

Abstract
The exhaustive trichlorosilylation of hexachloro‐1,3‐butadiene was achieved in one step by using a mixture of Si2Cl6 and [nBu4N]Cl (7:2 equiv) as the silylation reagent. The corresponding butadiene dianion salt [nBu4N]2[1] was isolated in 36 % yield after recrystallization. The negative charges of [1]2− are mainly delocalized across its two carbanionic (Cl3Si)2C termini (α‐effect of silicon) such that the central bond possesses largely C=C double‐bond character. Upon treatment with 4 equiv of HCl, [1]2− is converted into neutral 1,2,3,4‐tetrakis(trichlorosilyl)but‐2‐ene, 3. The Cl− acceptor AlCl3, induces a twofold ring‐closure reaction of [1]2− to form a six‐membered bicycle 4 in which two silacyclobutene rings are fused along a shared C=C double bond (84 %). Compound 4, which was structurally characterized by X‐ray crystallography, undergoes partial ring opening to a monocyclic silacyclobutene 2 in the presence of HCl, but is thermally stable up to at least 180 °C.
Keywords: Carbanions, Cyclization, Heterocycles, Silacyclobutenes, Silylation
Upcycling: A sixfold trichlorosilylated butadiene dianion is accessible in one step from hexachlorobutadiene and the Si2Cl6/Cl− system. Cl− abstraction with AlCl3 causes a double cyclization reaction and quantitatively forms a six‐membered double silacyclobutene.
Introduction
Functionalized π‐electron systems are cornerstones of organic synthesis. However, the field is still dominated by simple halogenated derivatives, whereas compounds carrying more sophisticated, value‐added functional groups are far less abundant. Two important classes of particularly versatile substituents are boronic acid esters (−B(OR)2) and silyl groups (−SiR3; R=halogeno, organyl, alkoxy). Corresponding derivatives serve as easy‐to‐handle precursors of various other functionalized species and indispensable reactants in metal‐mediated C−C coupling reactions (Suzuki‐ and Hiyama‐type protocols).1, 2 The Lewis‐acidic nature of boronic acid esters (imparted by their partially vacant boron pz orbitals) and organosilanes (imparted by their Si−R σ* orbitals) is the key asset for these transformations, which proceed via four‐coordinate borate and five‐ or six‐coordinate silicate intermediates. Also the Matteson homologation3 of R′−B(OR)2 species and the Peterson olefination4 of silylated carbanions [R′2C−SiR3]− depend on the electrophilicity of the heteroatom groups, which is, moreover, the reason why borylated or silylated alkyl carbon atoms can readily be deboronated or desilylated (as well as deprotonated) to generate synthetically useful carbanions.
To date, several universally applicable protocols for the synthesis of borylated aromatic hydrocarbons have been developed.5 Among the most powerful ones are Ir‐catalyzed C−H‐activation/borylation sequences using bis(pinacolato)‐diboron (B2pin2) as the boron source.6 Access to mono‐ and 1,2‐diborylalkenes is also granted through the hydroboration and diboration7 of alkynes, respectively. In stark contrast, routes to multiply borylated alkenes and gem‐diboronates8 are far less well established: It took until this year for the efficient synthesis of 1,1,2‐(Bpin)3‐alkenes through a Cu‐catalyzed triboration of terminal alkynes to be reported, as done by Marder and co‐workers.9
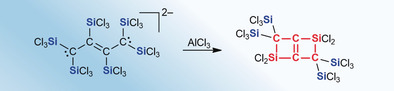
Our group recently discovered that treatment of tetrachloromethane (CCl4) or tetrachloroethene (C2Cl4) with Si2Cl6 in the presence of substoichiometric amounts of [nBu4N]Cl affords the mono‐ or dianion [A]− or [B]2− in excellent yields (Scheme 1).10 Proceeding from [A]− and [B]2−, various other multiply trichlorosilylated compounds HA–F are available.10 Compared to the common SiMe3 substituent, the SiCl3 group offers two important advantages: 1) It is the stronger Lewis acid and better π‐acceptor, and thus more prone to form hypercoordinate adducts and stabilize carbanions in α‐position. This pronounced α‐effect explains why we obtained the carbanions [A]− and [B]2− as primary reaction products. 2) Si−Cl bonds can be derivatized further to tailor the properties of the silyl group and to prepare novel organosilanes, organohydrosilanes, and (oligo)siloxanes.
Scheme 1.
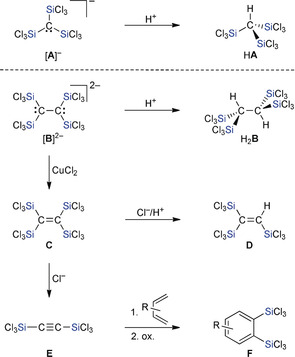
The monoanion [A]− and dianion [B]2− give access to the multiply trichlorosilylated compounds HA–F.
As a drawback, the conventional synthesis of organotrichlorosilanes from SiCl4 and organolithium or Grignard reagents suffers from a limited product selectivity and often furnishes mixtures of compounds RnSiCl4−n (n≥1). Our novel silylation strategy, based on the Si2Cl6/Cl− system, faithfully introduces exclusively SiCl3 moieties and consequently is a promising problem‐solving approach—provided the substrate scope can be significantly expanded. Herein, we describe the exhaustive trichlorosilylation of hexachloro‐1,3‐butadiene (C4Cl6, HCBD), which affords the corresponding dianion [1]2− in one step with good yields (Scheme 2). We subsequently succeeded in the conversion of [1]2− to the neutral 1,2,3,4‐tetrakis(trichlorosilyl)but‐2‐ene 3 and to the six‐membered bicycle 4 in which two silacyclobutene rings are fused along a shared C=C double bond. Compound 4 was isolated and fully characterized despite its ring strain (the neat carbonaceous analogue Δ1,4‐bicyclo[2.2.0]hexene polymerizes within seconds already at low temperatures11).
Scheme 2.
