

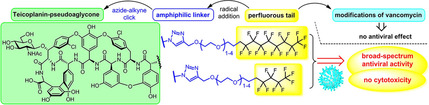
Abstract
The limited scope of antiviral drugs and increasing problem of antiviral drug resistance represent a global health threat. Glycopeptide antibiotics and their lipophilic derivatives have emerged as relevant inhibitors of diverse viruses. Herein, we describe a new strategy for the synthesis of dual hydrophobic and lipophobic derivatives of glycopeptides to produce selective antiviral agents without membrane‐disrupting activity. Perfluorobutyl and perfluorooctyl moieties were attached through linkers of different length to azido derivatives of vancomycin aglycone and teicoplanin pseudoaglycone, and the new derivatives were evaluated against a diverse panel of viruses. The teicoplanin derivatives displayed strong anti‐influenza virus activity at nontoxic concentrations. Some of the perfluoroalkylated glycopeptides were also active against a few other viruses such as herpes simplex virus or coronavirus. These data encourage further exploration of glycopeptide analogues for broad antiviral application.
Keywords: antiviral, coronavirus, glycopeptide antibiotic, influenza virus, perfluoroalkyl
Plan F: Perfluoroalkylated blocks were incorporated into azido glycopeptide derivatives of vancomycin and teicoplanin. The teicoplanin derivatives displayed strong antiviral activity against a diverse panel of viruses at nontoxic concentrations. The vancomycin derivatives showed cytotoxicity, while being inactive against influenza viruses. Perfluorobutylated teicoplanins displayed excellent antibacterial activity against a panel of Gram‐positive bacteria.
Introduction
Many biologically active small molecules contain one or more fluorine substituents, owing to the beneficial effect of fluorination on pharmacokinetic and pharmacodynamic properties.1, 2, 3, 4, 5, 6, 7 The use of fluorine substituents in medicinal chemistry has continuously increased,8, 9, 10 and currently more than 50 % of blockbuster drugs are fluorinated. In these compounds, the presence of a few fluorine atoms strongly modifies the biological activity and chemical reactivity, while having relatively low impact on the physical properties.11, 12 On the other hand, the physical characteristics of perfluorinated compounds are very different from those of the hydrogen‐containing analogues. Most importantly, perfluorination simultaneously enhances hydrophobicity and lipophobicity,13, 14 a unique characteristic that creates exceptional biomedical possibilities. Although the number of pharmacologically applicable molecules containing perfluoroalkyl substituents is currently still limited, polyfluorinated compounds have shown promise in several areas of medicinal chemistry.15, 16, 17
In recent years, we conducted a systematic study on the synthesis of lipophilic derivatives of the glycopeptide antibiotics vancomycin, teicoplanin and ristocetin, and this yielded several new antibiotics with promising antibacterial and antiviral activity.18, 19, 20, 21, 22 We demonstrated that the high lipophilicity of the side chains (C8−C10 alkyl groups) in these molecules is essential for antiviral activity against, among others, influenza virus. However, antiviral activity proved accompanied by high cytotoxicity, probably due to a membrane‐disrupting effect of the highly lipophilic side chains.22 In addition, we found that cytotoxicity was reduced by decreasing the overall lipophilicity of the compounds following incorporation of a tetra(ethylene glycol) linker between the peptide core and the lipophilic group.22 Hence, we assumed that attaching perfluoroalkyl groups that are highly hydro‐ and lipophobic might confer antiviral activity without creating a membrane‐disrupting effect. Namely, it has been shown that triphilic polymers bearing bulky and lipophobic perfluorinated blocks do not mix well with the hydrocarbon chains of lipids, explaining their low membrane partition coefficients.23, 24 This is the reason why fluorinated surfactants are non‐cytolytic, that is, they are unable to solubilize membranes, as opposed to their hydrogenated counterparts.25
Herein, we report on the synthesis and antiviral plus antibacterial evaluation of perfluoroalkyl derivatives of vancomycin aglycone and teicoplanin pseudoaglycone. Although synthetic modification of glycopeptide antibiotics is increasingly recognized in the context of antiviral26 and antibacterial27 drug design, our study is the first to explore conjugation of glycopeptides with perfluoroalkyl groups.
Results and Discussion
Synthesis
Our recent results on semisynthetic lipoglycopeptides revealed that the type and length of the linkers significantly modifies the antiviral activity and cytotoxicity.22 Herein, we have chosen the 1 a and 1 b allyl ethers of ethylene and tetra(ethylene glycol)s as linker chains between perfluoroalkyl substituents and the antibiotic molecules (Scheme 1). A light‐promoted atom‐transfer radical addition reaction28 of the commercially available perfluorobutyl iodide and perfluorooctyl iodide onto the double bond of 1 a and 1 b resulted in 2 a and 2 b, as well as 5 a and 5 b in good yields. Attempted reductive removal of iodo substituent with LiAlH4 proceeded with low efficacy. Fortunately, catalytic hydrogenation of the iodo derivatives gave 3 a and 3 b, as well as 6 a and 6 b with moderate to high yields. Finally, these derivatives were reacted with propargyl bromide, respectively, to produce propargylated compounds 4 a and 4 b and 7 a and 7 b that are suitable for the azide alkyne cycloaddition click reaction.
Scheme 1.
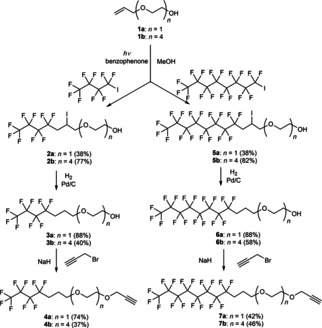
Assembly of the clickable perfluoroalkyl side chains 4 a, 4 b, 7 a and 7 b.
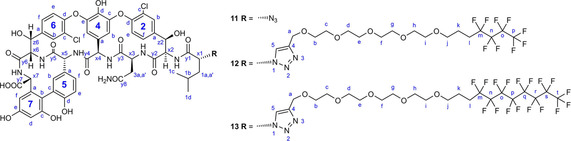
For the derivatization of vancomycin aglycon, an N‐terminal azido analogue 11 was prepared (Scheme 2). Vancomycin hexapeptide 8 obtained from vancomycin aglycone by Edman degradation29 was acylated with d‐azidoleucine succinimide ester 10 prepared from d‐azidoleucine 9.30 A copper(I)‐catalyzed 1,3‐dipolar cycloaddition reaction of 4 b or 7 b with 11 resulted in perfluorobutyl and perfluorooctyl derivatives 12 and 13 of vancomycin aglycone.
Scheme 2.
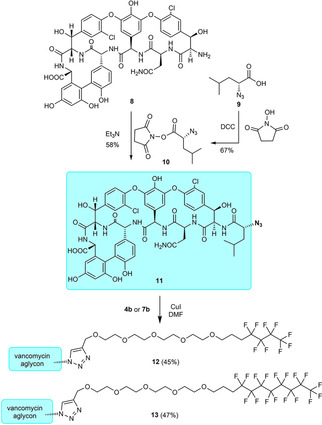
Synthesis of vancomycin aglycone azide 11 and its conjugation with fluorous side chains.
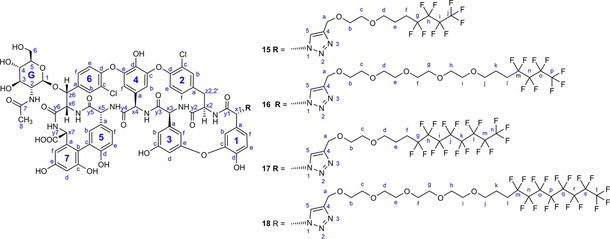
Similar click reactions of azido derivative of teicoplanin pseudoaglycone 14 18 with 4 a, 4 b, 7 a, or 7 b afforded perfluoroalkyl derivatives 15, 16, 17 and 18 with linker chains of different length (Scheme 3).
Scheme 3.
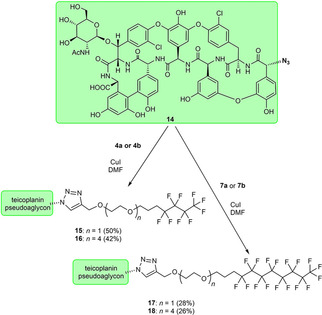
Attachment of fluorous side chains of different length to teicoplanin pseudoaglycone.
Structure elucidation and testing oligomerization
In addition to NMR structure validation (see the Supporting Information, including 13C HSQC, HMBC, 13C and 19F spectra and spectral assignments) we tested compound 17 for possible oligomerization. Strong head to tail dimers are commonly found of glycopeptide antibiotics in aqueous solutions, that is co‐operatively enhanced by ligand binding.31, 32, 33 As 17 is not soluble in water, it was dissolved in MeOD for NMR diffusion experiments.34 We used the internal mass standard method as described earlier.35 TMS (tetramethylsilane, MW=88.2) and β‐cyclodextrin (β‐CD, MW=1135) were used alternatively. About 20 mg of sample 17 was dissolved in 500 μL MeOD and measured at 300 K temperature. Spectra were recorded with 32 or 64 linear gradient steps and evaluated with topspin 2.1 software using inverse Laplace transformation to obtain diffusion domain. These experiments yielded MW between 7–8 kDa when referenced to internal TMS. (Figure S1). However, in repeated measurements with ∼3 mg of 17 dissolved in MeOD and referenced to internal β‐CD, DOSY yielded only 2.4 kDa mass for 17, that is closer to monomeric state (Figure S2). In control DOSY experiments of teicoplanin and vancomycin dissolved in [D6]DMSO with β‐CD mass reference, DOSY yielded the monomeric molecular masses as expected. Hence, we do not suppose the presence of oligomers in case of dilute MeOD solutions of 17.
Biological evaluation
Vancomycin aglycone derivatives 12 and 13 were inactive against various influenza strains and both were highly cytotoxic in MDCK cells (Table 1). Perfluorobutyl and perfluorooctyl derivatives 15, 16 and 17 of teicoplanin pseudoaglycone displayed robust activity against influenza A and B viruses with a favorable selectivity (i. e., ratio between cytotoxicity and activity). In contrast, the similar perfluorooctyl derivative 18 with a tetra(ethylene glycol) linker displayed high cytotoxicity without antiviral activity. Although this marked difference between vancomycin and teicoplanin derivatives may seem surprising, we previously demonstrated that even a slight difference in the peptide core can lead to very different antiviral properties.36
Table 1.
Cytotoxicity and anti‐influenza virus activity in MDCK[a] cell cultures.
|
Compound |
Cytotoxicity [μM] |
Antiviral EC50 [d] [μM] |
||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
A/H1N1 |
A/H3N2 |
Influenza B |
|||
|
|
MCC[b] |
CC50 [c] |
CPE |
MTS |
CPE |
MTS |
CPE |
MTS |
|
12 |
42 |
4 |
>100 |
>100 |
>100* |
>100* |
>100 |
>100 |
|
13 |
0.8 |
0.8 |
>100 |
>100 |
>100* |
>100* |
>100 |
>100 |
|
15 |
100 |
>100 |
7.7 |
7.2 |
2.3 |
1.9 |
8.9 |
11 |
|
16 |
100 |
97 |
5.6 |
1.6 |
2.3* |
2.5* |
5.6 |
6.5 |
|
17 |
100 |
44 |
6.8 |
8.6 |
1.2 |
1.6 |
4.0 |
3.4 |
|
18 |
– |
4.2 |
– |
>100 |
– |
>100* |
– |
>100 |
|
teicoplanin |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
|
14 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
|
vancomycin⋅HCl |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
|
zanamivir |
>100 |
>100 |
1.5 |
3.1 |
20 |
9.0 |
2 |
1.7 |
[a] Madin Darby canine kidney cells. Virus strains: A/H1 N1: A/Ned/378/05; A/H3 N2: A/HK/7/87* or A/Victoria/361/11; influenza B virus: B/Ned/537/05. [b] Minimum cytotoxic concentration, i. e., minimal compound concentration causing a microscopically detectable alteration in cell morphology. [c] 50 % cytotoxic concentration based on the formazan‐based MTS cell viability assay. [d] 50 % effective concentration, or concentration producing 50 % inhibition of virus‐induced cytopathic effect, as determined by visual CPE scoring (left column), or by measuring cell viability with the MTS assay (right column).
Besides, activity was noted against herpes simplex virus and vaccinia virus (compounds 15, 16, 17 and 18), adenovirus (15 and 17) and coronavirus (17 and 18; Table 2). Two other sensitive viruses were respiratory syncytial virus (EC50 values: 9.9 μM for 15; 11 μM for 16 and 4.6 μM for 17; MTS‐based CPE assay in HeLa cells) and Zika virus (EC50 for 15: 10 μM; MTS‐based CPE assay in Vero cells; Tables S1 and S2).
Table 2.
Cytotoxicity and antiviral activity in HEL[a] cell cultures.
|
Compound |
Cytotoxicity |
Antiviral EC50 [c] [μM] |
|||||
|---|---|---|---|---|---|---|---|
|
|
CC50 [b] [μM] |
HSV‐1 |
HSV‐1/TK− |
HSV‐2 |
Vaccinia |
AdV‐2 |
HCoV |
|
15 |
>100 |
54 |
39 |
2.0 |
10 |
42 |
>100 |
|
16 |
>100 |
33 |
41 |
25 |
48 |
>100 |
>100 |
|
17 |
>100 |
16 |
16 |
2.2 |
7.9 |
60 |
40 |
|
18 |
>100 |
9.8 |
19 |
14 |
17 |
>100 |
4.9 |
|
cidofovir |
>250 |
2.4 |
4.7 |
1.0 |
10 |
6.4 |
– |
|
acyclovir |
>250 |
2.4 |
146 |
0.05 |
>250 |
– |
– |
|
alovudine |
>250 |
– |
– |
– |
– |
5.9 |
– |
[a] HEL: human embryonic lung fibroblast cells. Viruses: herpes simplex virus type 1 (HSV‐1) or type 2 (HSV‐2); a thymidine‐kinase deficient (TK−) mutant of HSV‐1; vaccinia virus; human adenovirus type 2 (AdV‐2) and human coronavirus (HCoV) 229E. [b] 50 % Cytotoxic concentration based on the formazan‐based MTS cell viability assay. [c] 50 % Effective concentration, based on measuring cell viability with the MTS assay.
The broad activity of teicoplanin pseudoaglycone derivatives 15, 16 and 17 against different viruses is consistent with our hypothesis that these derivatives may act by disrupting the viral endocytosis process, similarly to what we reported for a glycopeptide active against influenza virus.37 Recently, it has been described that glycopeptide antibiotics like teicoplanin, dalbavancin, oritavancin and telavancin are able to prevent the host cell entry processes of Ebola virus, Middle East respiratory syndrome coronavirus (MERS‐CoV) and severe acute respiratory syndrome coronavirus (SARS‐CoV), which results also fit with our surmise.38, 39
Finally, the antibacterial activity was evaluated on a panel of Gram‐positive bacteria (Table 3). Vancomycin derivatives 12 and 13 displayed moderate activity. Perfluorooctyl derivatives 17 and 18 of teicoplanin pseudoaglycone were inactive, but perfluorobutyl compounds 15 and 16 had excellent antibacterial activity against sensitive and resistant staphylococci and good activity against resistant enterococci having vanA and vanB genes.
Table 3.
Antibacterial effects.
|
|
|
|
MIC[g] [μg/mL] |
|||||
|---|---|---|---|---|---|---|---|---|
|
|
TEI |
VAN |
12 |
13 |
15 |
16 |
17 |
18 |
|
Bacillus subtilis ATCC[a] 6633 |
0.5 |
0.5 |
32 |
16 |
0.5 |
2 |
128 |
4 |
|
Staphylococcus aureus MSSA[b] ATCC 29213 |
0.5 |
0.5 |
4 |
8 |
0.5 |
0.5 |
16 |
8 |
|
S. aureus MRSA[c] ATCC 33591 |
0.5 |
0.5 |
8 |
8 |
0.5 |
0.5 |
16 |
4 |
|
Staphylococcus epidermidis ATCC 35984 biofilm |
2 |
2 |
8 |
16 |
0.5 |
0.5 |
16 |
4 |
|
S. epidermidis mecA[d] |
16 |
4 |
8 |
8 |
0.5 |
0.5 |
256 |
8 |
|
Enterococcus faecalis ATCC 29212 |
2 |
1 |
8 |
4 |
0.5 |
1 |
256 |
16 |
|
E. faecalis 15376 VanA[e] |
256 |
256 |
16 |
256 |
1 |
2 |
256 |
32 |
|
E. faecalis ATCC 51299 VanB[f] |
4 |
128 |
16 |
32 |
4 |
1 |
256 |
32 |
[a] American Type Culture Collection. [b] Methicillin‐sensitive S. aureus. [c] Methicillin‐resistant S. aureus. [d] mecA gene expression in Staphylococcus. [e] vanA gene positive. [f] vanB gene positive. [g] Minimum inhibitory concentration. TEI: teicoplanin, VAN: vancomycin.
