

Abstract
Breast cancer risk is approximately twice as high in first‐degree relatives of female breast cancer cases than in women in the general population. Less than half of this risk can be attributed to the currently known genetic risk factors. Recessive risk alleles represent a relatively underexplored explanation for the remainder of familial risk. To address this, we selected 19 non‐BRCA1/2 breast cancer families in which at least three siblings were affected, while no first‐degree relatives of the previous or following generation had breast cancer. Germline DNA from one of the siblings was subjected to exome sequencing, while all affected siblings were genotyped using SNP arrays to assess haplotype sharing and to calculate a polygenic risk score (PRS) based on 160 low‐risk variants. We found no convincing candidate recessive alleles among exome sequencing variants in genomic regions for which all three siblings shared two haplotypes. However, we found two families in which all affected siblings carried the CHEK2*1100delC. In addition, the average normalized PRS of the “recessive” family probands (0.81) was significantly higher than that in both general population cases (0.35, P = .026) and controls (P = .0004). These findings suggest that the familial aggregation is, at least in part, explained by a polygenic effect of common low‐risk variants and rarer intermediate‐risk variants, while we did not find evidence of a role for novel recessive risk alleles.
Keywords: breast cancer, exome, polygenic, recessive, susceptibility
Short abstract
What's new?
To find new breast cancer susceptibility alleles, these authors tested families in which at least three affected siblings had non‐BRCA1/2 breast cancer. No new susceptibility alleles emerged, but the analysis did reveal that on average, women from these families who had cancer had significantly higher polygenic risk scores than either sporadic cases or controls. This result highlights the importance of moderate risk alleles acting together in familial breast cancer.
Abbreviations
- BCAC
Breast Cancer Association Consortium
- BED
browser extensible data
- CADD
combined annotation dependent depletion
- GATK
genome analysis toolkit
- GoNL
genome of the Netherlands
- IBD
identical by descent
- IGV
integrative genome viewer
- LUMC
Leiden University Medical Center
- NKI‐AvL
Netherlands Cancer Institute‐Antoni van Leeuwenhoek Ziekenhuis
- OR
odds ratio
- PRS
polygenic risk score
- PTV
protein‐truncating variant
- SNP
single nucleotide polymorphism
- VUS
variant of uncertain significance
1. INTRODUCTION
Breast cancer is the most common cancer in females in the Western world and has a complex etiology in which both genetic and environmental factors affect disease risk. Having a family member affected by the disease is one of the most important risk factors. 1 Pathogenic variants in the two most well‐known high‐risk breast cancer genes, BRCA1 and BRCA2, explain approximately 17% of the familial relative risk. 2 In addition, a number of less frequently mutated high‐risk genes (eg, TP53) and a number of genes in which pathogenic variants are associated with a more moderately increased risk (eg, CHEK2) together explain another 5%. Moreover, approximately 160 common polymorphisms have been associated with small increases in risk, which jointly explain about 18% of the excess familial risk. 3
Since the discovery of BRCA1 and BRCA2, several segregation studies have concluded that a polygenic model, or a model with a recessive allele would best explain the remaining familial risk.4, 5, 6, 7 Genetic searches for new loci, while successful, have focused on detecting rare dominant high‐risk alleles (by candidate gene re‐sequencing) or common low‐risk variants. Systematic searches for recessive alleles have not been conducted, despite evidence suggesting that such alleles could play a role in the genetic etiology of breast cancer. For example, a large meta‐analysis on familial breast cancer risk has shown that having a sister affected with breast cancer is associated with a stronger increase in risk than having a mother with breast cancer. 8 In addition, an increased breast cancer risk has been reported in the offspring of consanguineous parents. 9 Studies assessing regions of homozygosity in outbred populations have not shown more or larger regions of homozygosity in breast cancer cases, but some have suggested an increased frequency of homozygosity in specific genomic regions.10, 11
We performed a small‐scale search for recessive breast cancer risk alleles in families with at least three affected siblings and no other first or second‐degree relatives with early‐onset breast cancer. The regions in which all affected siblings shared two haplotypes, as determined by low‐density SNP arrays, were identified and used to filter the exome sequence data that was generated for one of the siblings. This approach significantly reduces the number of potentially interesting variants, allowing for less stringent filters on allele frequency and hence fewer assumptions about the characteristics of a novel breast cancer risk‐associated variant. In addition, we calculated a polygenic risk score based on 160 known breast cancer risk‐associated polymorphisms and assessed the contribution of exonic variants in known breast cancer susceptibility genes that were predicted to be damaging by in silico prediction algorithms.
2. METHODS
2.1. Selection of families
Families were ascertained through the clinical genetics centers of two Dutch hospitals, the Leiden University Medical Center (LUMC) and the Netherlands Cancer Institute Antoni van Leeuwenhoek Hospital (NKI‐AvL) and from a previously described set of breast cancer families collected throughout the Netherlands. 12 We enriched for families with a presumed recessive mode of inheritance by selecting families in which at least three siblings were affected with breast cancer at any age. Sib‐ships that had first‐degree relatives with breast cancer in the previous or following generation were excluded, as were families with second‐degree relatives with breast cancer diagnosed before age 50. DNA from blood lymphocytes had to be available for at least two affected siblings. Availability of DNA samples from parents or other family members was not a selection criterion. In every family, at least one affected individual had been extensively tested according to local testing standards for pathogenic variants in BRCA1 and BRCA2, and all families with a pathogenic variant or variant of uncertain significance in BRCA1 or BRCA2 were excluded.
