

Abstract
Objective
The objective of this study was to assess the efficacy and safety of nabilone, a synthetic tetrahydrocannabinol analogue, as a treatment for non‐motor symptoms (NMS) in Parkinson's disease (PD).
Methods
This was a phase II placebo‐controlled, double‐blind, parallel‐group, enriched enrollment randomized withdrawal trial conducted at the Medical University Innsbruck. A random sample of 47 patients with PD with stable motor disease and disturbing NMS defined by a score of ≥4 points on the Movement Disorder Society ‐ Unified PD Rating Scale‐I (MDS‐UPDRS‐I) underwent open‐label nabilone titration (0.25 mg once daily to 1 mg twice daily, phase I). Responders were randomized 1:1 to continue with nabilone or switch to placebo for 4 weeks (phase II). The primary efficacy criterion was the change of the MDS‐UPDRS‐I between randomization and week 4. Safety was analyzed in all patients who received at least one nabilone dose.
Results
Between October 2017 and July 2019, 19 patients received either nabilone (median dose = 0.75 mg) or placebo. At week 4, mean change of the MDS‐UPDRS‐I was 2.63 (95% confidence interval [CI] 1.53 to 3.74, p = 0.002, effect size = 1.15) in the placebo versus 1.00 (95% CI −0.16 to 2.16, p = 0.280, effect size = 0.42) in the nabilone‐group (difference: 1.63, 95% CI 0.09 to 3.18, p = 0.030, effect size = 0.66). Seventy‐seven percent of patients had adverse events (AEs) during open‐label titration, most of them were transient. In the double‐blind phase, similar proportions of patients in each group had AEs (42% in the placebo group and 32% in the nabilone group). There were no serious AEs.
Interpretation
Our results highlight the potential efficacy of nabilone for patients with PD with disturbing NMS, which appears to be driven by positive effects on anxious mood and night‐time sleep problems.
Trial registry: ClinicalTrials.gov (NCT03769896) and EudraCT (2017‐000192‐86). ANN NEUROL 2020;88:712–722
Non‐motor symptoms (NMS) in Parkinson's disease (PD) 1 include autonomic nervous system dysfunction (orthostatic hypotension and obstipation), olfactory loss, disorders of mood and cognition, as well as sleep problems, such as insomnia, daytime sleepiness, or rapid‐eye‐movement (REM) sleep behavior disorder. Many of these may antedate the typical motor symptoms of PD by years or even decades, 1 , 2 but the burden of NMS generally increases during the disease course. NMS are a major determinant of quality of life, progressive disability, and dependence in patients with PD 1 but there is a paucity of controlled clinical trial data concerning their treatment. 3 Available treatment options are limited and outcomes often unsatisfactory. The potential therapeutic effect of cannabinoids on motor and NMS in PD is a prominent topic and commonly raised by patients in the consulting room, 4 but there is limited evidence supporting their use in PD because available trials were either small‐sized 5 , 6 , 7 or uncontrolled. 7 , 8 , 9 , 10 , 11 , 12 , 13 The endocannabinoid system (ECS) plays a significant role in many physiological body functions, 7 , 14 although the exact details of the neural circuitry through which it modulates these functions remain uncertain. In this study, we aimed to investigate the effect of nabilone for the treatment of NMS in PD in a controlled fashion. Nabilone is a synthetic analogue of tetrahydrocannabinol, the psychoactive component of cannabis, with similar pharmacological properties. It acts as a partial agonist on both cannabinoid 1 and cannabinoid 2 receptors in humans, thus mimicking the effects of tetrahydrocannabinol with the advantage of more predictable side effects and less euphoria. 15 , 16 Given the data and possible modes of action of the ECS, we hypothesized that nabilone may improve NMS in patients with PD and have a favorable safety profile.
The outcome of this trial may contribute to a better understanding of the value of cannabinoids for treating NMS in patients with PD.
Methods
This single‐center, phase II, placebo‐controlled, double‐blind, parallel‐group study used an enriched enrollment randomized withdrawal (EERW) design to assess the efficacy and safety of nabilone for NMS in patients with PD. The PD diagnosis was based on standard criteria and NMS severity was assessed by the non‐motor section (part I) of the Movement Disorders Society ‐ Unified Parkinson's Disease Rating Scale (MDS‐UPDRS). For eligibility, male and female patients with PD had to be older than 30 years of age and have a score of ≥ 4 points on the MDS‐UPDRS‐I with ≥ 2 points in the item for anxiety (1.4) or pain (1.9). Stable motor disease with steady medication for > 30 days prior to screening was required. All participants had to abstain from recreational use of cannabinoids. Exclusion criteria included evidence of secondary or atypical parkinsonism, a Hoehn and Yahr stage > 3, disturbing motor fluctuations or dyskinesia (ie, a score ≥ 2 on one of the items of the MDS‐UPDRS part IV), neurosurgical intervention for PD, and evidence of disturbing impulse control disorders as defined by cut‐off values of the Questionnaire for Impulsive‐Compulsive Disorders in PD‐Rating Scale (QUIP‐RS, Gambling: 3, Sex: 5, Buying: 5, Eating: 4, Punding: 3, Hobbyism: 4, and PD Medication Use: 3 points 17 ). Patients with PD with symptomatic orthostatic hypotension, sinus tachycardia, and major psychiatric disorders were not allowed to participate in this study as they are more vulnerable to possible hazardous adverse reactions that may occur during nabilone intake. Patients with at least moderately impaired liver function and/or chronic alcohol or drug abuse were excluded (see related publication for full list of inclusion and exclusion criteria 18 ). The study was approved by the local ethics committee and the Austrian national regulatory authorities. All individuals gave written informed consent before participation. No participant received a stipend. All procedures were performed in accordance with the 1964 Declaration of Helsinki and its later amendments. Details of the study design have been previously published. 18
Procedures, Randomization, and Masking
Phase I of the trial was open‐label and nabilone was given orally daily starting with a dose of 0.25 milligrams (mg, 1 capsule) in the evening after the baseline visit and concluded with a twice daily dosing. Nabilone was titrated in 0.25 mg‐increments every 1‐to 4‐days after consultation with the study team during regular telephone calls. Dose adjustments were performed until patients met the responder criterion defined as a patient‐based rating of their NMS as “much improved” or “very much improved” on the 7‐point Clinical Global Impression of Improvement Scale (CGI‐I). Patients failing to meet this criterion at the maximum daily dose of 2 mg or patients with intolerable side effects related to the study drug were discontinued.
In phase II of the study, responders were randomly assigned (1:1) to either their optimal nabilone dose as established during phase I or placebo (corn starch) of matching color and shape and supplied in identical packaging. Randomization was performed with a computer‐generated schedule provided by the Department of Medical Statistics of the Medical University of Innsbruck. Respective medication boxes with either nabilone or placebo were labeled consecutively (1–48) according to the randomization list to ensure concealment. Neither a member of the study team nor the participants were informed about treatment assignment. 18 The 4‐week, double‐blind, withdrawal phase ended with a termination visit from which the amount of the drug was tapered. A safety follow‐up visit was performed after 2 weeks of discontinuation from the study drug (Fig 1). Assessments included the MDS‐UPDRS, NMS‐Scale (NMSS), Hospital Anxiety and Depression Scale (HADS), PD‐Questionnaire‐39 (PDQ‐39), Montreal Cognitive Assessment (MoCA), Epworth Sleepiness Scale (ESS), Fatigue Severity Scale (FSS), visual analogue scale (VAS) of pain, King's PD Pain Scale (KPPS), QUIP‐RS, and CGI‐I.
FIGURE 1.
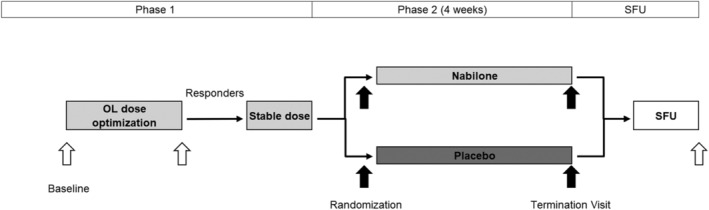
Schedule of trial activities. All patients received nabilone during phase I of the trial. The mean durations of phase I (including the open‐label titration phase and open‐label phase with stable nabilone dosage) and phase II (ie, double‐blind withdrawal phase) were 39.90 days ± 12.10 (median 37.00 days) and 28.37 days ± 3.23 (median 28.00 days), respectively. OL, open‐label; SFU, safety follow‐up.
Safety parameters were monitored throughout the study via telephone calls and at on‐site visits. Blood pressure was measured with the patient in supine position (after having been in this position for 10 minutes) and after 3 minutes in standing position after postural change. Blinded assessment of safety was performed during trial conduction via the safety data monitoring board (H.G.K., K.S., and M.P.).
Outcomes
The primary end point of the study was the change of the MDS‐UPDRS‐I score from randomization to the termination visit 4 weeks thereafter (0–52 points with higher scores indicating greater symptom severity). Secondary efficacy outcomes were changes from randomization to the termination visit in all other clinical scales and questionnaires. CGI‐I ratings were evaluated at week 4. As exploratory analyses, we also assessed treatment effects on the single items of the MDS‐UPDRS‐I and the NMSS domains, as well as changes of scales and questionnaires in the open‐label trial phase. Safety and tolerability were evaluated with reference to the number of subjects (%) who discontinued the study due to an adverse event (AE) or other reasons, AEs, clinical and laboratory measurements, urinalysis, electrocardiogram results, vital signs including orthostatic hypotension, Columbia‐Suicide Severity Rating Scale (C‐SSRS), and the hallucination, orthostatic hypotension, and day time sleepiness items of the MDS‐UPDRS‐I.
Statistical Analyses
We aimed to enroll 48 patients to account for dropouts and include 19 patients per treatment group in the randomized trial phase, which was considered sufficient to detect a treatment difference of 2.5 points in the primary end point (standard deviation 2.4) with 80% power and a 2‐sided α‐level of 0.05 (Mann–Whitney U test; nQuery Advisor version 7). 18 A descriptive analysis (χ2 test for categorical variables, Mann‐Whitney U test for continuous variables) of demographic and clinical data at baseline was performed using the full dataset of all patients that have ever taken nabilone in this trial (n = 47). Analyses of the efficacy end points included all randomized subjects with scoring of MDS‐UPDRS‐I at randomization and termination visit. Safety and tolerability summaries were based on the safety set, which includes patients receiving at least 1 dose of study medication during both trial phases. The changes of all outcome variables during phase I of the trial were assessed using a Wilcoxon matched‐pairs test for within‐group comparison. The primary, secondary, and exploratory end points of phase II were analyzed separately for the nabilone and placebo groups using a Wilcoxon matched‐pairs test for within‐group comparison (correction for multiple comparisons with a factor of 2) and a Mann–Whitney U test for between‐group comparisons. For all analyses, statistical significance was set at a 2‐sided 5% α‐level. Secondary and exploratory outcome analyses were not adjusted for multiple comparisons and used 5% as the nominal significance level. For CGI analysis, distributions of dichotomized ratings (deterioration vs no deterioration) in both groups at the termination visit were compared. Effect sizes for the different end points were calculated according to Cohen's D, 19 except for single MDS‐UPDRS‐I items and the CGI where rcontrast and φ coefficient 20 , 21 were used. Cohen's D of 0.2, 0.5, and 0.8 as well as rcontrast and φ coefficient of 0.1, 0.3, and 0.5 were considered “small,” “medium,” and “large” effect sizes. Spearman's rank‐order correlation (rs) was used to test for any relationship of nabilone dosage with number of AEs in the open‐label phase of the trial. To test for any dose–response relationship, we performed a linear regression analysis with change of MDS‐UPDRS‐I during the open‐label phase of the trial as dependent variable and nabilone dosage as well as MDS‐UPDRS‐I at baseline as independent predictors. As a sensitivity analysis, we fitted a repeated‐measures mixed model with MDS‐UPDRS‐I score as dependent variable and a factorial interaction between group assignment and time as an independent variable. We applied an unstructured within‐subject covariance matrix assuming that each timepoint and each pair of timepoints have their own variance and covariance, respectively. SPSS version 25.0 for Windows (SPSS, IBM Corporation, and other(s) 1989, 2017, Chicago, IL) was used to analyze data. This trial is registered with ClinicalTrials.gov (NCT03769896) and EudraCT (2017‐000192‐86).
Results
Between October 17, 2017, and July 15, 2019 (last patient last visit), 48 participants were screened. Demographic and clinical characteristics were balanced between treatment groups (Table 1). All patients were white. There was one screening failure due to the use of prohibited concomitant medication. During open‐label titration (phase I), 9 patients were either nonresponders as defined per protocol (n = 5, 10.42%) or discontinued (n = 4, 8.33%, 1 drop‐out, 3 due to AEs; Fig 2). Median daily dose of nabilone was 0.75 mg at randomization (range 0.25 to 1.75 mg; see Table 1). Table S1 summarizes the distribution of total daily doses at randomization. In phase I, both the MDS‐UPDRS‐I and NMSS decreased significantly in all patients (Table 2, Table S2). There was no relationship between nabilone dosage and MDS‐UPDRS‐I change during phase I (ß‐coefficient − 0.42, 95% confidence interval [CI] −3.47 to 2.64, p = 0.783).
TABLE 1.
Demographics and Results at Baseline
| Total (n = 47) | Placebo group (n = 19) | Nabilone group (n = 19) | |
|---|---|---|---|
| Age, yr | 65.05 ± 8.12 (66.83) | 63.95 ± 8.04 (65.92) | 65.38 ± 7.94 (66.83) |
| Women | 19 (40%) | 5 (26%) | 9 (47%) |
| Disease duration, yr | 7.86 ± 5.17 (7.00) | 7.39 ± 5.14 (5.75) | 7.83 ± 5.47 (7.25) |
| Daily nabilone dose, mg a at randomization | 0.86 ± 0.40 (0.75) | 0.80 ± 0.41 (0.75) | 0.91 ± 0.40 (1.00) |
| Charlson Comorbidity Index | 0.43 ± 0.77 (0.00) | 0.47 ± 0.84 (0.00) | 0.32 ± 0.75 (0.00) |
| Education, yr | 12.85 ± 2.71 (12.00) | 13.08 ± 3.19 (12.00) | 12.87 ± 2.78 (12.00) |
| H&Y scale |
1.89 ± 0.43 (2.00) (95% CI 1.77; 2.02) |
1.95 ± 0.41 (2.00) (95% CI 1.75; 2.14) |
1.84 ± 0.50 (2.00) (95% CI 1.60; 2.08) |
| MDS‐UPDRS‐I | 12.36 ± 4.92 (12.00) | 12.26 ± 5.85 (12.00) | 13.53 ± 4.39 (15.0) |
| MDS‐UPDRS‐II | 9.83 ± 5.12 (9.00) | 10.47 ± 4.50 (10.00) | 10.37 ± 6.24 (9.00) |
| MDS‐UPDRS‐III | 26.70 ± 11.22 (26.0) | 27.90 ± 9.98 (27.00) | 26.00 ± 13.25 (25.00) |
| MDS‐UPDRS‐IV | 1.68 ± 2.13 (0.00) | 1.42 ± 1.92 (0.00) | 2.16 ± 2.34 (2.00) |
| MDS‐UPDRS Total Score | 51.81 ± 18.88 (50.00) | 52.05 ± 14.75 (53.00) | 52.05 ± 22.97 (49.00) |
| MDS‐UPDRS Motor Score | 36.53 ± 14.79 (37.00) | 38.37 ± 11.93 (39.00) | 36.37 ± 18.64 (37.00) |
| MoCA | 27.94 ± 1.81 (28.0) | 27.95 ± 1.47 (28.00) | 28.11 ± 1.27 (28.00) |
| PDQ‐39 SI | 22.97 ± 15.41 (20.94) | 25.18 ± 16.38 (21.25) | 21.11 ± 11.69 (21.04) |
Data are given for all patients who have ever taken nabilone in this trial (full dataset). Data are presented as mean ± standard deviation (median) for continuous variables and number (percent) for categorical variables. For the H&Y scale, the 95% CI is also given. Higher Score values indicate worse outcome in all scales and questionnaires but in the MoCA.
Data on nabilone dose refers to the 38 randomized patients.
CI = confidence interval; H&Y = Hoehn and Yahr; MDS‐UPDRS = Movement Disorders Society ‐ Unified Parkinson's Disease Rating Scale; MoCA = Montreal Cognitive Assessment; PDQ‐39 = Parkinson's Disease Questionnaire‐39; SI = Summary Index.
FIGURE 2.
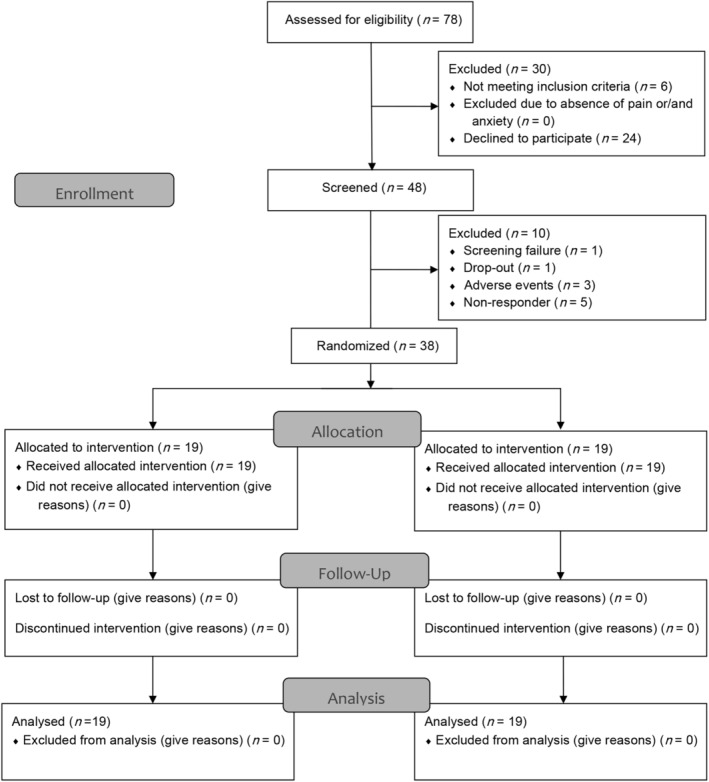
Flow chart (adapted from CONSORT 2010). n, number.
TABLE 2.
Change in End Point Scores During Open‐label Administration of Nabilone, Patients n = 38
| Baseline | Randomization | Mean change (95% CI) from BL to R | p value a | |
|---|---|---|---|---|
| MDS‐UPDRS‐I | 12.90 ± 5.14 | 9.11 ± 5.54 | −3.79 (−4.97; −2.61) | <0.001 |
| NMSS Total Score | 49.82 ± 31.03 | 39.79 ± 27.48 | −10.03 (−16.65; −3.40) | 0.002 |
| MDS‐UPDRS‐II | 10.42 ± 5.37 | 10.03 ± 5.09 | −0.40 (−1.42; 0.63) | 0.406 |
| MDS‐UPDRS‐III | 26.95 ± 11.61 | 24.71 ± 10.36 | −2.24 (−4.90; 0.42) | 0.058 |
| MDS‐UPDRS Motor Score | 37.37 ± 15.47 | 34.74 ± 13.95 | −2.63 (−5.72; 0.45) | 0.085 |
| ESS | 8.00 ± 3.95 | 8.47 ± 4.16 | 0.47 (−0.29; 1.24) | 0.308 |
| FSS | 34.08 ± 13.73 | 34.08 ± 11.05 | 0.00 (−3.22; 3.22) | 0.941 |
| HADS‐A | 5.39 ± 3.58 | 5.50 ± 3.53 | 0.26 (−0.48; 1.01) | 0.793 |
| HADS‐D | 5.05 ± 3.21 | 5.32 ± 3.47 | 0.11 (−0.70; 0.91) | 0.500 |
| MoCA | 28.03 ± 1.37 | 28.08 ± 2.11 | 0.05 (−0.48; 0.59) | 0.646 |
| PDQ‐39 SI | 23.14 ± 14.18 | 23.16 ± 14.02 | 0.02 (−2.42; 2.45) | 0.521 |
| KPPS Total Score | 21.24 ± 14.61 | 17.47 ± 13.65 | −3.76 (−7.33; −0.20) | 0.022 |
| QUIP‐RS Total Score | 0.71 ± 1.29 | 0.95 ± 2.04 | 0.24 (−0.38; 0.86) | 0.482 |
| VAS of pain, mm | 47.16 ± 21.92 | 35.05 ± 24.44 | −12.11 (−18.68; −5.53) | 0.001 |
Data of continuous variables are presented as mean ± standard deviation (end point scores at baseline and randomization) or mean (95% CI; change of end point scores). Higher Score values indicate worse outcome in all scales and questionnaires but in the MoCA.
Within‐group comparison. For all p values, significance level was set at p ≤ 0.05.
BL = baseline; CGI‐I = Clinical Global Impression – Improvement Scale; CI = confidence interval; ESS = Epworth Sleepiness Scale; FSS = Fatigue Severity Scale; HADS‐A/D = Hospital Anxiety and Depression Scale ‐ Anxiety/Depression; KPPS = King’s Parkinson's Disease Pain Scale; MDS‐UPDRS = Movement Disorder Society ‐ Unified Parkinson's Disease Rating Scale; MoCA = Montreal Cognitive Assessment; NMSS = Non‐Motor Symptoms Scale; PDQ‐39 = Parkinson's Disease Questionnaire‐39; QUIP‐RS = Questionnaire for Impulsive‐Compulsive Disorders in Parkinson's Disease ‐ Rating Scale; R = randomization; SI = Summary Index; VAS = Visual Analogue Scale.
Primary Outcome
Thirty‐eight patients entered phase II and were randomized to the nabilone or placebo arm (n = 19 each). No patient discontinued phase II and all patients were included in the final analysis (see Fig 2). Mean change of the MDS‐UPDRS‐I score during the randomized double‐blind phase was 2.63 points (95% CI 1.53 to 3.74, p = 0.002, effect size = 1.15) in the placebo versus 1.00 points (95% CI −0.16 to 2.16, p = 0.280, effect size = 0.42) in the nabilone group (difference between groups: 1.63, 95% CI 0.09 to 3.18, p = 0.030, effect size = 0.66; Fig 3A). The change of the MDS‐UPDRS‐I score was not significant in the nabilone group, which is reflected by the small effect size. The placebo group deteriorated significantly with a large effect size. There was a significant between‐group difference for the MDS‐UPDRS‐I with a medium effect size to the disadvantage of the placebo arm (Table S6). The sensitivity analyses (mixed model, as described above) showed a significant partial interaction between treatment and time (from randomization to the termination visit).
FIGURE 3.
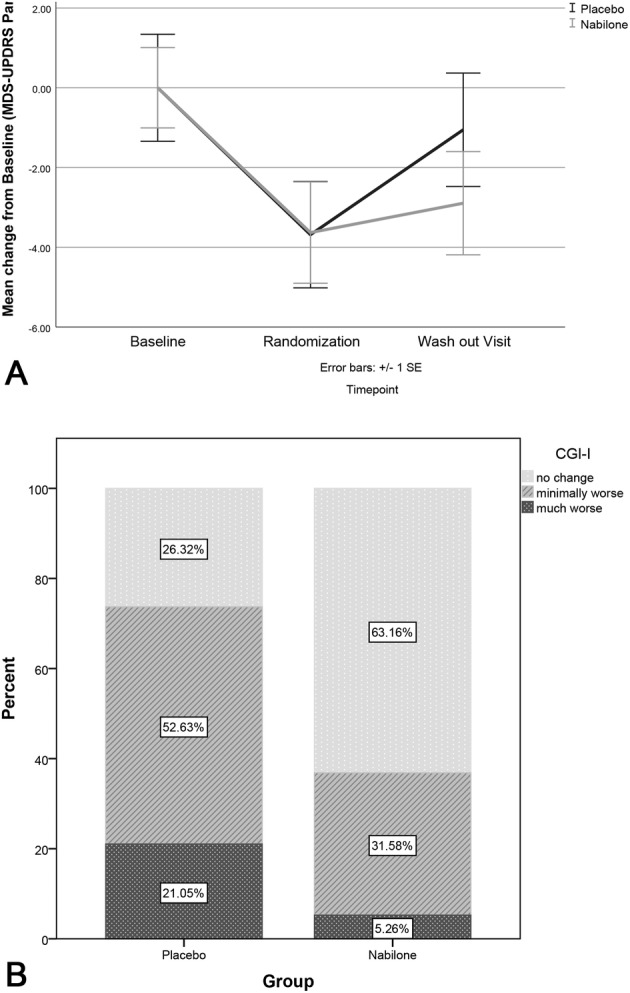
Data representing the change of MDS‐UPDRS‐I during the study and CGI‐I during double‐blind treatment. (A) Change of MDS‐UPDRS‐I during the study; (B) Change of CGI‐I during double‐blind treatment. CGI‐I, Clinical Global Impression – Improvement Scale; MDS‐UPDRS, Movement Disorder Society ‐ Unified Parkinson's Disease Rating Scale; SE, standard error.
Secondary and Exploratory Outcomes
Mean change of the NMSS at the termination visit was 11.00 points (95% CI 4.68 to 17.32, p = 0.004, effect size = 0.84) in the placebo versus 4.05 points (95% CI −0.65 to 8.75, p = 0.096, effect size = 0.42) in the nabilone group (difference between groups: 6.95, 95% CI −0.66 to 14.55, p = 0.147, effect size = 0.58). The change of the NMSS during the randomized double‐blind phase was not significant in the nabilone group. The placebo group, however, worsened significantly with a large effect size. Of note, the between‐group effect size of deterioration was also medium to the disadvantage of the placebo arm for the NMSS (see Table S6). In line with this, there was a significant deterioration with a medium effect size for the CGI‐I to the disadvantage of the placebo arm (between‐group difference: 0.53, 95% CI 0.09 to 0.96, p = 0.019). Fourteen patients in the placebo group (73.68%) rated themselves as worsened during the double‐blind period, compared with only 7 patients in the nabilone group (36.84%, p = 0.049; see Fig 3B), revealing a φ coefficient of 0.37 (medium effect size; see Table S6).
The MDS‐UPDRS‐III and the motor sum score worsened in the placebo arm, whereas the between‐group differences were not significant with medium effect size of 0.39 and 0.44 (see Table S6). None of the further secondary outcome measures showed significant within‐group or between‐group differences and no effect size exceeded 0.5. Although pain‐related end points (KPPS and VAS of pain) improved significantly during the open‐label trial phase (see Table 2), the change during the randomized phase was not significant (see Table S6). The exploratory analyses of the single MDS‐UPDRS‐I items revealed significant between‐group changes for items 1.4 (ie, anxious mood: 0.37, 95% CI −0.07 to 0.80, p = 0.044, rcontrast = 0.33, medium effect size) and 1.7 (ie, night‐time sleeping problems: 1.74, 95% CI 0.95 to 2.53, p < 0.001, rcontrast = 0.61, large effect size) to the advantage of the nabilone group. The deterioration of these 2 items in the placebo arm was significant only for item 1.7 (Table S7). In line with this, there was a worsening of the NMSS domain 2 (ie, Sleep/Fatigue) in the placebo group with a significant between‐group difference (7.53, 95% CI 1.86 to 13.19, p = 0.023, effect size = 0.81, large effect size; see Table S7). On the other hand, although there was a significant group‐effect for MDS‐UPDRS item 1.4 (ie, anxious mood), there was none for the NMSS domain 3 (ie, Mood/Apathy), most probably because this NMSS domain not only addresses anxiety but also includes items covering depressive mood and apathy.
Safety Analyses
During the open‐label phase of the trial, 1 patient declined further participation (2.08% of all 48 patients) and 3 patients (6.25% of all 48 patients) discontinued due to AEs (gonarthrosis leading to knee‐replacement surgery, migraine after intake of the first nabilone dose, and confusion). The latter 2 were of moderate severity. Only confusion was rated “possibly related” to the intake of study drug by the investigators. Common AEs (> 1 patient) are given in Table 3. There was no significant correlation between daily nabilone dose at randomization and number of AEs in the open‐label study phase (rs = −0.19, p = 0.264). During the open‐label phase, the most common treatment‐related AEs were fatigue, dizziness, dry mouth, and sleepiness. All but 2 treatment‐related AEs were transient during the open‐label titration phase, dry mouth in 2 patients persisted only upon completion of nabilone administration. During the randomized phase, the overall incidences of all‐cause AEs were similar between groups. There was only one possible treatment‐related AE of occurrence of a transient panic attack in the nabilone arm during the randomized controlled phase of the trial (see Table S4). MDS‐UPDRS items considered as safety parameters did not change during the randomized trial phase (see Table S7). There was, however, a medium effect size of 0.51 for the NMSS domain 1 (ie, Cardiovascular) to the disadvantage of the nabilone arm, although the difference was not significant. No severe AE (SAE), suspected unexpected serious adverse reaction, or suicidality (C‐SSRS) occurred in any patient during the study and follow‐up period. We did not record any clinically relevant changes in laboratory measures, blood pressure readings, or electrocardiogram recordings. There were neither significant within‐group nor between‐group differences in the postural changes of systolic and diastolic blood pressure readings after 3 minutes in the standing position (Table S3).
TABLE 3.
Safety Analysis
| Most common AEs (n > 1) during the open‐label phase | |||
|---|---|---|---|
| AE | total (n) | Severity of AE (n) | |
| Mild a | Moderate a | ||
| Fatigue | 17 | 15 (4/6/5/0) | 2 (1/0/1/0) |
| Dizziness | 9 | 8 (5/1/1/1) | 1 (0/1/0/0) |
| Daytime sleepiness | 5 | 4 (0/3/1/0) | 1 (0/1/0/0) |
| Upper respiratory tract infection | 5 | 4 (0/0/0/4) | 1 (0/0/0/1) |
| Dry mouth | 4 | 4 (0/3/1/0) | 0 |
| Confusion and disorientation | 3 | 1 (0/1/0/0) b | 2 (0/2/0/0) c |
| Gastroesophageal reflux | 2 | 2 (0/0/0/2) | 0 |
| Fall | 2 | 2 (0/0/0/2) | 0 |
| Headache | 2 | 1 (0/0/0/1) | 1 (0/0/0/1) d |
| Most common AEs (n > 1) during the double‐blind phase | |||
|---|---|---|---|
| AE | Total (n) | Nabilone (n) | Placebo (n) |
| Mild/Moderate | Mild/Moderate | ||
| Insomnia | 4 | 2/0 | 0/2 |
| Upper respiratory tract infection | 3 | 0/0 | 3/0 |
| Pain (including worsening) | 3 | 0/1 | 1/1 |
| Fall (including recurrent falls) | 2 | 1/0 | 1/0 |
| Syncope | 2 | 0/0 | 0/2 |
No severe AE or SAEs were reported during both phases of the trial. AEs during the double‐blind phase of the trial listed in the table were rated as unrelated to treatment.
Definitely related to treatment/ probably related/ possibly related/ not related.
Resolved after down‐titration from nabilone 0.25 mg b.i.d. to 0.25 mg q.d.
Confusion leading to study discontinuation of one patient (resolved after discontinuation), the other patient was a non‐responder suffering from confusion (resolved after discontinuation).
Migraine leading to study discontinuation of one patient.
AE = adverse event; SAE = serious adverse event.
Discussion
In this randomized placebo‐controlled, double‐blind, parallel‐group, EERW trial, we examined the efficacy and safety of the synthetic cannabinoid nabilone in patients with PD with troublesome NMS. The primary end point of the study, assessing differences in the change of NMS between the 2 treatment arms using part I of the MDS‐UPDRS, was met. NMS scores of the MDS‐UPDRS‐I deteriorated significantly less in the nabilone group compared with those switched to placebo with a medium effect size. Positive treatment effects of nabilone were also reflected in patient's self‐rating, as assessed with the CGI‐I. In line with this, there was also a deterioration of the NMSS with a medium effect to the disadvantage of the placebo compared with the nabilone arm, although the between‐group difference was not significant. Differences in the construct of the MDS‐UPDRS‐I and the NMSS 22 may explain that the between‐group difference was not significant for the NMSS, although it was for the MDS‐UPDRS‐I. Nevertheless, positive treatment effects of nabilone were reflected in medium effect sizes as assessed by both scales, most probably because a strong convergent validity between the MDS‐UPDRS‐I and NMSS has been reported. 22
Noteworthy, most patients with PD responded to a dose up to 1 mg of nabilone per day, indicating a benefit from even a small dose of cannabinoids. Observational studies assessing the use of non‐prescribed cannabis in patients with PD reported lower levels of disability and positive effects on mood, fatigue, sleep, and pain. 8 , 10 , 23 , 24 In line with this, we observed beneficial effects of nabilone on anxious mood and night‐time sleep problems in the double‐blind phase of the trial. On the other hand, postural dizziness was reported commonly as an AE and symptoms of postural dizziness worsened moderately on nabilone, as assessed with the NMSS, which is not surprising because orthostatic hypotension is a well‐known side effect of treatment with cannabinoids. 25 However, the assessment of active orthostatism was mostly unremarkable. Patients with PD with symptomatic orthostatic hypotension might possibly not be good candidates for the use of nabilone.
Overall, nabilone treatment was well tolerated. During the open‐label phase, the most common treatment‐related AEs were transient mild fatigue, postural dizziness, dry mouth, and somnolence, which is in line with information from the summary of product characteristics (SmPCs) and other controlled trials using nabilone. 5 , 6 In the randomized phase of the trial, no difference in AEs or tolerability issues was found between the 2 study arms. Most AEs were of mild severity and unrelated to the intake of the study drug. Two of the oldest patients with PD reported confusion and delusions during the titration phase of the trial. The first patient (aged 71.7 years) was maintained in the study after down‐titration to a single dose of nabilone 0.25 mg in the evening resolved the AE. The other patient (aged 74.5 years) was discontinued from further study participation and confusion resolved after 3 weeks. Some observational studies reported of confusion and hallucination after smoking cannabinoids in patients with PD. 8 Although extensive use of cannabis was reported to impair verbal and working memory as well as cognitive processing, 13 , 26 , 27 no worsening of cognitive function was observed subjectively or objectively (assessed with the MoCA) in our patients.
In this pilot trial, we also found a worsening of motor symptoms (MDS‐UPDRS‐III and the total motor score [parts II and III]) in the placebo group. Amelioration of tremor, bradykinesia, and rigidity in patients with PD has been described after smoking cannabis in a few small‐sized studies. 10 Trials in PD mainly focused on the effect of cannabinoids on levodopa‐induced dyskinesia (LID) given their interaction with the glutamatergic system. 28 , 29 As disturbing motor complications were an exclusion criterion in this study, we are not able to report about effects of nabilone on LID.
The ECS plays a significant part in motor control and the regulation of various non‐motor functions, including mood, attention and concentration, eating habits, sleep, and pain. 7 , 14 In animal models, a high amount of cannabinoid 1 receptors is found in presynaptic nerve terminals of gamma‐Aminobutyric acid‐ergic synapses and in cortical and limbic serotoninergic, noradrenergic, and dopaminergic neurons, as well as neurons with μ‐opioid receptors. Moreover, structures of the ECS co‐localize with nociceptive pathways in the spinal cord. Consequently, cannabinoids are believed to modulate monoaminergic, gamma‐Aminobutyric acid‐ergic, glutamatergic, and opioid signaling. 30 , 31 , 32 Data from animal and human positron emission tomography (PET) studies reveal a high density of cannabinoid receptors in the basal ganglia where the ECS is believed to function as a regulator of dopamine release and uptake. 33 , 34 , 35 , 36 Following the loss of dopamine, the ECS is overactive in the striatum of patients with PD with upregulation of its neurotransmitter and receptor levels possibly reflecting a compensational mechanism. 7 , 34 , 37 With respect to these findings, a positive effect of nabilone on NMS in patients with PD is not surprising. The few studies assessing the ECS and sleep showed that exogenous cannabinoids promote sleep, increase REM sleep, and the stability of non‐REM sleep. 38 In patients with PD, an influence of cholinergic neurons in brain areas involved in the regulation of sleep and sleep–wake cycle has been proposed as a possible underlying mechanism. 9 Besides symptomatic treatment, preclinical research revealing neuroprotective properties of cannabinoids gains interest in clinicians dealing with patients with movement disorders. 39 , 40 , 41
Our study has several strengths and limitations. All of our consecutively screened patients met the inclusion criteria of presence of significant anxiety and pain, indicating that these are common problems in patients with PD of a tertiary care center and that our criteria captured a representative sample. The study's withdrawal design is inevitably associated with a negative expectation related to receiving placebo. Indeed, the nonsignificant deterioration of single NMS with small effect sizes, as measured with the MDS‐UPDRS part I and the NMSS in the nabilone group, might be impacted by negative expectations related to receiving placebo (ie, “lessebo effect” 42 ). Selection of open‐label responders can raise concerns about generalizability of the results and thus affect external validity. In this study, most patients enrolled in the open‐label phase were responders, therefore, selection bias can be considered small. Inclusion of responders only can lead to overestimation of the effect of a novel treatment. However, restriction to responders reflects clinical practice by limiting long‐term treatment to patients who might benefit from it, in line with a personalized medicine approach. The enrichment design has been suggested to be sensitive and efficient for proof‐of‐concept studies of new treatment strategies in humans. 43 With our trial design, total exposure to placebo is reduced compared with a standard randomized controlled trial. The open‐label phase grants the assessment of a dose–response effect and provides a range of doses to be considered when planning a confirmatory study. Heterogeneity of response during the open‐label phase of the trial reflects individual treatment response, as seen in daily clinical routine.
Conclusions
To the best of our knowledge, this is the first study to evaluate the efficacy and safety of cannabinoids for the treatment of NMS in patients with PD, making this a unique pilot trial. Our findings show an improvement of overall NMS burden with nabilone, especially reflected by amelioration of anxiety and sleeping problems. The treatment was well tolerated. This study adds to the limited evidence of safety and efficacy of cannabinoid‐based treatment in patients with PD with troublesome NMS. Further and larger controlled trials assessing the effects of cannabinoids on PD symptoms are clearly needed.
Authors Contributions
M.P., K.S., R.S., W.P., and K.K. contributed to the conception and design of the study. M.P., K.S., A.D., M.W., F.C., P.E., B.H., K.M., H.S., G.K.W., D.V., H.G.K., F.K., G.G., and H.U. contributed to the acquisition and analysis of data. M.P., K.S., F.S., and W.P. contributed to drafting the text and preparing the figures.
Members of the Parkinson's Disease Working Group Innsbruck are listed in a Supplementary Table.
Potential Conflicts of Interest
M.P. received travel compensation for presentation of the study data after study end from AOP Orphan Pharmaceuticals AG, which manufactures the drug that is tested in this study. A.D. and B.H. received a travel grant from AOP Orphan Pharmaceuticals AG, which manufactures the study drug. K.K. is an employee at AOP Orphan Pharmaceuticals AG, which manufactured the study drug. K.S. reports personal fees from AOP Orphan Pharmaceuticals AG that manufactures the drug tested in this study. F.K., H.G.K., M.W., F.C., P.E., K.M., D.V., H.S., G.G., H.U., H.K., G.K.W., R.S., and W.P. have no conflict of interest to report.
Supporting information
Table S1. Dose and number of patients with the respective dose
Table S2. Change in endpoint subscores during open‐label administration of nabilone, patients n = 38
Table S3. Change in blood pressure readings during open‐label administration of nabilone and during double‐blind treatment
Table S4. Further Details of the Safety Analyses
Table S5. Members of the Parkinson's Disease Working Group Innsbruck
Table S6. Change in primary and secondary endpoint scores during double‐blind treatment
Table S7. Change in MDS‐UPDRS Subitems and NMSS domains during double‐blind treatment
Acknowledgments
The authors want to thank all participants for their voluntary consent to take part in this study. Additionally, we want to thank AOP Orphan Pharmaceuticals AG for providing the investigational medicinal product and placebo and for compensation of in‐person study visits and the independent monitoring conducted by the Clinical Trial Centre of the Medical University of Innsbruck.
References
- 1. Poewe W. Non‐motor symptoms in Parkinson's disease. Eur J Neurol 2008;15:14–20. [DOI] [PubMed] [Google Scholar]
- 2. Postuma RB, Iranzo A, Hogl B, et al. Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: a multicenter study. Ann Neurol 2015;77:830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seppi K, Ray Chaudhuri K, Coelho M, et al. Update on treatments for nonmotor symptoms of Parkinson's disease‐an evidence‐based medicine review. Mov Disord 2019;34:180–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bega D, Simuni T, Okun MS, et al. Medicinal cannabis for Parkinson's disease: practices, beliefs, and attitudes among providers at National Parkinson Foundation Centers of excellence. Mov Disord Clin Pract 2017;4:90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carroll CB, Bain PG, Teare L, et al. Cannabis for dyskinesia in Parkinson disease: a randomized double‐blind crossover study. Neurology 2004;63:1245–1250. [DOI] [PubMed] [Google Scholar]
- 6. Sieradzan KA, Fox SH, Hill M, et al. Cannabinoids reduce levodopa‐induced dyskinesia in Parkinson's disease: a pilot study. Neurology 2001;57:2108–2111. [DOI] [PubMed] [Google Scholar]
- 7. Kluger B, Triolo P, Jones W, Jankovic J. The therapeutic potential of cannabinoids for movement disorders. Mov Disord 2015;30:313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Balash Y, Bar‐Lev Schleider L, Korczyn AD, et al. Medical cannabis in Parkinson disease: real‐life patients' experience. Clin Neuropharmacol 2017;40:268–272. [DOI] [PubMed] [Google Scholar]
- 9. Chagas MH, Eckeli AL, Zuardi AW, et al. Cannabidiol can improve complex sleep‐related behaviours associated with rapid eye movement sleep behaviour disorder in Parkinson's disease patients: a case series. J Clin Pharm Ther 2014;39:564–566. [DOI] [PubMed] [Google Scholar]
- 10. Lotan I, Treves TA, Roditi Y, Djaldetti R. Cannabis (medical marijuana) treatment for motor and non‐motor symptoms of Parkinson disease: an open‐label observational study. Clin Neuropharmacol 2014;37:41–44. [DOI] [PubMed] [Google Scholar]
- 11. Venderova K, Ruzicka E, Vorisek V, Visnovsky P. Survey on cannabis use in Parkinson's disease: subjective improvement of motor symptoms. Mov Disord 2004;19:1102–1106. [DOI] [PubMed] [Google Scholar]
- 12. Zuardi AW, Crippa JA, Hallak JE, et al. Cannabidiol for the treatment of psychosis in Parkinson's disease. J Psychopharmacol 2009;23:979–983. [DOI] [PubMed] [Google Scholar]
- 13. Hill KP. Medical use of cannabis in 2019. JAMA 2019;322:974. [DOI] [PubMed] [Google Scholar]
- 14. Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron 2012;76:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Balter RE, Haney M. The synthetic analog of Δ9‐ tetrahydrocannabinol (THC): Nabilone. Pharmacology and clinical application In: Preedy VR, ed. Handbook of cannabis and related pathologies. London, San Diego: Cambridge, Kidlington Oxford (p2), Elsevier, 2017:821‐827. [Google Scholar]
- 16. Lemberger L, Rubin A, Wolen R, et al. Pharmacokinetics, metabolism and drug‐abuse potential of nabilone. Cancer Treat Rev. 1982;9:17–23. [DOI] [PubMed] [Google Scholar]
- 17. Probst CC, Winter LM, Moller B, et al. Validation of the questionnaire for impulsive‐compulsive disorders in Parkinson's disease (QUIP) and the QUIP‐rating scale in a German speaking sample. J Neurol 2014;261:936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peball M, Werkmann M, Ellmerer P, et al. Nabilone for non‐motor symptoms of Parkinson's disease: a randomized placebo‐controlled, double‐blind, parallel‐group, enriched enrolment randomized withdrawal study (the NMS‐nab study). J Neural Transm (Vienna) 2019;126:1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cohen J. The t test for means In: Cohen J, ed. Statistical power analysis for the behavioral sciences. New York, NY: Routledge, 1988:19‐74. [Google Scholar]
- 20. Rosnow RL. Effect sizes for experimenting psychologists. Can J Exp Psychol 2003;57:221–237. [DOI] [PubMed] [Google Scholar]
- 21. Fritz CO, Morris PE, Richler JJ. Effect size estimates: current use, calculations, and interpretation. J Exp Psychol Gen 2012;141:2–18. [DOI] [PubMed] [Google Scholar]
- 22. Martinez‐Martin P, Chaudhuri KR, Rojo‐Abuin JM, et al. Assessing the non‐motor symptoms of Parkinson's disease: MDS‐UPDRS and NMS scale. Eur J Neurol 2015;22:37–43. [DOI] [PubMed] [Google Scholar]
- 23. Kindred JH, Li K, Ketelhut NB, et al. Cannabis use in people with Parkinson's disease and multiple sclerosis: a web‐based investigation. Complement Ther Med 2017;33:99–104. [DOI] [PubMed] [Google Scholar]
- 24. Yust‐Katz S, Hershkovitz R, Gurevich T, Djaldetti R. Pain in extrapyramidal neurodegenerative diseases. Clin J Pain 2017;33:635–639. [DOI] [PubMed] [Google Scholar]
- 25. Whiting PF, Wolff RF, Deshpande S, et al. Cannabinoids for medical use: a systematic review and meta‐analysis. JAMA 2015;313:2456–2473. [DOI] [PubMed] [Google Scholar]
- 26. Schoeler T, Bhattacharyya S. The effect of cannabis use on memory function: an update. Subst Abuse Rehabil 2013;4:11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Volkow ND, Swanson JM, Evins AE, et al. Effects of cannabis use on human behavior, including cognition, motivation, and psychosis: a review. JAMA Psychiat 2016;73:292–297. [DOI] [PubMed] [Google Scholar]
- 28. Gubellini P, Picconi B, Bari M, et al. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci 2002;22:6900–6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol 2001;85:468–471. [DOI] [PubMed] [Google Scholar]
- 30. Chiou LC, Hu SS, Ho YC. Targeting the cannabinoid system for pain relief? Acta Anaesthesiol Taiwan 2013;51:161–170. [DOI] [PubMed] [Google Scholar]
- 31. Fitzgibbon M, Finn DP, Roche M. High times for painful blues: the endocannabinoid system in pain‐depression comorbidity. Int J Neuropsychopharmacol 2015;19:pyv095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang WJ, Chen WW, Zhang X. Endocannabinoid system: role in depression, reward and pain control (review). Mol Med Rep 2016. Oct;14:2899–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mursaleen LR, Stamford JA. Drugs of abuse and Parkinson's disease. Prog Neuropsychopharmacol Biol Psychiatry 2016;64:209–217. [DOI] [PubMed] [Google Scholar]
- 34. Brotchie JM. CB1 cannabinoid receptor signalling in Parkinson's disease. Curr Opin Pharmacol 2003;3:54–61. [DOI] [PubMed] [Google Scholar]
- 35. Melis M, Pistis M. Endocannabinoid signaling in midbrain dopamine neurons: more than physiology? Curr Neuropharmacol 2007;5:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Terry GE, Hirvonen J, Liow JS, et al. Imaging and quantitation of cannabinoid CB1 receptors in human and monkey brains using (18)F‐labeled inverse agonist radioligands. J Nucl Med 2010;51:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pisani A, Fezza F, Galati S, et al. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson's disease patients. Ann Neurol 2005;57:777–779. [DOI] [PubMed] [Google Scholar]
- 38. Pava MJ, Makriyannis A, Lovinger DM. Endocannabinoid Signaling regulates sleep stability. PLoS One 2016;11:e0152473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iannotti FA, Di Marzo V, Petrosino S. Endocannabinoids and endocannabinoid‐related mediators: targets, metabolism and role in neurological disorders. Prog Lipid Res 2016;62:107–128. [DOI] [PubMed] [Google Scholar]
- 40. Lastres‐Becker I, Molina‐Holgado F, Ramos JA, et al. Cannabinoids provide neuroprotection against 6‐hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson's disease. Neurobiol Dis 2005;19:96–107. [DOI] [PubMed] [Google Scholar]
- 41. Jin KL, Mao XO, Goldsmith PC, Greenberg DA. CB1 cannabinoid receptor induction in experimental stroke. Ann Neurol 2000;48:257–261. [PubMed] [Google Scholar]
- 42. Mestre TA, Shah P, Marras C, et al. Another face of placebo: the lessebo effect in Parkinson disease: meta‐analyses. Neurology 2014;82:1402–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hewitt DJ, Ho TW, Galer B, et al. Impact of responder definition on the enriched enrollment randomized withdrawal trial design for establishing proof of concept in neuropathic pain. Pain 2011;152:514–521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Dose and number of patients with the respective dose
Table S2. Change in endpoint subscores during open‐label administration of nabilone, patients n = 38
Table S3. Change in blood pressure readings during open‐label administration of nabilone and during double‐blind treatment
Table S4. Further Details of the Safety Analyses
Table S5. Members of the Parkinson's Disease Working Group Innsbruck
Table S6. Change in primary and secondary endpoint scores during double‐blind treatment
Table S7. Change in MDS‐UPDRS Subitems and NMSS domains during double‐blind treatment