhibitor (≥2‐fold increase in AUC), as per the US Food and Drug Administration Draft Guidance for Clinical Drug Interaction Studies, October 2017. 24 Estimates were back‐transformed to provide summaries on the original scale. The model included a fixed‐effect term for the PK assessment period. An unstructured covariance matrix was used. Kenward and Roger's method was used to calculate the denominator degrees of freedom for the fixed effects. The PK analyses were conducted using Phoenix WinNonlin.
For the descriptive statistics of PK parameters for CLB and N‐CLB, Cmax and AUCtau were dose‐normalized as Cmax and AUC divided by the dose (expressed in mg/kg).
Sample Size
There was no formal sample size calculation, and analyses were descriptive only. This trial was not designed to assess efficacy or exposure PK/pharmacodynamic relationships because of its short treatment duration (31 days), limited sample size (20 patients), and broad inclusion criteria for baseline seizure frequency and type (≥1 seizure of any type within 2 months before randomization).
Results
Subject Demographics
Patient disposition is summarized in Figure 1. A total of 20 patients were enrolled in a 4:1 ratio to receive CBD:placebo. Two patients withdrew from the trial (both taking CBD); 1 withdrew consent to participate after intolerable AEs, and the second was withdrawn by the investigator because of a serious AE of a cluster of 12 seizures. Seven patients (1 taking placebo and 6 taking CBD) were excluded from the PK analysis because of misdosing (n = 2), withdrawal of consent (n = 1), treatment‐emergent AEs (n = 2), and CLB dose reductions (n = 2).
Figure 1.
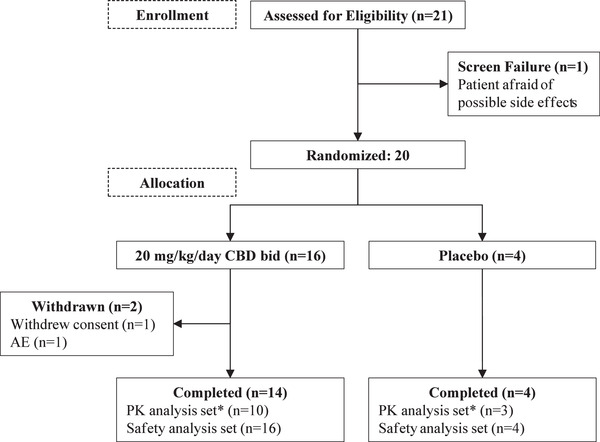
CONSORT flow chart of patient disposition.
*Included all patients who received ≥1 dose of study drug and provided sufficient PK data to derive PK parameters at Day 1 or 2 and at Day 33 or 34. Six CBD patients and 1 placebo patient were excluded owing to: CBD and CLB dose modifications (2 CBD patients), CLB dose modification (1 CBD patient), discontinued trial before the last visit (1 CBD patient), CBD dosed at 10 mg/kg/day instead of 20 mg/kg/day (1 CBD patient), stopped taking CBD before the last visit (1 CBD patient), and study drug taken after predose sampling (1 placebo patient). AE, adverse event; bid., twice daily; CBD, cannabidiol; CLB, clobazam; PK, pharmacokinetics.
The demographics were similar across the 2 treatment groups, with 10 male and 10 female subjects enrolled overall (8 of each sex in the CBD group and 2 of each sex in the placebo group). Most patients were white (19 [95%]), and 1 (5.0%) was Asian. Mean age was similar across the 2 treatment groups at 36.6 years in the CBD group and 37.6 years in the placebo group. Mean body mass index was 25.98 kg/m2 in the placebo group and 28.25 kg/m2 in the CBD group (Table 2).
Table 2.