2.2. Haplotype analysis
We genotyped all available DNA samples from the affected siblings using the HumanLinkage V Panel from Illumina. Sample preparation was done according to the manufacturer's protocol (Rev. B October 2010). Samples were hybridized to GoldenGate Universal‐32 BeadChip (Illumina) and chips were scanned using a Bead Array Reader (Illumina). The GenomeStudio software (version 2011.1, Illumina) was used to call genotypes. We used Merlin (v1.12) to calculate, for each sib pair and marker position, the probability that at this position the sib pair shared zero, one or two alleles identical by decent (IBD). 13 On average a sib pair is expected to share two haplotypes in 25% of their genomes. To decrease the chance of false‐negative regions, we set a probability cut‐off such that for all sib pairs at least 25% of the markers were selected as sharing two alleles IBD (cut‐off: P > .05). We then selected all positions in which all siblings shared two alleles IBD or, for the analysis allowing for one phenocopy, all positions in which all but one sib shared two alleles IBD. These positions were converted into a BED file describing the regions IBD for both haplotypes. Each of these regions started one base pair after the last upstream position for which the affected siblings did not share two alleles IBD and ran until one base pair before the first downstream position for which they did not share two alleles IBD.
2.3. Exome sequencing and analysis
From each family, one affected individual was selected for exome sequencing of germline DNA. In most instances, this was the individual with the youngest age of diagnosis; however, in two families, another individual was selected due to limited availability of DNA. Samples were prepared using Illumina's Paired‐End Library Preparation Kit, after which the coding regions of the genome were captured using SeqCap EZ Exome v3.0 (Nimblegen). Sequencing was done on a HiSeq 2000 (Illumina), generating 2×100 base pair reads. We used GATK for indel realignment, base recalibration and finally variant calling using Haplotypecaller. 14 These analyses were done according to the GATK best practices guidelines for DNA sequencing analysis. A detailed description of the settings and version numbers of the used software is given in Supporting Information.
2.4. Variant filtering and validation
Figure 1 outlines our strategy for identifying recessively predisposing genetic variants in the affected sib ships. We first selected, for each individual, variants in regions in which they shared two haplotypes IBD with their siblings, using the family‐specific BED files. We then annotated the variants using Seattleseq (138, v9.03). 15 Next, we selected all stop‐gained, frameshift and canonical splice site variants. These predicted protein‐truncating variants (PTV) could be either heterozygous or homozygous. We removed variants with an allele frequency >10% in either the exome variant server, Hapmap, 1000 genomes, ExAC or Genome of the Netherlands (GoNL) data.16, 17, 18, 19, 20 In addition, we removed all variants with an allele frequency of >30% in our dataset, since these are likely to be experiment‐specific artifacts. All remaining variants were manually inspected in the Integrative Genomics Viewer (IGV; v2.3.34) to remove any clear misalignments or other calling errors. 21 In the genes in which a heterozygous potential PTV was found, we searched for a “second hit”, defined as either another potential PTV or a missense variant, satisfying the same frequency cut‐off. When two (or more) “hits” in a gene were identified, these variants were validated using Sanger sequencing. Primer sequences are available upon request.
FIGURE 1.
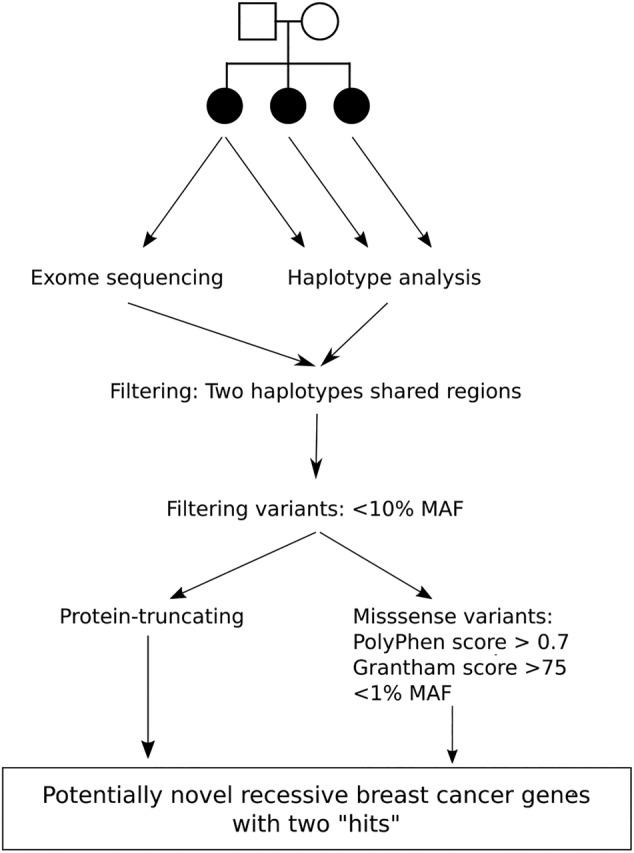
Strategy for the identification of recessively predisposing genetic variants. This overview presents our strategy for exome variant filtering to detect potential new breast cancer risk alleles with a recessive mode of inheritance
We also considered a scenario in which two missense changes in a gene on two haplotypes could cause a recessive inheritance (ie, either homozygous or compound heterozygous). For this, we selected all missense changes in the regions specified by the BED files, with allele frequencies <1% and in silico annotations suggestive of deleteriousness (PolyPhen score > 0.7; Grantham score > 75).
2.5. Variants in known and suspected breast cancer genes
We examined a set of 35 known and suspected breast cancer susceptibility genes (derived from commercially available multigene panels, Table S1) for genetic variants regardless of haplotype sharing. The genes were assigned into four categories, based on the level of evidence for being associated with breast cancer risk (strong to unlikely); a separate category consisted of “syndromic” genes, in which variants have been associated with a range of cancers typical of certain familial cancer syndromes (TP53, CDH1, PTEN). PTVs in level 1/2 genes were filtered on allele frequency in the general population (exome variant server, Hapmap, 1000 genomes or GoNL) with a cut‐off of 0.1% for the high‐risk genes (BRCA1, BRCA2, PALB2, TP53, PTEN and CDH1) and 2% for the moderate risk genes (ATM, CHEK2), allowing for the observation that some PTVs in moderate‐risk genes (such as the c.1100delC in CHEK2) occur at >0.5% allele frequencies in some populations. All missense variants in the 35 genes were selected if their allele frequency in the general population was <2% and they had either a CADD 22 score >20 or were found in one of the levels 1/2 genes. All selected variants were inspected manually in the IGV to remove misalignments. Variants that were both rare and not likely to result from a misalignment were then validated using Sanger sequencing.
2.6. Validation of potential recessive risk alleles
To further assess the association of selected variants with recessive breast cancer, we selected a set of 111 women diagnosed with breast cancer 35 or younger, through the clinical genetics center of the LUMC.
2.7. Polygenic risk score analysis
All affected sibs for whom DNA was available were genotyped using one of two SNP arrays partly designed to study SNPs associated with breast cancer risk: the iCOGs array and the OncoArray. To calculate polygenic risk scores, we selected all independent SNPs shown to be significantly (P < 5 × 10−8) associated with overall breast cancer by the Breast Cancer Association Consortium (BCAC), the largest case‐control study to date. 23 The selected SNPs and respective ORs are shown in Table S2. A small number of known low‐risk variants were not included on the arrays. These variants were imputed with the help of IMPUTE2 based on the genome of the Netherlands (GoNL release 5.3) and 1000 genomes (Phase 3) data (Supporting Information).18, 19, 24 Polygenic risk scores were calculated using:
where n ij is the number of risk alleles (0, 1 or 2) SNP i carried by individual j and OR i is the per‐allele odds ratio associated with SNP i (derived fromMichailidou et al 23 ; Table S2). We compared the PRS of the family probands (the same individuals subjected to exome sequencing) with 357 sporadic cases and 327 age‐matched controls from the ORIGO study. 25 These individuals were genotyped using the iCOGS array and imputed in the same way as the familial cases. The PRS was normalized based on the mean and SD of the ORIGO controls so that one unit in PRS corresponded to one SD. The odds ratio per unit SD of the PRS was obtained via univariate logistic regression within the ORIGO population. The null hypothesis of there not being a true difference in mean PRS between the “recessive” family probands, population cases and population controls was tested using a Welch two‐sample t‐test. All analyses were performed using R version 3.4.1.
All individuals provided informed consent and approval of the medical ethical committee at the LUMC was obtained.
3. RESULTS
3.1. Selected families and haplotype analysis
Nineteen families were selected for analysis (Figure S1). Samples were available from two affected siblings for three families, three affected siblings for 14 families and four affected siblings for two families. The average age at diagnosis of first primary breast cancer was 49.9. One family included a male breast cancer patient diagnosed at age 65. The “two haplotypes shared IBD” regions for each family covered on average 31.6%, 10.1% and 2.9% of the genome for families with two, three or four DNA samples available respectively. This is slightly higher than predicted proportions (25%, 6.25% and 1.6% respectively), but this was expected given our conservative IBD probability cut‐off (see Section 2).
3.2. Exome sequencing
Exome sequencing of one affected individual per family achieved 51× average on target coverage and detected on average 28 724 variants per individual. After filtering these variants based on the family‐specific haplotype sharing regions, an average of 10 775 (37.5%), 3222 (11.2%) and 734 (2.6%) variants remained in families with two, three or four individuals genotyped respectively. We first focused on variants that were predicted to result in a truncated protein. When a heterozygous protein‐truncating variant (PTV) was found, we assessed the gene for a second hit which could also be a missense variant (Table 1).
TABLE 1.
rare protein‐truncating and missense variants found in the regions where the sibships share two haplotypes
| Family | Gene | Variant (coding DNA) | Variant (protein) | Rs‐number | Co‐segregation a | Frequency in GoNL b (%) |
|---|---|---|---|---|---|---|
| RF1 | PDIA2 | c.442C>T | p.R148* | rs370453080 | 2/3 | 0 |
| c.1418G>A | p.R473Q | rs116969376 | 3/3 | 1.3 | ||
| RF4 | TLR5 | c.1174C>T | p.R392* | rs5744168 | 3/3 | 6.5 |
| c.541C>A | p.Q181K | rs45528236 | 3/3 | 6.5 | ||
| RF6 | TRPM1 | c.4240G>T | p.E1414* | rs3784589 | 2/3 | 4.9 |
| c.1930G>A | p.V644M | rs17815774 | 3/3 | 4.7 | ||
| RF13 | UNC93A | c.625+1G>C | p.? | rs113906647 | 1/3 | 3.3 |
| c.1159T>C | p.Y387H | rs663227 | 1/3 | 0.7 | ||
| RF14 | PLXNB3 | c.1629+2C>T | p.? | — | 1/3 | 0 |
| c.4787T>A | p.V1596E | rs146832392 | 3/3 | 6.0 | ||
| RF17 | CCHCR1 | c.121G>T | p.E41* | rs72856718 | 3/3 | 9.6 |
| c.2147G>A | p.R716Q | rs130072 | 3/3 | 9.6 | ||
| c.803T>A | p.L268Q | rs11540822 | 3/3 | 9.6 |
Indicates the number of siblings carrying the allele out of the total number of siblings from this family tested.
Frequency in Genome of the Netherlands: genome sequences of 998 independent Dutch individuals. 22 Accession numbers for the transcripts and protein sequences used to describe the variants: PDIA2: NM_006849.2, NP_006840.2; TLR5: NM_003268.5, NP_003259.2; TRPM: NM_001252020.1, NP_001238949.1; UNC93A: NM_018974.3, NP_061847.2; PLXNB3: NM_005393.2, NP_005384.2; CCHCR1: NM_001105564.1, NP_001099034.1.
We originally set the PTV allele frequency cut‐off relatively high (<10%) to allow for the possibility of a single variant that was homozygous in multiple families. No such variants were detected in our dataset, but we did find six genes with two or more heterozygous positions in six different families. For compound heterozygotes, we assumed that the allele frequency of a potentially causal variant was lower (<2%), rendering the variants in TLR5, TRPM1, UNC93A, PLXNB3 and CCHCR1 unlikely candidates. In the remaining gene, PDIA2, we identified a PTV p.R148* and a missense variant p.R473Q, shared IBD in one family. PDIA2 encodes an oxidoreductase involved in protein folding and specifically expressed in the pancreas.26, 27, 28 In addition, it binds estrogen (specifically 17β‐estradiol) and might buffer the local estrogen levels in the pancreas. 29 To further examine the possibility that variants in PDIA2 are associated with breast cancer, we genotyped a set of 111 patients diagnosed with breast cancer before the age of 35 for the two variants detected in family RF1. The PTV p.R148* was not observed, while the missense variant p.R473Q was detected twice (0.9%). The allele frequency of 1.3% in the Genome of the Netherlands, also suggests that this variant is not associated with breast cancer. 19
A similar filter for missense variants revealed two rare homozygous missense variants, SERINC2 p.R126W in family RF4 and ZNF717 p.H63L in family RF7 (Table S3).
SERINC2 regulates lipid biosynthesis and incorporates serine into membrane lipids, while the function of ZNF71 is unknown. The CADD scores for both variants were <20. Based on this, neither variant was considered as a serious candidate for follow‐up studies.
3.3. Analyses allowing for one phenocopy
Since breast cancer is a common disease, there is a high probability that a case in a family is not genetic (ie, a phenocopy). Therefore, we assessed the regions of the genome where only two out of three (or three out of four) affected sisters share two haplotypes. PTVs obtained in this way were then filtered as in the previous analysis (Table 2). Again, most variants were relatively common, but did not occur in multiple families. The only gene in which variants are rare enough to be a possible candidate was SLC26A10, with variants c.1206G>A (p.W402*) and c.1247T>G (p.L416R) found in family RF2. Both variants were shared by two of the three affected sisters. However, in GoNL, both variants were present in the same seven individuals and predicted to be on the same haplotype, excluding the possibility of compound heterozygosity. SCL26A10 has no known function and has been suggested to be an imprinted, maternally expressed, pseudogene.30, 31
TABLE 2.
Rare protein‐truncating and missense variants found in the regions where the sibships share two haplotypes, allowing for one phenocopy
| Family | Gene | Variant (coding DNA) | Variant (protein) | Rs‐number | Co‐segregation a | Frequency in GoNL b (%) |
|---|---|---|---|---|---|---|
| RF2 | ZAN | c.1249 + 1G>A | p.? | rs117406702 | 3/3 | 3.8 |
| c.8132C>T | p.P2711L | rs201771583 | 3/3 | 0 | ||
| SLC26A10 | c.1206G>A | p.W402* | rs113207856 | 2/3 | 0.7 | |
| c.1247T>G | p.L416R | rs111924104 | 2/3 | 0.7 | ||
| RF6 | CCHCR1 | c.121G>T | p.E41* | rs72856718 | 1/3 | 9.6 |
| c.803T>C | p.L232Q | rs11540822 | 1/3 | 9.6 | ||
| RF8 | PLA2G4C | c.893delC | p.P298fs | rs11564598 | 3/3 | 2.9 |
| c.452C>T | p.P151L | rs11564538 | 1/3 | 5.0 | ||
| RF14 | PKHD1L1 | c.7246 + 1G>C | p.? | rs17368310 | 3/3 | 4.5 |
| c.10310A>G | p.D3437G | rs118053060 | 2/3 | 2.5 |
Indicates the number of siblings carrying the allele out of the total number of siblings from this family tested.
Frequency in Genome of the Netherlands: genome sequences of 998 independent Dutch individuals. 22 Accession numbers for the transcripts and protein sequences used to describe the variants: ZAN: NM_003386.2, NP_003377.2; SLC26A10: NM_133489.2, NP_597996.2; CCHCR1: NM_001105564.1, NP_001099034.1; PLA2G4C, NM_003706.2, NP_003697.2; PKHD1L1: NM_177531.4, NP_803875.2.
3.4. Known and suspected moderate and high‐risk genes
We next examined 35 genes in which PTVs have been demonstrated or suspected to be associated with breast cancer risk (Tables 3 and S1). We found two rare missense variants in known high‐risk genes, one in PALB2 and one in BRCA2. ClinVar lists the variant in PALB2 as benign, the one in BRCA2 as variant of uncertain significance (VUS). Family RF17 was included in our study as being non‐BRCA1/2 because the sister not carrying the missense variant was the one tested in the clinical setting. No studies on the functional effects of this variant have been published to date, but the CADD score of 35 indicates that it might affect protein function. Therefore, it is possible that this family harbors a pathogenic BRCA2 variant.
TABLE 3.
Rare genetic variant in known and suspected breast cancer genes
| Gene | Family | Variant (coding DNA) | Variant (protein) | Rs‐number | Co‐segregation a | Frequency b (%) |
|---|---|---|---|---|---|---|
| ATM | RF6 | c.146C>G | p.S49C | rs1800054 | 2/3 | 1.7 |
| ATM | RF7 | c.2531G>A | p.G844E | rs587781808 | 2/3 | 0.002 |
| ATM | RF10 | c.2991A>G | p.(=) | rs1203368496 | 3/3 | 0 |
| ATM | RF18 | c.584C>T | p.T195I | rs1196611507 | 2/3 | — |
| ATM | RF20 | c.146C>G | p.S49C | rs1800054 | 3/3 | 1.7 |
| BRCA2 | RF17 | c.8290G>A | p.A2764T | rs786202189 | 2/3 | — |
| CDH1 | RF21 | c.1689C>T | p.(=) | rs587780786 | 2/2 | 0.007 |
| CHEK2 | RF4 | c.1100delC | p.T367fs | rs555607708 | 3/3 | 1 |
| CHEK2 | RF8 | c.1100delC | p.T367fs | rs555607708 | 3/3 | 1 |
| CHEK2 | RF14 | c.556A>C | p.N186H | rs146198085 | 1/3 | 0.01 |
| PALB2 | RF20 | c.150A>T | p.K50N | — | 1/2 | – |
| RAD51C | RF8 | c.790G>A | p.G264S | rs147241704 | 3/3 | 0.3 |
| RAD51C | RF19 | c.790G>A | p.G264S | rs147241704 | 1/2 | 0.3 |
Indicates the number of siblings carrying the allele out of the total number of siblings from this family tested.
Highest frequency in either ESP, ExAc, gnomAD, or GoNL; — if no entry listed; Accession numbers for the transcripts and protein sequences used to describe the variants: ATM: NM_000051.3, NP_000042.3; BRCA1: NM_007294.3, NP_009225.1; BRCA2: NM_000059.3, NP_000050.2; CDH1: NM_004360.3, NP_004351.1; CHEK2: NM_007194.3, NP_009125.1; PALB2: NM_024675.3, NP_078951.2; RAD51C: NM_058216.2, NP_478123.1.
The c.1100delC pathogenic variant in CHEK2, associated with an odds ratio (OR) of approximately 2.3, 32 was found in all affected individuals of families RF4 and RF8, with all individuals being heterozygous. We found several missense variants in the (suspected) moderate‐risk genes ATM, CHEK2 and RAD51C. The effect of missense changes in ATM and CHEK2 on breast cancer risk is, besides a few specific examples, largely uncertain.33, 34, 35 None of the variants listed in Table 3 belong to any of these exceptions, but some do have CADD scores >20 suggestive of pathogenicity. Two other variants have previously been associated with breast cancer risk, although association data have been conflicting. ATM c.146C>G (p.S49C) was detected in families RF6 and RF20; its associated breast cancer risk is unlikely to be larger than 1.5.34, 36, 37 Likewise, conflicting results were obtained for the breast and/or ovarian cancer risk of RAD51C c.790G>A (p.G264S) in families RF8 and RF19. 38 The contribution of these variants to breast cancer susceptibility, if any, is therefore uncertain.
3.5. Polygenic risk score analysis
Over 160 independent common SNPs have been found to be convincingly associated with breast cancer and can be combined into a PRS 23 (Table S2). To examine the effect of the PRS on the breast cancer cases in our families, we genotyped or imputed these SNPs for all individuals from whom DNA was available. The PRS was normalized such that the mean and SD of the population controls were 0 and 1, respectively. Figure 2 shows the difference in distribution between our familial cases and a set of population cases and controls, clearly showing a strong skewing toward PRS >0 for the familial cases. The odds ratio per unit SD of the PRS was 1.46. The average PRS of all the affected siblings in the families was 0.63, corresponding to an odds ratio (OR) of 1.27. The average score of the family probands (0.81, OR 1.36) was significantly higher than that in both population cases (0.35, OR 1.14, P = .026) and controls (P = .0004).
FIGURE 2.
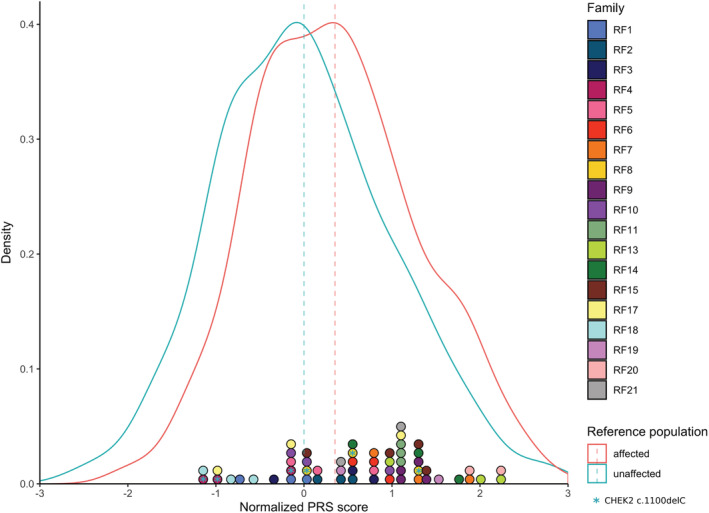
PRS scores for recessive families compared to population cases and controls. The blue and red line represent the density plots of PRS for population controls and cases, respectively. Colored circles at the ordinate each represent one individual from the 19 investigated families, circles with the same color belong to the same family. Circles with a blue star represent carriers of the CHEK2 c.1100delC variant. The dotted lines represent the mean PRS for the population controls and familial cases [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
In our study, we assessed whether breast cancer in families with at least three affected siblings, can be explained by a susceptibility gene with a recessive mode of inheritance. After a haplotype‐guided exome analysis, we identified no homozygous or compound‐heterozygous variants that were likely to explain the clustering of breast cancers in the selected families. We did identify two families in which all affected individuals carry the known moderate risk variant CHEK2*1100delC. Furthermore, we showed that on average, the affected women in these families had significantly higher PRS than both sporadic cases and population controls. Together, these results indicate that our selection criteria enrich for these factors and suggest that, rather than being caused by a single highly penetrant variant, increased breast cancer risk in some of these families may be due to the combined effect of multiple rare and common genetic variants with varying effect‐sizes, and perhaps other nongenetic risk factors as well.
Due to a few limitations of our study, we cannot completely rule out that some of our families are nonetheless explained by recessive risk alleles. First, some of the variants we identified (eg, PDIA2 p.R148*) are so rare in the general population that they would require very large case‐control populations to assess their association with breast cancer. As they grow in size, publicly available reference datasets and databases in which variants in potential disease‐associated genes can be reported are becoming very valuable for this purpose. Second, a recessive risk allele might be located outside the protein‐coding regions of the genome and thus not be captured by an exome sequencing approach. Moreover, structural variation, affecting more than a few base pairs, is mostly undetectable with the methods used in our study. Whole‐genome sequencing would identify these, but their mostly poor genomic annotation will make their filtering for follow‐up analyses very hard.
Third, our family selection has led to many sibships that could also be explained by a dominant allele with incomplete penetrance. While our study design had advantages for the variant filtering, there are alternative ways to enrich for recessive alleles, such as population‐based sib pairs or early‐onset cases with unaffected parents. Such studies have not yet been published for breast cancer but would probably also suffer from severe genetic model heterogeneity. Thus, the existence of recessive breast cancer alleles remains possible, although it is remarkable in this regard, that only a handful of the >160 common breast cancer loci derived from population‐based genome‐wide association studies affect risk in a recessive mode, rather than in a co‐dominant way. 25
Nonetheless, our results are in agreement with previous exome sequencing studies in non‐BRCA1/2 familial breast cancer cases. Although more than 20 such studies have been published, only two new breast cancer genes suggested by these studies were replicated independently: FANCM and RECQL.39, 40, 41, 42 Most of these studies, however, reported pathogenic variants in known moderate‐risk genes. Studies employing gene panel sequencing in a large numbers of familial breast cancer cases suggest that approximately 4% carry a pathogenic or likely pathogenic variant in a breast cancer gene other than BRCA1 or BRCA2.43, 44, 45 We found two index cases carrying the CHEK2*1100delC pathogenic variant (consistent with high frequency of this variant in the Dutch population), and four possibly pathogenic variants in other susceptibility genes. At least for CHEK2*1100delC it has been shown that the risk associated with this pathogenic variant and the risk associated with a PRS combine multiplicatively. 46 With regard to the common low‐risk variants, our results are consistent with studies which have found that non‐BRCA1/2 familial breast cancer cases have a higher PRS than both cases from the general population and cases who carry a BRCA1 or BRCA2 pathogenic variant.47, 48, 49, 50 Whether the prevalence of rare missense variants in the known breast cancer genes we observed in our families is causally linked to breast cancer, will need very large case‐control studies to substantiate further.
The enrichment of moderate and low‐risk alleles among the cases of at least part of the families in our study adds to a growing body of evidence on the importance of this type of risk alleles in causing familial breast cancer. Multigene panel sequencing has rendered the detection of rare variation in known risk genes standard clinical genetic practice, but the genotyping of the many common low‐risk alleles is not yet routinely performed in this setting. Nonetheless, the risks associated with the PRS and the likely multiplicative way in which it combines with those of pathogenic variants in moderate‐risk genes argue for a more comprehensive approach to genetic testing and counseling. This calls for the development of integrative risk prediction models, including the effect of mammographic density, lifestyle and environmental risk factors.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
ETHICS STATEMENT
All individuals provided informed consent and approval of the medical ethical committee at the LUMC was obtained (protocols P49/99 and P09.203).
Supporting information
Data S1 Supporting Information.
ACKNOWLEDGEMENTS
This work was supported by the Dutch Cancer Society [KWF UL 2009‐4388], CR‐UK, Genome Canada and NIH/NCI. Our study makes use of data generated by the Genome of the Netherlands Project. A full list of the investigators is available from www.nlgenome.nl. Funding for the project was provided by the Netherlands Organization for Scientific Research under award number 184021007, dated July 9, 2009, and made available as a Rainbow Project of the Biobank and Biomolecular Research Infrastructure Netherlands (BBMRI‐NL). The sequencing was carried out in collaboration with the Beijing Institute for Genomics (BGI).
Hilbers FS, van ‘t Hof PJ, Meijers CM, et al. Clustering of known low and moderate risk alleles rather than a novel recessive high‐risk gene in non‐BRCA1/2 sib trios affected with breast cancer. Int. J. Cancer. 2020;147:2708–2716. 10.1002/ijc.33039
Funding information Cancer Research UK; KWF Kankerbestrijding, Grant/Award Number: KWF UL 2009‐4388; Genome Canada
DATA ACCESSIBILITY
The data that support the findings of our study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Collaborative Group on Hormonal Factors in Breast Cancer . Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet. 2001;358:1389‐1399. [DOI] [PubMed] [Google Scholar]
- 2. Easton DF, Pharoah PDP, Antoniou AC, et al. Gene‐panel sequencing and the prediction of breast‐cancer risk. N Engl J Med. 2015;372:2243‐2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mavaddat N, Michailidou K, Dennis J, et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet. 2019;104:21‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Antoniou AC, Pharoah PD, McMullan G, Day NE, Ponder BA, Easton D. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population‐based study. Genet Epidemiol. 2001;21:1‐18. [DOI] [PubMed] [Google Scholar]
- 5. Antoniou AC, Pharoah PDP, McMullan G, et al. A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br J Cancer. 2002;86:76‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cui J, Antoniou AC, Dite GS, et al. After BRCA1 and BRCA2‐what next? Multifactorial segregation analyses of three‐generation, population‐based Australian families affected by female breast cancer. Am J Hum Genet. 2001;68:420‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaufman DJ, Beaty TH, Struewing JP. Segregation analysis of 231 Ashkenazi Jewish families for evidence of additional breast cancer susceptibility genes. Cancer Epidemiol Biomarkers Prev. 2003;12:1045‐1052. [PubMed] [Google Scholar]
- 8. Pharoah PD, Day NE, Duffy S, Easton DF, Ponder BA. Family history and the risk of breast cancer: a systematic review and meta‐analysis. Int J Cancer. 1997;71:800‐809. [DOI] [PubMed] [Google Scholar]
- 9. Liede A, Malik IA, Aziz Z, de los Rios PP, Kwan E, Narod SA. Contribution of BRCA1 and BRCA2 mutations to breast and ovarian cancer in Pakistan. Am J Hum Genet. 2002;71:595‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Assié G, LaFramboise T, Platzer P, Eng C. Frequency of germline genomic homozygosity associated with cancer cases. JAMA. 2008;299:1437‐1445. [DOI] [PubMed] [Google Scholar]
- 11. Enciso‐Mora V, Hosking FJ, Houlston RS. Risk of breast and prostate cancer is not associated with increased homozygosity in outbred populations. Eur J Hum Genet. 2010;18:909‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oldenburg RA, Kroeze‐Jansema KHG, Houwing‐Duistermaat JJ, et al. Genome‐wide linkage scan in Dutch hereditary non‐BRCA1/2 breast cancer families identifies 9q21‐22 as a putative breast cancer susceptibility locus. Genes Chromosomes Cancer. 2008;47:947‐956. [DOI] [PubMed] [Google Scholar]
- 13. Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97‐101. [DOI] [PubMed] [Google Scholar]
- 14. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.SeattleSeq Variation Annotation. http://snp.gs.washington.edu/SeattleSeqAnnotation138/index.jsp. Accessed May 1, 2015.
- 16.Exome Variant Server. http://evs.gs.washington.edu/EVS/. Accessed June 1, 2015.
- 17. International HapMap Consortium . A haplotype map of the human genome. Nature. 2005;437:1299‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. 1000 Genomes Project Consortium , Abecasis GR, Altshuler D, et al. A map of human genome variation from population‐scale sequencing. Nature. 2010;467:1061‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Genome of the Netherlands Consortium . Whole‐genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet. 2014;46:818‐825. [DOI] [PubMed] [Google Scholar]
- 20. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Michailidou K, Lindström S, Dennis J, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet. 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Michailidou K, Hall P, Gonzalez‐Neira A, et al. Large‐scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353‐361. 361e1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Desilva MG, Lu J, Donadel G, et al. Characterization and chromosomal localization of a new protein disulfide isomerase, PDIp, highly expressed in human pancreas. DNA Cell Biol. 1996;15:9‐16. [DOI] [PubMed] [Google Scholar]
- 27. Fu X‐M, Zhu BT. Human pancreas‐specific protein disulfide‐isomerase (PDIp) can function as a chaperone independently of its enzymatic activity by forming stable complexes with denatured substrate proteins. Biochem J. 2010;429:157‐169. [DOI] [PubMed] [Google Scholar]
- 28. Klappa P, Stromer T, Zimmermann R, Ruddock LW, Freedman RB. A pancreas‐specific glycosylated protein disulphide‐isomerase binds to misfolded proteins and peptides with an interaction inhibited by oestrogens. Eur J Biochem. 1998;254:63‐69. [DOI] [PubMed] [Google Scholar]
- 29. Fu X‐M, Zhu BT. Human pancreas‐specific protein disulfide isomerase homolog (PDIp) is an intracellular estrogen‐binding protein that modulates estrogen levels and actions in target cells. J Steroid Biochem Mol Biol. 2009;115:20‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alper SL, Sharma AK. The SLC26 gene family of anion transporters and channels. Mol Aspects Med. 2013;34:494‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17:1723‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. CHEK2 Breast Cancer Case‐Control Consortium . CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet. 2004;74:1175‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kleiblova P, Stolarova L, Krizova K, et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int J Cancer. 2019;145:1782‐1797. [DOI] [PubMed] [Google Scholar]
- 34. Fletcher O, Johnson N, dos Santos SI, et al. Missense variants in ATM in 26,101 breast cancer cases and 29,842 controls. Cancer Epidemiol Biomarkers Prev. 2010;19:2143‐2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Foo TK, Tischkowitz M, Simhadri S, et al. Compromised BRCA1‐PALB2 interaction is associated with breast cancer risk. Oncogene. 2017;36:4161‐4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buchholz TA, Weil MM, Ashorn CL, et al. A Ser49Cys variant in the ataxia telangiectasia, mutated, gene that is more common in patients with breast carcinoma compared with population controls. Cancer. 2004;100:1345‐1351. [DOI] [PubMed] [Google Scholar]
- 37. Stredrick DL, Garcia‐Closas M, Pineda MA, et al. The ATM missense mutation p.Ser49Cys (c.146C>G) and the risk of breast cancer. Hum Mutat. 2006;27:538‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sopik V, Akbari MR, Narod SA. Genetic testing for RAD51C mutations: in the clinic and community. Clin Genet. 2015;88:303‐312. [DOI] [PubMed] [Google Scholar]
- 39. Kiiski JI, Pelttari LM, Khan S, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple‐negative breast cancer. Proc Natl Acad Sci U S A. 2014;111:15172‐15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peterlongo P, Catucci I, Colombo M, et al. FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum Mol Genet. 2015;24:5345‐5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun J, Wang Y, Xia Y, et al. Mutations in RECQL gene are associated with predisposition to breast cancer. PLoS Genet. 2015;11:e1005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cybulski C, Carrot‐Zhang J, Kluźniak W, et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat Genet. 2015;47:643‐646. [DOI] [PubMed] [Google Scholar]
- 43. Tung N, Battelli C, Allen B, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next‐generation sequencing with a 25‐gene panel. Cancer. 2015;121:25‐33. [DOI] [PubMed] [Google Scholar]
- 44. Thompson ER, Rowley SM, Li N, et al. Panel testing for familial breast cancer: calibrating the tension between research and clinical care. J Clin Oncol. 2016;34:1455‐1459. [DOI] [PubMed] [Google Scholar]
- 45. Susswein LR, Marshall ML, Nusbaum R, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next‐generation cancer panel testing. Genet Med. 2016;18:823‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Muranen TA, Greco D, Blomqvist C, et al. Genetic modifiers of CHEK2*1100delC‐associated breast cancer risk. Genet Med. 2016;19:599‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mavaddat N, Pharoah PDP, Michailidou K, et al. Prediction of breast cancer risk based on profiling with common genetic variants. J Natl Cancer Inst. 2015;107:djv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muranen TA, Mavaddat N, Khan S, et al. Polygenic risk score is associated with increased disease risk in 52 Finnish breast cancer families. Breast Cancer Res Treat. 2016;158:463‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sawyer S, Mitchell G, McKinley J, et al. A role for common genomic variants in the assessment of familial breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30:4330‐4336. [DOI] [PubMed] [Google Scholar]
- 50. Lakeman IMM, Hilbers FS, Rodriguez‐Girondo M, et al. Addition of a 161‐SNP polygenic risk score to family history‐based risk prediction: impact on clinical management in non‐BRCA1/2 breast cancer families. J Med Genet. 2019;56:581‐589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting Information.
Data Availability Statement
The data that support the findings of our study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.