Conclusion
We have designed and synthesized the first members of fluoroglycopeptides, a novel type of glycopeptide antibiotic derivatives bearing perfluoroalkyl side chains. Such substituents are not only hydrophobic, but at the same time lipophobic endowing the antibiotic molecules with unique physicochemical properties that are worth exploring and exploiting from an antimicrobial point of view. As the lipophobic perfluoroalkyl blocks are known to have no membrane activity, low cytotoxicity of the perfluoroalkylated glycopeptides were expected. However, the vancomycin derivatives and one of the teicoplanin molecules showed cytotoxicity on canine kidney cells whereas they were inactive against influenza viruses. At the same time, three of the new teicoplanin derivatives displayed high activity against the influenza strains studied, two were active against human corona and adenoviruses, and all teicoplanins proved to be active against vaccinia and herpes viruses without showing cytotoxicity. Moreover, perfluorobutyl derivatives of teicoplanin pseudoaglycone displayed excellent antibacterial activity against a panel of Gram‐positive bacteria.
We hope that these results can open a new way in finding more effective antivirals based on glycopeptide antibiotics.
Experimental Section
General information
Compounds 1 b,40 8,29 9 30 and 14 18 were synthesized according to the literature. Compound 1 a was purchased from Sigma‐Aldrich, nonafluoro‐1‐iodobutane from TCI and heptadecafluoro‐1‐iodooctane from Alfa Aesar. TLC was performed on Kieselgel 60 F254 (Merck) with detection either by immersing into ammonium molybdate‐sulfuric acid solution followed by heating or by using Pauly's reagent for detection. Flash column chromatography was performed using Silica gel 60 (Merck 0.040–0.063 mm). The photoinitiated reactions were carried out in a borosilicate vessel by irradiation with a Hg lamp giving maximum emission at 365 nm. The 1H NMR (500, 400 and 360 MHz) 13C NMR (125, 100 and 90 MHz) and 2D NMR spectra were recorded with a Bruker DRX‐360, Bruker DRX‐400 and Bruker Avance II 500 spectrometer at 298 or 300 K. For DOSY (Diffusion Ordered Spectroscopy) experiments, Bruker AVANCE‐II, 500 MHz spectrometer was applied, with manufacturer‘s “ledbpgp2 s” pulse sequence. Chemical shifts are referenced to Me4Si (0.00 ppm for 1H) and to the solvent residual signals. MALDI‐TOF MS analyses of the compounds were carried out in the positive reflectron mode (20 kV) using a BIFLEX III mass spectrometer (Bruker) equipped with delayed‐ion extraction. A nitrogen laser (337 nm, 3 ns pulse width, 106–107 W/cm2) operating at 4 Hz was applied to produce laser desorption. 2,5‐Dihydroxybenzoic acid (DHB) was used as matrix and F3CCOONa as cationizing agent in DMF. ESI‐QTOF MS measurements were carried out on a maXis II UHR ESI‐QTOF MS instrument (Bruker), in positive ionization mode. The following parameters were applied for the electrospray ion source: capillary voltage: 3.5 kV; end plate offset: 500 V; nebulizer pressure: 0.8 bar; dry gas temperature: 200 °C and dry gas flow rate: 4.5 L/min. Constant background correction was applied for each spectrum, the background was recorded before each sample by injecting the blank sample matrix (solvent). Na‐formate calibrant was injected after each sample, which enabled internal calibration during data evaluation. Mass spectra were recorded by otofControl version 4.1 (build: 3.5, Bruker) and processed by Compass DataAnalysis version 4.4 (build: 200.55.4). The antibacterial evaluations were carried out as it was described in our previous publication.27b
Table 4.
NMR data of vancomycin derivatives.
|
| ||||||
|---|---|---|---|---|---|---|
|
|
11 |
12 |
13 |
|||
|
|
1H |
13C |
1H |
13C |
1H |
|
|
y1 |
– |
177.2113 |
– |
173.99 |
– |
174.55 |
|
y8 (Asp) |
– |
173.9445 |
– |
171.2 |
– |
171.52 |
|
y2 |
– |
172.0452 |
– |
169.9 |
– |
170.39 |
|
y3 |
– |
171.4567 |
– |
169.09 |
– |
169.55 |
|
y4 |
– |
171.3606 |
– |
169.02 |
– |
168.79 |
|
y5 |
– |
169.1608 |
– |
168.25 |
– |
166.96 |
|
y7 |
– |
n.d. |
– |
166.35 |
– |
157.41 |
|
y6 |
– |
n.d. |
– |
166.27 |
– |
155.81 |
|
7e |
– |
157.5295 |
– |
157.03 |
– |
155.15 |
|
7c |
– |
156.6308 |
– |
155.36 |
– |
150.35 |
|
5d |
– |
155.5733 |
– |
154.75 |
– |
148.80 |
|
2d |
– |
151.3203 |
– |
150.02 |
– |
147.79 |
|
4e |
– |
150.7161 |
– |
148.4 |
– |
147.61 |
|
4c |
– |
149.3844 |
– |
147.45 |
– |
147.51 |
|
6d |
– |
148.395 |
– |
147.16 |
– |
143.94 |
|
6a |
– |
141.2641 |
– |
143.63 |
– |
143.94 |
|
2a |
– |
139.7893 |
– |
142.25 |
– |
142.57 |
|
7a |
– |
138.83 |
– |
139.28 |
– |
139.48 |
|
5b |
7.09 |
136.83 |
7.22 |
135.81 |
7.23 |
136.30 |
|
4d |
– |
135.07 |
|
133.6 |
– |
133.94 |
|
4a |
– |
129.33 |
|
128.42 |
– |
128.85 |
|
2b |
7.2 |
129.91 |
7.42 |
128.24 |
7.4 |
128.84 |
|
6b |
7.56 |
128.57 |
7.89 |
127.28 |
7.88 |
127.74 |
|
6f |
7.56 |
128.57 |
7.46 |
126.94 |
7.47 |
127.45 |
|
2c |
– |
128.77 |
|
126.51 |
– |
127.01 |
|
2f |
7.22 |
128.18 |
7.52 |
126.94 |
7.48 |
127.44 |
|
5a |
– |
127.16 |
|
125.81 |
– |
126.86 |
|
6c |
– |
126.82 |
|
125.64 |
– |
126.19 |
|
5f |
6.87 |
127 |
6.75 |
124.51 |
6.72 |
124.97 |
|
2e |
n.d. |
n.d. |
7.23 |
124.09 |
7.25 |
124.50 |
|
6e |
n.d. |
n.d. |
7.23 |
122.74 |
7.24 |
123.30 |
|
5c |
– |
123.31 |
|
122.36 |
– |
126.08 |
|
7b |
– |
119.11 |
|
117.21 |
– |
122.71 |
|
5e |
6.82 |
118.44 |
6.68 |
115.62 |
6.69 |
116.01 |
|
4f |
5.49 |
106.59 |
5.6 |
106.82 |
5.57 |
107.11 |
|
7f |
6.38 |
108.63 |
n.d. |
n.d. |
6.6 |
108.20 |
|
4b |
5.26 |
105.23 |
5.21 |
104.12 |
5.21 |
104.49 |
|
7d |
6.36 |
103.3 |
6.27 |
101.23 |
6.29 |
101.63 |
|
z6 |
5.18 |
72.76 |
5.18 |
71.01 |
5.15 |
71.35 |
|
z2 |
5.3 |
72.24 |
5.12 |
70.72 |
5.15 |
71.06 |
|
x6 |
4.09 |
63.7 |
4.17 |
61.52 |
4.18 |
61.90 |
|
x1 |
3.81 |
61.54 |
5.73 |
60.67 |
5.76 |
60.97 |
|
x2 |
4.33 |
60.43 |
4.73 |
58.99 |
4.73 |
59.45 |
|
x7 |
4.42 |
55.28 |
4.34 |
58.99 |
4.33 |
59.29 |
|
x4 |
5.96 |
55.28 |
5.7 |
54.41 |
5.71 |
54.73 |
|
x5 |
4.72 |
52.72 |
4.48 |
53.39 |
4.48 |
53.41 |
|
x3 |
n.d. |
n.d. |
4.33 |
50.75 |
4.35 |
51.15 |
|
1a,a’ |
2.36 |
39.26 |
1.92/2.04 |
39.96 |
1.91, 2.03 |
40.08 |
|
3a,a’ |
n.d. |
n.d. |
2.16/2.58 |
36.26 |
2.89 |
35.61 |
|
1b |
1.75 |
24.64 |
1.23 |
23.96 |
1.18 |
24.36 |
|
1c |
n.d. |
n.d. |
0.9 |
21.04 |
0.82 |
22.60 |
|
1d |
n.d. |
n.d. |
0.83 |
22.15 |
0.89 |
21.30 |
|
side chain |
|
|
|
|
|
|
|
triazole CH (5) |
|
|
8.18 |
122.61 |
8.2 |
123.13 |
|
triazole C (4) |
|
|
– |
118.32 |
– |
118.01 |
|
OCH2 (a) |
|
|
4.55 |
63.15 |
4.52 |
63.51 |
|
OCH2 |
|
|
3.52 |
69.41 |
3.5 |
69.80 |
|
OCH2 |
|
|
3.54 |
69.36 |
3.5 |
69.72 |
|
OCH2 |
|
|
3.52 |
69.11 |
3.5 |
69.49 |
|
OCH2 |
|
|
3.58 |
68.62 |
3.56 |
69.00 |
|
|
|
|
3.5 |
68.11 |
3.48 |
68.50 |
|
CH2 (l) |
|
|
2.29 |
26.45 |
2.26 |
26.9 |
|
CH2 (k) |
|
|
1.77 |
19.92 |
1.75 |
20.30 |
n.d.: not determined .
2‐((4,4,5,5,6,6,7,7,7‐Nonafluoro‐2‐iodoheptyl)oxy)ethan‐1‐ol (2 a)
Nonafluoro‐1‐iodobutane (2.1 mL, 4.15 g, 12 mmol) and benzophenone (20 mg, 0.11 mmol) were added to a solution of 2‐(allyloxy)ethan‐1‐ol (1 a; 1.02 g, 10 mmol) in methanol (15 mL). Argon gas was bubbled through the solution and then irradiation occurred for 10 min. The solvent was evaporated, and the product was purified by flash column chromatography (hexane/acetone 8 : 2) to yield 2 a (1.7 g, 38 %) as a colorless liquid. R f=0.34 (hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3): δ=4.44–4.35 (m, 1H, CIH), 3.83–3.59 (m, 6H, 3CH2), 3.13–2.94 (m, 1H, CH2), 2.84–2.64 (m, 1H, CH2), 2.12 ppm (s, 1H, OH); 13C NMR (100 MHz, CDCl3): δ=117.7, 106.7 (4 C, CF2 and CF3), 75.8, 72.4, 61.8 (3 C, CH2), 37.8 (t, 1 C, CH2), 14.9 ppm (1 C, CHI); HRMS (ESI): m/z calcd for C9H10F9IO2+Na+: 470.9479 [M+Na]+; found: 470.9474.
16,16,17,17,18,18,19,19,19‐Nonafluoro‐14‐iodo‐3,6,9,12‐tetraoxanonadecan‐1‐ol (2 b)
Nonafluoro‐1‐iodobutane (0.944 mL, 1.9 g, 5.5 mmol) and benzophenone (10 mg, 0.055 mmol) were added to a solution of 3,6,9,12‐tetraoxapentadec‐14‐en‐1‐ol (1 b; 1.17 g, 5 mmol) in methanol (12 mL). Argon gas was bubbled through the solution and then irradiation occurred for 10 min. The solvent was evaporated, and the product was purified by flash column chromatography (hexane/acetone 7 : 3) to yield 2 b (2.09 g, 77 %) as a colorless liquid. R f=0.29 (hexane/acetone 7 : 3); 1H NMR (360 MHz, CDCl3): δ=4.41–4.31 (m, 1H, CIH), 3.84–3.56 (m, 18H, 9CH2), 3.20–3.01 (m, 1H, CH2), 2.93–2.85 (m, 1H, OH), 2.77–2.57 ppm (m, 1H, CH2); 13C NMR (90 MHz, CDCl3): δ=76.2, 72.6, 70.7, 70.65, 70.6, 70.5, 70.4, 61.7 (9 C, CH2), 37.4 (t, 1 C, CH2), 14.5 ppm (1 C, CHI); HRMS (ESI): m/z calcd for C15H22F9IO5+Na+: 603.0266 [M+Na]+; found: 603.0260.
2‐((4,4,5,5,6,6,7,7,7‐Nonafluoroheptyl)oxy)ethan‐1‐ol (3 a)
To the solution of 2 a (0.896 g, 2 mmol) in methanol (15 mL) 10 % palladium on activated charcoal (270 mg) and NaHCO3 (420 mg, 5 mmol) were added. The reaction mixture was stirred overnight under H2 atmosphere, then filtered through Celite, and the solvent was evaporated. The residue was dissolved in dichloromethane (50 mL), and the solution was washed with distilled water (10 mL) two times, dried with anhydrous Na2SO4, filtered, and the solvent was evaporated in vacuum. The product was purified by flash column chromatography (hexane/acetone 8 : 2) to yield 3 a (571 mg, 88 %) as a colorless liquid. R f=0.50 (hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3): δ=3.75 (s, 2H, CH2), 3.62–3.53 (m, 4H, 2CH2), 2.32–2.11 (m, 3H, CH2, OH), 1.98–1.86 ppm (m, 2H, CH2); 13C NMR (100 MHz, CDCl3): δ=72.2, 69.7, 61.9 (3 C, CH2), 27.9 (t, 1 C, CH2), 20.9 ppm (1 C, CH2).
16,16,17,17,18,18,19,19,19‐Nonafluoro‐3,6,9,12‐tetraoxanonadecan‐1‐ol (3 b)
LiAlH4 (304 mg, 8 mmol) was added under argon to a solution of 2 b (2.32 g, 4 mmol) in abs. THF (20 mL), and the reaction mixture was stirred overnight. Then 10 % Na2SO4 solution (5 mL) was added, and the mixture was stirred for 30 min. After filtration through Celite, the solvent was evaporated, and the product was purified by flash column chromatography (hexane/acetone 7 : 3) to yield 3 b (728 mg, 40 %) as a colorless liquid. R f=0.40 (hexane/acetone 7 : 3); 1H NMR (360 MHz, CDCl3): δ=3.77–3.52 (m, 18H, 9CH2), 2.28–2.10 (m, 2H, CH2), 1.95–1.83 ppm (m, 2H, CH2); 13C NMR (90 MHz, CDCl3): δ=72.7, 70.7, 70.6, 70.4, 70.3, 69.8, 61.8 (9 C, CH2), 27.9 (t, 1 C, CH2), 20.8 ppm (1 C, CH2); HRMS (ESI): m/z calcd for C15H23F9O5+Na+: 477.1299 [M+Na]+; found: 477.5.
Table 5.
NMR data of teicoplanin derivatives.
|
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
|
15 |
16 |
17 |
18 |
||||
|
|
1H |
13C |
1H |
13C |
1H |
13C |
1H |
13C |
|
y1 |
– |
174.36 |
– |
173.79 |
– |
n.d. |
– |
173.47 |
|
y7 |
– |
170.02 |
– |
169.90 |
– |
169.67 |
– |
169.94 |
|
y4 |
– |
169.68 |
– |
169.44 |
– |
169.67 |
– |
169.37 |
|
y2 |
– |
169.36 |
– |
169.25 |
– |
169.17 |
– |
169.19 |
|
y5 |
– |
169.00 |
– |
168.87 |
– |
168.58 |
– |
168.83 |
|
y3 |
– |
168.33 |
– |
168.22 |
– |
167.16 |
– |
168.17 |
|
y6 |
– |
167.46 |
– |
167.30 |
– |
166.53 |
– |
167.30 |
|
C=O(NAc) G7 |
– |
165.94 |
– |
165.58 |
– |
n.d. |
– |
165.59 |
|
3c |
– |
158.56 |
– |
158.46 |
– |
159.90 |
– |
158.45 |
|
7e |
– |
157.75 |
– |
157.65 |
– |
158.51 |
– |
157.60 |
|
3e |
– |
156.87 |
– |
156.75 |
– |
158.24 |
– |
156.73 |
|
7c |
– |
155.75 |
– |
155.62 |
– |
157.05 |
– |
155.67 |
|
5d |
– |
155.61 |
– |
155.49 |
– |
155.95 |
– |
155.44 |
|
2d |
– |
151.16 |
– |
151.07 |
– |
155.95 |
– |
151.04 |
|
6d |
– |
149.21 |
– |
149.10 |
– |
151.15 |
– |
149.08 |
|
4c |
– |
148.96 |
– |
148.83 |
– |
149.53 |
– |
148.77 |
|
4e |
– |
148.02 |
– |
147.93 |
– |
148.13 |
– |
147.91 |
|
1d |
– |
147.49 |
– |
147.42 |
– |
147.60 |
– |
147.40 |
|
triazole‐q (4) |
– |
143.78 |
– |
143.76 |
– |
143.59 |
– |
143.75 |
|
6a |
– |
142.74 |
– |
142.60 |
– |
142.19 |
– |
142.61 |
|
1c |
– |
140.94 |
– |
140.89 |
– |
139.09 |
– |
140.83 |
|
3a |
– |
139.49 |
– |
139.68 |
– |
138.17 |
– |
139.37 |
|
7a |
– |
138.62 |
– |
138.64 |
– |
135.18 |
– |
138.60 |
|
5b |
7.12 |
136.1 |
7.12 |
136.01 |
7.17. 7.09 |
136.3 |
7.11 |
135.97 |
|
2a |
– |
135.41 |
– |
135.3 |
– |
135.18 |
– |
135.30 |
|
4d |
– |
134.43 |
– |
134.74 |
– |
n.d. |
– |
134.31 |
|
1a |
– |
n.d. |
– |
134.36 |
– |
n.d. |
– |
n.d. |
|
2b |
7.21 |
131 |
7.2 |
130.91 |
7.15 |
130.24 |
7.21 |
130.94 |
|
2f |
7.94 |
130.6 |
7.89 |
130.46 |
7.72 |
131.25 |
7.92 |
130.44 |
|
4a |
– |
127.22 |
– |
127.13 |
– |
126.81 |
– |
128.6 |
|
6c |
– |
126.7 |
– |
126.59 |
– |
126.71 |
– |
128 |
|
6f |
7.26 |
127.81 |
7.28 |
127.8 |
7.25 |
127.74 |
7.27 |
128 |
|
6b |
7.86 |
128.49 |
7.87 |
128.35 |
7.81 |
128.29 |
7.87 |
128.37 |
|
2c |
– |
125.43 |
– |
125.33 |
– |
n.d. |
– |
127.13 |
|
5a |
– |
125.43 |
– |
125.33 |
– |
n.d. |
– |
126.59 |
|
5f |
6.67 |
125.12 |
6.66 |
125.29 |
6.65 |
125.02 |
6.66 |
125.32 |
|
1f |
6.9 |
125.59 |
6.9 |
125.48 |
7.04 |
124.33 |
6.9 |
125.48 |
|
2e |
7.19 |
124.84 |
7.18 |
124.72 |
7.08 |
127.15 |
7.19 |
124.73 |
|
6e |
6.99 |
119.0 |
6.96 |
118.94 |
7.24 |
123 |
7.27 |
123.05 |
|
triazole‐CH (5) |
7.74 |
123.98 |
7.71 |
123.81 |
7.7 |
123.88 |
7.72 |
123.78 |
|
5c |
– |
122.17 |
– |
122.19 |
– |
122.2 |
– |
122.06 |
|
1e |
7.26 |
123.24 |
7.25 |
123.06 |
6.89 |
118.61 |
7.05 |
119.37 |
|
7b |
– |
117.82 |
– |
117.71 |
– |
117.87 |
– |
117.71 |
|
1b |
7.04 |
119.49 |
7.04 |
119.39 |
6.94 |
119.6 |
6.97 |
118.91 |
|
5e |
6.65 |
116.45 |
6.64 |
116.38 |
6.65 |
116.32 |
6.64 |
116.37 |
|
3b |
6.34 |
110.03 |
6.34 |
109.97 |
6.32 |
109.53 |
6.34 |
109.97 |
|
4b |
5.5 |
108.05 |
5.56 |
107.92 |
5.54 |
107.65 |
5.56 |
108 |
|
7f |
6.5 |
107.86 |
6.52 |
107.92 |
6.53 |
107.73 |
6.48 |
107.68 |
|
3d |
6.38 |
105.1 |
6.37 |
104.97 |
6.34 |
104.17 |
6.37 |
104.99 |
|
4f |
5.1 |
104.59 |
5.1 |
104.46 |
5.09 |
104.26 |
5.1 |
104.47 |
|
3f |
6.55 |
104.02 |
6.55 |
103.84 |
6.57 |
103.39 |
6.55 |
103.85 |
|
7d |
6.32 |
101.68 |
6.3 |
101.51 |
6.31 |
101.52 |
6.3 |
101.56 |
|
G1 |
4.38 |
99.27 |
4.4 |
98.73 |
4.37 |
99.42 |
4.42 |
98.57 |
|
G5 |
3.1 |
76.79 |
3.11 |
76.77 |
3.09 |
76.44 |
3.11 |
76.77 |
|
G3 |
5.4 |
76.14 |
5.45 |
75.58 |
5.35 |
76.2 |
5.44 |
75.43 |
|
G4 |
3.39 |
73.38 |
3.4 |
73.4 |
3.4 |
72.89 |
3.41 |
73.4 |
|
z6 |
3.22 |
70.02 |
3.25 |
69.97 |
3.26 |
69.13 |
3.23 |
70.05 |
|
x6 |
4.16 |
60.94 |
4.15 |
60.88 |
4.2 |
60.46 |
4.14 |
60.87 |
|
G6 |
3.62 |
60.25 |
3.6 |
60.29 |
3.39 |
60.1 |
3.6 |
60.38 |
|
x3 |
5.4 |
58.31 |
5.41 |
58.21 |
5.36 |
58.41 |
5.41 |
58.17 |
|
x1 |
4.33 |
59.28 |
4.33 |
59.22 |
4.28 |
59.08 |
4.36 |
58.9 |
|
x7 |
7.12 |
64.04 |
7.09 |
64.03 |
7.06 |
64.18 |
7.1 |
63.98 |
|
G2 |
3.52 |
55.8 |
3.52 |
55.9 |
3.59 |
55.23 |
3.51 |
55.98 |
|
x4 |
5.65 |
54.67 |
5.63 |
54.65 |
5.6 |
54.27 |
5.64 |
54.64 |
|
x2 |
4.87 |
55.46 |
4.89 |
55.37 |
4.97 |
55 |
4.88 |
55.38 |
|
x5 |
4.37 |
53.61 |
4.37 |
53.53 |
4.38 |
53.22 |
4.36 |
53.54 |
|
z2,2’ |
|
|
n.d. |
n.d. |
|
|
3.32, 2.92 |
36.18 |
|
Side chain |
|
|
|
|
|
|
|
|
|
OCH2 (a) |
4.45 |
63.29 |
4.45 |
63.25 |
4.45 |
63.04 |
4.45 |
63.23 |
|
OCH2 |
3.48 |
69.34 |
3.48 |
69.32 |
3.48 |
69.07 |
3.5 |
69.67 |
|
OCH2 |
3.51 |
68.9 |
3.51 |
69.08 |
3.51 |
68.62 |
3.5 |
69.38 |
|
OCH2 |
3.44 |
68.48 |
3.44 |
68.5 |
3.44 |
68.24 |
3.46 |
68.48 |
|
|
|
|
|
|
|
|
3.5 |
68.39 |
|
CH2 (e) |
1.73 |
20.25 |
|
|
1.73 |
20.26 |
|
|
|
CH2 (f) |
2.24 |
26.76 |
|
|
2.23 |
26.87 |
|
|
|
CH2 (k) |
|
|
1.76 |
20.23 |
|
|
1.76 |
20.24 |
|
CH2 (l) |
|
|
2.23 |
26.86 |
|
|
2.28 |
26.85 |
2‐((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11‐Heptadecafluoro‐2‐iodoundecyl)oxy)ethan‐1‐ol (5 a)
Heptadecafluoro‐1‐iodooctane (3.17 mL, 6.55 g, 12 mmol) and benzophenone (20 mg, 0.11 mmol) were added to a solution of 2‐(allyloxy)ethan‐1‐ol (1 a; 1.02 g, 10 mmol) in methanol (15 mL). Argon gas was bubbled through the solution and then irradiation occurred for 10 min. The solvent was evaporated, and the product was purified by flash column chromatography (hexane/acetone 8 : 2) to yield 5 a (2.07 g, 38 %) as a colorless liquid. R f=0.43 (hexane/acetone 7 : 3); 1H NMR (360 MHz, CDCl3): δ=4.46–4.33 (m, 1H, CIH), 3.83–3.61 (m, 6H, 3CH2), 3.16–2.91 (m, 1H, CH2), 2.87–2.62 (m, 1H, CH2), 2.28–2,14 ppm (m, 1H, OH); 13C NMR (90 MHz, CDCl3): δ=117.9, 111.2 (CF2 and CF3), 75.8, 72.4, 61.8 (3 C, CH2), 37.9 (t, 1 C, CH2), 15.0 ppm (1 C, CHI); HRMS (ESI): m/z calcd for C13H10F17IO2+Na+: 670.9352 [M+Na]+; found: 670.9346.
16,16,17,17,18,18,19,19,20,20,21,21,22,22,23,23,23‐Heptadecafluoro‐14‐iodo‐3,6,9,12‐tetraoxatricosan‐1‐ol (5 b)
Heptadecafluoro‐1‐iodooctane (1.46 mL, 3.0 g, 5.5 mmol) and benzophenone (10 mg, 0.055 mmol) were added to a solution of 3,6,9,12‐tetraoxapentadec‐14‐en‐1‐ol (1 b; 1.17 g, 5 mmol) in methanol (12 mL). Argon gas was bubbled through the solution and then irradiation occurred for 10 min. The solvent was evaporated, and the product was purified by flash column chromatography (hexane/acetone 7 : 3) to yield 5 b (3.2 g, 82 %) as a colorless liquid. R f=0.40 (hexane/acetone 7 : 3); 1H NMR (360 MHz, CDCl3): δ=4.42–4.32 (m, 1H, CIH), 3.85–3.56 (m, 18H, 9CH2), 3.20–2.91 (m, 2H, CH2, OH), 2.78–2.58 ppm (m, 1H, CH2); 13C NMR (90 MHz, CDCl3): δ=117.8, 114.3, 111.2, 110.8 (8 C, CF2, CF3), 76.2, 72.7, 70.7, 70.66, 70.6, 70.5, 70.3, 61.7 (9 C, CH2), 37.5 (t, 1 C, CH2), 14.5 ppm (1 C, CHI); HRMS (ESI): m/z calcd for C19H22F17IO5+Na+: 803.0138 [M+Na]+; found: 803.0133.
2‐((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11‐Heptadecafluoroundecyl)oxy)ethan‐1‐ol (6 a)
To a solution of 5 a (1.296 g, 2 mmol) in methanol (15 mL) 10 % palladium on activated charcoal (270 mg) and NaHCO3 (420 mg, 5 mmol) were added. The reaction mixture was stirred overnight under H2 atmosphere, then filtered through Celite, and the solvent was evaporated. The residue was dissolved in dichloromethane (50 mL) and the solution was washed with distilled water (10 mL) two times, dried with anhydrous Na2SO4, filtered and the solvent was evaporated in vacuum. The product was purified by flash column chromatography (hexane/acetone 8 : 2) to yield 6 a (922 mg, 88 %) as a colorless liquid. R f=0.58 (hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3): δ=3.78–3.72 (m, 2H, CH2), 3.61–3.53 (m, 4H, 2CH2), 2.29–2.04 (m, 3H, CH2, OH), 1.96–1.86 ppm (m, 2H, CH2); 13C NMR (100 MHz, CDCl3): δ=118.7, 115.9, 110.9 (8 C, CF2, CF3); 72.2, 69.8, 61.9 (3 C, CH2), 28.1 (t, 1 C, CH2), 20.9 ppm (1 C, CH2); HRMS (ESI): m/z calcd for C13H11F17O2+Na+: 545.0385 [M+Na]+; found: 545.0380.
16,16,17,17,18,18,19,19,20,20,21,21,22,22,23,23,23‐Heptadecafluoro‐3,6,9,12‐tetraoxatricosan‐1‐ol (6 b)
LiAlH4 (304 mg, 8 mmol) was added under argon to a solution of 5 b (3.12 g, 4 mmol) in abs. THF (20 mL), and the reaction mixture was stirred overnight. Then 10 % Na2SO4 solution (5 mL) was added and the mixture was stirred for 30 min. After filtration through Celite, the solvent was evaporated, and the product was purified by flash column chromatography (hexane/acetone 7 : 3) to yield 6 b (1.52 g, 58 %) as a colorless liquid. R f=0.30 (hexane/acetone 7 : 3); 1H NMR (360 MHz, CDCl3): δ=3.76–3.51 (m, 18H, 9CH2), 2.28–2.10 (m, 2H, CH2), 1.95–1.83 ppm (m, 2H, CH2); 13C NMR (90 MHz, CDCl3): δ=121.5, 118.8, 114.3, 110.8, 108.2 (8 C, CF2, CF3), 72.7, 70.6, 70.58, 70.3, 70.2, 69.7, 61.7 (9 C, CH2), 27.9 (t, 1 C, CH2), 20.7 ppm (1 C, CH2); HRMS (ESI): m/z calcd for C19H23F17O5+Na+: 677.1172 [M+Na]+; found: 677.1166.
General procedure for propargylation (4 a, 4 b, 7 a and 7 b)
NaH (60 % dispersion in mineral oil; 2 mmol) was washed with hexane, abs. THF (10 mL) was added and 3 a, 3 b, 6 a or 6 b (1 mmol) was dissolved in it. After 30 min of stirring, propargyl bromide (80 % solution in toluene; 1.2 mmol) was added, and the reaction mixture was stirred for 3 h. Then ethyl acetate (5 mL) and methanol (1 mL) were added to the mixture, and it was stirred for 15 min. The solvent was evaporated, the residue was dissolved in dichloromethane and the solution was washed with distilled water (3x15 mL). The organic phase was dried on anhydrous Na2SO4, then it was filtered and evaporated in vacuum. The product was purified by flash column chromatography (hexane/acetone 8 : 2) to yield 4 a, 4 b, 7 a or 7 b as yellowish liquids.
1,1,1,2,2,3,3,4,4‐Nonafluoro‐7‐(2‐(prop‐2‐yn‐1‐yloxy)ethoxy)heptane (4 a): Yield 267 mg (74 %); R f=0.58 (hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3): δ=4.21 (d, J=2.4 Hz, 2H, CH2 propargyl), 3.73–3.67 (m, 2H, CH2), 3.66–3.61 (m, 2H, CH2), 3.56 (t, J=6.1 Hz, 2H, CH2), 2.43 (t, J=2.4 Hz, 1H, CH propargyl), 2.29–2.12 (m, 2H, CH2), 1.95–1.85 ppm (m, 2H, CH2); 13C NMR (100 MHz, CDCl3): δ=79.6 (1 C, Cq propargyl), 74.6 (1 C, CH propargyl), 70.2, 69.9, 69.2 (3 C, CH2), 58.5 (1 C, CH2 propargyl) 28.0 (t, 1 C, CH2), 20.8 ppm (1 C, CH2).
20,20,21,21,22,22,23,23,23‐Nonafluoro‐4,7,10,13,16‐pentaoxatricos‐1‐yne (4 b): Yield 180 mg (37 %); R f=0.46 (hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3): δ=4.20 (d, J=2.4 Hz, 2H, CH2 propargyl), 3.74–3.57 (m, 16H, CH2), 3.55 (t, J=6.1 Hz, 2H, CH2), 2.43 (t, J=2.4 Hz, 1H, CH propargyl), 2.27–2.10 (m, 2H, CH2), 1.93–1.83 ppm (m, 2H, CH2); 13C NMR (100 MHz, CDCl3): δ=74.6 (1 C, CH propargyl), 70.7, 70.5, 70.3, 69.8, 69.2 (9 C, CH2), 58.5 (1 C, CH2 propargyl) 27.9 (t, 1 C, CH2), 20.8 ppm (1 C, CH2); HRMS (ESI): m/z calcd for C18H25F9O5+Na+: 515.1456 [M+Na]+; found: 515.1451.
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8‐Heptadecafluoro‐11‐(2‐(prop‐2‐yn‐1‐yloxy)ethoxy)undecane (7 a): Yield 234 mg (42 %); R f=0.64 (hexane/acetone 7 : 3); 1H NMR (400 MHz, CDCl3): δ=4.21 (d, J=2.4 Hz, 2H, CH2 propargyl), 3.73–3.67 (m, 2H, CH2), 3.66–3.61 (m, 2H, CH2), 3.56 (t, J=6.1 Hz, 2H, CH2), 2.43 (t, J=2.4 Hz, 1H, CH propargyl), 2.29–2.12 (m, 2H, CH2), 1.95–1.85 ppm (m, 2H, CH2); 13C NMR (100 MHz, CDCl3): δ=118.7, 116.2, 111.0 (8 C, CF2, CF3); 79.7 (1 C, Cq propargyl), 74.6 (1 C, CH propargyl), 70.2, 69.9, 69.2 (3 C, CH2), 58.6 (1 C, CH2 propargyl) 28.1 (t, 1 C, CH2), 20.9 ppm (1 C, CH2); HRMS (ESI): m/z calcd for C16H13F17O2+Na+: 583.0542 [M+Na]+; found: 583.0535.
20,20,21,21,22,22,23,23,24,24,25,25,26,26,27,27,27‐Heptadecafluoro‐4,7,10,13,16‐pentaoxaheptacos‐1‐yne (7 b): Yield 318 mg (46 %); R f=0.42 (hexane/acetone 7 : 3); Compound 7 b was reacted with the azido glycopeptide derivatives without characterization by NMR; HRMS (ESI): m/z calcd for C22H25F17O5+Na+: 715.1328 [M+Na]+; found: 715.1323.
2,5‐Dioxopyrrolidin‐1‐yl (R)‐2‐azido‐4‐methylpentanoate (10)
2‐Azido‐4‐methylpentanoic acid (9; 1.256 g, 8 mmol) and N‐hydroxysuccinimide (1.013 g, 8.8 mmol) were dissolved in abs. dichloromethane (50 mL) and after cooling in an ice bath, dicyclohexylcarbodiimide (1.76 g, 8.5 mmol) was added. The reaction mixture was stirred overnight at room temperature, then filtered through Celite and the solvent was evaporated in vacuum, and co‐evaporated with chloroform (2×50 mL) two times. Then it was dissolved in acetonitrile, filtered through Celite and after evaporation the product was purified by flash column chromatography (hexane/ethyl acetate 8 : 2) to yield 10 (1.1 g, 67 %). R f= (hexane/ethyl acetate 8 : 2); 1H NMR (400 MHz, CDCl3): δ=4.17 (dd, J=8.8, 5.8 Hz, 2H, CH), 2.86 (s, 4H, CH2), 1.99–1.77 (m, 3H, CH, CH2), 1.01 ppm (dd, J=9.5, 6.4 Hz, 6H, CH3); 13C NMR (100 MHz, CDCl3): δ=168.7, 166.9 (3 C, C=O), 58.3 (1 C, CH), 39.9 (1 C, CH2), 25.6 (2 C, CH2) 24.9, 22.7, 21.5 ppm (3 C, CH, CH3); MALDI‐TOF MS: [M+Na]+=277.204 m/z. Calcd (C10H14N4O4Na) 277.091 m/z.
Compound 11
To the solution of vancomycin aglycone hexapeptide (8; 1.5 g, 1.48 mmol) in abs. N,N‐dimethylformamide (50 mL) triethylamine (206 μL, 1.48 mmol) and 10 (753 mg, 2.96 mmol) were added. The reaction mixture was stirred for 3 h, the solvent was evaporated, and the product was purified by flash column chromatography (acetonitrile/water 9 : 1) to yield 11 (986 mg, 58 %) as a yellow powder. R f=0.34 (acetonitrile/water 9 : 1); NMR data can be found in Table 4.; MALDI‐TOF MS: [M+Na]+=1177.089 m/z. Calcd (C52H48Cl2N10O17Na) 1177.247 m/z.
General procedure for click reaction (12, 13, 15, 16, 17 and 18)
To the solution of azide (11 or 14; 0.1 mmol) in N,N‐dimethylformamide (2 mL) triethylamine (14 μL, 0.1 mmol), alkyne (4 a, 4 b, 7 a or 7 b; 0.12 mmol) and CuI iodide (10 mg) were added. The reaction mixture was stirred overnight, then the solvent was evaporated and the product was purified by flash column chromatography (toluene/methanol 7 : 3→6 : 4 →1 : 1 for 12 and 13 or acetonitrile/water 9 : 1 for 15, 16, 17, and 18) to yield 12, 13, 15, 16, 17 or 18. After lyophilization all of the products were yellow solid foams.
12: Yield 71 mg (45 %); R f=0.53 (toluene/methanol 6 : 4); NMR data can be found in Table 4; HRMS (ESI): m/z calcd for C70H73Cl2F9N10O22+Na+: 1669.4032 [M+Na]+; found: 1669.4020.
13: Yield 87 mg (47 %); R f=0.58 (toluene/methanol 6 : 4); NMR data can be found in Table 4; HRMS (ESI): m/z calcd for C74H73Cl2F17N10O22+Na+: 1869.3904 [M+Na]+; found: 1869.3873.
15: Yield 90 mg (50 %); R f=0.49 (acetonitrile/water 85 : 15); NMR data can be found in Table 5; HRMS (ESI): m/z calcd for C78H69Cl2F9N10O25+Na+: 1809.3566 [M+Na]+; found: 1809.3549.
16: Yield 80 mg (42 %); R f=0.14 (acetonitrile/water 9 : 1); NMR data can be found in Table 5.; HRMS (ESI): m/z calcd for C84H80Cl2F9N10O28Na+Na+: 1963,4554 [M−H+2Na]+; found: 1963.4150.
17: Yield 56 mg (28 %); R f=0.50 (acetonitrile/water 85 : 15); NMR data can be found in Table 5.; HRMS (ESI): m/z calcd for C82H67Cl2F17N10O25Na2+Na+: 2053.3078 [M−2H+3Na]+; found: 2053.3078.
18: Yield 52 mg (26 %); R f=0.23 (acetonitrile/water 9 : 1); NMR data can be found in Table 5.; HRMS (ESI): m/z calcd for C88H81Cl2F17N10O28+Na+: 2141.4225 [M+Na]+; found: 2141.4199.
Antiviral procedures
The CPE reduction assay for influenza virus was described in full detail in previous publications.41 The virus strains were: A/PR/8/34 (A/H1 N1); A/Virginia/ATCC3/2009 (A/H1 N1pdm); A/HK/7/87 (A/H3 N2); B/Ned/537/05; and B/HK/5/72. On day 0, Madin‐Darby canine kidney (MDCK) cells in 96‐well plates were infected with influenza virus at a multiplicity of infection (MOI) of 0.0004 plaque forming units (PFU) per cell. After three days incubation at 35 °C, virus‐induced CPE and compound cytotoxicity were scored by microscopy, after which the data were confirmed by formazan‐based MTS cell viability assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay from Promega). The antiviral effect was expressed as the compound concentration producing 50 % inhibition of the virus‐induced CPE (EC50). Compound cytotoxicity was expressed as the compound concentration causing minimal changes in cell morphology (MCC), and 50 % cytotoxic concentration (CC50) based on MTS assay.42
Inhibitory effect against human coronavirus 229E was determined using a CPE reduction assay in human embryonic lung fibroblast (HEL) 299 cells, described in full detail elsewhere.43 We also reported the detailed methodology for the other DNA and RNA viruses in the test panel.44 The EC50, CC50 and MCC values were calculated as described.42
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the European Regional Development Fund under the projects GINOP‐2.3.2‐15‐2016‐00044, GINOP‐2.3.2‐15‐2016‐00008 and GINOP‐2.3.3‐15‐2016‐00004 and by the European Social Fund under the project EFOP‐3.6.3‐VEKOP‐16‐2017‐00009. This research was also funded by the National Research, Development and Innovation Office of Hungary (K119509).
I. Bereczki, M. Csávás, Z. Szűcs, E. Rőth, G. Batta, E. Ostorházi, L. Naesens, A. Borbás, P. Herczegh, ChemMedChem 2020, 15, 1661.
Contributor Information
Prof. Anikó Borbás, Email: borbas.aniko@pharm.unideb.hu.
Prof. Pál Herczegh, Email: herczeghp@gmail.com.
References
- 1. Böhm H. J., Banner D., Bendels S., Kansy M., Kuhn B., Müller K., Obst-Sander U., Stahl M., ChemBioChem 2004, 5, 637–643. [DOI] [PubMed] [Google Scholar]
- 2. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 3. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- 4. Hagmann W. K., J. Med. Chem. 2008, 51(15), 4359–4369. [DOI] [PubMed] [Google Scholar]
- 5. Ojima I., Fluorine in Medicinal Chemistry and Chemical Biology, Blackwell Publishing Ltd ; 2009. [Google Scholar]
- 6. Gouverneur V., Müller K., Fluorine in Pharmaceutical and Medicinal Chemistry, Imperial College Press, Singapore: 2012. [Google Scholar]
- 7. Haufe G., Leroux F., Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals: Progress in Fluorine Science Series, 1st ed., Academic Press ; 2018. [Google Scholar]
- 8. Wang J., Sanchez-Rosellj M., Aceña J. L., del Pozo C., Sorochinsky A. E., Fustero S., Soloshonok V. A., Liu H., Chem. Rev. 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- 9. Zhou Y., Wang J., Gu Z., Wang S., Zhu W., Aceña J. L., Soloshonok V. A., Izawa K., Liu H., Chem. Rev. 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- 10. Mei H., Han J., Fustero S., Medio-Simon M., Sedgwick D. M., Santi C., Ruzziconi R., Soloshonok V. A., Chem. Eur. J. 2019, 25, 11797–11819. [DOI] [PubMed] [Google Scholar]
- 11. Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J., Meanwell N. A., J. Med. Chem. 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 12. Meanwell N. A., J. Med. Chem. 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 13. Bégué J.-P., Bonnet-Delpon D., Bioorganic and Medicinal Chemistry of Fluorine, Wiley ; 2008. [Google Scholar]
- 14. Jeffries B., Wang Z., Felstead H. R., Le Questel J.-Y., Scott J. S., Chiarparin E., Graton J., Linclau B., J. Med. Chem. 2020, 63, 1002–1031. [DOI] [PubMed] [Google Scholar]
- 15. Prchalová E., Štěpánek O., Smrček S., Kotora M., Future Med. Chem. 2014, 6, 1201–1229. [DOI] [PubMed] [Google Scholar]
- 16. Saloutin V. I., Il'enkoI V. I., Piterskikh A., PlatonovI V. G., Kiseleva V., Pashkevich K. I., Pharm. Chem. J. 1986, 20, 501–504. [Google Scholar]; Translated from Khim.-Farm. Zh. 1986, 20(7), 847–851. [Google Scholar]
- 17. Bassetto M., Ferla S., Pertusati F., Future Med. Chem. 2015, 7(4), 527–546. [DOI] [PubMed] [Google Scholar]
- 18. Pintér G., Batta G., Kéki S., Mándi A., Komáromi I., Takács-Novák K., Sztaricskai F., Rőth E., Ostorházi E., Rozgonyi F., Naesens L., Herczegh P., J. Med. Chem. 2009, 52(19), 6053–6061. [DOI] [PubMed] [Google Scholar]
- 19. Szűcs Z., Bereczki I., Csávás M., Rőth E., Borbás A., Batta G., Ostorházi E., Szatmári R., Herczegh P., J. Antibiot. 2017, 70, 664–670. [DOI] [PubMed] [Google Scholar]
- 20. Szűcs Z., Csávás M., Rőth E., Borbás A., Batta G., Perret F., Ostorházi E., Szatmári R., Vanderlinden E., Naesens L., Herczegh P., J. Antibiot. 2017, 70, 152–157. [DOI] [PubMed] [Google Scholar]
- 21. Szűcs Z., Ostorházi E., Kicsák M., Nagy L., Borbás A., Herczegh P., J. Antibiot. 2019, 72, 524–534. [DOI] [PubMed] [Google Scholar]
- 22. Szűcs Z., Kelemen V., Thai S. L., Csávás M., Rőth E., Batta G., Stevaert A., Vanderlinden E., Naesens L., Herczegh P., Borbás A., Eur. J. Med. Chem. 2018, 157, 1017–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amadoa E., Kressler J., Soft Matter 2011, 7, 7144–7149. [Google Scholar]
- 24. Amado E., Blumeand A., Kressler J., React. Funct. Polym. 2009, 69, 450–456. [Google Scholar]
- 25. Park K.-H., Berrier C., Lebaupain F., Pucci B., Popot J.-L., Ghazi A., Zito F., Biochem. J. 2007, 403, 183–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Colson P., Raoult D., Int. J. Antimicrob. Agents 2016, 48, 349–352; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Li G., De Clercq E., Nat. Rev. Drug Discovery 2020, 19, 149–150. [DOI] [PubMed] [Google Scholar]
- 27.Recent examples for semisynthetic glycopeptides with high antibacterial activity:
- 27a. Yarlagadda V., Akkapeddi P., Manjunath G. B., Haldar J., J. Med. Chem. 2014, 57, 4558–4568; [DOI] [PubMed] [Google Scholar]
- 27b. Szűcs Z., Bereczki I., Csávás M., Rőth E., Borbás A., Batta G., Ostorházi E., Szatmári R., Herczegh P., J. Antibiot. 2017, 70, 664–670; [DOI] [PubMed] [Google Scholar]
- 27c. Szűcs Z., Ostorházi E., Kicsák M., Nagy L., Borbás A., Herczegh P., J. Antibiot. 2019, 72, 524–534; [DOI] [PubMed] [Google Scholar]
- 27d. Vimberg V., Gazak R., Szűcs Z., Borbás A., Herczegh P., Cavanagh J. P., Zieglerova L., Závora J., Adámková V., Balikova Novotna G., J. Antibiot. 2019, 72, 114–117. [DOI] [PubMed] [Google Scholar]
- 28. Beniazza R., Atkinson R., Absalon C., Castet F., Denisov S. A., McClenaghan N. D., Lastécouères D., Vincent J.-M., Adv. Synth. Catal. 2016, 358, 2949–2961. [Google Scholar]
- 29. Booth P. M., Stone D. J. M., Williams D. H., J. Chem. Soc. Chem. Commun. 1987, 1694–1695. [Google Scholar]
- 30. Valverde I. E., Bauman A., Kluba C. A., Vomstein S., Walter M. A., Mindt T. L., Angew. Chem. Int. Ed. 2013, 52(34), 8957–8960. [DOI] [PubMed] [Google Scholar]
- 31. Batta G., Sztaricskai F., Kövér K. E., Rüdel C., Berdnikova T. F., J. Antibiot. 1991, 44, 1208–1221. [DOI] [PubMed] [Google Scholar]
- 32. Prowse W. G., Kline A. D., Skelton M. A., Loncharich R. J., Biochemistry 1995, 34, 9632–9644. [DOI] [PubMed] [Google Scholar]
- 33. Allen N. E., Nicas T. I., Microbiol. Rev. 2003, 26, 511–532. [DOI] [PubMed] [Google Scholar]
- 34. Stilbs P., Prog. Nucl. Magn. Reson. Spectrosc. 1987, 19, 1–45. [Google Scholar]
- 35. Orosz L., Batta G., Kéki S., Nagy M., Deák G., Zsuga M., Carbohydr. Res. 2007, 342, 1323–1328. [DOI] [PubMed] [Google Scholar]
- 36. Bereczki I., Mándi A., Rőth E., Borbás A., Fizil Á., Komáromi I., Sipos A., Kurtán T., Batta G., Ostorházi E., Rozgonyi F., Vanderlinden E., Naesens L., Sztaricskai F., Herczegh P., Eur. J. Med. Chem. 2015, 94, 73–86. [DOI] [PubMed] [Google Scholar]
- 37. Vanderlinden E., Vanstreels E., Boons E., ter Veer W., Huckriede A., Daelemans D., Van Lommel A., Roth E., Sztaricskai F., Herczegh P., Naesens L., J. Virol. 2012, 86, 9416–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou N., Pan T., Zhang J., Li Q., Zhang X., Bai C., Huang F., Peng T., Zhang J., Liu C., Tao L., Zhang H., J. Biol. Chem. 2016, 291, 9218–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Y., Cui R., Li G., Gao Q., Yuan S., Altmeyer R., Zou G., Antiviral Res. 2016, 125, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.
- 40a. Voerman D., Schluck M., Weiden J., Joosten B., Eggermont L. J., van den Eijnde T., Ignacio B., Cambi A., Figdor C. G., Kouwer P. H. J., Verdoes M., Hammink R., Rowan A. E., Biomacromolecules 2019, 20, 2587–2597; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40b. Zona C., D'Orazio G., La Ferla B., Synlett 2013, 24, 709–712. [Google Scholar]
- 41. Cihan-Üstündag G., Zopun M., Vanderlinden E., Ozkirimli E., Persoons L., Capan G., Naesens L., Bioorg. Med. Chem. 2020, 28, 115130. [DOI] [PubMed] [Google Scholar]
- 42. Vrijens P., Noppen S., Boogaerts T., Vanstreels E., Ronca R., Chiodelli P., Laporte M., Vanderlinden E., Liekens S., Stevaert A., Naesens L., J. Gen. Virol. 2019, 100, 583–601. [DOI] [PubMed] [Google Scholar]
- 43. Apaydin Ç. B., Cesur N., Stevaert A., Naesens L., Cesur Z., Arch. Pharm. (Weinheim) ; 2019, 352:e1800330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rogolino D., Carcelli M., Bacchi A., Compari C., Contardi L., Fisicaro E., Gatti A., Sechi M., Stevaert A., Naesens L., J. Inorg. Biochem. 2015, 150, 9–17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary