

Abstract
[FeFe] hydrogenases are the most active H2 converting catalysts in nature, but their extreme oxygen sensitivity limits their use in technological applications. The [FeFe] hydrogenases from sulfate reducing bacteria can be purified in an O2‐stable state called Hinact. To date, the structure and mechanism of formation of Hinact remain unknown. Our 1.65 Å crystal structure of this state reveals a sulfur ligand bound to the open coordination site. Furthermore, in‐depth spectroscopic characterization by X‐ray absorption spectroscopy (XAS), nuclear resonance vibrational spectroscopy (NRVS), resonance Raman (RR) spectroscopy and infrared (IR) spectroscopy, together with hybrid quantum mechanical and molecular mechanical (QM/MM) calculations, provide detailed chemical insight into the Hinact state and its mechanism of formation. This may facilitate the design of O2‐stable hydrogenases and molecular catalysts.
Keywords: hydrogenase, protein structures, sulfide, vibrational spectroscopy, x-ray absorption spectroscopy
Caught in the Hinact: [FeFe] hydrogenases are highly efficient catalysts for the interconversion of H2 and H+. However, they are highly O2 sensitive. Here, the X‐ray crystal structure of an O2‐stable state of [FeFe] hydrogenase, along with comprehensive spectroscopic analysis and molecular calculations reveals the presence of a sulfur ligand bound to the active site. These results provide crucial insight into O2 stability mechanisms in proteins.
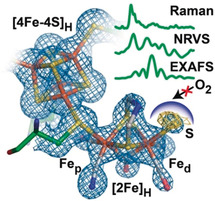
Introduction
Hydrogen is a promising green energy carrier for the future because it can easily be produced by water electrolysis using renewable energy and, later used in a fuel cell to generate energy producing only water as a byproduct.1 Currently, H2 is produced mostly from fossil fuels or to a small extent by water electrolysis using expensive noble metal catalysts. In nature, efficient and reversible H2 conversion is performed by a group of metalloenzymes called hydrogenases.2 These biocatalysts use earth abundant metals such as nickel and/or iron in their active site.3 Of the three groups of hydrogenases ([NiFe] hydrogenases, [FeFe] hydrogenases and [Fe] hydrogenases), the [FeFe] hydrogenases are the most active (100 000 s−1 in H2 oxidation and up to 10 000 s−1 in H+ reduction).4 However, these enzymes are extremely oxygen sensitive.5 Vigorous efforts have been made in order to protect [FeFe] hydrogenases, and hydrogenases in general, from oxygen.6 Although various oxygen inactivation mechanisms have been proposed,7 there is still a lack of understanding on how exactly O2 attacks their active site. Such insights may help in designing strategies to protect hydrogenases and molecular catalysts from O2 damage.
The active site of the [FeFe] hydrogenases, the H‐cluster, consists of a binuclear [2Fe] sub‐cluster ([2Fe]H) covalently attached by a cysteine sulfur to a [4Fe–4S] cluster ([4Fe–4S]H).8 [2Fe]H contains two irons bridged by the thiol groups of an aza‐propane 1,3‐dithiolate (ADT) ligand,9 a bridging CO ligand, with an additional CN− and CO ligated to each iron. The (proximal) iron (Fep) directly bound to the [4Fe–4S]H sub‐cluster is always coordinatively saturated, while the distal iron (Fed) possesses an open coordination site in most catalytic states, where substrates (H2 and H+) and inhibitors (including CO and O2) can bind. The nitrogen atom in the ADT bridge serves as a base and Fed acts as a Lewis acid, together forming a frustrated Lewis pair, which is essential to heterolytically split H2 at Fed.3 The catalytic cycle of these enzymes has been extensively studied through different spectroscopic techniques.10
When purified aerobically from the native organism, the [FeFe] hydrogenase from Desulfovibrio desulfuricans remains in an inactive oxygen‐stable state called Hinact (or Hox air), which can be reactivated upon reduction.4, 11 This state is thought to be “overoxidized” with an FeIIFeII configuration at the binuclear site and an additional ligand bound to Fed.11 The reduction of Hinact to an intermediate state Htrans is reversible while the further conversion of Htrans to Hox is thought to be irreversible, involving the release of the putative ligand from Fed.11 The nature of this putative ligand in the Hinact state has remained a mystery for more than two decades. Despite considerable spectroscopic analysis,12 new approaches are clearly needed to define the electronic and geometric configuration of the H‐cluster, and identify the nature of the exogenous ligand. Theoretical calculations have suggested that the extra ligand could be H2O or OH−.13 Interestingly, an [FeFe] hydrogenase from Clostridium beijerinckii has been shown to convert into the Hinact state in a highly reversible fashion, but the presence of an extra ligand bound in this state is so far unknown and its formation mechanism remains elusive.14
Recently, we showed that the Hinact state is formed upon oxidation of DdHydAB in the presence of sulfide (Na2S). Based on this result, we suggested that the extra ligand bound to the open coordination site might be a sulfur species, possibly SH−.15 However, we were unable to identify whether sulfide was directly bound to the H‐cluster, in what configuration, and whether there were any additional changes to the enzyme during Hinact formation. In this work, we identify the nature of the additional ligand as SH− through combined crystallographic and spectroscopic investigations. These results together with hybrid QM/MM calculations provide deeper understanding on the formation mechanism of this state and how it is protected against O2. This new insight may allow the general protection of metalloenzymes against oxygen, enabling their implementation in fuel cells and ultimately, it may provide design principles for developing O2‐stable bio‐inspired molecular catalysts.
Results and Discussion
Crystal Structure of DdHydAB in the Hinact State
DdHydAB in the Hinact state was crystallized under aerobic conditions at 12 °C. Brown crystals (indicating the presence of iron–sulfur clusters) were observed within three days and retained their dark color for at least two weeks. IR spectra of crystals taken from the same drop confirmed that the DdHydAB was in the Hinact state (Figure S1 in the Supporting Information). DdHydAB in the Hinact state crystallized in an orthorhombic space group P212121, and the asymmetric unit contains one biological assembly. In contrast, the previously reported structure was obtained from crystals with the space group P21212 and the asymmetric unit contained two biological assemblies. The structure of Hinact was solved using molecular replacement with the structure published by Nicolet et al.8b (PDB ID 1HFE) as a starting model, and was refined to a resolution of 1.65 Å (crystal parameters and refinement statistics in Table S1). The structure by Nicolet et al. is the only available structure of DdHydAB and the redox state of the enzyme in these crystals was not defined but assumed to be a mixture of the Hox and Hred states.
The overall architecture of DdHydAB in the Hinact state is essentially identical to the starting model with a root mean square deviation (RMSD) of 0.631 Å (calculated for all Cα atoms of residues 2–397 without outlier rejection, Figure 1 A). The electron density for the H‐cluster in the active site is well‐defined (Figure 2 A); however, the occupancy of the [2Fe] sub‐cluster had to be reduced to 0.6 to fit the experimental data. The low [2Fe] content indicates the presence of some apo protein in the preparation, partly a limitation of the artificial maturation procedure (see methods in the Supporting Information),16 and partly from some decomposition of the H‐cluster.15 A more detailed analysis of the atomic coordinates at the [2Fe] sub‐site shows that there are a few small differences in the atomic positions, in particular at the bridging ligands, ADT and CO (Figure 1 B), which likely, arise from the restraints introduced by the ligand library (crystallographic information file).17
Figure 1.
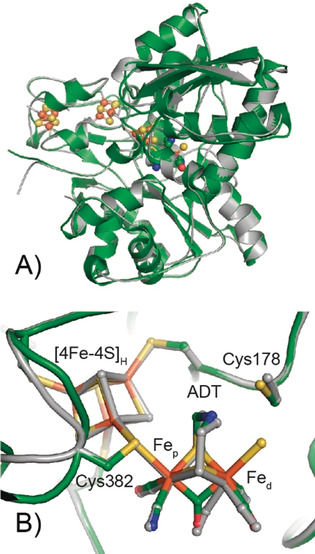
Superposition of the whole structure (A) and the active site H‐cluster region (B) of DdHydAB in the Hinact state (green) and a partially reduced state from PDB ID 1 HFE[[8b](gray). The H‐cluster, cluster ligating cysteines and C178 are shown in the sticks representation, and the protein backbone is shown in the cartoon representation. The overall architecture of DdHydAB in both states is virtually identical with an RMSD of 0.631 Å (calculated for all Cα atoms of residues 2–397).
Figure 2.
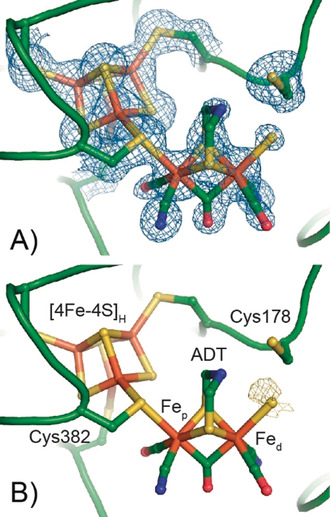
Crystal structure around the H‐cluster in the Hinact state of DdHydAB. (A) The protein backbone is presented in the cartoon representation (green), and amino acid side chains and the H‐cluster are shown in the stick representation. A 2Fo − Fc electron density map (blue mesh, contoured at 1.0 σ) is shown for Cys 178, all Cys ligating the [4Fe–4S]H sub‐cluster and the H‐cluster. An omit map generated from a model lacking the [2Fe]H sub‐site and additional S ligand is shown in Figure S2. (B) Anomalous difference map (yellow mesh, contoured at 2.0 σ) is shown for the additional ligand at the apical position on Fed.
The more pronounced difference in the location and orientation of the bridging CO also arises from the different ligand restraints. While Nicolet et al. modelled the CO ligand as non‐bonded; we used the ligand library also employed by Duan et al. for the [FeFe] hydrogenase from Clostridium pasteurianum (CpHydA1).18 Notably, the temperature values of the bridging CO in our structure are lower than in the one reported by Nicolet et al., in agreement with our structure being that of a unique state, while Nicolet's structure was suggested to be a mixture of oxidized and reduced active states.8b No significant oxidative damage during crystallization or radiation damage to the accessory iron–sulfur clusters during the measurement were observed (Figure S3).
The electron density map reveals that the H‐cluster is intact and contains two CN− ligands, two terminal CO ligands, the bridging ADT ligand, as well as the bridging CO ligand (Figure 2 A). In addition, the electron density in the active site clearly shows the presence of an additional ligand in the apical position on Fed at a distance of 2.4 Å (see Figure S4). The ligand consists of only one non‐hydrogen atom, in agreement with the presence of a (hydro)sulfide, hydroxide or oxo ligand. Modelling of the sulfur ligand with the same occupancy as that of the [2Fe] subcluster (0.6) resulted in a good fit, but with a high B‐factor (45 Å2), indicating some intrinsic disorder of the exogenous ligand. Modelling with an oxygen ligand gave a similar occupancy, but a slightly lower B‐factor (38 Å2). Thus, anomalous scattering on exactly the same crystal was utilized to provide further information about the nature of the additional ligand. By measuring diffraction data at 6 keV, the anomalous signal from the iron atoms is suppressed, while that from other heavy atoms (such as S or Cl) is enhanced. The resulting anomalous electron density map shows clear evidence for anomalous density at the apical position on Fed (Figure 2 B and Figure S5 A and B). To further support this observation, anomalous diffraction measurements were also performed before native data collection on a second crystal obtained under identical conditions (see Supporting Information, Table S1). The anomalous density map of the second crystal also showed distinct anomalous density at the apical position on Fed. While this strongly supports that the additional ligand is actually a sulfur species, we cannot exclude the possibility of a Cl− ligand. Interestingly, Cl− has been suggested to bind to the H‐cluster under certain circumstances.19 Hence, we investigated the spectroscopic properties of the Hinact state to provide further insight.
Characterization of the Hinact State by X‐Ray Absorption Spectroscopy
X‐ray absorption spectroscopy (XAS) on the Hinact state (containing the additional ligand) and the well‐characterized Hox state (lacking the additional ligand) were measured for comparison. Figure 3 shows the Fourier‐transformed (FT) spectrum of the extended X‐ray absorption fine structure (EXAFS) region for Hox (Figure 3 A) and Hinact (Figure 3 B) after subtraction of the [4Fe–4S] cluster contribution (see discussion in the Supporting Information). The presented data thus correspond to the average environment around the two iron atoms of the H‐cluster. Comparison of the FTs clearly shows that the H‐cluster of Hinact has greater amplitude than that of Hox, consistent with the presence of an additional heavy scatterer in the first coordination sphere of Hinact. Hox is best fit with 3 Fe−C scatterers at 1.80 Å (from the terminal CN, terminal CO and the bridging CO), 2.5 Fe−S scatterers at 2.26 Å (from ADT ligand sulfurs and the Fep‐bound cysteine sulfur), and an Fe−Fe scattering path at 2.60 Å (Table S4). In addition, Fe−C−O/N multiple scattering paths have to be included in the fit. The Fe−C−O/N multiple scattering paths and the Fe−Fe scattering path are highly correlated, resulting in a somewhat larger error in the fit of the Fe−Fe distance.
Figure 3.
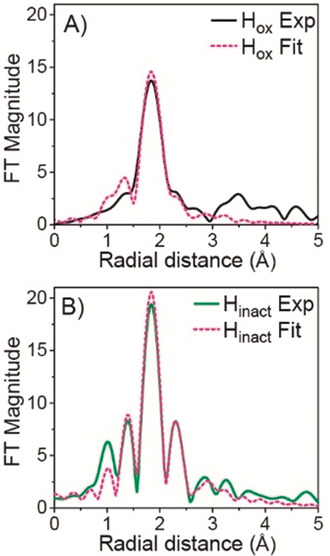
Non phase‐shifted Fourier transform of k3‐EXAFS of DdHydAB for Hox (black solid line) and Hinact (green solid line). Corresponding fits shown as pink dashed lines. See Figure S12 for data before Fourier transformation and Table S4 and S5 for fitting parameters.
For Hinact, the first shell consists of the same scattering paths (Fe−C, Fe−S, Fe−Fe and Fe−C−O multiple scattering) with the same degeneracy for every path as for Hox except for the Fe−S path, for which the degeneracy needed to be increased to N=3 (see EXAFS Discussion and Table S5 in the Supporting Information). This is consistent with the presence of an additional S ligand in Hinact coordinated to one of the H−cluster Fe atoms. Attempts to separate the Fe−S contributions into shorter and longer Fe−S distances (as observed in the crystal structure) resulted in the fit paths coalescing to the same distance. This suggests that the separation of the two sulfur contributions is beyond the resolution of our data (≈0.16 Å). The ability to fit unique Fe−S contributions is further complicate by the strong correlation of the various scattering paths in our system. Further, we note that similar to the protein crystallography, the EXAFS cannot distinguish between Cl or S as the additional ligand. Nevertheless, the first shell distances extracted from the EXAFS fits are in reasonable agreement, within the associated errors (≈0.1 Å), with the crystal structure (Table S5). For various crystal structures obtained with similar resolution diffraction data errors in the positions of the atoms, and hence bond lengths also, of up to 0.1 Å have been determined.20 Figure 4 A presents the Fe K‐edge XAS spectra of DdHydAB in the Hox and Hinact states (with the [4Fe–4S] cluster contribution subtracted, see Supporting Information for details). The shift of the rising edge toward higher energy for Hinact is consistent with a more oxidized binuclear site (homovalent FeIIFeII in Hinact vs. mixed‐valent FeIFeII in Hox). The differences in the pre‐edge region (7110–7115 eV) suggest a different coordination environment of the [2Fe] sub‐cluster in the two states.
Figure 4.
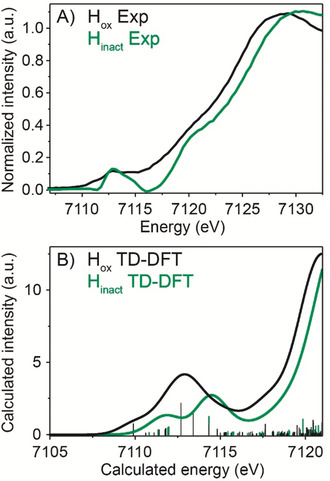
Fe K‐edge XAS experimental and calculated spectra. Fe K‐edge XAS spectra were measured in the partial fluorescence yield mode on 3 mm DdHydAB samples at 10 K. (A) Fe K‐edge XAS difference spectra of DdHydAB Hox (black trace) and Hinact (green trace) both apo subtracted. (B) TD‐DFT calculated Fe K‐edge XAS spectra using the QM/MM Hox model (black trace) and the Hinact‐SH model (green trace), applying a 2 eV (FWHM) broadening and an energy shift of 30.2 eV.
To understand the features in the experimental XAS spectra and to gain insight into the origins of the observed changes, time dependent density functional theory (TDDFT) calculations were performed with Hox and Hinact‐SH quantum mechanics/molecular mechanics (QM/MM) models (see Supporting Information Figure S7). The TDDFT calculated pre‐edge spectra (Figure 4 B) reproduce the general experimental trends in terms of the pre‐edge energies, intensity distributions and the onset of the rising edge. This indicates that the QM/MM models are consistent with the XAS data. Overall, these results support an oxidized FeIIFeII sub‐cluster for Hinact with a bound ligand at Fed. We note, however, that the XAS edges are not sensitive to the exact nature of the apical ligand (see Supporting Information).
Characterization of the Hinact State by Vibrational Spectroscopy
Figure 5 shows experimental and calculated IR spectra of the Hox and Hinact states. In the experimental spectra, all the bands of Hinact are shifted toward higher energy with respect to Hox. This is consistent with a more oxidized [2Fe] sub‐cluster in Hinact, which leads to reduced backbonding into the π* orbitals of the ligands resulting in shorter CO and CN bonds.21 The calculated IR spectra for the Hox and Hinact‐SH QM/MM models show that the calculated frequencies are in reasonable agreement (see Supporting Information for more details). The magnitudes of the shifts are underestimated, especially for the terminal CO groups, suggesting that the experimental change in back‐bonding upon oxidation is not quite reproduced by the calculations (even though the Hinact‐SH model is oxidized). Importantly, the calculated shift of the bridging CO, which should be sensitive to the addition of a new ligand is consistent with the experimental shift, albeit slightly underestimated as well.
Figure 5.
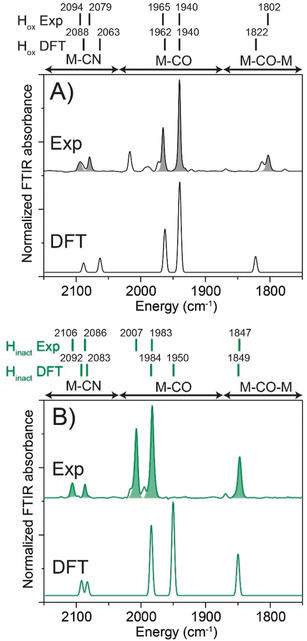
Experimental (above) and calculated (below) IR spectra of DdHydAB in (A) the Hox state and (B) the Hinact state. The experimental IR spectra were measured at 25 °C and 2 cm−1 resolution. For the calculated IR spectra, the QM/MM Hox and Hinact‐SH models were used. The experimental intensities were used in the plotted DFT spectra. Scaling factors for calculated frequencies were used (see Supporting Information).
Nuclear resonance vibrational spectroscopy (NRVS) measures vibrational sidebands coupled to nuclear transitions for Mössbauer‐active nuclei, including 57Fe.22 NRVS has already been used to study states of [FeFe] hydrogenases, including the catalytic intermediate Hhyd.23 Artificial maturation with 57Fe‐labelled [2Fe] precursors results in selectively labeled [257Fe] sub‐clusters.24 As such, predominantly vibrations associated with the [2Fe] sub‐cluster are observed. Figure 6 A presents the experimental NRVS spectra of Hox and Hinact, where clear differences can be observed. Low energy features in the 150–400 cm−1 region emerge primarily from Fe−S vibrations (bending and stretching motions). Bands around 450 cm−1 are mostly due to Fe−CN motion, while the strong bands between 500–600 cm−1 arise predominantly from Fe−CO bending and stretching modes. By correlating the experimental spectra to QM/MM NRVS calculations, the most important differences in the spectra can be interpreted. The calculated NRVS spectra (B) using the Hox and Hinact‐SH models correlate well with the experimental. Plots C and D in Figure 6 highlight the Fe−S region of the NRVS spectra. Compelling evidence for an extra sulfur bound to Hinact arise from the peak at ≈350 cm−1 (356.52 cm−1 in the calculated spectrum) in the Fe−S region (marked with an asterisk). We note that the absolute prediction of complete NRVS spectra from theoretical calculations is a challenge due to the densely populated spectra and the nature of the low‐energy modes involved, which are sensitive to the computational model. It is, therefore, advantageous to focus on the difference between Hox and Hinact and on the Fe−S region.
Figure 6.
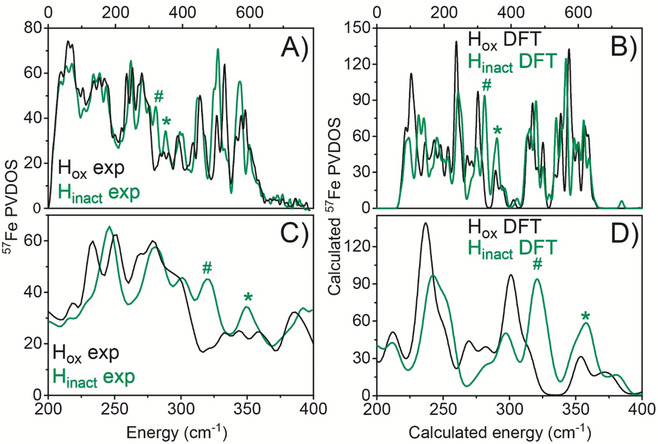
Experimental and calculated NRVS data of DdHydAB in the Hox and Hinact states. The spectra were measured on 3 mm DdHydAB samples at 40–70 K. (A) Experimental NRVS data of Hox (black trace) and Hinact (green trace). (B) Calculated NRVS data using the Hox QM/MM model (black trace) and the Hinact‐SH QM/MM model (green trace). C and D are enlargements of the Fe−S vibrational region of A and B, respectively. The asterisk indicates the peak assigned primarily to a terminal −SH group in Hinact while the hashtag indicates other Fe−S modes (associated with ADT ligand and Fep‐Cys) that shift upon oxidation.
Calculations reveal that the increased intensity in this region of the Hinact‐SH model (compared to Hox) arises from the Fe−S stretching vibration associated with an SH ligand. This feature is reproduced by the Hinact‐SH model but cannot be reproduced in models with lighter ligands such as OH− (see Supporting Information, Figure S23). The calculations cannot exclude a Cl− ligand bound to Fed due to its similar mass and covalency, which gives a comparable spectrum in this region (see Supporting Information, Figure S23). However, the Hinact state can be formed in the strict absence of chloride (Figure S20), suggesting that a Cl− bound to the open coordination site is unlikely. Furthermore, the experimental peak at ≈322 cm−1 (324 cm−1 in the calculated spectra) in Hinact (marked with a hashtag) is assigned to Fe−S modes from the ADT ligand and cysteine, which are shifted to higher energy (compared to Hox) due to a more oxidized [2Fe] sub‐cluster. The calculations demonstrate the sensitivity of NRVS spectra with respect to cluster oxidation state, but also with respect to the light vs. heavy atom nature of the ligand.
In order to directly identify the exogenous sulfur ligand, we compared NRVS spectra of Hinact samples prepared using natural abundance (95 % 32S) and 34S‐labelled sodium sulfide. We observed small differences between the 32S and 34S spectra, particularly in the 340–360 cm−1 region (Figure 7 B and Figure S24). Similar results were obtained with resonance Raman (RR) spectroscopy (Figure 7 A and Figure S25), where small changes can be observed in the 340–360 cm−1 region when going from the 32S spectrum to the 34S spectrum, coinciding with the expected lower frequencies of vibrations related to a heavier atom. Calculations support the assignment of two peaks in this region to Fe−S stretching modes from exogenously bound SH− (Figure 7 C and Table S12).
Figure 7.
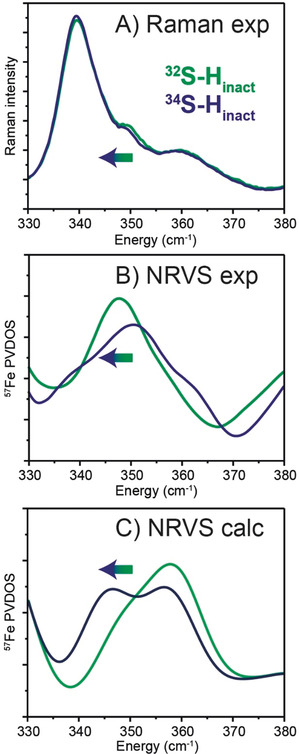
32S/34S isotope related shifts in the vibrational spectra of the Hinact state. A) Resonance Raman spectra were measured on 3 mm DdHydAB samples at 80 K using 514 nm excitation. Experimental spectra of Hinact prepared with natural abundance Na2S (green trace) and 34S‐labelled Na2S (blue) in the Fe−S region between 330 and 380 cm−1 (see Figure S25 for the full spectrum). The spectra are normalized to modes at 622 cm−1 and 644 cm−1 corresponding to the amino acid side chains phenylalanine and tyrosine, respectively.30 Experimental (B) and QM/MM‐calculated 57Fe NRVS data (C) The corresponding spectra of DdHydAB in the Hinact state with 32S (green) and 34S (blue) ligand are also displayed in the region between 330 and 380 cm−1 (see Figure S24 for the full spectra and a wider view of the Fe−S region). Calculated band positions using the Hinact‐SH QM/MM model with 32S and 34S bound at open coordination site of Fed are presented in Table S12 and shown as Movies S1 and S2.
Mechanism of Hinact Formation and Implications
Using our QM/MM model, we calculated the binding of H2S to the Hox state, (see Scheme in Figure S26).15 Sulfur likely reaches the H‐cluster via diffusion through the same hydrophobic gas channels used by H2, CO and O2. Thus, protonation to H2S (pK a≈7)25 in solution facilitates this process. Interestingly, partial formation of a very similar Hinact state with Na2Se could be achieved (Figure S27), but only at pH 4, where the enzyme is not very stable. This supports the idea that the neutral species (H2S or H2Se) are involved, as H2Se has a much lower pK a (3.89)25 than H2S. Direct H2S binding to Hox is calculated to be thermoneutral (ΔG=+0.2 kcal mol−1), while deprotonation of bound H2S by the NH group of the ADT is quite favorable (ΔG=−4.9 kcal mol−1). This leads to an Fe−SH bound intermediate that is favorable with respect to free H2S (ΔG=−4.7 kcal mol−1). The H‐cluster is subsequently oxidized to give an FeIIFeII binuclear sub‐cluster, a process driven by application of oxidizing potentials. This likely proceeds via proton‐coupled electronic rearrangement, whereby the electron is first transferred from the binuclear sub‐cluster to the [4Fe–4S] sub‐cluster, followed by its oxidation. The oxidation steps take place by one‐electron transfers from the [4Fe–4S]H subcluster, via the F‐clusters, to the available high potential oxidant (including oxygen). Calculations performed with our Hinact‐OH QM/MM model suggest that H2O binds effectively at Hox but will not deprotonate via the ADT ligand, due to the larger pK a difference than for H2S (see Supporting Information).
A number of other metalloenzymes bind sulfide under similar conditions. Both [NiFe] hydrogenase and Ni‐dependent CO dehydrogenase are inhibited by sulfide at high applied potentials, presumably by sulfide binding in a bridging position between Ni and Fe.26 Recent studies suggest that, in nitrogenase, reductive displacement of an active site “belt” sulfide could be important for binding of substrates/inhibitors.27 Thus, binding of additional sulfides to compensate for increased positive charge on oxidized metal ions could be a common theme among enzymes, highlighting the importance of understanding how sulfide interacts with metals in nature. Diiron model complexes including thioether groups are also involved in oxidation state dependent sulfur coordination from the S group.28 However, in none of these cases has additional oxygen protection due to sulfur binding been demonstrated, as is observed in [FeFe] hydrogenase.
Handling air‐sensitive enzymes such as [FeFe] hydrogenases under air has definite advantages, particularly, for crystallization and manipulation of crystals. Our DdHydAB Hinact structure is the first [FeFe] hydrogenase structure for which the redox state has been defined using single crystal spectroscopy, as previously demonstrated for [NiFe] hydrogenases.29 This provides the opportunity to directly correlate structural and spectroscopic properties of the H‐cluster. Interestingly, there are very few differences in the structure of the H‐cluster compared with previously published structures, suggesting an H‐cluster environment that minimizes redox state dependent structural changes, lowering reorganization energy and enhancing catalysis. Air‐stable [FeFe] hydrogenases may also be industrially useful for example, in fuel cells. Fuel cells containing [FeFe] hydrogenase embedded in a redox polymer have been prepared under strict anaerobic conditions,6 but could be prepared under air with the Hinact state, simplifying the process and increasing the scalability.
The Relevance of O2 Protection via Hinact In Vivo
The bacterium Desulfovibrio desulfuricans has evolved in anaerobic environments and, therefore, its hydrogenase is extremely oxygen sensitive, becoming inactivated irreversibly even by traces of O2.11, 31 Although the mechanism of oxygen inactivation is not yet completely understood, O2 is believed to attack the active site by binding to the open coordination site on Fed.30 If the bound‐O2 cannot be reduced to water, it may form reactive oxygen species, which could destroy the active site. As the open coordination site is blocked by sulfide in the Hinact state, this prevents O2 binding and destruction of the active site.
A plausible scenario is that in vivo, DdHydAB is constantly exposed to H2S (since Desulfovibrio desulfuricans reduces sulfate to sulfide). Under reductive conditions H2S is displaced by H2, binding to the H‐cluster. In the presence of oxygen, however, H2S becomes locked to the H‐cluster forming the Hinact state to protect the enzyme from oxygen inactivation. It is interesting that the [FeFe] hydrogenase from Clostridium beijerinckii (CbH5A) can form the Hinact state without exogenous sulfide.14 How, the Hinact state in this enzyme differs structurally from that in DdHydAB is not known, but it seems likely that, in the absence of available sulfide in this organism, an endogenous sulfur ligand, such as a cysteine nearby the active site, has evolved to play a role. Another important difference between these two enzymes is that DdHydAB functions as a periplasmic H2 uptake enzyme with extremely high activity,32 while CbH5A shows a strong bias for H2 production.14 This lack of activity in H2 uptake may be due to spontaneous formation of Hinact at high potentials. As DdHydAB requires exogenous sulfide, it will only be inactive when sulfide levels are high, which may serve to regulate metabolism.
Conclusion
Here, we provide direct structural and spectroscopic evidence for an exogenously bound sulfur in the apical coordination site of the [FeFe] hydrogenase from Desulfovibrio desulfuricans in the O2‐stable Hinact state. In our previous work, we showed that exogenous sulfide was required for Hinact formation, but we were unable to demonstrate if and how sulfide binds to the H‐cluster. The 1.65 Å crystal structure shows electron density at the apical position on the distal Fe and anomalous diffraction suggests this is consistent with sulfur. EXAFS shows an additional sulfur in the Fe‐coordination environment of Hinact compared with Hox. Fe K‐edge XAS data reveal a more oxidized [2Fe] sub‐cluster in Hinact compared to Hox and a different coordination environment of the Fe ions in the [2Fe] subcluster. Comparison of Hinact and Hox NRVS spectra, as well as 32S/34S isotope‐labelling in both NRVS and resonance Raman spectroscopy, provide additional compelling evidence for an exogenous sulfur ligand. Calculations with an Hinact‐SH model provide close agreement to all the experimental data and shed light on the mechanism of forming Hinact. In particular, the most likely pathway involved H2S binding at the open coordination site, followed by proton transfer via the ADT ligand to the proton transfer channel. Hinact formation is then completed upon proton coupled electronic reconfiguration of the H‐cluster and oxidation of [4Fe–4S]H. Since we previously demonstrated that this in vitro Hinact approach works for other [FeFe] hydrogenases such as Chlamydomonas reinhardtii (CrHydA1), it demonstrates the wider applicability of this method. Thus, it would be interesting to perform similar structural and spectroscopic studies of Hinact in other enzymes, including CbH5A and CrHydA1. Our highly complementary structural, spectroscopic and theoretical approach represents a major advance for the understanding the function of this O2‐stable state and its mechanism of formation, as well as a possible implementation of these enzymes in biotechnological applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
The authors would like to thank Nina Breuer and Patricia Malkowski for help with preparation of DdHydAB samples. Dr. George E. Cutsail III, Dr. Justin Travis Henthorn, Dr. Alexander Gutt and Dr. Casey Van Stappen are gratefully thanked for helpful assistance with X‐ray data collection and for fruitful discussions. Dr. Justin Travis Henthorn and Ms. Rebeca G. Castillo are also thanked for helping to prepare the Na2 34S and to fit the pre‐edge region for Hox and Hinact, respectively. We acknowledge Dr. Casey Van Stappen for his help in preparing XAS plots. We are also grateful to Dr. Erik Jonathan Nelson and Dr. Matthew J. Latimer at SSRL for providing assistance in using beamline 9‐3 and to Dr. Kenji Tamasaku for assistance in NRVS measurements in Spring‐8. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank Anja Burkhardt, Olga Lorbeer, and Eva Crosas for assistance in using the Bioimaging and diffraction beamline P11. The authors would also like to express their gratitude to Dr. Nipa Chongdar for critical reading of the manuscript. ((Et4N)2[57Fe2(ADT)(CO)4(CN)2] was a kind gift from Prof. Thomas B. Rauchfuss, University of Illinois Urbana‐Champaign, IL, USA. NRVS data were collected during beam times 2018B1379 and 2019A1259 at BL19LXU at SPring‐8, Japan. This work was financially supported by the Max Planck Society (P.R‐M., S.D., J.A.B.), the Deutsche Forschungsgemeinschaft (DFG) Priority Programme “Iron‐Sulfur for Life: Cooperative Function of Iron‐Sulfur Centers in Assembly, Biosynthesis, Catalysis and Disease” (SPP 1927) Projects DE 1877/1‐1 (S.D.), BI 2198/1‐1 (J.A.B.), IS 1476/4‐1 (I.S.) and ZE 510/2‐2 (I.Z., C.L.); further funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2008–390540038—UniSysCat (I.Z., C.L.), and the National Institutes of Health NIH GM‐65440 (S.P.C.). S.P.C. and I.Z. are grateful to the Einstein Foundation Berlin for funding (grant number EVF‐2016‐277). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE‐AC02‐76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). Open access funding enabled and organized by Projekt DEAL.
P. Rodríguez-Maciá, L. M. Galle, R. Bjornsson, C. Lorent, I. Zebger, Y. Yoda, S. P. Cramer, S. DeBeer, I. Span, J. A. Birrell, Angew. Chem. Int. Ed. 2020, 59, 16786.
Contributor Information
Prof. Dr. Serena DeBeer, Email: serena.debeer@cec.mpg.de.
Prof. Dr. Ingrid Span, Email: ingrid.span@hhu.de.
Dr. James A. Birrell, Email: james.birrell@cec.mpg.de.
References
- 1. Armaroli N., Balzani V., ChemSusChem 2011, 4, 21–36. [DOI] [PubMed] [Google Scholar]
- 2. Vincent K. A., Parkin A., Armstrong F. A., Chem. Rev. 2007, 107, 4366–4413. [DOI] [PubMed] [Google Scholar]
- 3. Lubitz W., Ogata H., Rüdiger O., Reijerse E., Chem. Rev. 2014, 114, 4081–4148. [DOI] [PubMed] [Google Scholar]
- 4. Martin W. G., Glick B. R., Martin S. M., Can. J. Microbiol. 1980, 26, 1214–1223. [DOI] [PubMed] [Google Scholar]
- 5. Armstrong F. A., Hirst J., Proc. Natl. Acad. Sci. USA 2011, 108, 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oughli A. A., Conzuelo F., Winkler M., Happe T., Lubitz W., Schuhmann W., Rüdiger O., Plumeré N., Angew. Chem. Int. Ed. 2015, 54, 12329–12333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12506–12510. [Google Scholar]
- 7.
- 7a. Stripp S. T., Goldet G., Brandmayr C., Sanganas O., Vincent K. A., Haumann M., Armstrong F. A., Happe T., Proc. Natl. Acad. Sci. USA 2009, 106, 17331–17336; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Swanson K. D., Ratzloff M. W., Mulder D. W., Artz J. H., Ghose S., Hoffman A., White S., Zadvornyy O. A., Broderick J. B., Bothner B., King P. W., Peters J. W., J. Am. Chem. Soc. 2015, 137, 1809–1816; [DOI] [PubMed] [Google Scholar]
- 7c. Kubas A., Orain C., De Sancho D., Saujet L., Sensi M., Gauquelin C., Meynial-Salles I., Soucaille P., Bottin H., Baffert C., Fourmond V., Best R. B., Blumberger J., Léger C., Nat. Chem. 2017, 9, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Peters J. W., Lanzilotta W. N., Lemon B. J., Seefeldt L. C., Science 1998, 282, 1853–1858; [DOI] [PubMed] [Google Scholar]
- 8b. Nicolet Y., Piras C., Legrand P., Hatchikian C. E., Fontecilla-Camps J. C., Structure 1999, 7, 13–23. [DOI] [PubMed] [Google Scholar]
- 9. Silakov A., Wenk B., Reijerse E., Lubitz W., Phys. Chem. Chem. Phys. 2009, 11, 6592–6599. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Adamska A., Silakov A., Lambertz C., Rüdiger O., Happe T., Reijerse E., Lubitz W., Angew. Chem. Int. Ed. 2012, 51, 11458–11462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11624–11629; [Google Scholar]
- 10b. Sommer C., Adamska-Venkatesh A., Pawlak K., Birrell J. A., Rüdiger O., Reijerse E. J., Lubitz W., J. Am. Chem. Soc. 2017, 139, 1440–1443; [DOI] [PubMed] [Google Scholar]
- 10c. Mulder D. W., Guo Y., Ratzloff M. W., King P. W., J. Am. Chem. Soc. 2017, 139, 83–86; [DOI] [PubMed] [Google Scholar]
- 10d. Katz S., Noth J., Horch M., Shafaat H. S., Happe T., Hildebrandt P., Zebger I., Chem. Sci. 2016, 7, 6746–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roseboom W., de Lacey A. L., Fernández V. M., Hatchikian C., Albracht S. P. J., J. Biol. Inorg. Chem. 2006, 11, 102–118. [DOI] [PubMed] [Google Scholar]
- 12. Pereira A. S., Tavares P., Moura I., Moura J. J. G., Huynh B. H., J. Am. Chem. Soc. 2001, 123, 2771–2782. [DOI] [PubMed] [Google Scholar]
- 13. Greco C., Bruschi M., De Gioia L., Ryde U., Inorg. Chem. 2007, 46, 5911–5921. [DOI] [PubMed] [Google Scholar]
- 14. Morra S., Arizzi M., Valetti F., Gilardi G., Biochemistry 2016, 55, 5897–5900. [DOI] [PubMed] [Google Scholar]
- 15. Rodríguez-Maciá P., Reijerse E. J., van Gastel M., DeBeer S., Lubitz W., Rüdiger O., Birrell J. A., J. Am. Chem. Soc. 2018, 140, 9346–9350. [DOI] [PubMed] [Google Scholar]
- 16. Birrell J. A., Wrede K., Pawlak K., Rodriguez-Maciá P., Rüdiger O., Reijerse E. J., Lubitz W., Isr. J. Chem. 2016, 56, 852–863. [Google Scholar]
- 17.
- 17a. Brown I. D., McMahon B., Acta Crystallogr. Sect. B 2002, 58, 317–324; [DOI] [PubMed] [Google Scholar]
- 17b. Hall S. R., Allen F. H., Brown I. D., Acta Crystallogr. Sect. A 1991, 47, 655–685. [Google Scholar]
- 18. Duan J., Senger M., Esselborn J., Engelbrecht V., Wittkamp F., Apfel U.-P., Hofmann E., Stripp S. T., Happe T., Winkler M., Nat. Commun. 2018, 9, 4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. del Barrio M., Sensi M., Fradale L., Bruschi M., Greco C., de Gioia L., Bertini L., Fourmond V., Léger C., J. Am. Chem. Soc. 2018, 140, 5485–5492. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Guss J. M., Bartunik H. D., Freeman H. C., Acta Crystallogr. Sect. B 1992, 48, 790–811; [DOI] [PubMed] [Google Scholar]
- 20b. Solomon E. I., Szilagyi R. K., DeBeer George S., Basumallick L., Chem. Rev. 2004, 104, 419–458. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Siebel J. F., Adamska-Venkatesh A., Weber K., Rumpel S., Reijerse E., Lubitz W., Biochemistry 2015, 54, 1474–1483; [DOI] [PubMed] [Google Scholar]
- 21b. Rodríguez-Maciá P., Reijerse E., Lubitz W., Birrell J. A., Rüdiger O., J. Phys. Chem. Lett. 2017, 8, 3834–3839. [DOI] [PubMed] [Google Scholar]
- 22. Wang H., Alp E. E., Yoda Y., Cramer S. P., in Metalloproteins: Methods and Protocols (Eds.: J. C. Fontecilla-Camps, Y. Nicolet), Humana Press, Totowa, NJ, 2014, pp. 125–137. [Google Scholar]
- 23. Pelmenschikov V., Birrell J. A., Pham C. C., Mishra N., Wang H., Sommer C., Reijerse E., Richers C. P., Tamasaku K., Yoda Y., Rauchfuss T. B., Lubitz W., Cramer S. P., J. Am. Chem. Soc. 2017, 139, 16894–16902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gilbert-Wilson R., Siebel J. F., Adamska-Venkatesh A., Pham C. C., Reijerse E., Wang H., Cramer S. P., Lubitz W., Rauchfuss T. B., J. Am. Chem. Soc. 2015, 137, 8998–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bjerrum J., Sillén L. G., Schwarzenbach G. K., Berecki-Biedermann C., Stability constants of metal-ion complexes, with solubility products of inorganic substances. 2 : Inorganic ligands, Chemical society, London, 1958. [Google Scholar]
- 26.
- 26a. Vincent K. A., Cracknell J. A., Clark J. R., Ludwig M., Lenz O., Friedrich B., Armstrong F. A., Chem. Commun. 2006, 5033–5035; [DOI] [PubMed] [Google Scholar]
- 26b. Wang V. C. C., Can M., Pierce E., Ragsdale S. W., Armstrong F. A., J. Am. Chem. Soc. 2013, 135, 2198–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Sippel D., Rohde M., Netzer J., Trncik C., Gies J., Grunau K., Djurdjevic I., Decamps L., Andrade S. L. A., Einsle O., Science 2018, 359, 1484–1489; [DOI] [PubMed] [Google Scholar]
- 27b. Spatzal T., Perez K. A., Einsle O., Howard J. B., Rees D. C., Science 2014, 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Razavet M., Borg S. J., George S. J., Best S. P., Fairhurst S. A., Pickett C. J., Chem. Commun. 2002, 700–701. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Ilina Y., Lorent C., Katz S., Jeoung J.-H., Shima S., Horch M., Zebger I., Dobbek H., Angew. Chem. Int. Ed. 2019, 58, 18710–18714; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 18883–18887; [Google Scholar]
- 29b. Siebert E., Rippers Y., Frielingsdorf S., Fritsch J., Schmidt A., Kalms J., Katz S., Lenz O., Scheerer P., Paasche L., Pelmenschikov V., Kuhlmann U., Mroginski M. A., Zebger I., Hildebrandt P., J. Phys. Chem. B 2015, 119, 13785–13796. [DOI] [PubMed] [Google Scholar]
- 30. Paulo F. M. B., Freire T. C., Lima J. A., Melo F. E. A., Filho J. M., in Raman Spectroscopy and Applications, Vol. Open access peer-reviewed Edited Volume, IntechOpen, 2017. [Google Scholar]
- 31. Vincent K. A., Parkin A., Lenz O., Albracht S. P. J., Fontecilla-Camps J. C., Cammack R., Friedrich B., Armstrong F. A., J. Am. Chem. Soc. 2005, 127, 18179–18189. [DOI] [PubMed] [Google Scholar]
- 32. Hatchikian E. C., Forget N., Fernandez V. M., Williams R., Cammack R., Eur. J. Biochem. 1992, 209, 357–365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary