

Abstract
Objective
Calcitonin gene–related peptide (CGRP) pathway inhibitors are emerging treatments for migraine. CGRP‐mediated vasodilation is, however, a critical rescue mechanism in ischemia. We, therefore, investigated whether gepants, small molecule CGRP receptor antagonists, worsen cerebral ischemia.
Methods
Middle cerebral artery was occluded for 12 to 60 minutes in mice. We compared infarct risk and volumes, collateral flow, and neurological deficits after pretreatment with olcegepant (single or 10 daily doses of 0.1–1mg/kg) or rimegepant (single doses of 10–100mg/kg) versus vehicle. We also determined their potency on CGRP‐induced relaxations in mouse and human vessels, in vitro.
Results
Olcegepant (1mg/kg, single dose) increased infarct risk after 12‐ to 20‐minute occlusions mimicking transient ischemic attacks (14/19 vs 6/18 with vehicle, relative risk = 2.21, p < 0.022), and doubled infarct volumes (p < 0.001) and worsened neurological deficits (median score = 9 vs 5 with vehicle, p = 0.008) after 60‐minute occlusion. Ten daily doses of 0.1 to 1mg/kg olcegepant yielded similar results. Rimegepant 10mg/kg increased infarct volumes by 60% after 20‐minute ischemia (p = 0.03); 100mg/kg caused 75% mortality after 60‐minute occlusion. In familial hemiplegic migraine type 1 mice, olcegepant 1mg/kg increased infarct size after 30‐minute occlusion (1.6‐fold, p = 0.017). Both gepants consistently diminished collateral flow and reduced reperfusion success. Olcegepant was 10‐fold more potent than rimegepant on CGRP‐induced relaxations in mouse aorta.
Interpretation
Gepants worsened ischemic stroke in mice via collateral dysfunction. CGRP pathway blockers might thus aggravate coincidental cerebral ischemic events. The cerebrovascular safety of these agents must therefore be better delineated, especially in patients at increased risk of ischemic events or on prophylactic CGRP inhibition. ANN NEUROL 2020;88:771–784
Introduction
Migraine is a common paroxysmal neurovascular disorder, typically characterized by disabling attacks of headache, associated autonomic features, and in one‐third of patients, aura. Calcitonin gene–related peptide (CGRP) is an important neurotransmitter within the migraine headache–generating trigeminovascular system and believed to play a crucial role in migraine pathophysiology. 1 Gepants, short‐acting small molecule CGRP receptor antagonists, and long‐acting monoclonal antibodies targeting CGRP or the CGRP receptor have recently emerged as promising acute and prophylactic therapeutic options for migraine. 2 Analysts expect these drugs to be used by millions of migraineurs by 2027 in the G7 countries alone. 3
CGRP is also among the most potent vasodilators in animals and humans. 4 It induces endothelium‐independent vasodilation via direct action on vascular smooth muscle cells in cerebral and coronary vascular beds. Cerebral blood flow (CBF) autoregulation is in part mediated by CGRP. 5 Several lines of evidence suggest that CGRP‐mediated vasodilation is a rescue mechanism in brain ischemia. 6 CGRP released from trigeminal perivascular nerves 7 is a potent direct vascular smooth muscle dilator counteracting vasoconstriction and hypoperfusion in the cerebrovascular bed. 5 , 8 , 9 Low pH‐ and ischemia‐induced CGRP release from perivascular C‐fibers results in coronary and cerebrovascular vasodilation and restoration of circulation. 10 , 11 , 12 Treatment with CGRP improves and genetic ablation of CGRP worsens blood flow and outcomes after focal cerebral arterial occlusion in experimental animals. 13 , 14 , 15 Administration of CGRP has shown beneficial effects in peripheral and coronary artery disease 16 , 17 , 18 and ameliorated experimental cerebral and cardiac ischemia by preserving blood flow and the blood–brain barrier. 15 , 19 , 20 , 21 Consequently, inhibition of the CGRP system with gepants or monoclonal antibodies might theoretically worsen the outcome of coincidental coronary and cerebral ischemic events. 6
Despite these obvious concerns, remarkably few studies have addressed the cardiovascular safety of inhibitors of CGRP or its receptor. As far as we know, their cerebrovascular safety has never been studied. Patients with important vascular risk factors were even excluded from most clinical trials. 22 , 23 This is all the more surprising because migraine with aura is an established risk factor for ischemic stroke, especially in women. 24 The higher stroke risk is likely due to increased risk of experiencing cerebral ischemic events 25 in combination with enhanced susceptibility of brain tissue to infarction if and when such events occur. 26 , 27 We, therefore, investigated whether gepants worsen cerebral ischemic outcomes, and the mechanism of this effect, in established mouse models of transient ischemic attack (TIA) and stroke.
Materials and Methods
Experimental Animals and Design
Experiments were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and carried out in accordance with the Guide for Care and Use of Laboratory Animals (NIH Publication No. 85‐23, 1996). In addition to wild‐type mice (C57BL/6J), we also used heterozygous transgenic knock‐in mice expressing the human familial hemiplegic migraine type 1 (FHM1) S218L missense mutation in the α1A subunit of CaV2.1 voltage‐gated Ca2+ channels and their wild‐type littermates. 28 Mice were housed with their littermates, in cages with standard embedding and enrichment, reversed light/dark cycle, and food and water ad libitum. Whereas migraine is most prevalent among women aged <50 years, the vast majority of non–migraine‐related strokes occur in the elderly, including men. We, therefore, studied both young and aged mice (2–16 months), and both males and females. A priori sample size determinations aimed to achieve 80% power to detect a 33% effect size on infarct volume with 20% standard deviation of the mean (α = 0.05, β = 0.20). Animals were randomized to treatment arms using an online tool (CoinTosser, Hello Heydays and Excel 2016, Microsoft, Redmond, WA). Each treatment group had its own control group. Group sizes, exclusions, early mortality, and age and sex distributions for each experiment are shown in Table S1. All investigators were blinded to the treatment group during the surgical procedures, data collection, and analysis.
Treatments
Animals were treated with the small molecule CGRP receptor antagonists olcegepant (BIBN4096, 0.1 or 1mg/kg, 97.5% purity, intravenous in acute and intraperitoneal in chronic treatment; Tocris Bioscience, Bristol, UK) or rimegepant (BMS‐927711, 10 or 100mg/kg, intraperitoneal; 99.08% purity; MedChemExpress, Monmouth Junction, NJ), either with a single dose 10 minutes before ischemia onset (olcegepant and rimegepant) or with 10 once‐daily doses over 2 weeks followed by a single dose 10 minutes before ischemia onset (olcegepant only). These commercial vendors were selected based on previous literature. 29 , 30 , 31 Stock solutions were prepared in dimethyl sulfoxide and diluted as needed in saline (0.02–10mg/ml) to administer the target dose in 4 to 10μl volume per gram of body weight. Control groups received the same volume of identical vehicle via the same route. The selected olcegepant doses were within the previously reported dose range (1μg/kg–30mg/kg) in experimental models in rodents. 29 , 30 , 32 , 33 , 34 , 35 , 36 , 37 In rats, 1mg/kg olcegepant, which we used herein, has yielded a peak plasma concentration of 0.8μM compared with peak plasma concentrations of approximately 0.15μM after a 10mg dose in clinical studies. 34 , 38 , 39 , 40 , 41 Unfortunately, plasma concentrations of rimegepant have not been reported in rodents or in humans. Rimegepant doses were selected based on a single report in the marmoset, 42 taking into account (1) the significantly lower affinity of small molecule CGRP receptor antagonists in rodents compared with primates, 43 as we confirmed herein using isolated vessels; (2) in vitro data showing significantly lower binding affinity for rimegepant than olcegepant in isolated vessels 31 ; and (3) the 7‐fold higher efficacious human plasma concentrations of rimegepant compared with olcegepant in clinical trials. 38 , 44 , 45 Buprenorphine (0.08mg/kg, subcutaneous; Patterson Veterinary, Devens, MA) was given as analgesic prior to surgery.
Focal Cerebral Ischemia
All experiments were carried out under isoflurane anesthesia (3% induction, 1.5% maintenance in 70% N2O and 30% O2). Rectal temperature was maintained at 36.7 ± 0.3°C using a thermostatic heating pad (FHC, Bowdoinham, ME). Focal ischemia was induced using transient middle cerebral artery occlusion (MCAO) by a nylon monofilament (701723Re, Doccol Corporation, Sharon, MA). After a ventral midline neck incision (1–2cm), common carotid artery bifurcation was gently dissected free of connected tissue, taking care to preserve the vagus nerve. A closed system around the bifurcation was created, and the filament was inserted into the external carotid artery, retrogradely guided into the internal carotid artery, and advanced until the origin of the middle cerebral artery. After 12 to 60 minutes of occlusion, the filament was removed to achieve reperfusion. CBF was monitored (% of baseline) using laser Doppler flowmetry in ischemic core (PeriFlux System 5000, Perimed, Järfälla‐Stockholm, Sweden). In experiments with short occlusion times mimicking TIA in wild‐type mice, and with 30‐minute ischemia in FHM1 mutant mice, CBF was recorded throughout the MCAO and reperfusion. In 60‐minute ischemia experiments, animals were allowed to recover from anesthesia shortly after MCAO, and reanesthetized for the reperfusion procedure; therefore, in this cohort CBF monitoring was discontinued after MCAO. After the surgical procedure, mice were placed in a temperature‐controlled heating chamber (~32°C) for 2 hours. At 24 hours after MCAO, we quantified the sensorimotor deficits using a scale ranging from 3 (normal) to 18 as previously described with minor modifications. 46 We then harvested the brain to measure the infarct volume (mm3) by integrating the infarct area (ImageJ, NIH, Bethesda, MD) on 1mm‐thick coronal slices stained with 2,3,5‐triphenyltetrazolium chloride, while correcting for ischemic swelling using the following formula:
With A = direct infarct (mm2), B = total ipsilateral hemisphere (mm2), C = total contralateral hemisphere (mm2), and n = slice number. Exclusion criteria were incomplete MCAO (residual CBF > 30% after anoxic depolarization) and subarachnoid hemorrhage.
Resting CBF, Blood Pressure, and Heart Rate
A separate group of naive mice (male, C57BL/6J, 2–3 months old) were anesthetized using 0.4ml of 20mg/ml Avertin in saline solution (2,2,2‐tribromoethanol, Sigma‐Aldrich, St Louis, MO). To examine resting CBF, laser Doppler flow probes were placed bilaterally at 2mm posterior, 2mm lateral from the bregma. To examine blood pressure and heart rate, a femoral artery catheter was placed in an additional group of naive mice (male, C57BL/6J, 2–3 months old). After 5 minutes of baseline recordings under isoflurane anesthesia (3% induction, 1.5% maintenance in 70% N2O and 30% O2), vehicle or olcegepant (1mg/kg) was administered via the tail vein, and measurements continued for 30 minutes. All data were averaged in 5‐minute bins to plot the time course of changes.
Olcegepant and Rimegepant in Human and Mouse Isolated Arteries
Mice (n = 11; 5 males, 6 females; 3–5 months of age) were sacrificed at the Leiden University Medical Center in accordance with the local committee for animal health, ethics, and research. Aortas were isolated and immediately transported to the Erasmus Medical Center, Rotterdam at 4°C in Medium 199 (Lonza Bioscience, Basel, Switzerland; transportation time ~ 1 hour). Upon arrival, aortas were stored in an oxygenated and carbonated Krebs solution (118mM NaCl, 4.7mM KCl, 2.5mM CaCl2, 1.2mM MgSO4, 1.2mM KH2PO4, 25mM NaHCO3, and 8.3mM glucose, pH = 7.4) overnight. Human coronary arteries were isolated from hearts of 7 heart valve donors (4 males and 3 females) aged 40 ± 5 years (mean ± standard error of the mean [SEM]). The hearts were provided by ETB‐BISLIFE, Multi Tissue Bank (Beverwijk, the Netherlands), from Dutch post‐mortem donors, after donor mediation via the Dutch Transplant Foundation (Leiden, the Netherlands), following removal of the aortic and pulmonary valves for homograft valve transplantation. All donors gave permission for research. Immediately after circulatory arrest, the hearts were generally stored at 4°C in a sterile organ‐protecting solution and brought to the laboratory within the first 24 hours after death. Distal portions of the left anterior descending coronary artery were isolated and subsequently stored in Krebs solution at 4°C until the start of the experiment. Human middle meningeal arteries were obtained from 4 individuals (2 males, 2 females, aged 52 ± 9 years) undergoing neurosurgical procedures at the Erasmus Medical Center, Rotterdam, the Netherlands. Middle meningeal arteries were stored in Medium 199 and transported to the laboratory immediately. Subsequently, surrounding tissue was removed and the artery was stored in a cold oxygenated Krebs solution with a high glucose content, as described before 47 (119mM NaCl, 4.7mM KCl, 1.25mM CaCl2, 1.2mM MgSO4, 1.2mM KH2PO4, 25mM NaHCO3, and 11.1mM glucose, pH = 7.4) at 4°C overnight. For functional experiments, human coronary arteries and mouse aorta were cut into 2mm segments and mounted in Mulvany myograph organ baths (Danish Myo Technology, Aarhus, Denmark), using Ø 40μm stainless‐steel wires. Organ baths were filled with oxygenated Krebs solution at 37°C. Vessel segments were allowed to equilibrate. Next, the segments were stretched to a tension normalized to 0.9 times the estimated diameter at 100mmHg transmural pressure. 48 Data were recorded using LabChart data acquisition (AD Instruments, Oxford, UK). First, all segments were exposed to 30mM KCl, followed by 100mM KCl to determine the maximum contraction. Next, vessel segments were incubated with and without olcegepant (1μM, 3μM, or 10μM, dissolved in 1N HCl and diluted in distilled water; Tocris Bioscience) or rimegepant (BMS‐927711, 3μM, 10μM, or 30μM in mouse aorta, 1nM, 10nM, 100nM, or 1μM in human coronary arteries, 1nM, 10nM, or 100nM in human middle meningeal arteries, dissolved in dimethyl sulfoxide and diluted in distilled water; MedChemExpress) for 30 minutes before a concentration–response curve to human α‐CGRP (0.01nM–1μM, half logarithmic steps, human coronary artery and human middle meningeal artery) or rat α‐CGRP (0.01nM–3μM, half logarithmic steps, mouse aorta) was constructed. Precontraction was established using 30mM KCl, and the relaxation responses to CGRP were expressed as a percentage of precontraction. Sigmoidal curves were constructed using non‐linear regression. The pEC50 values were used to calculate dose ratios for Schild plots. Using linear regression, the pA2 and/or pKb values of the different antagonists in both human and mouse tissue were determined.
Statistical Analysis
Primary outcome variables were infarct presence (TIA experiment) or volume (stroke experiments). Secondary outcome variables were neurologic deficit score, CBF, heart rate, and blood pressure. Neurological deficit scores were log‐transformed to achieve normality. Statistical tests were chosen based on the experimental design. In the TIA experiments, we used multiple logistic regression (independent variables: treatment, ischemia duration; dependent variable: infarct presence) and Fisher exact test (independent variable: treatment; dependent variable: infarct presence). In stroke experiments using wild‐type mice, we used 2‐way analysis of variance (ANOVA; independent variables: treatment, sex; dependent variables: infarct volume, neurological deficits, CBF; interaction terms: treatment and sex when applicable). In stroke experiments using FHM1 mice, we used multiple linear regression (independent variables: treatment, age, sex; dependent variable: infarct volume). For systemic physiology and CBF, we used 2‐way repeated measures ANOVA (independent variables: time and treatment; dependent variables: blood pressure, heart rate, and CBF). Grubbs' method was used to identify outliers. In isolated vessel experiments, we used 1‐way ANOVA for repeated measures (independent variable: CGRP concentrations; dependent variable: relaxation) or mixed‐effect model in the case of missing values. All statistical tests, variables, sample sizes, point estimates including 95% confidence intervals (CIs), and exact 2‐sided p values (prespecified α = 0.05) are provided throughout the article where data are presented. Statistical tests were carried out in Prism 8 (GraphPad Software, San Diego, CA) and SPSS 20.0 (IBM, Armonk, NY).
Results
We first investigated whether CGRP receptor antagonism increased the infarct risk after brief MCAO mimicking a TIA. To this end, we treated mice (C57BL/6J, male, 2–3 months old) with a single dose of olcegepant (1mg/kg, n = 19) or vehicle (n = 18) 10 minutes before brief MCAO (12, 15, or 20 minutes; Fig 1). Manifest infarcts often involved subcortical tissues (blue arrows). Multiple logistic regression revealed significantly higher infarct risk associated with olcegepant treatment (parameter estimate b = 2.44, odds ratio [OR] = 11.4, 95% CI = 2.0–109.0, p = 0.013). Three‐quarters of all animals treated with olcegepant developed an infarct (14/19) compared with only 33% of vehicle‐treated animals (6/18) when all TIA durations were pooled (relative risk = 2.21, 95% CI = 1.17–4.70, p < 0.022, Fisher exact test). The increase in infarct risk by olcegepant was most conspicuous with the shortest MCAO time (12 minutes, p = 0.009, Fisher exact test). As expected, infarct risk also showed a direct relationship to ischemia duration (b = 0.48, OR = 1.6, 95% CI = 1.2–2.4, p = 0.005). Given the known vasodilator role of CGRP in cerebral vessels, we examined the CBF changes during MCAO and found lower residual CBF (% of baseline) after olcegepant treatment (1mg/kg, n = 19) compared with vehicle (n = 18), which was apparent at every successive stage of the occlusion procedure (p = 0.029; 2‐way repeated measures ANOVA).
FIGURE 1.
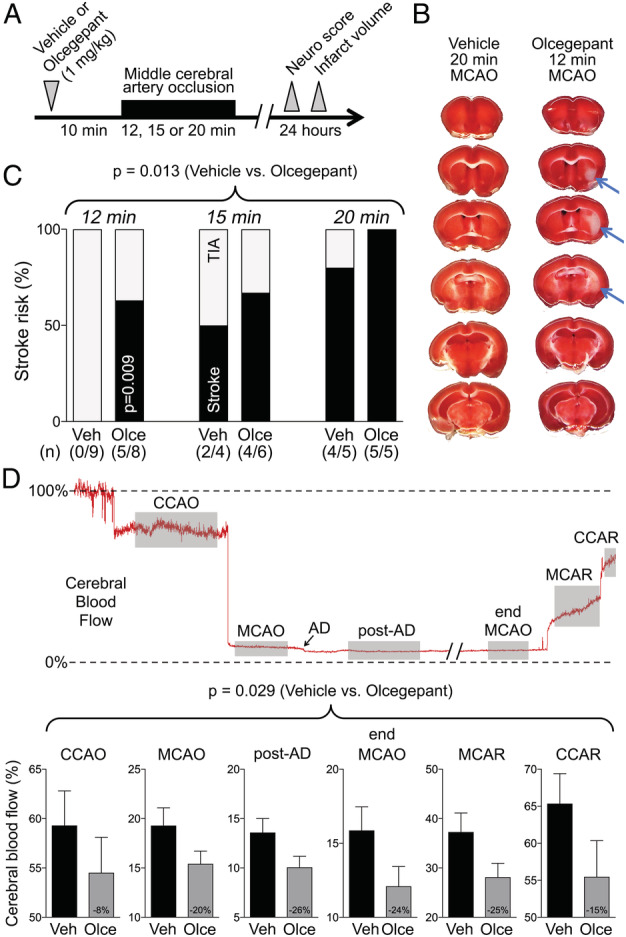
Olcegepant worsens the cerebral blood flow deficit and outcome of 12‐, 15‐, or 20‐minute focal cerebral ischemia. (A) Experimental timeline. (B) 2,3,5‐Triphenyltetrazolium chloride–stained coronal brain sections showing the infarct (blue arrows) after 12‐minute middle cerebral artery occlusion (MCAO) after pretreatment with a single dose of olcegepant (1mg/kg), and normal brain 20‐minute MCAO with vehicle injection. (C) Stroke risk (%) in vehicle (Veh) and olcegepant (Olce) groups after 12‐, 15‐, and 20‐minute MCAO. Data represent the percentage of infarct absence (transient ischemic attack [TIA], gray portion) or presence (stroke, black portion). Actual numbers of stroke/total are also provided in parentheses under the bars. Data were analyzed using multiple logistic regression (independent variables: treatment, ischemia duration; dependent variable: infarct presence) for the entire cohort, as well as using Fisher exact test in the 12‐minute MCAO group. (D) Representative cerebral blood flow tracing during MCAO and reperfusion measured by laser Doppler flowmetry. Gray shades represent typical time segments used to quantify the CBF during ischemia and after reperfusion. Lower panel shows the residual CBF (% of baseline) during different stages of MCAO in vehicle (n = 18) and olcegepant (1mg/kg, n = 19) arms (p = 0.029, 2‐way repeated measures analysis of variance). Data are from pooled 12‐, 15‐, and 20‐minute MCAO experiments. Numbers within the gray bars represent the relative difference between treatment arms calculated as (CBFOlce/CBFVeh) − 1. AD = anoxic depolarization; CCAO = common carotid artery occlusion; CCAR = common carotid artery reperfusion; MCAR = middle cerebral artery reperfusion. Seven mice in the vehicle arm and 1 mouse in the olcegepant arm were excluded from analyses based on predetermined criteria (see Materials and Methods). There was no mortality.
After showing that olcegepant promotes transformation of brief ischemia (TIAs) into infarcts, we next tested olcegepant in a more severe stroke model induced by longer occlusion time (Fig 2). Sixty‐minute MCAO induced infarcts in all mice, male and female, but these were >2‐fold larger (p < 0.001) and associated with more severe neurological deficits (median deficit score = 9.0 vs 5.0, p = 0.008) after a single dose of olcegepant (1mg/kg, n = 9) compared with vehicle (n = 8, 2‐way ANOVA, independent variables: treatment and sex). Sex did not affect the infarct volume (p = 0.509). Absence of an interaction between treatment and sex for infarct volume (p = 0.579) and neurological deficits (p = 0.470) suggested that olcegepant worsened outcomes in both sexes. The lower residual CBF in the olcegepant arm did not reach statistical significance in this experiment (p = 0.223, 2‐way ANOVA for repeated measures).
FIGURE 2.
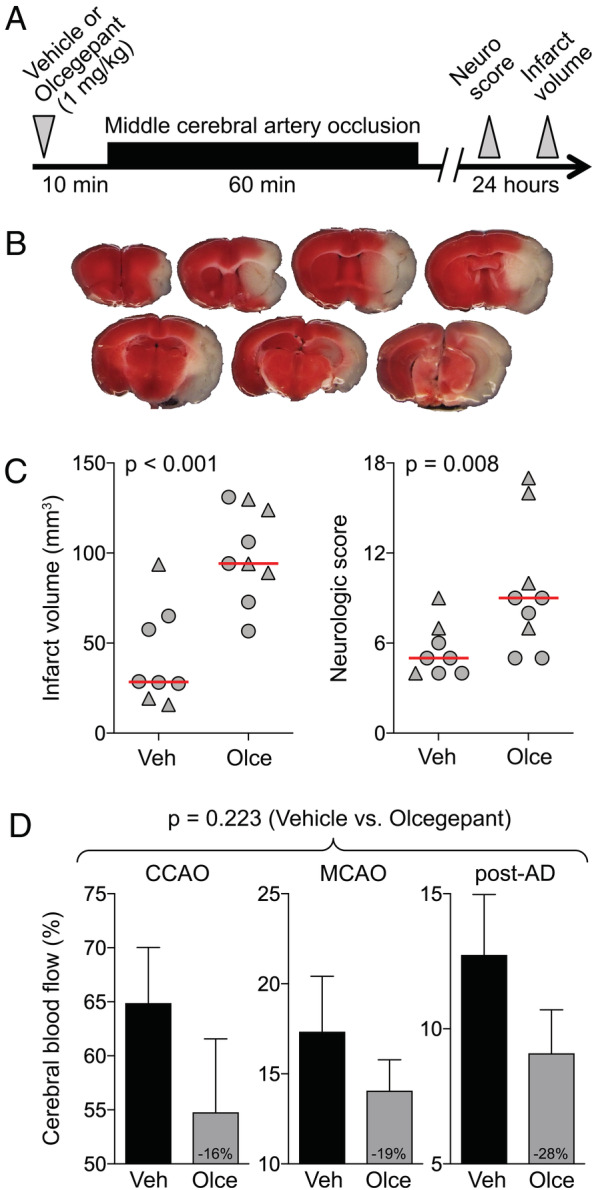
Olcegepant worsens the outcomes after 60‐minute focal cerebral ischemia. (A) Experimental timeline. (B) 2,3,5‐Triphenyltetrazolium chloride–stained coronal brain sections showing a typical infarct (unstained tissue) after 60‐minute middle cerebral artery occlusion (MCAO). (C) Infarct volume (mm3) and neurologic deficit score (p = 0.001 and p = 0.011, respectively, 2‐way analysis of variance [ANOVA]). Data from individual animals are shown (triangles = male; circles = female) along with their group median (red lines). Mean ages were 5.3 ± 1.4 and 5.7 ± 1.5 months in the vehicle (Veh) and olcegepant (Olce; 1mg/kg) groups, respectively (all wild type; mean ± standard deviation). (D) Residual cerebral blood flow (CBF; % of baseline) during MCAO (p = 0.223, 2‐way repeated measures ANOVA). Numbers within the gray bars represent the relative difference between treatment arms calculated as (CBFOlce/CBFVeh) − 1. Two mice in the vehicle arm and 1 mouse in the olcegepant arm were excluded from analyses based on predetermined criteria (see Materials and Methods). There was no mortality.
In the third series of experiments, we wanted to assess whether the ischemia worsening effect is specific to olcegepant or rather a class effect of CGRP receptor antagonists. We, therefore, studied rimegepant, a recently developed next generation small molecule CGRP antagonist. First, we tested rimegepant (100mg/kg) in the 60‐minute MCAO model but encountered 75% (6/8) mortality compared with none (0/8) in the vehicle group (p = 0.007, Fisher exact test; Fig 3). The only 2 surviving mice in the rimegepant group had infarct volumes of 85 and 109mm3 and neurological deficit scores of 13 and 16, compared with a median infarct volume of 62mm3 and neurological deficit score of 11 in the vehicle group. Once again, the residual collateral CBF in rimegepant‐treated animals was lower than in the vehicle group (p = 0.014, 2‐way ANOVA for repeated measures). Because very high mortality precluded the assessment of infarct volume, we next examined the effects of a 10‐fold lower dose of rimegepant (10mg/kg) versus vehicle in a 20‐minute TIA model (Fig 4; one outlier excluded from the rimegepant arm per Grubbs' method). All animals survived and developed an infarct. In rimegepant‐treated mice, infarcts were approximately 60% larger than in vehicle‐treated mice (p = 0.030, unpaired t‐test), and residual CBF tended to be lower (p = 0.135, 2‐way ANOVA for repeated measures).
FIGURE 3.
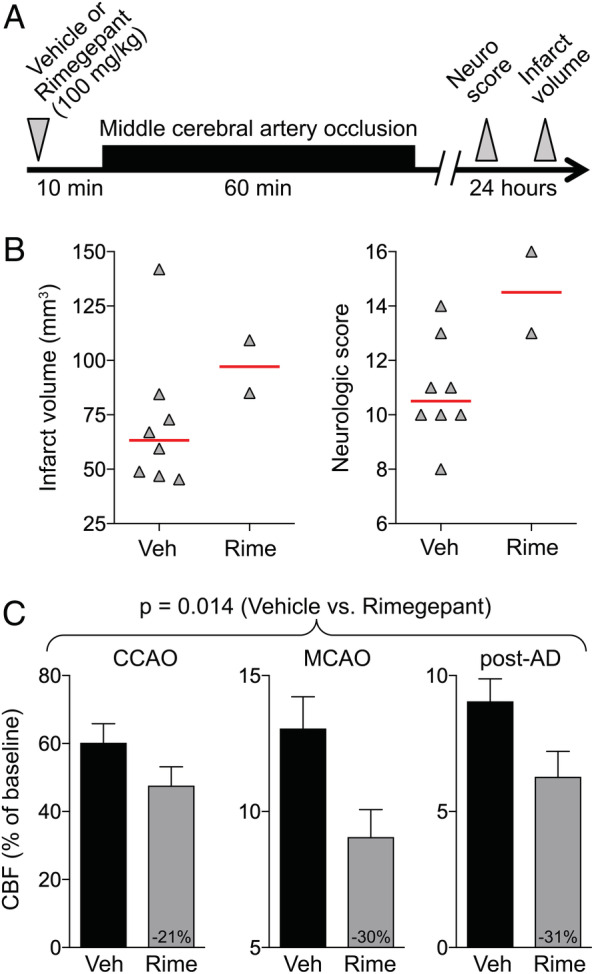
Rimegepant worsens the outcomes after 60‐minute focal cerebral ischemia. (A) Experimental timeline. (B) Infarct volume (mm3) and neurologic deficit scores. Data from individual animals are shown along with their group median (red lines). Mean ages were 2.2 ± 0.2 and 2.3 ± 0.3 months in vehicle (Veh) and rimegepant (Rime; 100mg/kg) groups, respectively (all male, wild type; mean ± standard deviation). (C) Residual cerebral blood flow (CBF; % of baseline) during middle cerebral artery occlusion (MCAO; p = 0.014, 2‐way repeated measures analysis of variance). Numbers within the gray bars represent the relative difference between treatment arms calculated as (CBFOlce/CBFVeh) − 1. One mouse each in the vehicle and rimegepant arms were excluded from analyses based on predetermined criteria (see Materials and Methods). Six mice died prior to outcome assessments in the rimegepant arm. AD = anoxic depolarization; CCAO = common carotid artery occlusion. [Color figure can be viewed at www.annalsofneurology.org]
FIGURE 4.
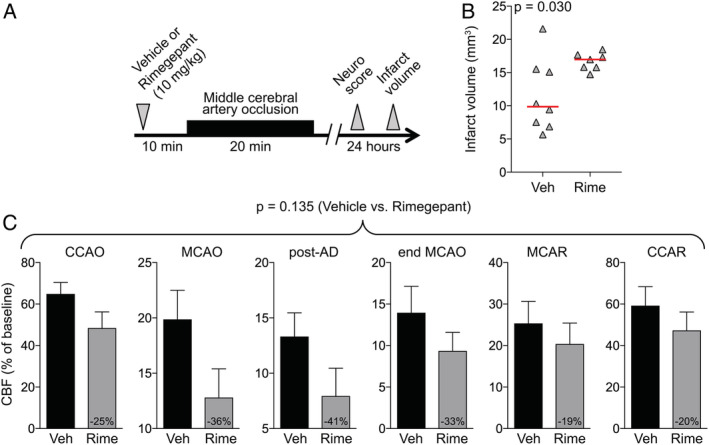
Rimegepant worsens the outcomes after 20‐minute focal cerebral ischemia. (A) Experimental timeline. (B) Infarct volume (mm3; p = 0.030, t test). Data from individual animals are shown along with their group median (red lines). Mean ages were 2.5 ± 0.3 and 2.4 ± 0.2 months in vehicle (Veh) and rimegepant (Rime; 10mg/kg) groups, respectively (all male, wild type; mean ± standard deviation). (C) Residual cerebral blood flow (CBF; % of baseline) during middle cerebral artery occlusion (MCAO; p = 0.135, 2‐way repeated measures analysis of variance). Numbers within the gray bars represent the relative difference between treatment arms calculated as (CBFOlce/CBFVeh) − 1. There was no mortality. AD = anoxic depolarization; CCAO = common carotid artery occlusion; CCAR = common carotid artery reperfusion; MCAR = middle cerebral artery reperfusion. [Color figure can be viewed at www.annalsofneurology.org]
In the next series of experiments, we treated mice with 10 once‐daily doses of olcegepant (0.1 or 1mg/kg) or vehicle over a 2‐week period (Fig 5A), mimicking daily dosing in migraine prophylaxis. Compared with vehicle, both chronic dose levels increased the infarct volumes and worsened the neurological deficit scores to a similar extent as a single dose (see Fig 5B). These data suggested that, within the 2‐week timeframe tested, chronic CGRP blockade does not aggravate the adverse effect, or ameliorate it by inducing alternative compensatory mechanisms restoring collateral perfusion.
FIGURE 5.
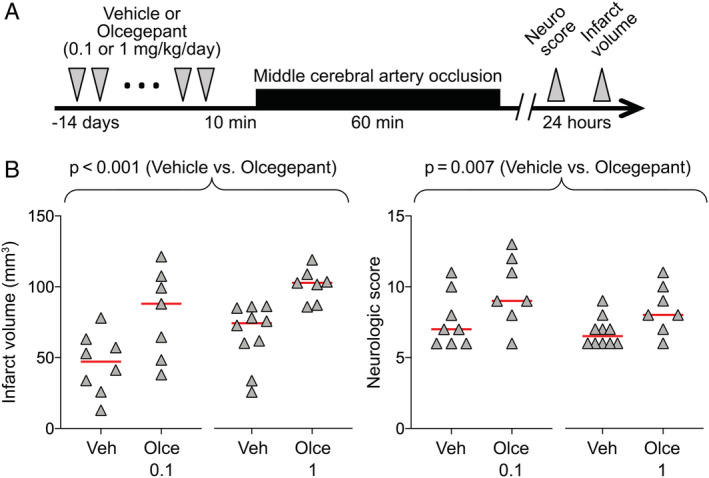
Prolonged treatment with olcegepant worsens the outcomes after 60‐minute focal cerebral ischemia. (A) Experimental timeline. (B) Infarct volume (mm3) and neurologic deficit scores (p < 0.001 and p = 0.007, respectively, 2‐way analysis of variance). Data from individual animals are shown along with their group mean for infarct and median for neurologic score (red lines). All mice were 2 months old, male, and wild type. Two mice in the vehicle (Veh) and 3 mice in the olcegepant (Olce) 0.1mg/kg and olcegepant 1mg/kg arms died prior to outcome assessments. There were no exclusions. [Color figure can be viewed at www.annalsofneurology.org]
In a fifth set of experiments, we investigated whether the ischemia‐aggravating effect of CGRP‐receptor antagonists might be even greater in FHM1 mice. These mice have been shown to develop worse outcomes after cerebral ischemia due to enhanced susceptibility to anoxic and peri‐infarct spreading depolarizations. 26 To that end, we tested the effects of a single dose of olcegepant (0.1 or 1mg/kg) versus vehicle 10 minutes before MCAO. To avoid a ceiling effect for infarct volume and high mortality, we employed a 30‐minute MCAO model (Fig 6A).
FIGURE 6.
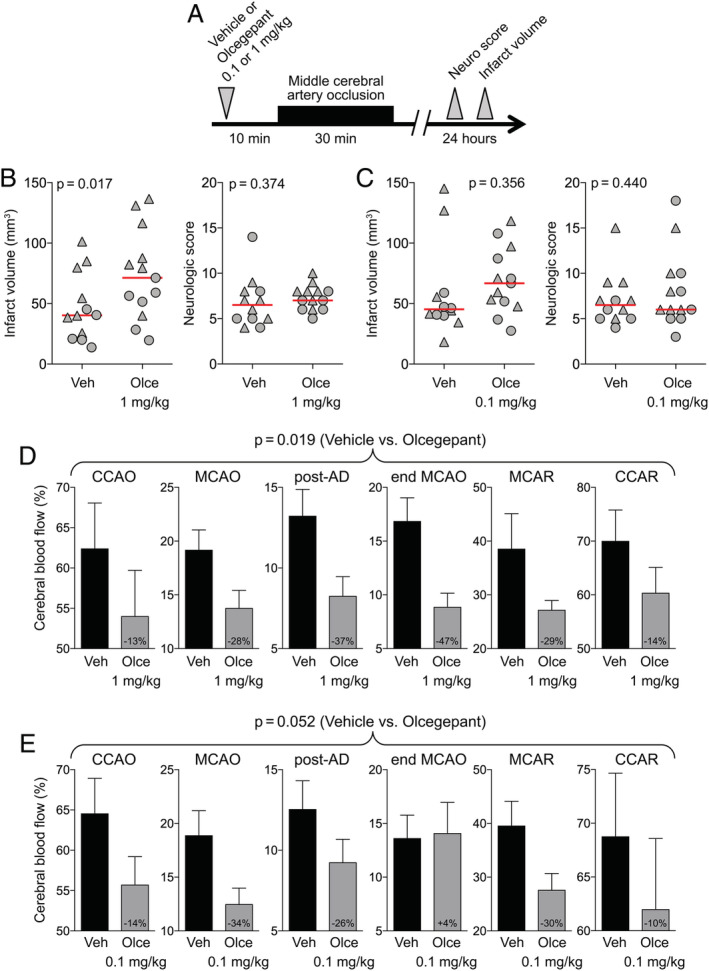
Stroke outcome in familial hemiplegic migraine type 1 (FHM1) mice after a single dose of olcegepant. (A) Experimental timeline. (B) Infarct volume (mm3) and neurologic deficit scores in experiment testing olcegepant 1mg/kg (p = 0.017 and p = 0.374, respectively, 2‐way analysis of variance [ANOVA]). Data from individual animals are shown (triangles = male; circles = female) along with their group median (red lines). Mean ages were 6.4 ± 4.5 and 7.0 ± 6.5 months in vehicle (Veh) and olcegepant (Olce) groups, respectively (all FHM1 mutants; mean ± standard deviation [SD]). (C) Infarct volume (mm3) and neurologic deficit scores in experiment testing olcegepant 0.1mg/kg (p = 0.356 and p = 0.440, respectively, 2‐way ANOVA). Data from individual animals are shown (triangles = male; circles = female) along with their group median (red lines). (D) Residual cerebral blood flow (CBF; % of baseline) during middle cerebral artery occlusion (MCAO; p = 0.019, 2‐way repeated measures ANOVA). Mean ages were 13.2 ± 3.9 and 12.3 ± 2.3 months in the vehicle and olcegepant (1mg/kg) groups, respectively (all FHM1 mutants; mean ± SD). (E) Residual CBF (% of baseline) during MCAO (p = 0.052, 2‐way repeated measures ANOVA). Numbers within the gray bars represent the relative difference between treatment arms calculated as (CBFOlce/CBFVeh) − 1. Seven mice in the vehicle, 3 mice in the olcegepant 0.1mg/kg, and 6 in the olcegepant 1mg/kg arms were excluded from analyses based on predetermined criteria (see Materials and Methods). Two mice in the vehicle, 1 in olcegepant 0.1mg/kg, and 2 in the olcegepant 1mg/kg arms died prior to outcome assessments. AD = anoxic depolarization; CCAO = common carotid artery occlusion; CCAR = common carotid artery reperfusion; MCAR = middle cerebral artery reperfusion. [Color figure can be viewed at www.annalsofneurology.org]
As the age range among the available FHM1 mice bred in our facility was wider than for the wild‐type mice obtained from commercial vendors, we introduced age as an independent variable in addition to sex and treatment in a general linear model and determined the contribution of each independent variable to stroke outcome. Whereas treatment (parameter estimate, β = 28.5, 95% CI = 6.3–50.6, p = 0.014) and sex (β = 40.6, 95% CI = 18.0–63.3, p = 0.001) significantly affected the infarct volumes, age did not (β = −0.15, 95% CI = −2.15 to 1.84, p = 0.875). In FHM1 mice, a single dose of olcegepant (1mg/kg, n = 13) increased infarct volumes by 75% compared with vehicle (n = 12, p = 0.017; see Fig 6B). The effect of olcegepant, however, did not appear to be stronger in mutant than in wild‐type mice (see Fig 2). As in wild‐type mice, olcegepant (1mg/kg) reduced collateral CBF in FHM1 mutants (p = 0.019, 2‐way ANOVA for repeated measures; see Fig 6). A separate group treated with the lower dose of olcegepant (0.1mg/kg) developed 50% larger infarcts compared with vehicle, although this was not statistically significant (p = 0.356). Tissue perfusion once again tended to be worse (p = 0.052). Altogether, these data reproduced the harmful effect of olcegepant on ischemic outcome but did not support a higher sensitivity of FHM1 mutants to CGRP blockade with olcegepant.
In a separate cohort, we sought to rule out systemic hemodynamic changes as a potential explanation for the effect of CGRP antagonism on collateral CBF. Because accurate measurements of systemic blood pressure in mice requires an indwelling arterial (often femoral) catheter, to avoid excess morbidity we could not perform these experiments in ischemic animals. Therefore, we studied olcegepant (1mg/kg) in separate groups of naive nonischemic mice and found, consistent with previous reports, 37 , 49 no effect on systemic blood pressure, heart rate, or resting CBF (Fig 7).
FIGURE 7.
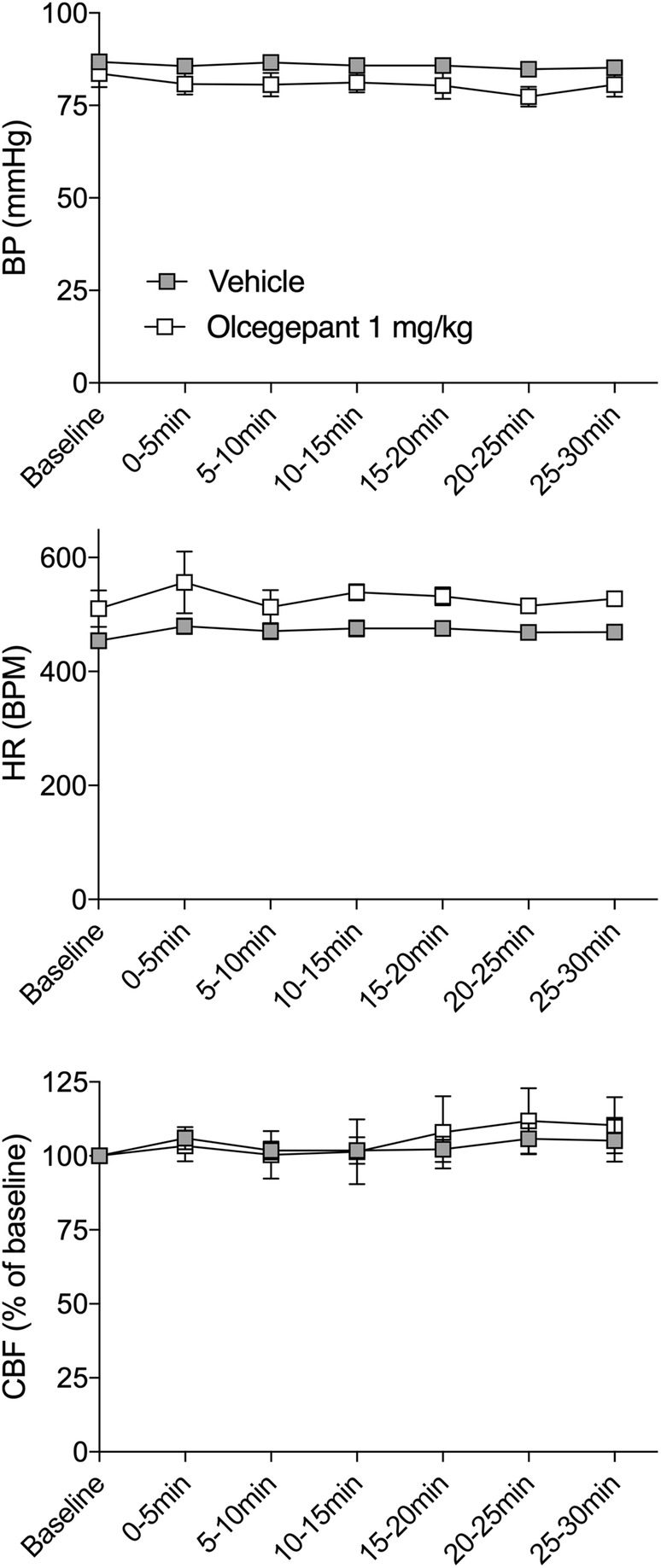
Baseline hemodynamic parameters in young male wild‐type mice after a single dose of olcegepant. Baseline blood pressure (BP; p = 0.177), heart rate (HR; p = 0.917), and cerebral blood flow (CBF; p = 0.441) were measured over a period of 30 minutes after vehicle or olcegepant (1mg/kg) treatment (both groups: 2.6 ± 0.2 months of age, all male, wild‐type mice). Two‐way repeated measures analysis of variance, n = 5 per group. BPM = beats per minute.
Finally, we sought to examine the relative potencies of olcegepant and rimegepant in isolated mouse and human blood vessels to confirm the dose ranges we selected based on previous work (Fig 8). 32 , 33 , 34 , 35 , 36 , 37 , 42 , 43 To this end, we tested these drugs. In mouse aorta, the 1μM, 3μM, and 10μM concentrations of olcegepant all significantly shifted the concentration–response curve to rat α‐CGRP (control pEC50 = 8.02 ± 0.08, 1μM pEC50 = 6.55 ± 0.16, 3μM pEC50 = 6.35 ± 0.11, 10μM pEC50 = 5.41 ± 0.18, p < 0.001, mean ± SEM, 1‐way ANOVA for repeated measures). The Schild plot slope of 1.16 (95% CI = 0.65–1.66) did not differ significantly from unity. As an estimate of the potency of olcegepant in mouse aorta, the pA2 was 7.22 ± 0.20 (n = 4–5). Rimegepant concentrations of 3μM, 10μM, and 30μM shifted the concentration–response curve to rat α‐CGRP in mouse aorta as well (control pEC50 = 8.21 ± 0.08, 3μM pEC50 = 7.56 ± 0.11, 10μM pEC50 = 7.35 ± 0.22, 30μM pEC50 = 6.96 ± 0.17, p < 0.001). The slope of the Schild plot (0.71, 95% CI = 0.16–1.25) did not differ from unity. The corresponding pA2 value for rimegepant in mouse aorta was 6.24 ± 0.27 (n = 4). Similar experiments were performed in human coronary arteries, in which lower concentrations of rimegepant already significantly shifted the concentration–response curve to human α‐CGRP (control pEC50 = 8.51 ± 0.08, 1nM pEC50 = 7.74 ± 0.11, 10nM pEC50 = 7.22 ± 0.14, 100nM pEC50 = 6.77 ± 0.13, 1μM pEC50 = 6.08 ± 0.30, p < 0.001). The Schild plot slope of 0.56 was significantly smaller than unity (95% CI = 0.40–0.71); therefore, we did not calculate a pA2 value. Instead, pKb values were calculated for each individual concentration of rimegepant to determine its potency in human coronary arteries (1nM: 9.74 ± 0.05, 10nM: 9.25 ± 0.15, 100nM: 8.71 ± 0.16, 1μM: 8.43 ± 0.25). In human middle meningeal arteries, all rimegepant concentrations visibly shifted the concentration–response curve to CGRP, although only the 10nM and 100nM concentrations reached statistical significance (control pEC50 = 8.22 ± 0.15, 1nM pEC50 = 7.63 ± 0.22, 10nM pEC50 = 6.62 ± 0.28, 100nM pEC50 = 5.71 ± 0.17, p = 0.002, n = 3–4). The Schild plot slope of 0.91 did not differ from unity (95% CI = 0.60–1.22), resulting in a pA2 value of 10.02 ± 0.33.
FIGURE 8.
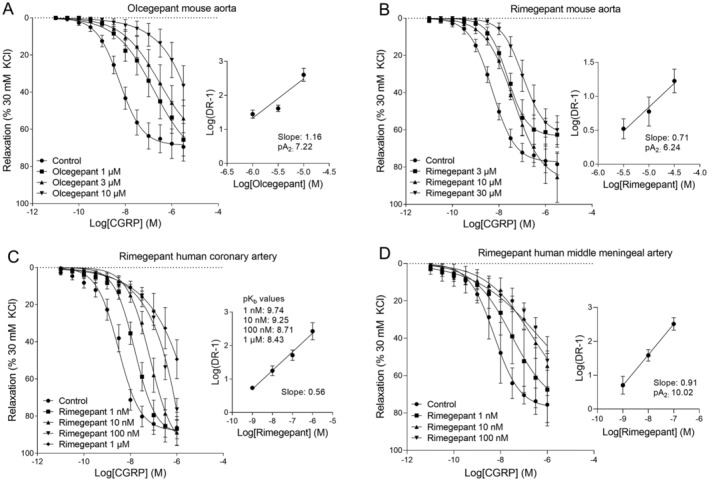
Pharmacological characterization of olcegepant and rimegepant in mouse aortas, human coronary arteries, and human middle meningeal arteries. (A) Concentration–response curves to rat α–calcitonin gene–related peptide (CGRP) in mouse aortas in the absence or presence of olcegepant (1μM, 3μM, and 10μM; 3–6 females and 3–5 males) with the corresponding Schild plot (pA2 = 7.22, 2 females and 3 males). (B) Concentration–response curves to rat α‐CGRP in mouse aortas in the absence or presence of rimegepant (3μM, 10μM, 30μM; 2–5 females and 2–4 males) with the corresponding Schild plot (pA2 = 6.24, 3 females and 4 males). (C) Concentration–response curves to human α‐CGRP in human coronary arteries in the absence or presence of rimegepant (1nM, 10nM, 100nM, 1μM; 2–3 females and 3–4 males) with the corresponding Schild plot and pKb values (3 females and 4 males). (D) Concentration–response curves to human α‐CGRP in human middle meningeal arteries in the absence or presence of rimegepant (1nM, 10nM, 100nM; 2 females and 1–2 males) with the corresponding Schild plot (pA2 = 10.02; 2 females and 2 males). All data are represented as mean ± standard error of the mean. DR‐1 = Dose Ratio–1; KCL = kaliumchloride; M = Molar
Discussion
CGRP receptor antagonists and antibodies targeting CGRP or the CGRP receptor are highly publicized as novel, effective, and reportedly safe acute and prophylactic treatments for migraine. 3 , 22 CGRP‐mediated collateral vasodilation, however, is an important rescue mechanism in cerebral ischemia, for which migraine is a risk factor. Here, we show that single and chronic doses of 2 different gepants aggravate experimental cerebral ischemia in standard mouse models of cerebral ischemia. Both agents transformed TIA‐like mild ischemic events into full‐blown infarcts and caused established infarcts to grow larger, with more severe neurological deficits and higher mortality. These findings call for a more careful assessment of the vascular safety of CGRP inhibitors. This seems all the more pressing for patients who are at increased risk of experiencing coincident ischemic events, and patients who are using these agents prophylactically, blocking CGRP pathways for prolonged periods of time. Such patients are at increased risk of experiencing ischemic events while their physiological protection system may be dysfunctional.
Thus far, no vascular safety issues have been reported in clinical gepant and antibody trials, 2 , 22 although a few cases with vascular events while on treatment have appeared. 22 , 50 , 51 Although this is reassuring, it is important to note that the clinical trials to date were not designed to detect the type of risk increase our data suggest: not an increase in the risk of ischemic events, but an increase in the risk of worse outcomes if and when ischemic events happen while on treatment. The studies were far too small and of too short a duration to detect uncommon events such as stroke. Even more importantly, patients with recent comorbid cardiovascular and cerebrovascular disease, who are at increased risk of experiencing ischemic events, were excluded from participation in virtually all of these trials. Remarkably, despite serious theoretical and experimentally driven concerns, 4 , 6 only a few studies have specifically addressed the cardiovascular safety of CGRP inhibitors, and only after single doses. 52 None of these studies, however, was conducted in the presence of contemporaneous acute focal arterial occlusion. We are also not aware of any study on the safety of long‐term inhibition for prophylactic purposes or specifically investigating cerebrovascular safety. Single doses of CGRP inhibitors did not seem to exacerbate exercise‐induced ischemic symptoms and signs in patients with stable angina 53 , 54 and appeared safe in a small cohort of migraine patients with comorbid coronary artery disease. 55 However, design caveats diminished the predictive value of these studies for cardiovascular safety. 56 First, these studies were primarily conducted in males, rather than females, which is surprising, because migraine has a 3:1 female preponderance and the increased cardiovascular risk in migraineurs especially affects women. Second, in the only study specifically assessing the cardiovascular safety of a CGRP receptor antibody, 53 , 54 the cardiovascular system was challenged and examined probably too soon after administration of the antibody, when vascular CGRP receptor blockade had likely not yet been fully achieved. 56 A potentially harmful effect of CGRP receptor blockade could have been missed. Remarkably, despite this conspicuous lack of formal safety data, comorbid vascular disease has not been listed as a formal contraindication for CGRP pathway inhibitors.
To inhibit CGRP‐mediated collateral vasodilation in vivo after systemic administration, CGRP receptor blockers must gain access from the blood vessel lumen across the endothelium to the CGRP receptors on the smooth muscle layer. There is major debate whether, and to what extent, gepants and monoclonal antibodies cross the blood–brain barrier under physiological conditions. 45 , 57 , 58 , 59 Our data show that, in mice, systemic administration of gepants impairs the ability of the cerebral vasculature to redirect collateral CBF to ischemic brain tissue upon focal arterial occlusion. Thus, under ischemic conditions, small molecule CGRP receptor antagonists may cross the endothelial blood vessel wall to reach cerebrovascular CGRP receptors in concentrations sufficient to prevent pial vasodilation normally resulting from ischemia‐induced local release of endogenous CGRP. Whether this also applies to human cerebral vasculature and to anti‐CGRP or CGRP receptor antibodies remains to be investigated. Currently available antibodies, however, are human (erenumab) or humanized (eptinezumab, fremanezumab, galcanezumab) and, except for galcanezumab, 60 either do not bind or have not been tested against rodent targets, hampering studies in experimental animals. Although species differences must always be considered when interpreting experimental work, to date there are no data to suggest functionally important differences in the CGRP system and ischemia‐compensating mechanisms between rodent and human cerebral vasculature.
Taken together, our data indicate that the doses employed in the current study are relevant, because the 10‐fold difference in dosing for olcegepant (1mg/kg) and rimegepant (10mg/kg) matched their relative potencies in blocking α‐CGRP–induced relaxation of isolated mouse aortas. For both antagonists, the slope of the Schild plot did not differ from unity, suggesting a competitive type of antagonism in mouse aorta. However, the shift of the concentration–response curve by increasing concentrations of olcegepant to some extent appeared to be biphasic, similar to what was observed for olcegepant in human coronary arteries. 61 A second CGRP receptor, such as the amylin type 1 receptor, may be responsible for the potentially biphasic effect as observed in the Schild plot. 62 Both olcegepant and rimegepant were less potent in mouse aorta than in human coronary artery. The shift in the concentration–response curve to α‐CGRP induced by rimegepant appeared to decrease for increasing concentrations of the antagonist, which is again suggestive of involvement of multiple receptors. Therefore, the pKb of the lowest concentration of rimegepant (1nM) that induced a shift in the concentration–response curve to α‐CGRP was used as a measure of potency, as it likely represents the potency at the canonical CGRP receptor. The pKb at 1nM rimegepant in human coronary artery and the pA2 in mouse aorta together suggested ~3,000‐fold higher potency in human coronary arteries compared with mouse aorta. Rimegepant was even more potent in human middle meningeal arteries compared with human coronary arteries (~2‐fold). Olcegepant was also more potent in human coronary arteries compared to mouse aorta (~200‐fold) based on the previously published pKb of 9.56 ± 0.22 for 1nM olcegepant. 61 Olcegepant was shown to be slightly more potent in human middle meningeal artery as well (10‐fold, pA2 = 10.59 ± 0.54). 63 Although the potency of CGRP receptor antagonists may vary across different vascular beds, as shown here and in our previous work with telcagepant, 64 , 65 the interspecies differences are much larger.
Residual blood flow within ischemic core is critically dependent on pial collaterals, the magnitude of which determines how long the tissue survives during the ischemic event before irreversible injury and infarction. The absolute differences in residual CBF between gepant and vehicle groups in ischemic core, where residual CBF was already <20% of baseline in most animals, were small but important, and likely reflected poor collateral function and larger perfusion defects after treatment with gepants. Interestingly, reduced CBF in the gepant arms always got worse after anoxic depolarization, suggesting that endogenous CGRP opposes the vasoconstrictive coupling during anoxic depolarization, consistent with CGRP‐mediated vasodilation during spreading depolarizations. 66 Last but not the least, recanalization was also less effective in restoring CBF in gepant arms, suggesting that CGRP antagonism worsens the no‐reflow phenomenon. These findings indicate that CGRP antagonism abrogates an important endogenous defense mechanism to counteract low perfusion pressure and high vascular tone that develops in the ischemic brain, and thereby renders the brain more susceptible to infarction upon arterial occlusions.
Hypothetically, long‐term blockade of CGRP pathways could lead to upregulation of alternative rescue mechanisms in cerebral ischemia. For the brain, we did not find any indication that alternative mechanisms take over the anti‐ischemic effects of CGRP‐mediated collateral vasodilation. After 2 weeks of daily treatment with olcegepant, infarcts and neurological deficits remained negatively affected by gepants. This is well in line with worse stroke outcomes reported in CGRP knockout mice. 14
Lastly, we advise caution when extrapolating our experimental data to the clinical setting. Clearly, the murine dose ranges, pharmacokinetics, formulations, and delivery routes of olcegepant and rimegepant used in our study were different from those in humans. Guided by previously published studies, 29 , 30 , 31 we obtained the drugs from commercial vendors (Tocris Bioscience, MedChemExpress), rather than the pharmaceutical companies that developed the clinical preparations, and relied upon the product data sheets and purity reports provided by these vendors, all of which are publicly available. Although we have picked the dose ranges from the existing literature, 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 42 , 43 , 44 , 45 as a caveat we have not measured the plasma or brain drug levels to inform us about the relevance of the dose–route combination used in our study to clinical dose–route combinations. However, as both antagonists we tested showed much higher potency in human compared with mouse vessels, lower plasma levels may be sufficient to observe the same effect on stroke outcomes in humans. Lastly, the widely used middle cerebral artery occlusion procedure by an intraluminal filament might have facilitated drug access into the brain. Future studies using other models are needed to eliminate this potential caveat.
In summary, CGRP receptor antagonists and, by inference, likely also CGRP or CGRP receptor antibodies may worsen the outcome of coincidental cerebral ischemic events by blocking CGRP‐mediated vascular rescue mechanisms. Many migraineurs are soon expected to be using these agents. Therefore, their cerebrovascular safety must be urgently defined in migraineurs with comorbid vascular disease who are at risk of experiencing ischemic events, and in prophylactic CGRP inhibition where long‐term exposure occurs.
Author Contributions
I.A.M., T.d.V., A.M., A.M.J.M.v.d.M., M.J.H.W., M.D.F., and C.A. contributed to the conception and design of the study. I.A.M., M.L., T.d.V., T.Q., T.Y., and K.S. contributed to the acquisition and analysis of data. I.A.M., T.d.V., A.M., and C.A. contributed to the statistical analysis. I.A.M., T.d.V., A.v.d.B., A.H.J.D., M.J.H.W., A.M., A.M.J.M.v.d.M., M.D.F., and C.A. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
A.M. has received research grants and/or consultant fees from Amgen/Novartis, Lilly, and Teva, which develop anti‐CGRP and/or antimigraine therapeutics. C.A. has received research grants from electroCore, which develops an antimigraine neuromodulation device. M.D.F. receives consultant fees from Company Medtronic, Novartis, Lilly, and Teva, which develop anti‐CGRP and/or antimigraine therapeutics. All companies listed above may be affected by the study.
Supporting information
TABLE S1. Overview of all in vivo experimental groups including sample sizes, exclusions, early mortality, and age and sex distributions. BP = blood pressure; CBF = cerebral blood flow; d = days; F = female; HR = heart rate; M = male; MCAO = middle cerebral artery occlusion; Olce = olcegepant; Rime = rimegepant; SD = standard deviation.
Acknowledgment
The statistical work described was conducted with support from Harvard Catalyst, Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, NIH award UL 1TR002541) and financial contributions from Harvard University and its affiliated academic health care centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University, and its affiliated academic health care centers, or the NIH. Funding was provided by the NIH National Institute of Neurological Disorders and Stroke (R01NS102969 to C.A.), International Headache Society 2017 Fellowship Award (to I.A.M.), Royal Netherlands Academy of Arts and Sciences Van Leersum grant 2017 (to I.A.M.), Netherlands Organization for Scientific Research Vidi grant 917.113.349 (to A.M.), Center for Medical Systems Biology in the framework of the Netherlands Genomics Initiative and Marie Curie IAPP Program BRAINPATH (No. 612360 to A.M.J.M.v.d.M.), Dutch Research Council (NWO), The Netherlands Organisationfor Health Research and Development (ZonMW), European Community, Dutch Heart Foundation, and Spinoza 2009 award from the Netherlands Organization for Scientific Research (to M.D.F.).
References
- 1. Iyengar S, Johnson KW, Ossipov MH, Aurora SK. CGRP and the trigeminal system in migraine. Headache 2019;59:659–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Edvinsson L, Haanes KA, Warfvinge K, Krause DN. CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat Rev Neurol 2018;14:338–350. [DOI] [PubMed] [Google Scholar]
- 3. Sparrow AM, Searles JW. The market for migraine drugs. Nat Rev (2019). [DOI] [PubMed] [Google Scholar]
- 4. Russell FA, King R, Smillie SJ, et al. Calcitonin gene‐related peptide: physiology and pathophysiology. Physiol Rev 2014;94:1099–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hong KW, Pyo KM, Lee WS, et al. Pharmacological evidence that calcitonin gene‐related peptide is implicated in cerebral autoregulation. Am J Physiol 1994;266(pt 2:H11–H16. [DOI] [PubMed] [Google Scholar]
- 6. MaassenVanDenBrink A, Meijer J, Villalon CM, Ferrari MD. Wiping out CGRP: potential cardiovascular risks. Trends Pharmacol Sci 2016;37:779–788. [DOI] [PubMed] [Google Scholar]
- 7. Edvinsson L, Ekman R, Jansen I, et al. Peptide‐containing nerve fibers in human cerebral arteries: immunocytochemistry, radioimmunoassay, and in vitro pharmacology. Ann Neurol 1987;21:431–437. [DOI] [PubMed] [Google Scholar]
- 8. Edvinsson L, Jansen Olesen I, Kingman TA, et al. Modification of vasoconstrictor responses in cerebral blood vessels by lesioning of the trigeminal nerve: possible involvement of CGRP. Cephalalgia 1995;15:373–383. [DOI] [PubMed] [Google Scholar]
- 9. McCulloch J, Uddman R, Kingman TA, Edvinsson L. Calcitonin gene‐related peptide: functional role in cerebrovascular regulation. Proc Natl Acad Sci U S A 1986;83:5731–5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Franco‐Cereceda A, Liska J. Potential of calcitonin gene‐related peptide in coronary heart disease. Pharmacology 2000;60:1–8. [DOI] [PubMed] [Google Scholar]
- 11. Geppetti P, Del Bianco E, Patacchini R, et al. Low pH‐induced release of calcitonin gene‐related peptide from capsaicin‐sensitive sensory nerves: mechanism of action and biological response. Neuroscience 1991;41:295–301. [DOI] [PubMed] [Google Scholar]
- 12. Macfarlane R, Moskowitz MA, Tasdemiroglu E, et al. Postischemic cerebral blood flow and neuroeffector mechanisms. Blood Vessels 1991;28:46–51. [DOI] [PubMed] [Google Scholar]
- 13. Du Z, Zhang H, Chen Q, et al. Intranasal calcitonin gene‐related peptide protects against focal cerebral ischemic injury in rats through the Wnt/beta‐catenin pathway. Med Sci Monit 2018;24:8860–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhai L, Sakurai T, Kamiyoshi A, et al. Endogenous calcitonin gene‐related peptide suppresses ischemic brain injuries and progression of cognitive decline. J Hypertens 2018;36:876–891. [DOI] [PubMed] [Google Scholar]
- 15. Zhang JY, Yan GT, Liao J, et al. Leptin attenuates cerebral ischemia/reperfusion injury partially by CGRP expression. Eur J Pharmacol 2011;671:61–69. [DOI] [PubMed] [Google Scholar]
- 16. Brain SD, Grant AD. Vascular actions of calcitonin gene‐related peptide and adrenomedullin. Physiol Rev 2004;84:903–934. [DOI] [PubMed] [Google Scholar]
- 17. Bunker CB, Reavley C, O'Shaughnessy DJ, Dowd PM. Calcitonin gene‐related peptide in treatment of severe peripheral vascular insufficiency in Raynaud's phenomenon. Lancet 1993;342:80–83. [DOI] [PubMed] [Google Scholar]
- 18. Shawket SA, Brown MJ. Pathogenetic and therapeutic implications of calcitonin‐gene‐related peptide in the cardiovascular system. Trends Cardiovasc Med 1991;1:211–215. [DOI] [PubMed] [Google Scholar]
- 19. Homma S, Kimura T, Sakai S, et al. Calcitonin gene‐related peptide protects the myocardium from ischemia induced by endothelin‐1: intravital microscopic observation and (31)P‐MR spectroscopic studies. Life Sci 2014;118:248–254. [DOI] [PubMed] [Google Scholar]
- 20. Lei J, Zhu F, Zhang Y, et al. Transient receptor potential vanilloid subtype 1 inhibits inflammation and apoptosis via the release of calcitonin gene‐related peptide in the heart after myocardial infarction. Cardiology 2016;134:436–443. [DOI] [PubMed] [Google Scholar]
- 21. Liu Z, Liu Q, Cai H, et al. Calcitonin gene‐related peptide prevents blood‐brain barrier injury and brain edema induced by focal cerebral ischemia reperfusion. Regul Pept 2011;171:19–25. [DOI] [PubMed] [Google Scholar]
- 22. Dodick DW, Goadsby PJ, Silberstein SD, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene‐related peptide, for the prevention of frequent episodic migraine: a randomised, double‐blind, placebo‐controlled, exploratory phase 2 trial. Lancet Neurol 2014;13:1100–1107. [DOI] [PubMed] [Google Scholar]
- 23. Sacco S, Bendtsen L, Ashina M, et al. European headache federation guideline on the use of monoclonal antibodies acting on the calcitonin gene related peptide or its receptor for migraine prevention. J Headache Pain 2019;20:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurth T, Chabriat H, Bousser MG. Migraine and stroke: a complex association with clinical implications. Lancet Neurol 2012;11:92–100. [DOI] [PubMed] [Google Scholar]
- 25. Mawet J, Kurth T, Ayata C. Migraine and stroke: in search of shared mechanisms. Cephalalgia 2015;35:165–181. [DOI] [PubMed] [Google Scholar]
- 26. Eikermann‐Haerter K, Lee JH, Yuzawa I, et al. Migraine mutations increase stroke vulnerability by facilitating ischemic depolarizations. Circulation 2012;125:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mawet J, Eikermann‐Haerter K, Park KY, et al. Sensitivity to acute cerebral ischemic injury in migraineurs: a retrospective case‐control study. Neurology 2015;85:1945–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van den Maagdenberg AM, Pizzorusso T, Kaja S, et al. High cortical spreading depression susceptibility and migraine‐associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol 2010;67:85–98. [DOI] [PubMed] [Google Scholar]
- 29. Pinho‐Ribeiro FA, Baddal B, Haarsma R, et al. Blocking neuronal signaling to immune cells treats streptococcal invasive infection. Cell 2018;173:1083–1097.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aubdool AA, Graepel R, Kodji X, et al. TRPA1 is essential for the vascular response to environmental cold exposure. Nat Commun 2014;5:5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheykhzade M, Amandi N, Pla MV, et al. Binding and functional pharmacological characteristics of gepant‐type antagonists in rat brain and mesenteric arteries. Vascul Pharmacol 2017;90:36–43. [DOI] [PubMed] [Google Scholar]
- 32. Aviles‐Rosas VH, Rivera‐Mancilla E, Marichal‐Cancino BA, et al. Olcegepant blocks neurogenic and non‐neurogenic CGRPergic vasodepressor responses and facilitates noradrenergic vasopressor responses in pithed rats. Br J Pharmacol 2017;174:2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hershey JC, Corcoran HA, Baskin EP, et al. Investigation of the species selectivity of a nonpeptide CGRP receptor antagonist using a novel pharmacodynamic assay. Regul Pept 2005;127:71–77. [DOI] [PubMed] [Google Scholar]
- 34. Hirsch S, Corradini L, Just S, et al. The CGRP receptor antagonist BIBN4096BS peripherally alleviates inflammatory pain in rats. Pain 2013;154:700–707. [DOI] [PubMed] [Google Scholar]
- 35. Michot B, Bourgoin S, Viguier F, et al. Differential effects of calcitonin gene‐related peptide receptor blockade by olcegepant on mechanical allodynia induced by ligation of the infraorbital nerve vs the sciatic nerve in the rat. Pain 2012;153:1939–1948. [DOI] [PubMed] [Google Scholar]
- 36. Munro G, Petersen S, Jansen‐Olesen I, Olesen J. A unique inbred rat strain with sustained cephalic hypersensitivity as a model of chronic migraine‐like pain. Sci Rep 2018;8:1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petersen KA, Birk S, Doods H, et al. Inhibitory effect of BIBN4096BS on cephalic vasodilatation induced by CGRP or transcranial electrical stimulation in the rat. Br J Pharmacol 2004;143:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iovino M, Feifel U, Yong CL, et al. Safety, tolerability and pharmacokinetics of BIBN 4096 BS, the first selective small molecule calcitonin gene‐related peptide receptor antagonist, following single intravenous administration in healthy volunteers. Cephalalgia 2004;24:645–656. [DOI] [PubMed] [Google Scholar]
- 39. Tvedskov JF, Tfelt‐Hansen P, Petersen KA, et al. CGRP receptor antagonist olcegepant (BIBN4096BS) does not prevent glyceryl trinitrate‐induced migraine. Cephalalgia 2010;30:1346–1353. [DOI] [PubMed] [Google Scholar]
- 40. Petersen KA, Birk S, Lassen LH, et al. The CGRP‐antagonist, BIBN4096BS does not affect cerebral or systemic haemodynamics in healthy volunteers. Cephalalgia 2005;25:139–147. [DOI] [PubMed] [Google Scholar]
- 41. Petersen KA, Lassen LH, Birk S, et al. BIBN4096BS antagonizes human alpha‐calcitonin gene related peptide‐induced headache and extracerebral artery dilatation. Clin Pharmacol Ther 2005;77:202–213. [DOI] [PubMed] [Google Scholar]
- 42. Luo G, Chen L, Conway CM, et al. Discovery of (5S,6S,9R)‐5‐amino‐6‐(2,3‐difluorophenyl)‐6,7,8,9‐tetrahydro‐5H‐cyclohepta[b]pyri din‐9‐yl 4‐(2‐oxo‐2,3‐dihydro‐1H‐imidazo[4,5‐b]pyridin‐1‐yl)piperidine‐1‐carboxylate (BMS‐927711): an oral calcitonin gene‐related peptide (CGRP) antagonist in clinical trials for treating migraine. J Med Chem 2012;55:10644–10651. [DOI] [PubMed] [Google Scholar]
- 43. Mallee JJ, Salvatore CA, LeBourdelles B, et al. Receptor activity‐modifying protein 1 determines the species selectivity of non‐peptide CGRP receptor antagonists. J Biol Chem 2002;277:14294–14298. [DOI] [PubMed] [Google Scholar]
- 44. Marcus R, Goadsby PJ, Dodick D, et al. BMS‐927711 for the acute treatment of migraine: a double‐blind, randomized, placebo controlled, dose‐ranging trial. Cephalalgia 2014;34:114–125. [DOI] [PubMed] [Google Scholar]
- 45. Olesen J, Diener HC, Husstedt IW, et al. Calcitonin gene‐related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med 2004;350:1104–1110. [DOI] [PubMed] [Google Scholar]
- 46. Chen J, Zhang C, Jiang H, et al. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab 2005;25:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rubio‐Beltrán E, Chan KY, Danser AJ, et al. Characterisation of the calcitonin gene‐related peptide receptor antagonists ubrogepant and atogepant in human isolated coronary, cerebral and middle meningeal arteries. Cephalalgia 2020;40:357–366. [DOI] [PubMed] [Google Scholar]
- 48. Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res 1977;41:19–26. [DOI] [PubMed] [Google Scholar]
- 49. Petersen KA, Nilsson E, Olesen J, Edvinsson L. Presence and function of the calcitonin gene‐related peptide receptor on rat pial arteries investigated in vitro and in vivo. Cephalalgia 2005;25:424–432. [DOI] [PubMed] [Google Scholar]
- 50. Ashina M, Dodick D, Goadsby PJ, et al. Erenumab (AMG 334) in episodic migraine: interim analysis of an ongoing open‐label study. Neurology 2017;89:1237–1243. [DOI] [PubMed] [Google Scholar]
- 51. Aradi S, Kaiser E, Cucchiara B. Ischemic stroke associated with calcitonin gene‐related peptide inhibitor therapy for migraine: a case report. J Stroke Cerebrovasc Dis 2019;28:104286. [DOI] [PubMed] [Google Scholar]
- 52. Hargreaves R, Olesen J. Calcitonin gene‐related peptide modulators—the history and renaissance of a new migraine drug class. Headache 2019;59:951–970. [DOI] [PubMed] [Google Scholar]
- 53. Chaitman BR, Ho AP, Behm MO, et al. A randomized, placebo‐controlled study of the effects of telcagepant on exercise time in patients with stable angina. Clin Pharmacol Ther 2012;91:459–466. [DOI] [PubMed] [Google Scholar]
- 54. Depre C, Antalik L, Starling A, et al. A randomized, double‐blind, placebo‐controlled study to evaluate the effect of erenumab on exercise time during a treadmill test in patients with stable angina. Headache 2018;58:715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ho TW, Ho AP, Chaitman BR, et al. Randomized, controlled study of telcagepant in patients with migraine and coronary artery disease. Headache 2012;52:224–235. [DOI] [PubMed] [Google Scholar]
- 56. Maassen van den Brink A, Rubio‐Beltran E, Duncker D, Villalon CM. Is CGRP receptor blockade cardiovascularly safe? Appropriate studies are needed. Headache 2018;58:1257–1258. [DOI] [PubMed] [Google Scholar]
- 57. Maasumi K, Michael RL, Rapoport AM. CGRP and migraine: the role of blocking calcitonin gene‐related peptide ligand and receptor in the management of migraine. Drugs 2018;78:913–928. [DOI] [PubMed] [Google Scholar]
- 58. Edvinsson L, Nilsson E, Jansen‐Olesen I. Inhibitory effect of BIBN4096BS, CGRP(8‐37), a CGRP antibody and an RNA‐Spiegelmer on CGRP induced vasodilatation in the perfused and non‐perfused rat middle cerebral artery. Br J Pharmacol 2007;150:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grell AS, Haanes KA, Johansson SE, et al. Fremanezumab inhibits vasodilatory effects of CGRP and capsaicin in rat cerebral artery—potential role in conditions of severe vasoconstriction. Eur J Pharmacol 2019;864:172726. [DOI] [PubMed] [Google Scholar]
- 60. Benschop RJ, Collins EC, Darling RJ, et al. Development of a novel antibody to calcitonin gene‐related peptide for the treatment of osteoarthritis‐related pain. Osteoarthritis Cartilage 2014;22:578–585. [DOI] [PubMed] [Google Scholar]
- 61. Gupta S, Mehrotra S, Villalón CM, et al. Characterisation of CGRP receptors in human and porcine isolated coronary arteries: evidence for CGRP receptor heterogeneity. Eur J Pharmacol 2006;530:107–116. [DOI] [PubMed] [Google Scholar]
- 62. Haanes KA, Chan KY, MaassenVanDenBrink A. Comment on "A second trigeminal CGRP receptor: function and expression of the AMY1 receptor". Ann Clin Transl Neurol 2016;3:307–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gupta S, Mehrotra S, Avezaat CJ, et al. Characterisation of CGRP receptors in the human isolated middle meningeal artery. Life Sci 2006;79:265–271. [DOI] [PubMed] [Google Scholar]
- 64. Chan KY, Edvinsson L, Eftekhari S, et al. Characterization of the calcitonin gene‐related peptide receptor antagonist telcagepant (MK‐0974) in human isolated coronary arteries. J Pharmacol Exp Ther 2010;334:746–752. [DOI] [PubMed] [Google Scholar]
- 65. Edvinsson L, Chan KY, Eftekhari S, et al. Effect of the calcitonin gene‐related peptide (CGRP) receptor antagonist telcagepant in human cranial arteries. Cephalalgia 2010;30:1233–1240. [DOI] [PubMed] [Google Scholar]
- 66. Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015;95:953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. Overview of all in vivo experimental groups including sample sizes, exclusions, early mortality, and age and sex distributions. BP = blood pressure; CBF = cerebral blood flow; d = days; F = female; HR = heart rate; M = male; MCAO = middle cerebral artery occlusion; Olce = olcegepant; Rime = rimegepant; SD = standard deviation.