Demographics and Baseline Characteristics; Safety Analysis Set
| Placebo (n = 4) | CBD (n = 16) | Total (n = 20) | |
|---|---|---|---|
| Number of Patients (%) | |||
| Sex | |||
| Male | 2 (50.0) | 8 (50.0) | 10 (50.0) |
| Female | 2 (50.0) | 8 (50.0) | 10 (50.0) |
| Race | |||
| White | 4 (100) | 15 (93.8) | 19 (95.0) |
| Asian | 0 | 1 (6.3) | 1 (5.0) |
| Mean (Standard Deviation) | |||
| Age, y | 37.57 (10.7) | 36.60 (8.5) | 36.79 (8.7) |
| BMI, kg/m2 | 25.98 (5.8) | 28.25 (5.2) | 27.80 (5.3) |
BMI, body mass index; CBD, cannabidiol.
All patients had a history of seizures, most commonly partial complex or focal onset with impaired awareness (15 patients overall [75%]), followed by secondarily generalized tonic‐clonic convulsions (6 patients overall [30%]). All patients had abnormal electroencephalogram or a history of abnormal neuroimaging at baseline.
Concomitant Medication
All patients were taking concomitant AEDs at baseline, as well as other medications. Per protocol, all patients were taking CLB (10‐20 mg/day in the CBD group; 5‐20 mg/day in the placebo group). The next most common AED was levetiracetam, taken by 9 patients overall (45%). Concomitant AEDs ongoing at baseline and taken during the trial are summarized in Table 3. Fifteen patients (75%) were taking other medications. The most common classes of other medication were antihistamines for systemic use (5 patients [25%]), followed by antianemic preparations (3 patients [15%]). The most common other concomitant medication was folic acid (3 patients [15%]).
Table 3.
Ongoing Antiepileptic Drugs at Trial Baseline and Taken During the Trial; Safety Analysis Set
| Antiepileptic Drug | Placebo (n = 4) | CBD (n = 16) | Total (n = 20) |
|---|---|---|---|
| Number of Patients (%) | |||
| Clobazam | 4 (100) | 16 (100) | 20 (100) |
| Levetiracetam | 2 (50.0) | 7 (43.8) | 9 (45.0) |
| Carbamazepine | 1 (25.0) | 4 (25.0) | 5 (25.0) |
| Lacosamide | 2 (50.0) | 3 (18.8) | 5 (25.0) |
| Lamotrigine | 1 (25.0) | 4 (25.0) | 5 (25.0) |
| Valproic acid | 0 | 3 (18.8) | 3 (15.0) |
| Eslicarbazepine | 0 | 3 (18.8) | 3 (15.0) |
| Oxcarbazepine | 1 (25.0) | 2 (12.5) | 3 (15.0) |
| Perampanel | 1 (25.0) | 0 | 1 (5.0) |
| Phenobarbital | 0 | 1 (6.3) | 1 (5.0) |
CBD, cannabidiol.
Pharmacokinetics
CLB and N‐CLB Pharmacokinetic Data
CLB and N‐CLB PK parameters are presented in Table 4. Plasma concentration‐by‐time profiles for CLB and N‐CLB are shown in Figure 2a and Figure 2b, respectively.
Table 4.
Absolute and Dose‐Normalized Pharmacokinetic Parameters; Pharmacokinetic Analysis Set
| Placebo | CBD | Placebo | CBD | ||
|---|---|---|---|---|---|
| (n = 3) | (n = 10) | (n = 3) | (n = 10) | ||
| Geometric Mean (CV%)a | |||||
| Parameter | Day | CLB | CLB Dose‐Normalized | ||
| Cmax (ng/mL) | Day 1 | 440 (29.9) | 330 (40.4) | 22.0 (29.9) | 19.3 (31.4) |
| Day 33 | 461 (102.3) | 329 (54.8) | 23.1 (102.3) | 19.2 (44.7) | |
| AUCtau (ng·h/mL) | Day 1 | 3320 (67.5) | 2690 (52.9) | 166 (67.5) | 157 (47.1) |
| Day 33 | 3310 (102.5) | 2840 (46.2) | 165 (102.5) | 166 (34.7) | |
| tmax (h) | Day 1 | 1.2 (1.0‐1.5) | 1.0 (0.8‐4.0) | – | – |
| Day 33 | 1.6 (1.5‐2.0) | 1.9 (0.5‐4.0) | – | – | |
| N‐CLB | N‐CLB Dose‐Normalized | ||||
|---|---|---|---|---|---|
| Cmax (ng/mL) | Day 1 | 1130 (82.1) | 2060 (138.4) | 56.6 (82.1) | 121 (125.1) |
| Day 33 | 1320 (134.1) | 4570 (54.0) | 66.1 (134.1) | 267 (38.6) | |
| AUCtau (ng·h/mL) | Day 1 | 11 400 (63.7) | 18 300 (124.2) | 571 (63.7) | 1070 (105.1) |
| Day 33 | 11 500 (79.1) | 48 400 (53.9) | 573 (79.1) | 2830 (38.3) | |
| tmax (h) | Day 1 | 2.0 (0.0‐12.0) | 1.5 (0.3‐6.0) | — | — |
| Day 33 | 1.0 (0.3‐2.0) | 3.0 (0.0‐11.1) | — | — | |
AUCtau, area under the plasma concentration‑time curve over a dosing interval, where tau is the dosing interval; CBD, cannabidiol; CLB, clobazam; Cmax, maximum measured plasma concentration; CV, coefficient of variation; N‐CLB, N‐desmethylclobazam; tmax, time to maximum plasma concentration.
AUCtau and Cmax values were dose‐normalized by dividing the parameters by the dose expressed in mg/kg.
Except for tmax, which is presented as median (range).
Figure 2.
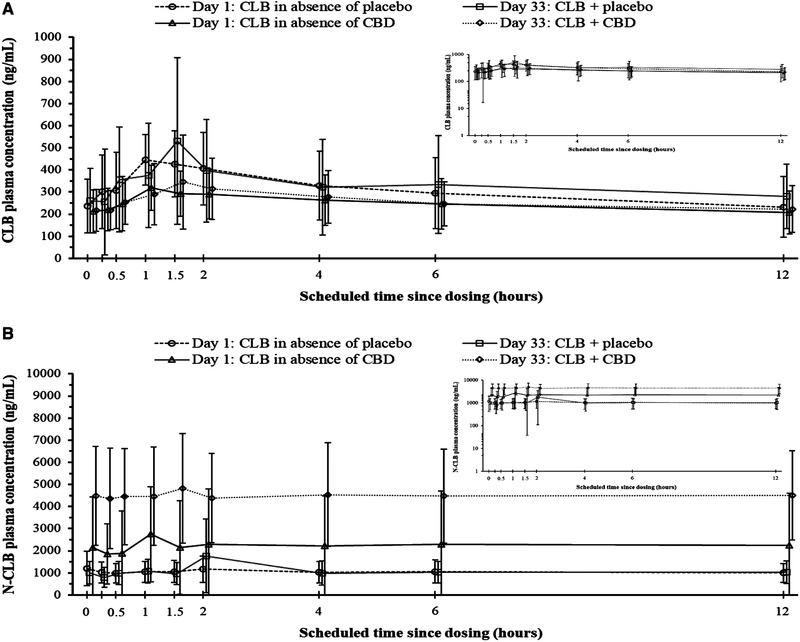
Mean (SD) (A) clobazam (CLB) and (B) N‐desmethylclobazam (N‐CLB) plasma concentrations versus time on Day 1 and Day 33 (linear scale; log‐linear scale values are shown in the insets) in the absence and presence of placebo or cannabidiol (CBD) coadministration; pharmacokinetics (PK) analysis set.
Concomitant administration of either placebo or CBD with CLB had no effect on CLB plasma concentrations (Figure 2a). Dose‐normalized mean values for exposure (Cmax and AUCtau) to CLB were unchanged between days 1 and 33 in both the placebo and CBD groups. Geometric mean ratios for CLB PK parameters showed no effect of CBD or placebo on CLB exposure on day 33 compared with day 1 (Table 5).
Table 5.
Geometric Mean Ratios of CLB and N‐CLB Pharmacokinetic Parameters on Day 33 Compared With Day 1; Pharmacokinetic Analysis Set
| Placebo | CBD | |
|---|---|---|
| (n = 3) | (n = 10) | |
| Day 33:Day 1 Ratio (90%CI) | ||
| Parameter | CLB | |
| Cmax | 1.1 (0.4‐2.7) | 1.0 (0.8‐1.2) |
| AUCtau | 1.0 (0.7‐1.5) | 1.1 (0.9‐1.2) |
| N‐CLB | ||
|---|---|---|
| Cmax | 1.2 (0.6‐2.2) | 2.2 (1.4‐3.5) |
| AUCtau | 1.0 (0.8‐1.3) | 2.6 (2.0‐3.6) |
AUCtau, area under the plasma concentration‑time curve over a dosing interval, where tau is the dosing interval; CBD, cannabidiol; CI, confidence interval; CLB, clobazam; Cmax, maximum measured plasma concentration; N‐CLB, N‐desmethylclobazam.
On day 1, the median CLB tmax was 1.2 hours (range, 1.0‐1.5 hours) in the placebo group and 1.0 hour (range, 0.8‐4.0 hours) in the CBD group. On day 33, the median tmax increased in both groups, to 1.6 hours (range, 1.5‐2.0 hours) in the placebo group and 1.9 hours (range, 0.5‐4.0 hours) in the CBD group.
There was little change in N‐CLB plasma concentrations between days 1 and 33 in the placebo group. In the CBD group, increases in N‐CLB plasma concentrations were observed between days 1 and 33 (Figure 2b).
In the CBD group, dose‐normalized mean values for exposure to N‐CLB were higher on day 33 than on day 1. Values were unchanged in the placebo group. Geometric mean day 33:day 1 ratios for CLB PK parameters showed evidence of a DDI between CBD and N‐CLB; ratios for Cmax and AUCtau, all >2 with CBD treatment, and the 90%CIs were outside the no‐effect boundary of 0.5 and 2.024 (Table 6).
Table 6.
Pharmacokinetic Parameter Data for CBD and Its Major Metabolites on Day 33; Pharmacokinetic Analysis Set (n = 10)
| Analyte | tmax (h)a | Cmax (ng/mL)b | AUCtau (ng·h/mL)b |
|---|---|---|---|
| CBD | 3.1 (1.5‐11.0) | 546 (54.0) | 3560 (45.2) |
| 6‐OH‐CBD | 4.0 (2.0‐11.0) | 15.5 (30.8) | 118 (29.6) |
| 7‐COOH‐CBD | 4.0 (0.5‐6.2) | 17 800 (56.6) | 165 000 (77.9) |
| 7‐OH‐CBD | 4.0 (2.0‐11.0) | 211 (44.8) | 1550 (32.2) |
6‐OH‐CBD, 6‐hydroxy‐cannabidiol; 7‐COOH‐CBD, 7‐carboxy‐cannabidiol; 7‐OH‐CBD, 7‐hydroxy‐cannabidiol; AUCtau, area under the plasma concentration‑time curve over a dosing interval, where tau is the dosing interval; CBD, cannabidiol; Cmax, maximum measured plasma concentration; CV, coefficient of variation; tmax, time to maximum plasma concentration.
Median and range.
Geometric mean and CV%.
The median N‐CLB tmax was 2.0 hours (range, 0.0‐12.0 hours) in the placebo group and 1.5 hours (range, 0.3‐6.0 hours) in the CBD group on day 1. On day 33, the median tmax was 1.0 hour (range, 0.3‐2.0 hours) in the placebo group and increased to 3.0 hours (range, 0.0‐11.1 hours) in the CBD group.
On day 1, geometric mean N‐CLB:CLB ratios for Cmax and AUCtau were 6.3 and 6.8, respectively, in the CBD group, and 2.6 and 3.4, respectively, in the placebo group. On day 33, the Cmax and AUCtau ratios increased to 13.9 and 17.1, respectively, in the CBD group and to 2.9 and 3.5, respectively, in the placebo group. As shown, in the placebo group, mean N‐CLB:CLB exposure ratios were similar on days 1 and 33.
CBD and Metabolite Pharmacokinetic Data
PK parameters of CBD and its major metabolites are summarized in Table 4. Following 21 days of maintenance dosing of 20 mg/kg/day CBD taken concomitantly with CLB, geometric mean Cmax values were 546 ng/mL for CBD, 15.5 ng/mL for 6‐OH‐CBD, 17 800 ng/mL for 7‐COOH‐CBD, and 211 ng/mL for 7‐OH‐CBD. Geometric mean AUCtau values were 3560 ng/mL for CBD, 118 ng/mL for 6‐OH‐CBD, 165 000 ng/mL for 7‐COOH‐CBD, and 1550 ng/mL for 7‐OH‐CBD.
The tmax values were similar across the major CBD metabolites, 6‐OH‐CBD, 7‐COOH‐CBD, and 7‐OH‐CBD (median tmax, 4.0 hours for all metabolites; range, 0.5‐11.0 hours across metabolites). CBD had an earlier tmax (median, 3.1 hours; range, 1.5‐11.0 hours). The most abundant metabolite was 7‐COOH‐CBD (metabolite‐to‐parent ratios [MR] for Cmax and AUCtau were 32.6 and 51.5, respectively), followed by 7‐OH‐CBD (MR for Cmax and AUCtau were both 0.4), and 6‐OH‐CBD being the least abundant (MR for Cmax and AUCtau were both 0.03).
THC Plasma Concentrations
After 21 days of maintenance dosing of 20 mg/kg/day CBD taken concomitantly with CLB, plasma concentrations of THC were below the limit of quantification (<0.125 ng/mL) for all patients in the CBD group predose and for most patients (9 of 10 [90%]) up to 12 hours postdose. Trace levels of THC were observed (range, 0.1‐0.4 ng/mL) in 6 of 10 patients (60%) 2 hours postdose and 6 of 10 patients (60%) 4 hours postdose.
Interpatient Variability
Interpatient variability (CV%) in exposure parameters (Cmax and AUC) for all analytes investigated ranged from moderate (29.9%) to high (138.4%) throughout the trial (Table 4).
Safety
All AEs experienced by >1 patient overall using the Medical Dictionary for Regulatory Activities Preferred Terms are presented in Table 7. Thirteen patients taking CBD (81.3%) and 2 patients taking placebo (50%) experienced 1 or more AEs. In the CBD group, most AEs were mild (5 [31.3%]) or moderate (7 [43.8%]), and 1 (6.3%) was severe. All AEs in the placebo group were mild. There were no deaths or pregnancies. Diarrhea, nausea, vomiting, sedation, somnolence, and dizziness were the most common AEs reported overall. One patient taking CBD (6.3%) experienced a serious AE of seizure cluster that led to withdrawal from the trial. In the CBD group, during the final trial visit, 2 patients (12.5%) had AEs related to raised transaminases, including alanine aminotransferase >3× the upper limit of normal; 1 of these patients was taking VPA concomitantly. Neither patient showed symptoms or signs of clinical hepatitis, and both completed the double‐blind phase of the trial without meeting Hy's Law criteria. Both patients were withdrawn from the follow‐up OLE study, and the AEs recovered following discontinuation of CBD. No clear trends in changes in other laboratory parameters were identified.
Table 7.
All‐Causality Treatment‐Emergent Adverse Events Experienced by >1 Patient Overall, by MedDRA System Organ Class and Preferred Terms; Safety Analysis Set
| System Organ Class | Placebo | CBD | Total |
|---|---|---|---|
| Preferred Term | (n = 4), n (%) | (n = 16), n (%) | (n = 20), n (%) |
| Subjects experiencing any AEs | 2 (50.0) | 13 (81.3) | 15 (75.0) |
| Gastrointestinal disorders | 1 (25.0) | 8 (50.0) | 9 (45.0) |
| Diarrheaa | 1 (25.0) | 6 (37.5) | 7 (35.0) |
| Nausea | 0 | 3 (18.8) | 3 (15.0) |
| Vomiting | 0 | 3 (18.8) | 3 (15.0) |
| Nervous system disorders | 0 | 9 (56.3) | 9 (45.0) |
| Dizziness | 0 | 2 (12.5) | 2 (10.0) |
| Sedation | 0 | 2 (12.5) | 2 (10.0) |
| Somnolence | 0 | 2 (12.5) | 2 (10.0) |
| Skin and subcutaneous tissue disorders | 0 | 6 (37.5) | 6 (30.0) |
| Dermatitis | 0 | 2 (12.5) | 2 (10.0) |
AE, treatment‐emergent adverse event; CBD, cannabidiol; MedDRA, Medical Dictionary for Regulatory Activities.
MedDRA preferred term “diarrhoea.”
There were AEs in 3 patients (18.9%) leading to reductions in CBD dose. These events included a rash, diarrhea, and multiple events of sedation, slurred speech, and word‐finding difficulties. All resolved following reduction in the CBD dose, and all patients remained on CBD after resolution of symptoms. One patient in the CBD group (6.3%) had a dose reduction in CLB from 15 to 5 mg/day because of sedation. AEs relating to vital signs were few and not clinically important.
Results of the C‐SSRS identified no treatment‐emergent suicidal ideation or behavior in patients taking CBD during the trial. There was no evidence of abuse liability or signs of withdrawal between the treatment groups.
Seizure activity was monitored as a safety end point: 9 patients in the CBD group (56.3%) reported improvement in seizure activity compared with 1 patient in the placebo group (25.0%).
Discussion
This is the first randomized, controlled trial to investigate the DDI between this oral CBD formulation and CLB in patients with epilepsy. The trial was designed as a standard intrapatient DDI assessment, whereby each patient served as his or her own control. Therefore, even with a relatively low number of patients enrolled, there was adequate power to describe the magnitude of the DDI. Patients enrolled in the trial were taking stable doses of CLB and no more than 2 other AEDs to limit the influence of other extrinsic factors on the specific DDI under investigation.
Effect of CBD on CLB and N‐CLB
CLB and N‐CLB plasma concentrations in the absence (predose on day 1) and presence (predose on day 33) of CBD demonstrated a PK interaction between CBD and N‐CLB, but not between CBD and CLB. These differences were reflected in the exposures (Cmax and AUCtau) for CLB and N‐CLB on day 33 compared with day 1; N‐CLB Cmax increased by 2.2‐fold and AUCtau increased by 2.6‐fold in the CBD group. These findings are consistent with those from a noncomparative, investigator‐led trial in children with refractory epilepsies, which reported a nonsignificant mean increase in CLB and a significant increase in N‐CLB (of 500% ± 300%) after 4 weeks of 20 mg/kg/day CBD treatment compared with pretreatment. 19 Similar findings were reported in a randomized, controlled trial in pediatric patients with DS who took multiple CBD doses of 5, 10, or 20 mg/kg/day. In that trial, there were no relevant changes in CLB plasma exposures with CBD treatment; however, for N‐CLB, there was a mean increase of >166% in all dose groups. 3 The current trial findings, along with reported literature data, provide evidence of a DDI between CBD and N‐CLB, most likely resulting from inhibition of CYP2C19 by CBD. 2 , 3 , 6 , 19 Administration of CLB with other drugs that inhibit CYP2C19 has also been shown to result in increased exposure to N‐CLB, 16 and increased exposure to N‐CLB may also contribute to an increased incidence of certain AEs, such as somnolence. 3
Although the present trial did not assess the effect of CLB on CBD and its metabolites, a recent trial assessing the bidirectional DDI between CBD and CLB in healthy volunteers found that CLB also increased exposure of CBD's active metabolite, 7‐OH‐CBD. 25 This bidirectional interaction is important because increases in the 7‐OH‐CBD metabolite could also contribute to efficacy and adverse effects.
CBD and Metabolite Plasma Concentrations
Following 20 mg/kg/day dosing with CBD, plasma concentrations of CBD, 6‐OH‐CBD, 7‐COOH‐CBD, and 7‐OH‐CBD were measured on day 33 at steady state. The 7‐COOH‐CBD metabolite was the major circulating product, followed by CBD and 7‐OH‐CBD, with relatively low concentrations of the 6‐OH‐CBD metabolite being observed in plasma (consistently <10% that of CBD). These findings are consistent with previously reported trials with CBD. 3 , 26
The moderate‐to‐high interpatient variability seen in this trial—possibly related to intrinsic and extrinsic factors not fully evaluated in this trial—is common with cannabinoids and has been previously documented. 27
THC Plasma Concentrations
Because GW's formulation of CBD contains trace THC of not more than 0.1% weight for weight active pharmaceutical ingredient, plasma concentrations of THC and its metabolites were also monitored in this trial. Throughout the trial, THC exposure was low, with plasma concentrations below the limit of quantification in most patients at most times.
Safety
CBD at 20 mg/kg/day coadministered with CLB had an acceptable safety profile in most patients. During the trial, there were more AEs in patients taking CBD than placebo. One patient in the CBD group withdrew because of a serious AE of seizure cluster that subsequently resolved. The safety findings in this trial are consistent with other trials with this formulation of CBD in patients with certain forms of epilepsy. 1 , 3 , 19 , 28
Most patients attained the target dose and completed the trial, and there were no deaths during the trial. Most patients in the CBD group (9 [56%]) reported ≥25% improvement in seizures, compared with 1 patient in the placebo group (25%).
Two patients taking CBD experienced AEs related to increased hepatic transaminases. Hepatic transaminase‐related AEs have been reported previously in both open‐label and controlled trials of CBD for severe refractory epilepsies. 1 , 3 , 28 Importantly, none of the transaminase elevations in this trial met the criteria for Hy's Law.
Conclusions
When 20 mg/kg/day CBD was added to CLB in patients with uncontrolled epilepsy, there was no evidence of a DDI between parent compounds CBD and CLB; however, systemic exposure of N‐CLB (the major CLB metabolite) increased by 2‐ to 3‐fold. CBD had an acceptable safety profile at 20 mg/kg/day when coadministered with CLB in adult patients with epilepsy, but a reduction in dosage of CLB may be needed if adverse reactions known to occur with CLB are experienced.
Conflicts of Interest
K.V.L. is an employee of Greenwich Biosciences Inc. and owns share options in the company. J.C. and G.M. are employees of GW Research Ltd. and own share options in the company. L.T. was an employee of GW Research Ltd. when the trial was conducted.
Funding
This trial was sponsored by GW Research Ltd.
Data Accessibility
This trial is registered at www.clinicaltrials.gov. The identifier is: NCT02565108. Data will be made available on request.
Acknowledgments
The authors thank the patients who took part in the trial, as well as the staff that assisted with the trial at each site. In addition, the authors thank Graham Blakey of consult2deliver Ltd (Nottingham, UK) for his critical appraisal and review of this article, Nateisha Williams of Greenwich Biosciences (Carlsbad, California) for her medical writing support, Laura Riordan of L.E. Riordan Editorial Services (Chicago, Illinois) for her editorial support, and Angus Clayton of GW Research Ltd. (Cambridge, UK) for his contributions as the clinical study manager.
References
- 1. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011‐2020. [DOI] [PubMed] [Google Scholar]
- 2. Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox‐Gastaut syndrome. N Engl J Med. 2018;378(20):1888‐1897. [DOI] [PubMed] [Google Scholar]
- 3. Devinsky O, Patel AD, Thiele EA, et al. Randomized, dose‐ranging safety trial of cannabidiol in Dravet syndrome. Neurology. 2018;90(14):e1204‐e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greenwich Biosciences . EPIDIOLEX® (cannabidiol) oral solution prescribing information. Carlsbad, CA: Greenwich Biosciences; 2018.
- 5. GW Research . EPIDYOLEX® (cannabidiol oral solution) summary of product characteristics. B.V. Amersfoort, Nederlands: GW Research; 2019.
- 6. Thiele EA, Marsh ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox‐Gastaut syndrome (GWPCARE4): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2018;391(10125):1085‐1096. [DOI] [PubMed] [Google Scholar]
- 7. Henstridge CM, Balenga NA, Kargl J, et al. Minireview: recent developments in the physiology and pathology of the lysophosphatidylinositol‐sensitive receptor GPR55. Mol Endocrinol. 2011;25(11):1835‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ibeas Bih C, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. 2015;12(4):699‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sylantyev S, Jensen TP, Ross RA, Rusakov DA. Cannabinoid‐ and lysophosphatidylinositol‐sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc Natl Acad Sci U S A. 2013;110(13):5193‐5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McPartland JM, Duncan M, Di Marzo V, Pertwee RG. Are cannabidiol and Δ(9) ‐tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol. 2015;172(3):737‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89(5‐6):165‐170. [DOI] [PubMed] [Google Scholar]
- 12. Zendulka O, Dovrtelova G, Noskova K, et al. Cannabinoids and cytochrome P450 interactions. Curr Drug Metab. 2016;17(3):206‐226. [DOI] [PubMed] [Google Scholar]
- 13. Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46(1):86‐95. [DOI] [PubMed] [Google Scholar]
- 14. Giraud C, Tran A, Rey E, Vincent J, Treluyer JM, Pons G. In vitro characterization of clobazam metabolism by recombinant cytochrome P450 enzymes: importance of CYP2C19. Drug Metab Dispos. 2004;32(11):1279‐1286. [PubMed] [Google Scholar]
- 15. Volz M, Christ O, Kellner HM, et al. Kinetics and metabolism of clobazam in animals and man. Br J Clin Pharmacol. 1979;7(suppl 1):41S‐50S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lundbeck . ONFI® (clobazam): prescribing information. Deerfield, IL: Lundbeck; 2016.
- 17. Friedman D, Cilio MR, Tilton N, et al. The effect of Epidiolex (cannabidiol) on serum levels of concomitant anti‐epileptic drugs in children and young adults with treatment‐resistant epilepsy in an expanded access program. Paper presented at: American Epilepsy Society; December 5‐9, 2014; Seattle, WA. Abstract No. 2.309.
- 18. Gaston TE, Bebin EM, Cutter GR, Liu Y, Szaflarski JP, Program UC. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. 2017;58(9):1586‐1592. [DOI] [PubMed] [Google Scholar]
- 19. Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug‐drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015;56(8):1246‐1251. [DOI] [PubMed] [Google Scholar]
- 20. World Medical Association . World Medical Association Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. https://www.wma.net/what-we-do/medical-ethics/declaration-of-helsinki/doh-oct2000/. Published 2000. Accessed October 25, 2019.
- 21. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guidelines: Guidelines for Good Clinical Practice E6(R1) European Medicines Agency (EMA), Amsterdam, the Netherlands; 1996. https://www.ich.org/page/efficacy-guidelines. [Google Scholar]
- 22. Committee for Medicinal Products for Human Use . Guideline on Clinical Investigation of Medicinal Products in the Treatment of Epileptic Disorders. European Medicines Agency (EMA), Amsterdam, the Netherlands; 2010. [Google Scholar]
- 23. Center for Drug Evaluation and Research . Guidelines for the Clinical Evaluation of Antiepileptic Drugs (Adults and Children). Food and Drug Administration (FDA). Rockville, MD; 1981. [Google Scholar]
- 24. Center for Drug Evaluation and Research . Clinical Drug Interaction Studies — Study Design, Data Analysis, and Clinical Implications Guidance for Industry. Food and Drug Administration (FDA). Silver Spring, MD; 2017. [Google Scholar]
- 25. Morrison G, Crockett J, Blakey G, Sommerville K. A phase 1, open‐label, pharmacokinetic trial to investigate possible drug‐drug interactions between clobazam, stiripentol, or valproate and cannabidiol in healthy subjects. Clin Pharmacol Drug Dev. 2019;8(8):1009‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double‐blind, placebo‐controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32(11):1053‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770‐1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment‐resistant epilepsy: an open‐label interventional trial. Lancet Neurol. 2016;15(3):270‐278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This trial is registered at www.clinicaltrials.gov. The identifier is: NCT02565108. Data will be made available on request.