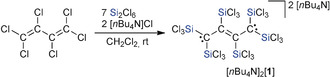
Synthesis of [nBu4N]2[1] through a one‐step reaction of hexachloro‐1,3‐butadiene (C4Cl6) with Si2Cl6/[nBu4N]Cl.
Results and Discussion
The actual reactive species generated from a mixture of Si2Cl6 and [nBu4N]Cl in CH2Cl2 is the chimeric [SiCl3]− ion,12, 13 which can react with electron‐poor perhalogenated alkenes in three principal ways:10, 14 1) A single electron is transferred from the anion to the organic substrate. 2) [SiCl3]− reacts as a silanide nucleophile and attacks either the carbon atoms or the chloro substituents of the organic substrate R−Cl. In the latter case, the resulting carbanion [R]− would have to react again with the byproduct SiCl4 (or residual Si2Cl6) to establish a C−SiCl3 bond. 3) [SiCl3]− loses a Cl− ligand and liberates the silylene [SiCl2], which inserts into a R−Cl bond. Finally coming back to the contrasting juxtaposition of Bpin and SiCl3 substituents, we emphasize that the liberation of [SiCl3]− through the Cl−‐induced heterolytic cleavage of Si2Cl6 is equivalent to the generation of transient [Bpin]− nucleophiles via the alkoxide‐mediated B−B‐bond heterolysis of B2pin2.7b, 15
Like tetrachloroethylene (C2Cl4), hexachloro‐1,3‐butadiene (C4Cl6) reacts with the Si2Cl6/[nBu4N]Cl system (7:2 equiv) in one pot to furnish a dianion salt ([nBu4N]2[1]; Scheme 2). This first key compound was isolated in 36 % yield after recrystallization from warm CH2Cl2. The formation of the exhaustively trichlorosilylated [1]2− is remarkable given that C4Cl6 is inert toward common acids and non‐nucleophilic bases and therefore widely used as a solvent in chlorination reactions.16
In a similar manner as in the case of the successful double C‐protonation of the ethylene dianion [B]2−, which led to the corresponding ethane H2 B,10 we next attempted to synthesize the hexakis(trichlorosilyl)but‐2‐ene H2 1 by treating [nBu4N]2[1] with 2 equiv of trifluoromethanesulfonic acid (HOTf). Surprisingly, the open‐chain target compound H2 1 was not obtained. The reaction rather afforded the silacyclobutene 2 in 92 % yield (Scheme 3). Even though 2 contains only one H atom, the quantitative conversion of [1]2− to 2 nevertheless requires the addition of a second equiv of HOTf. If a smaller amount of the acid was used (approximately 1.8 equiv), the reaction solution still contained a well‐defined second species together with the final product 2; 1 equiv of HOTf transforms [1]2− exclusively to this other species—obviously a reaction intermediate. According to NMR spectroscopy (1H, 13C{1H}, 29Si), the intermediate possesses C 1 symmetry, a partly unsaturated four‐carbon backbone, four kinds of magnetically non‐equivalent SiCl3 substituents, and a hydrogen atom.
Scheme 3.
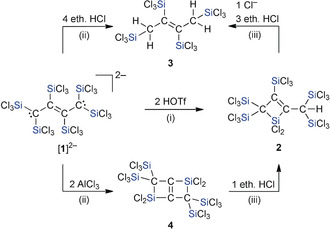
Synthesis of 2 and 3 via the reaction of [1]2− with HOTf or ethereal HCl; synthesis of 4 through the reaction of [1]2− with AlCl3; i) n‐hexane, room temperature; ii) CH2Cl2, room temperature; iii) CD2Cl2, room temperature.
Altogether, this points toward a monoprotonated compound [H1]−. Since several isomers are conceivable for [H1]−, a definite structural assignment has to await an X‐ray crystal structure analysis, even though quantum‐chemical calculations provide some further indications: An internally protonated open‐chain isomer can be excluded in terms of the relative Gibbs free energy (32.4 kcal mol−1 higher than that of the most favorable considered isomer). The same is true for a cyclic isomer by a calculated 29Si NMR chemical shift of −107.7 ppm for the endocyclic silicon atom that is not observed in the experiment (see the Supporting Information for more details).17 It is, moreover, safe to state that the second equivalent of HOTf is required as a Cl−‐abstracting reagent and that the concomitant liberation of HCl drives the cyclization reaction. We confirmed in separate experiments that 2 behaves inert towards ethereal HCl, but is selectively transformed back to [H1]− upon the addition of 1 equiv of [nBu4N]Cl.
A different product emerged upon treatment of [nBu4N]2[1] with ethereal HCl. The cleanest conversion was achieved by using 4 equiv of the acid, which generated the tetrakis(trichlorosilyl)but‐2‐ene 3 via a double C‐protonation and twofold protodesilylation (Scheme 3; yield: 44 % after workup).18 Compound 3 also formed quantitatively from 2, 3 equiv of ethereal HCl, and 1 equiv of [nBu4N]Cl (which is not consumed, but acts as a catalyst). These results underscore the prime importance of the choice of acids with either non‐nucleophilic (TfO−) or nucleophilic (Cl−) counter anions for the reaction outcome.10
Similar to the proton, the Lewis acid AlCl3 can, in principle, interact both with the carbanion centers19 and Cl atoms of [1]2−.20 Since Cl− abstraction would produce the stable and weakly coordinating [AlCl4]− anion, no accompanying desilylation should occur. Indeed, a quantitative twofold cyclization reaction took place when [nBu4N]2[1] was combined with 2 equiv of AlCl3. The resulting product 4 (yield: 84 %) consists of two silacyclobutene rings fused at their C=C double bonds and thus experiences considerable ring strain (Scheme 3). As a consequence, the formation of 4 is reversible and [1]2− can be recovered through mere addition of 2 equiv of [nBu4N]Cl, whereas HCl induces the opening of only one of the four‐membered rings to afford 2.
The reaction of [1]2− with only 1 equiv of AlCl3 is also highly selective: in situ NMR spectroscopy pointed toward the formation of a silacyclobutene [5]− with a carbanionic sidechain (Scheme 4). It is thus apparent that the cyclization of [1]2− can be induced not only through protonation, but also by providing a Cl−‐abstracting reagent. To further support our structural proposal for [5]−, we added 1 equiv of Cl− ions to the reaction mixture, whereupon the starting material [1]2− was fully regenerated. Quantitative conversions of [5]− also occurred in the presence of HOTf (1 equiv) or AlCl3 (1 equiv) and furnished the neutral silacyclobutenes 2 or 4, respectively (Scheme 4). The anion [5]− is thus the likely intermediate on the way from [1]2− to 4 in the presence of 2 equiv of AlCl3.
Scheme 4.
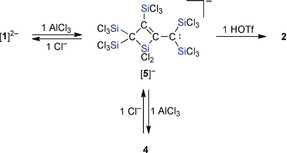
Formation of [5]− as the key intermediate in the reaction from [1]2− to 4 via Cl− abstraction with AlCl3; transformation of [5]− to the neutral compound 2 via protonation with HOTf; reaction conditions: CD2Cl2, room temperature.
Despite their strained molecular frameworks, 2 and 4 are thermally stable up to at least 150 and 180 °C, respectively. At these temperatures, endothermic events with no accompanying mass losses are apparent from DTA/TG measurements. The underlying phase transitions are reversible and do not correspond to melting of the sample, as confirmed by X‐ray powder diffractometry (see the Supporting Information for more details). Significant decomposition of 2 and 4 with overall mass losses of 54 % and 68 % sets in at T=230 °C (2) and 260 °C (4).
Differences and similarities in the molecular structures of [1]2−, 3, and [B]2− provide useful information to assess the degree of π‐delocalization in [1]2− (Figure 1). Unfortunately, the dianion of [nBu4N]2[1] is located on a center of inversion in the crystal lattice and the two central C atoms are disordered over two positions. The problem was solved by changing the counter cation to [Ph4P]+: in [Ph4P]2[1], [1]2− still possesses inversion symmetry, but now the internal C atoms are fully ordered with C−C bond lengths of 1.501(2) Å (terminal) and 1.353(2) Å (internal). The first value is even larger than the typical length of an isolated C(sp2)−C(sp2) single bond (1.47 Å), the second falls in the usual range of C=C double bonds (1.34 Å).21 Moreover, the planes spanned by the C4 chain on the one hand and each Si2C terminus on the other are mutually orthogonal (84.6(2)°). The C(1)−Si(1)/Si(2) bonds possess lengths of 1.774(1)/1.777(1) Å, similar to those in the ethylene dianion [B]2−,10 but remarkably shorter than the C(2)−Si(3) bond of [1]2− (1.882(1) Å). Thus, the degree of π‐delocalization is significantly large between the planar carbanionic centers and their attached SiCl3 substituents, but small along the carbon backbone of [1]2−. This conclusion gains further support from the solid‐state structure of the Ci‐symmetric trans‐but‐2‐ene 3, which features a central C(2)=C(2A) double bond (1.35(1) Å) and internal C(2)−Si(2) bonds (1.881(6) Å) of comparable lengths as [1]2− (Figure 1).
Figure 1.
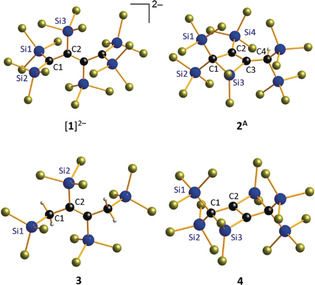
Molecular structures of [Ph4P]2[1], 2A, 3, and 4 in the solid state. [Ph4P]+ ions are omitted for clarity; compound 2 crystallizes with two crystallographically independent molecules, 2A and 2B, in the asymmetric unit.
By comparing 2 and 4, we will next discuss the impact of the edge‐fused additional four‐membered ring on the silacyclobutene scaffold (Figure 1). Compound 2 crystallizes with two independent molecules, 2 A and 2 B, in the asymmetric unit. Given that all key structural parameters of the two molecules are identical within the experimental error margins, only 2 A will be considered here. The molecule shows endocyclic bond lengths of Si(3)−C(1)/(C(3)=1.922(9)/1.847(9) Å and C(2)−C(3)=1.36(1) Å, as well as a C(1)‐Si(3)‐C(3) bond angle of 78.4(4)°. The double silacyclobutene 4 possesses an inversion‐symmetric, planar molecular framework with endocyclic bond lengths that do not vary appreciably from those of 2A [4: Si(3)−C(1)/(C(2A)=1.943(1)/1.841(2) Å, C(2)−C(2A)=1.366(3) Å; Figure 1). Conversely, however, the bond angle C(1)‐C(2)‐Si(3A)=159.3(1)° in the six‐membered bicycle is greatly expanded compared to the analogous angles C(1)‐C(2)‐Si(4)=126.9(6)° and C(4)‐C(3)‐Si(3)=140.2(7)° in the single silacyclobutene 2A. Thus, 4 suffers from additional ring strain, as is also indicated by our previous observations that 4 undergoes rapid ring opening in the presence of ethereal HCl, whereas the cyclic structure of 2 remains intact under these conditions. To further assess ring strain effects, we performed calculations on hypohomodesmotic bicyclization reactions as suggested by Wheeler et al.22 (Scheme 5). A smaller reaction enthalpy, and thus reduced ring‐strain energy (SE), upon inclusion of silicon atoms into the bicycle (SE=ΔH R=52.6 kcal mol−1) is observed compared to the carbonaceous compound Δ1,4‐bicyclo[2.2.0]hexene (ΔH R=84.6 kcal mol−1, in good agreement with the literature11). The results indicate a mitigating effect on the ring strain by including the larger and more flexibly binding silicon into the bicycle. This is in good accordance with prior works on silacarbocycles by Gordon et al. and Inagaki et al.23
Scheme 5.
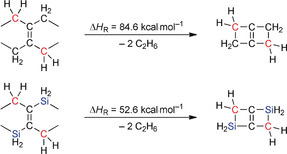
Hypohomodesmotic cyclization reactions calculated using the PBE0‐D4/def2‐QZVPP//PBE0‐D4(COSMO(CH2Cl2))/def2‐TZVP24 method.
All NMR spectra, except those of [nBu4N]2[1] (in [D8]THF), were recorded in CD2Cl2. The resonances of the new compounds have been assigned by 13C HSQC, 13C HMBC, and 29Si HMBC NMR spectra as well as by comparison with the NMR spectra of compounds [B]2−, H2 B, [HB]−, and C.10 The individual data are compiled in Figure 2; for facility of inspection, the chemical shift values have been rounded up to integer numbers. The 1H NMR spectrum of [nBu4N]2[1] shows exclusively the resonances of the ammonium cation. The 13C signals of the [1]2− anion appear at 159.7 ppm (internal C atoms; C: 178.4 ppm) and 50.2 ppm (terminal C atoms; [B]2−: 39.9 ppm). Our assignment of the 29Si resonances of [1]2− [−13.1 (terminal SiCl3), −15.8 ppm (internal SiCl3)] relies on the corresponding integral values of approximately 2:1 (note that the 29Si NMR signals of the neutral ethylene C at −12.2 ppm and ethylene dianion [B]2− at −15.5 ppm are also very similar to each other).
Figure 2.
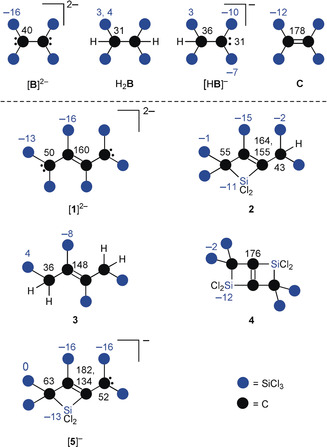
Overview of the 13C (black) and 29Si (blue) NMR chemical shift values of the new C4 compounds [1]2−–[5]− in comparison with the trichlorosilylated C2 species [B]2−, H2 B, [HB]−, and C.
It is also revealing to compare [1]2− with its doubly protonated and desilylated analogue 3, which represents a clear‐cut trans‐but‐2‐ene with no carbon‐based electron lone pairs (Figure 2): The signal of the olefinic C atoms in 3 possesses a chemical shift value of δ(13C)=147.8 ppm and the attached SiCl3 substituents resonate at δ(29Si)=−7.8 ppm, which is still close to the corresponding values of [1]2−. For the (Cl3Si)CH2 termini of 3, 1H, 13C, and 29Si NMR signals were detected at 3.49, 36.4, and 4.1 ppm, respectively. The downfield shift of the 29Si resonances upon going from [1]2− to 3 nicely reflects the removal of negative charge, which can no longer be π‐delocalized across the SiCl3 groups (α‐effect;1c see [B]2− vs. H2 B: δ(29Si)=−15.5 vs. 3.0/4.1 ppm10). The presence of CH2 moieties in 3 was confirmed by means of a 13C{1H} DEPT135 experiment and the corresponding released SiCl3 fragments reappeared as SiCl4 in the reaction mixture. The data acquired for [1]2− provide no evidence of extensive π‐delocalization along the C4 backbone or a significant intramolecular σ‐donor/acceptor equilibrium between the terminal carbanions and internal SiCl3 substituents.
Among the silacyclobutenes, we will first consider the C 2h‐symmetric bicyclic species 4. Compound 4 showed only the 13C resonance of its olefinic C atoms (175.6 ppm) whereas no signal belonging to the aliphatic C atoms was detectable. Using the SO‐ZORA‐PBE0(COSMO(CH2Cl2))/ZORA/QZ4P//PBE0‐D4(COSMO(CH2Cl2))/def2‐TZVP25 method, the corresponding chemical shifts are calculated to be 185.7 and 59.7 ppm, respectively. In contrast, both expected 29Si signals could be found and appeared at −2.3 (SiCl3; higher intensity) and −11.9 ppm (SiCl2; lower intensity). The corresponding resonances of the monocyclic congener 2 emerge at δ(13C)=163.8/155.3 (olefinic C atoms) and 55.0 ppm (endocyclic aliphatic C atom) as well as at δ(29Si)=−0.7 (2× exocyclic SiCl3) and −10.6 (endocyclic SiCl2); we assign a signal at δ(29Si)=−14.5 ppm to the third SiCl3 substituent of the silacyclobutene ring. The attached (Cl3Si)2CH fragment gives rise to 1H, 13C, and 29Si NMR signals at 4.00, 42.5, and −2.0 ppm, respectively.26 A 27Al NMR spectrum recorded on the reaction mixture of [nBu4N]2[1] and AlCl3 (1:1) in CD2Cl2 exclusively showed the prominent and sharp signal of the [AlCl4]− anion at 103.8 ppm.19 Four 13C and four 29Si NMR resonances testify to the low symmetry of the organosilicon product obtained (see [5]−; Scheme 4). Importantly, identical signal sets were recorded on solutions containing equimolar amounts of 4 and [nBu4N]Cl, which should give deprotonated 2 (≡[5]−; see the successful transformation of 4 to 2 with ethereal HCl). The NMR data of [5]− agree well with those of [1]2− and 2, the only exception being the signals for the olefinic C atoms ([5]−: 181.9, 134.0 ppm; 2: 163.8, 155.3 ppm; Figure 2).
The ease with which the single and double silacyclobutenes 2 and 4 are formed is remarkable. Still in 2006, the authors of a comprehensive review article on silacyclobutenes complained about the poor accessibility of these compounds: “They can be prepared on a laboratory scale, but only with a great effort and in comparably low yields by complicated synthetic methods or by gas phase pyrolysis of suitable precursors”.27 Among the most popular synthesis routes are [2+2] cycloadditions between alkynes and species containing highly reactive Si=C double bonds,28 such as silenes29 or Ni‐stabilized 1‐silapropadienes.30 Auner et al. prepared silacyclobutenes from Cl3Si−C(H)=CH2 and tBuLi in the presence of internal alkynes, RC≡CR′.31, 32 The initially generated α‐lithio adduct Cl3Si−C(H)Li−CH2 tBu nucleophilically adds to the alkyne, and the resulting γ‐lithiated species undergoes an intramolecular cyclization to form the four‐membered ring with elimination of LiCl. According to quantum‐chemical calculations, a silene intermediate is likely not involved in the overall scenario.33 The nucleophilic substitution reaction at the SiCl3 group in the last step of the reaction pathway provides a plausible mechanistic model also for the formation of 2 and 4 from [1]2−. Moreover, Auner's and our works provide extremely rare protocols for the synthesis of 1,1‐dichlorosilacyclobutenes,34, 35 which offer a multitude of options for subsequent Si derivatization.31, 34, 36, 37, 38, 39 Appropriately derivatized silacyclobutenes can be introduced into the polymer main chains of polysilanes, carbosilanes, and siloxanes by taking advantage of the functional groups at the Si centers or by performing ring‐opening polymerization reactions; silacyclobutenes are also promising building blocks for organic–inorganic optoelectronic materials.27, 30, 37, 38, 39, 40, 41
The six‐membered bicyclic scaffold of 4 has little precedence in the literature: A close relative G (Figure 3) stems from Weidenbruch's group and can be regarded as a sterically protected, desilylated version of 4. Molecule G was prepared from the photogenerated silylene [Si(tBu)2] and the 1,3‐diyne tBuC≡C−C≡CtBu via the rearrangement of a bis(silirene) intermediate.42 Isomers G′ and G′′ have been assessed by experimental and/or theoretical means.43 Compounds G′′ also contain the structural motif of a disilacyclobutene—a famous substance class derived from the cycloaddition of disilenes with alkynes.44 We finally note that all structures G–G′′ represent isomers of disila(Dewar‐benzene).
Figure 3.
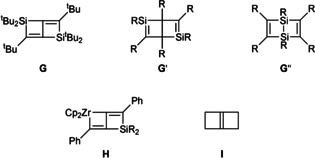
Known compounds G–I with six‐membered bicyclic scaffolds.
The formal exchange of one Si(tBu)2 moiety in G by a ZrCp2 unit yields compounds of type H (Figure 3), which have been employed by Takahashi et al. as precursors of various silacyclobutenes.45 The actual synthesis of H was achieved by intramolecular C−C coupling of bis(alkynyl)silanes with low‐valent zirconocenes. Finally, the hydrocarbon analogue I of 4 (Figure 3) has been described as “one of the most reactive of the alkenes”;46 the neat compound polymerizes already at −23 °C with a half‐time of <10 s.11a
Conclusion
To conclude, we have prepared the unique persilylated butadiene dianion [1]2− in one step from inexpensive hexachloro‐1,3‐butadiene and the Si2Cl6/Cl− silylation system. Contrary to other butadiene dianions,47 the carbon‐based lone pairs of electrons in [1]2− are not stabilized through metal–cation coordination, but mainly through the pronounced α‐effect of their SiCl3 substituents. Under inert conditions, [1]2− is stable over days in THF solution and months in the solid state. Yet, it readily reacts with Brønsted (HOTf) and Lewis (AlCl3) acids to furnish silacyclobutenes, such as the edge‐fused six‐membered double silacyclobutene 4. Especially the rich follow‐up chemistry of the strained C=C double bond of the carbonaceous analogue I of 4, which encompasses cycloadditions, carbometallations, skeletal rearrangements, and radical reactions,48 make us optimistic that also compound 4 will shape up as a versatile platform of further transformations in the future.49
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to the Evonik Resource Efficiency GmbH, Rheinfelden (Germany), for the generous donation of Si2Cl6. I.G. wishes to thank the Evonik Foundation for a Ph.D. grant. This work was partially funded by the Bundesministerium für Wirtschaft und Energie through the WIPANO grant number 03THW10K19. The German Research Foundation (DFG) is gratefully acknowledged for financial support through a Gottfried Wilhelm Leibniz prize to S.G.
I. Georg, M. Bursch, J. B. Stückrath, E. Alig, M. Bolte, H.-W. Lerner, S. Grimme, M. Wagner, Angew. Chem. Int. Ed. 2020, 59, 16181.
In memory of Professor Suning Wang
References
- 1.
- 1a. Brown H. C., Singaram B., Pure Appl. Chem. 1987, 59, 879–894; [Google Scholar]
- 1b. Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials (Ed.: D. G. Hall), Wiley-VCH, Weinheim, 2011; [Google Scholar]
- 1c. Brook M. A., Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000; [Google Scholar]
- 1d. Colvin E. W., Silicon in Organic Synthesis, Butterworths, London, 1981. [Google Scholar]
- 2.
- 2a. Suzuki A., Angew. Chem. Int. Ed. 2011, 50, 6722–6737; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6854–6869; [Google Scholar]
- 2b. Hiyama T., J. Organomet. Chem. 2002, 653, 58–61. [Google Scholar]
- 3.
- 3a. Matteson D. S., Chem. Rev. 1989, 89, 1535–1551; [Google Scholar]
- 3b. Matteson D. S., J. Org. Chem. 2013, 78, 10009–10023; [DOI] [PubMed] [Google Scholar]
- 3c. Leonori D., Aggarwal V. K., Acc. Chem. Res. 2014, 47, 3174–3183. [DOI] [PubMed] [Google Scholar]
- 4. van Staden L. F., Gravestock D., Ager D. J., Chem. Soc. Rev. 2002, 31, 195–200. [DOI] [PubMed] [Google Scholar]
- 5. von Grotthuss E., John A., Kaese T., Wagner M., Asian J. Org. Chem. 2018, 7, 37–53. [Google Scholar]
- 6.
- 6a. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931; [DOI] [PubMed] [Google Scholar]
- 6b. Hartwig J. F., Acc. Chem. Res. 2012, 45, 864–873. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Synthesis and Application of Organoboron Compounds (Eds.: E. Fernández, A. Whiting), Springer International Publishing, Cham, 2015; [Google Scholar]
- 7b. Dewhurst R. D., Neeve E. C., Braunschweig H., Marder T. B., Chem. Commun. 2015, 51, 9594–9607; [DOI] [PubMed] [Google Scholar]
- 7c. Neeve E. C., Geier S. J., Mkhalid I. A. I., Westcott S. A., Marder T. B., Chem. Rev. 2016, 116, 9091–9161. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Endo K., Hirokami M., Shibata T., Synlett 2009, 1331–1335; [Google Scholar]
- 8b. Takaya J., Kirai N., Iwasawa N., J. Am. Chem. Soc. 2011, 133, 12980–12983; [DOI] [PubMed] [Google Scholar]
- 8c. Li H., Shangguan X., Zhang Z., Huang S., Zhang Y., Wang J., Org. Lett. 2014, 16, 448–451; [DOI] [PubMed] [Google Scholar]
- 8d. Palmer W. N., Zarate C., Chirik P. J., J. Am. Chem. Soc. 2017, 139, 2589–2592; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Wen H., Zhang L., Zhu S., Liu G., Huang Z., ACS Catal. 2017, 7, 6419–6425; [Google Scholar]
- 8f. Procter R. J., Uzelac M., Cid J., Rushworth P. J., Ingleson M. J., ACS Catal. 2019, 9, 5760–5771. [Google Scholar]
- 9. Liu X., Ming W., Friedrich A., Kerner F., Marder T. B., Angew. Chem. Int. Ed. 2020, 59, 304–309; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 311–316. [Google Scholar]
- 10. Georg I., Teichmann J., Bursch M., Tillmann J., Endeward B., Bolte M., Lerner H.-W., Grimme S., Wagner M., J. Am. Chem. Soc. 2018, 140, 9696–9708. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Casanova J., Bragin J., Cottrell F. D., J. Am. Chem. Soc. 1978, 100, 2264–2265; [Google Scholar]
- 11b. Wiberg K. B., Bonneville G., Dempsey R., Isr. J. Chem. 1983, 23, 85–92; [Google Scholar]
- 11c. Wiberg K. B., Angew. Chem. Int. Ed. Engl. 1986, 25, 312–322; [Google Scholar]; Angew. Chem. 1986, 98, 312–322; [Google Scholar]
- 11d. Khoury P. R., Goddard J. D., Tam W., Tetrahedron 2004, 60, 8103–8112; [Google Scholar]
- 11e. Akhmetshina E. S., Khursan S. L., Thermochim. Acta 2020, 685, 178541. [Google Scholar]
- 12. Teichmann J., Bursch M., Köstler B., Bolte M., Lerner H.-W., Grimme S., Wagner M., Inorg. Chem. 2017, 56, 8683–8688. [DOI] [PubMed] [Google Scholar]
- 13. Teichmann J., Wagner M., Chem. Commun. 2018, 54, 1397–1412. [DOI] [PubMed] [Google Scholar]
- 14.In the absence of other trapping reagents, the Si2Cl6/[nBu4N]Cl mixture generates chain-like, cyclic, or cluster-type (“silafulleranes”) oligosilanes:
- 14a. Tillmann J., Meyer L., Schweizer J. I., Bolte M., Lerner H.-W., Wagner M., Holthausen M. C., Chem. Eur. J. 2014, 20, 9234–9239; [DOI] [PubMed] [Google Scholar]
- 14b. Tillmann J., Moxter M., Bolte M., Lerner H.-W., Wagner M., Inorg. Chem. 2015, 54, 9611–9618; [DOI] [PubMed] [Google Scholar]
- 14c. Tillmann J., Wender J. H., Bahr U., Bolte M., Lerner H.-W., Holthausen M. C., Wagner M., Angew. Chem. Int. Ed. 2015, 54, 5429–5433; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5519–5523; [Google Scholar]
- 14d.see Ref. [13].
- 15.The Si−Si bond in Si2Cl6 is particularly easy to activate, whereas the much more challenging cleavage of Si2Me6 usually requires specifically designed catalysts:
- 15a. Ansell M. B., Roberts D. E., Cloke F. G. N., Navarro O., Spencer J., Angew. Chem. Int. Ed. 2015, 54, 5578–5582; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5670–5674. However, Hiyama et al. have reported that hexaalkyldisilanes Si2R6 react also with [nBu4N]F in hexamethylphosphoric triamide (HMPA) to produce metal-free silyl anions [SiR3]− and FSiR3: [Google Scholar]
- 15b. Hiyama T., Obayashi M., Mori I., Nozaki H., J. Org. Chem. 1983, 48, 912–914. [Google Scholar]
- 16.“Chlorinated Hydrocarbons”: Rossberg M., Lendle W., Pfleiderer G., Tögel A., Dreher E.-L., Langer E., Rassaerts H., Kleinschmidt P., Strack H., Cook R., Beck U., Lipper K.-A., Torkelson T. R., Löser E., Beutel K. K., Mann T. in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2006. [Google Scholar]
- 17.Gibbs free energies and NMR parameters were calculated for several structure candidates using the methods PBE0-D4+COSMO-RS(CH2Cl2)/def2-QZVPPD//PBE0-D4(COSMO(CH2Cl2))/def2-TZVPD and SO-ZORA-PBE0(COSMO(CH2Cl2))/ZORA/QZ4P//PBE0-D4(COSMO(CH2Cl2))/def2-TZVPD, respectively. See the Supporting Information for more details.
- 18.A mixture of [nBu4N]2[1] with 1 equiv of ethereal HCl furnished mainly [nBu4N][H1]; in the presence of 2 equiv of the acid, the formation of [nBu4N][H1] and 3 was observed. Both mixtures were quantitatively converted to 3 upon addition of more HCl (3 and 2 equiv, respectively).
- 19. Teichmann J., Kunkel C., Georg I., Moxter M., Santowski T., Bolte M., Lerner H.-W., Bade S., Wagner M., Chem. Eur. J. 2019, 25, 2740–2744. [DOI] [PubMed] [Google Scholar]
- 20.Mixtures of AlCl3 and CH2Cl2 have been used as strong oxidizing agents. However, in our chemistry we never observed indications of oxidation reactions: Bock H., Lechner-Knoblauch U., J. Organomet. Chem. 1985, 294, 295–304. [Google Scholar]
- 21. Fox M. A., Whitesell J. K., Organic Chemistry, Jones and Bartlett Publishers, Burlington, 2004. [Google Scholar]
- 22. Wheeler S. E., Houk K. N., Schleyer P. v. R., Allen W. D., J. Am. Chem. Soc. 2009, 131, 2547–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Boatz J. A., Gordon M. S., J. Phys. Chem. 1988, 92, 3037–3042; [Google Scholar]
- 23b. Naruse Y., Ma J., Inagaki S., Tetrahedron Lett. 2001, 42, 6553–6556. [Google Scholar]
- 24.
- 24a. Adamo C., Barone V., J. Chem. Phys. 1999, 110, 6158–6170; [Google Scholar]
- 24b. Klamt A., Schüürmann G., J. Chem. Soc. Perkin Trans. 2 1993, 799–805; [Google Scholar]
- 24c. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305; [DOI] [PubMed] [Google Scholar]
- 24d. TURBOMOLE V7.3.1 2018, a Development of the University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com;
- 24e. Furche F., Ahlrichs R., Hättig C., Klopper W., Sierka M., Weigend F., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 91–100; [Google Scholar]
- 24f. Caldeweyher E., Bannwarth C., Grimme S., J. Chem. Phys. 2017, 147, 034112–034117; [DOI] [PubMed] [Google Scholar]
- 24g. Caldeweyher E., Ehlert S., Hansen A., Neugebauer H., Spicher S., Bannwarth C., Grimme S., J. Chem. Phys. 2019, 150, 154122. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. van Lenthe E., Baerends E. J., Snijders J. G., J. Chem. Phys. 1993, 99, 4597–4610; [Google Scholar]
- 25b. van Lenthe E., Baerends E. J., Snijders J. G., J. Chem. Phys. 1994, 101, 9783–9792; [Google Scholar]
- 25c. van Lenthe E., Snijders J. G., Baerends E. J., J. Chem. Phys. 1996, 105, 6505–6516; [Google Scholar]
- 25d. Pye C. C., Ziegler T., Theor. Chem. Acc. 1999, 101, 396–408; [Google Scholar]
- 25e. van Lenthe E., Baerends E. J., J. Comput. Chem. 2003, 24, 1142–1156; [DOI] [PubMed] [Google Scholar]
- 25f.ADF 2019.3, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com.
- 26.A justification of the assignment is provided in the Supporting Information.
- 27. Mohseni-Ala J., Auner N., Inorg. Chim. Acta 2006, 359, 4677–4697. [Google Scholar]
- 28. Milnes K. K., Jennings M. C., Baines K. M., J. Am. Chem. Soc. 2006, 128, 2491–2501. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Ottosson H., Eklöf A. M., Coord. Chem. Rev. 2008, 252, 1287–1314; [Google Scholar]
- 29b. Fischer R. C., Power P. P., Chem. Rev. 2010, 110, 3877–3923; [DOI] [PubMed] [Google Scholar]
- 29c. Zhang H.-J., Priebbenow D. L., Bolm C., Chem. Soc. Rev. 2013, 42, 8540–8571. [DOI] [PubMed] [Google Scholar]
- 30. Ishikawa M., Naka A., Kobayashi H., Coord. Chem. Rev. 2017, 335, 58–75. [Google Scholar]
- 31. Auner N., Seidenschwarz C., Herdtweck E., Angew. Chem. Int. Ed. Engl. 1991, 30, 1151–1152; [Google Scholar]; Angew. Chem. 1991, 103, 1172–1173. [Google Scholar]
- 32. Auner N., Heikenwaelder C. R., Wagner C., Organometallics 1993, 12, 4135–4140. [Google Scholar]
- 33. Müller T., Bendikov M., Auner N., Apeloig Y., Organometallics 2001, 20, 598–600. [Google Scholar]
- 34. Kang K.-T., Song H.-Y., Seo H.-C., Chem. Lett. 1985, 14, 617–620. [Google Scholar]
- 35. Wrackmeyer B., Khan E., Bayer S., Shahid K., Z. Naturforsch. B 2007, 62, 1174–1182. [Google Scholar]
- 36. Kang K.-T., Yoon U. C., Seo H. C., Kim K. N., Song H. Y., Lee J. C., Bull. Korean Chem. Soc. 1991, 12, 57–60. [Google Scholar]
- 37. Auner N., Nuyken O., Biebl B., J. Macromol. Sci. Pure Appl. Chem. 1997, 34, 225–234. [Google Scholar]
- 38. Yan D., Bolte M., Auner N., J. Organomet. Chem. 2008, 693, 908–916. [Google Scholar]
- 39. Yan D., Mohsseni-Ala J., Auner N., Bolte M., Bats J. W., Chem. Eur. J. 2007, 13, 7204–7214. [DOI] [PubMed] [Google Scholar]
- 40. Auner N., J. Prakt. Chem. 1995, 337, 79–92. [Google Scholar]
- 41. Yan D., Thomson M. D., Backer M., Bolte M., Hahn R., Berger R., Fann W., Roskos H. G., Auner N., Chem. Eur. J. 2009, 15, 8625–8645. [DOI] [PubMed] [Google Scholar]
- 42.
- 42a. Kirmaier L., Weidenbruch M., Marsmann H., Peters K., von Schnering H. G., Organometallics 1998, 17, 1237–1240; [Google Scholar]
- 42b. Ostendorf D., Kirmaier L., Saak W., Marsmann H., Weidenbruch M., Eur. J. Inorg. Chem. 1999, 2301–2307; [Google Scholar]
- 42c. Ostendorf D., Saak W., Weidenbruch M., Marsmann H., Organometallics 2000, 19, 4938–4940; [Google Scholar]
- 42d. Ostendorf D., Saak W., Haase D., Weidenbruch M., J. Organomet. Chem. 2001, 636, 7–11; [Google Scholar]
- 42e. Ostendorf D., Saak W., Weidenbruch M., Marsmann H., Organometallics 2002, 21, 636–640. [Google Scholar]
- 43.
- 43a. Rich J. D., West R., J. Am. Chem. Soc. 1982, 104, 6884–6886; [Google Scholar]
- 43b. Kabe Y., Ohkubo K., Ishikawa H., Ando W., J. Am. Chem. Soc. 2000, 122, 3775–3776; [Google Scholar]
- 43c. Kang S.-Y., Yoshizawa K., Yamabe T., Naka A., Ishikawa M., J. Organomet. Chem. 2000, 611, 280–287; [Google Scholar]
- 43d. Priyakumar U. D., Saravanan D., Sastry G. N., Organometallics 2002, 21, 4823–4832; [Google Scholar]
- 43e. Deva Priyakumar U., Punnagai M., Sastry G. N., J. Organomet. Chem. 2004, 689, 1284–1287; [Google Scholar]
- 43f. Nakata N., Oikawa T., Matsumoto T., Kabe Y., Sekiguchi A., Organometallics 2005, 24, 3368–3370; [Google Scholar]
- 43g. Nakata N., Oikawa T., Matsumoto T., Kabe Y., Sekiguchi A., Organometallics 2006, 25, 5850–5851; [Google Scholar]
- 43h. Oikawa T., Nakata N., Matsumoto T., Kabe Y., Sekiguchi A., Heteroat. Chem. 2008, 19, 87–92. [Google Scholar]
- 44.
- 44a. Atwell W. H., Uhlmann J. G., J. Organomet. Chem. 1973, 52, C21–C23; [Google Scholar]
- 44b. Sakurai H., Kobayashi T., Nakadaira Y., J. Organomet. Chem. 1978, 162, C43–C47; [Google Scholar]
- 44c. Fink M. J., DeYoung D. J., West R., Michl J., J. Am. Chem. Soc. 1983, 105, 1070–1071; [Google Scholar]
- 44d. Schäfer A., Weidenbruch M., Pohl S., J. Organomet. Chem. 1985, 282, 305–313; [Google Scholar]
- 44e. DeYoung D. J., West R., Chem. Lett. 1986, 15, 883–884; [Google Scholar]
- 44f. Ando W., Shiba T., Hidaka T., Morihashi K., Kikuchi O., J. Am. Chem. Soc. 1997, 119, 3629–3630; [Google Scholar]
- 44g. Kang S.-Y., Yamabe T., Naka A., Ishikawa M., Yoshizawa K., Organometallics 2002, 21, 150–160; [Google Scholar]
- 44h. Gottschling S. E., Milnes K. K., Jennings M. C., Baines K. M., Organometallics 2005, 24, 3811–3814; [Google Scholar]
- 44i. Bejan I., Inoue S., Ichinohe M., Sekiguchi A., Scheschkewitz D., Chem. Eur. J. 2008, 14, 7119–7122; [DOI] [PubMed] [Google Scholar]
- 44j. Scheschkewitz D., Chem. Eur. J. 2009, 15, 2476–2485; [DOI] [PubMed] [Google Scholar]
- 44k. Majumdar M., Bejan I., Huch V., White A. J. P., Whittell G. R., Schäfer A., Manners I., Scheschkewitz D., Chem. Eur. J. 2014, 20, 9225–9229; [DOI] [PubMed] [Google Scholar]
- 44l. Präsang C., Scheschkewitz D., Chem. Soc. Rev. 2016, 45, 900–921; [DOI] [PubMed] [Google Scholar]
- 44m. Milnes K. K., Pavelka L. C., Baines K. M., Chem. Soc. Rev. 2016, 45, 1019–1035; [DOI] [PubMed] [Google Scholar]
- 44n. Henry A. T., Bourque J. L., Vacirca I., Scheschkewitz D., Baines K. M., Organometallics 2019, 38, 1622–1626. [Google Scholar]
- 45.
- 45a. Takahashi T., Xi Z., Obora Y., Suzuki N., J. Am. Chem. Soc. 1995, 117, 2665–2666; [Google Scholar]
- 45b. Xi Z., Fischer R., Hara R., Sun W.-H., Obora Y., Suzuki N., Nakajima K., Takahashi T., J. Am. Chem. Soc. 1997, 119, 12842–12848. [Google Scholar]
- 46. Wiberg K. B., Matturro M. G., Okarma P. J., Jason M. E., Dailey W., Burgmaier G. J., Bailey W. F., Warner P., Tetrahedron 1986, 42, 1895–1902. [Google Scholar]
- 47. Gardiner M. G., Raston C. L., Cloke F. G. N., Hitchcock P. B., Organometallics 1995, 14, 1339–1353. [Google Scholar]
- 48.
- 48a. Wiberg K. B., Matturro M. G., Okarma P. J., Jason M. E., J. Am. Chem. Soc. 1984, 106, 2194–2200; [Google Scholar]
- 48b. Zhang Y., Smith J., Lemal D. M., J. Am. Chem. Soc. 1996, 118, 9454–9455. [Google Scholar]
- 49. Deposition Numbers 1999414 (for [nBu4N]2[1]), 1999415 (for [Ph4P]2[1]), 1999416 (for 2), 1999417 (for 3), and 1999418 (for 4) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary