

Abstract
Background
N8‐GP (turoctocog alfa pegol; Esperoct®, Novo Nordisk A/S, Bagsvaerd, Denmark) is a glycoPEGylated human recombinant factor VIII with a half‐life of ~1.6‐fold of standard FVIII products. pathfinder2 (NCT01480180) was a multi‐national, open‐label trial of N8‐GP in previously treated adolescent and adult patients with severe hemophilia A.
Objective
We report end‐of‐trial efficacy and safety of N8‐GP from pathfinder2.
Methods
pathfinder2 main phase and extension phase part 1 results have been previously reported. During extension phase part 2, patients could switch from N8‐GP prophylaxis 50 IU/kg every fourth day (Q4D) or 75 IU/kg once weekly (Q7D), depending on bleeding status. Extension phase part 2 collected long‐term safety and efficacy data for all regimens until trial end (first patient in main phase, 30 January 2012; trial end, 10 December 2018).
Results
Overall, 186 patients were exposed to N8‐GP for up to 6.6 years (median 5.4 years). The estimated annualized bleeding rate (ABR) was 2.14 (median 0.84) for the Q4D prophylaxis arm and 1.31 (median 1.67) for the Q7D prophylaxis arm. Nearly 30% of patients experienced zero bleeds throughout the entire duration of the trial, the hemostatic response was 83.2% across all treatment arms, and patient‐reported outcomes were maintained or slightly improved. No safety concerns were detected.
Conclusion
Data from the completed pathfinder2 trial, one of the largest and longest‐running clinical trials to investigate treatment of severe hemophilia A, demonstrate the efficacy and safety of N8‐GP in previously treated adolescent and adult patients.
Keywords: clinical trial, factor VIII, hemophilia A, turoctocog alfa pegol
ESSENTIALS.
Previously treated adolescent and adult patients with hemophilia A were treated with N8‐GP.
Overall, 186 patients were exposed to N8‐GP for a median of 5.4 years (6.6 years max).
~30% of patients on every fourth day prophylaxis experienced zero bleeds during the duration of the trial.
Median annualized bleeding rate for Q4D prophylaxis was 0.84 for the completed trial.
1. BACKGROUND
Patients with severe hemophilia A typically experience recurrent and spontaneous bleeding in joints, muscles, and soft tissues, which may lead to chronic arthropathy, muscular atrophy, and other disabilities. To prevent bleeding, the current standard of care is factor VIII (FVIII) replacement therapy. 1 , 2 , 3 While FVIII replacement therapy is generally effective, standard replacement products have short half‐lives in adults (~12 hours 4 ), and patients require regular intravenous injections. 5 In order to reduce the injection burden for patients, manufacturers have developed extended half‐life (EHL) coagulation factors. 6
N8‐GP (turoctocog alfa pegol; Esperoct®; Novo Nordisk A/S) is a glycoPEGylated human recombinant (r)FVIII. An additional 40‐kDa polyethylene glycol (PEG) moiety on the truncated B‐domain of turoctocog alfa (NovoEight®; Novo Nordisk A/S) extends the half‐life of FVIII (1.6‐fold in adults 4 and 1.9‐fold in children 7 ). Thrombin cleaves the B‐domain of N8‐GP (with the attached PEG moiety) to activate the molecule, leaving the native structure of activated FVIII intact.
The safety and efficacy of N8‐GP has been demonstrated in both adults 4 , 8 , 9 and children 7 during an extensive clinical trials program involving more than 270 patients, some for >5 years, 10 , 11 and N8‐GP has been approved for the clinical management of patients with hemophilia A. The pathfinder2 trial, a phase III, multi‐national, open‐label trial in previously treated adults and adolescents with hemophilia A, was a key element of the clinical trials program and consisted of a main phase followed by two extension phases. The main phase of pathfinder2 followed patients for up to 19 months 8 and investigated the efficacy, safety, and pharmacokinetics of N8‐GP dosed at 50 IU/kg every 4 days (Q4D). 8 After the main phase, a subset of patients with low bleeding status were randomized into 50 IU/kg Q4D or 75 IU/kg once‐weekly (Q7D) dosing during extension phase part 1. 9 Non‐randomized patients continued on the 50 IU/kg Q4D regimen. After 24 weeks, patients from extension phase part 1 continued into the non‐randomized extension part 2 until the end of trial at which point patients could switch from Q4D to Q7D regimen and vice versa depending on bleeding status. Here, we report end‐of‐trial efficacy and safety of N8‐GP from the completed pathfinder2, including combined data from the main phase and both extension phases.
2. METHODS
2.1. Patients and trial design
pathfinder2 (ClinicalTrials.gov identifier: NCT01480180) was a multi‐national, open‐label, phase III trial evaluating the safety, efficacy, and pharmacokinetics of N8‐GP treatment in previously treated patients ≥12 years of age with severe hemophilia A.
pathfinder2 comprised a non‐randomized main phase, a randomized extension phase part 1 performed in a subset of patients with low bleeding status, and a non‐randomized extension phase part 2 (first patient in main phase, 30 January 2012; trial end, 10 December 2018). The full details of the main phase 8 and extension phase part 1 9 inclusion/exclusion criteria, trial objectives, and endpoints have been described previously. Briefly, this trial included previously treated patients with ≥150 exposure days (EDs) to any FVIII‐containing product. Patients with any history of significant thromboembolic events, FVIII inhibitors, or a current FVIII inhibitor >0.6 Bethesda units (BU) at screening were excluded.
Treatment regimens and trial design are described in Figure 1. The main phase examined the efficacy and safety of N8‐GP 50 IU/kg Q4D prophylaxis. Twice‐weekly dosing was permitted at the discretion of the investigator. Patients were offered the option of on‐demand treatment. Extension phase part 1 aimed to assess the safety and efficacy of Q7D N8‐GP prophylaxis. A subset of patients was randomized 2:1 to Q7D (75 IU/kg) or Q4D (50 IU/kg) treatment. Eligibility for randomization required ≤2 bleeding episodes on Q4D prophylaxis during the 6 months prior to entering the extension phase. Extension phase part 1 comprised 24 weeks of treatment, after which patients transferred to extension phase part 2. The aim of extension phase part 2 was to collect long‐term safety and efficacy data up to the trial end. During this phase, patients on N8‐GP prophylaxis with ≤2 bleeding episodes within the last 6 months on Q4D regimen could switch to Q7D dosing. If a patient on Q7D treatment experienced ≥2 spontaneous bleeding episodes or one severe bleeding episode requiring hospitalization over an 8‐week period, they were switched back to Q4D treatment. Bleeding episodes were treated with 20‐75 IU/kg N8‐GP.
Figure 1.
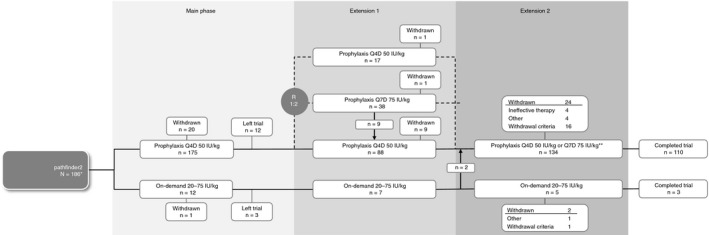
Trial design, treatment regimen, and disposition of patients throughout pathfinder2 *215 patients were screened, 186 (161 adults, 25 adolescents) were treated with N8‐GP during pathfinder2. **patients with ≤2 bleeding episodes within the last 6 months on the Q4D regimen could switch to Q7D dosing. Patients on Q7D treatment who experienced ≥2 spontaneous bleeds or 1 severe bleed requiring hospitalization over an 8‐week period were switched back to Q4D treatment. Q4D, every fourth day treatment; Q7D, weekly treatment; R, randomization
The pathfinder2 trial was approved according to local regulations by the appropriate ethics committees or institutional review boards and conducted in accordance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice. Written informed consent was obtained from all participants (or from the patient's legal guardian) prior to any trial‐related activities.
2.2. Objectives, endpoints, and assessments
Detailed methods for efficacy and safety assessments have been described previously. 8 , 9 Briefly, the two co‐primary endpoints of pathfinder2 were the annualized bleeding rate (ABR) of patients receiving prophylaxis and the incidence of FVIII‐inhibitors ≥0.6 BU. To detect FVIII inhibitors, a Nijmegen‐modified Bethesda assay with heat inactivation of residual FVIII activity was used, 12 , 13 with a positive inhibitor test result defined as ≥0.6 BU in two consecutive samples. 8
The key secondary objectives of extension phase part 2 and the end‐of‐trial analysis were to assess long‐term efficacy and safety of N8‐GP prophylaxis. Efficacy endpoints included ABR, the hemostatic effect of N8‐GP when used to treat a bleeding episode, and patient‐reported outcomes (PROs). The hemostatic effect of N8‐GP was assessed using a previously described 4‐point scale, 8 comprising the following ratings: “excellent,” “good,” “moderate,” and “none.” Excellent and good responses were considered successes, while moderate, none, and missing responses were considered failures.
Detailed methods and PRO results for the main phase have been previously reported. 14 PROs for this end‐of‐trial analysis were assessed as change in total patient scores in both prophylaxis arms using the HAEM‐A‐QOL, Hemo‐Sat, 14 the EQ‐5D‐VAS, and the ED‐5D over time from visit 1 (baseline) to predefined treatment durations (1 to <2 years, 2 to <3 years, 3 to <4 years, 4 to <5 years 5 to <6 years, and 6 to <7 years). PROs were assessed in this analysis only for patients who participated in extension phase part 2 and completed a questionnaire in the given year.
FVIII activity measurements were performed pre‐dose and post‐dose throughout all phases of the trial using a Coasys® Plus C analyzer (Roche Diagnostics) and a chromogenic assay (Chromogenix Coamatic® Factor VIII [diaPharma]) with a N8‐GP product‐specific standard for calibration.
Secondary safety endpoints included the incidence of adverse events (AEs); serious adverse events (SAE); and medical events of special interest (MESI), which included medication errors, inhibitor formation against FVIII, allergic reaction, thromboembolic events, and suspected transmission of an infectious agent via the trial product.
Exploratory endpoints included the incidence of N8‐GP binding and anti‐PEG antibodies. Detailed methods for antibody analysis in pathfinder2 have been previously published. 8
2.3. Statistical analyses
The safety analysis set and the full analysis set comprised all patients exposed to N8‐GP. Methods for statistical analyses in pathfinder2, including calculations of power and sample size, have been previously described in detail. 8 , 9
The rate of inhibitors was reported and a 1‐sided 97.5% upper confidence limit provided based on an exact calculation for a binomial distribution. For the calculation of the inhibitor rate, all patients with inhibitors were divided by all patients with a minimum of 50 EDs plus any patients with <50 EDs but with inhibitors. Adequate safety with regard to inhibitors could be concluded if the upper one‐sided 97.5% confidence limit was below 6.8%.
For the post‐hoc analysis of the proportion of patients who experienced no bleeding episodes in a given year, only patients who completed the study were included. As patients could switch treatment regimens, only the initial and first consecutive prophylaxis regimen was used for calculations. Thus, for patients who switched from Q4D treatment to Q7D treatment and back to Q4D treatment, only the first two treatment periods were used for analysis.
Observed ABRs were used for analysis. For calculation of Poisson estimate, see Giangrande et al 8 for full details. Briefly, a prophylactic effect of N8‐GP could be concluded if the ABR was significantly below 8.5, analyzed using a Poisson regression model on number of bleeds per patient allowing for over‐dispersion (using Pearson's Chi‐square divided by the degrees of freedom) and log planned observation duration as an offset. Estimates of ABR are provided with 95% confidence intervals (CI).
Hemostatic effect success rate was calculated using a logistic regression model (see Giangrande et al 8 for full details). Efficacy based on hemostatic response could be concluded if the one‐sided lower 97.5% confidence limit for the success rate was above 65%.
3. RESULTS
3.1. Patients
A total of 215 patients were screened in pathfinder2, and 186 (161 adults, 25 adolescents) were treated with N8‐GP during the main phase of the trial, of whom 175 received N8‐GP prophylaxis and 12 on‐demand treatment. One patient switched from on‐demand treatment to prophylaxis during the main phase, and two patients switched from on‐demand to prophylaxis after the main phase. These patients are counted in both treatment arms.
In total, 165 patients completed the main phase of the trial, 150 of whom continued into the extension phase part 1 and 139 continued into part 2 (Figure 1). After treatment‐arm transfers at entry to extension phase part 2, there were 94 patients receiving Q4D prophylaxis, 40 receiving Q7D prophylaxis, and 5 receiving on‐demand treatment. During this final phase, there were 15 occurrences of a patient transferring from Q4D to Q7D prophylaxis and 25 occurrences of a patient transferring from Q7D to Q4D prophylaxis. The reasons for transfer to Q4D during extension part 2 included ≥2 spontaneous bleeding episodes over 8 weeks on Q7D (n = 14), increased physical activity (n = 3), increased bleeding (n = 3), “other reason” (n = 3; one reason was “pain in both ankles”), investigator's discretion (n = 1), and AEs (n = 1).
Overall, at some point during extension part 2, 110 patients received Q4D and 52 patients received Q7D prophylaxis; 5 patients received on‐demand treatment. In total, 113 patients completed the trial, with 87 patients on 50 IU/kg Q4D, 23 patients on 75 IU/kg Q7D, and 3 patients on 20‐75 IU/kg on‐demand treatment. A total of 58 patients withdrew from the trial, 21 during the main phase (19 of which were reported in Giangrande et al 8 ), 11 during extension phase part 1, and 26 during extension phase part 2 (Figure 1).
As this trial was part of a wider concurrent clinical trials program, patients could be transferred to and back from other trials. A total of 36 patients were temporarily transferred from pathfinder2 to the pathfinder3 surgery trial (NCT01489111), 15 of whom 35 underwent surgery; all 36 of these patients subsequently returned to the pathfinder2 trial. Ten patients had surgery on more than one occasion, with a maximum of four occasions. Another 22 patients transferred from pathfinder2 to a pharmacokinetics trial (pathfinder7; NCT02920398; 21 of whom participated in the trial) and back to pathfinder2. Patient demographic and baseline characteristics have been previously reported and were representative of the severe hemophilia A population. 7 , 8
3.2. Exposure and consumption
Across the entire trial, 186 patients were exposed to N8‐GP for 66 577 EDs, with 172 patients achieving ≥50 EDs. The total median time on regimen in the trial across all treatment regimens was 1974 (range 5‐2397) days for a total mean of 358 (1‐697) EDs per patient. The median time on the Q4D prophylaxis regimen during the trial was 1479 (5‐2269) days, with a mean of 326 (range 1‐697) EDs. The median time on the Q7D prophylaxis regimen was 798 (18‐1689) days and 119 (4‐250) EDs.
The mean annual consumption of N8‐GP per patient (including prophylaxis, treatment of bleeds, minor surgeries, and PK doses) was 4829 IU/kg/year in the Q4D prophylaxis arm, 4242 IU/kg/year in the Q7D prophylaxis arm, and 1562 IU/kg/year in the on‐demand arm, with no apparent differences between adults and adolescents.
3.3. Pharmacokinetics
The overall mean FVIII activity trough level in patients on Q4D prophylaxis was 3.1 IU/dL (95% CI: 2.7; 3.6). Of the 3012 trough values measured, 17.9% were below the lower limit of quantification. In patients on Q7D prophylaxis, the overall mean FVIII activity trough level was 0.9 IU/dL (95% CI: 0.7; 1.2). Of the 858 trough values measured, 55.8% were below the lower limit of quantification.
3.4. Annualized bleeding rates
The estimated ABR for all patients in the Q4D prophylaxis arm was 2.14 (95% CI: 1.73; 2.65; P < .001) with a median ABR of 0.84 (interquartile range [IQR]: 0.00; 2.41). The estimated ABR for all the patients in the Q7D prophylaxis arm was 1.31 (95% CI: 0.89; 1.92; P < .001), with a median ABR of 1.67 (IQR: 0.43; 6.41), demonstrating the prophylactic effect of both regimens (Table 1). Imputed ABRs are reported in Table S1 in supporting information. The estimated ABR for spontaneous bleeds was 1.20 in the N8‐GP Q4D prophylaxis arm and 0.76 in the Q7D prophylaxis arm.
Table 1.
Cumulative annualized bleeding rates during the pathfinder2 completed trial
|
N8‐GP prophylaxis 50 IU/kg Q4D |
N8‐GP prophylaxis 75 IU/kg Q7D |
N8‐GP on‐demand 20‐75 IU/kg |
|
|---|---|---|---|
| Patients a , N | 177 | 61 | 12 |
| Mean treatment period, b years | 3.73 | 4.17 | 3.53 |
| ABR, observed | |||
| Poisson estimate (95% CI) | 2.14 (1.73; 2.65) | 1.31 (0.89; 1.92) | — |
| Median (IQR) | 0.84 (0.00; 2.41) | 1.67 (0.43; 6.41) | 30.98 (22.72; 39.82) |
| Spontaneous ABR, observed | |||
| Poisson estimate (95% CI) | 1.20 (0.93; 1.54) | 0.76 (0.44; 1.34) | — |
| Median (IQR) | 0.35 (0.00; 1.29) | 0.71 (0.00; 3.84) | 19.55 (13.76; 24.62) |
| Traumatic ABR, observed | |||
| Poisson estimate (95% CI) | 0.93 (0.72; 1.21) | 0.53 (0.35; 0.78) | — |
| Median (IQR) | 0.19 (0.00; 0.95) | 0.00 (0.00; 1.15) | 3.96 (1.26; 9.32) |
Abbreviations: ABR, annualized bleeding rate; CI, confidence interval; IQR, interquartile range; Q4D, every fourth day treatment; Q7D, weekly treatment.
Several patients changed treatment regimen during the trial; therefore, a patient may be included in more than one treatment arm, but only counted once in the total.
For patients withdrawing prematurely, the planned treatment duration was used; for completers, the actual treatment duration was used.
In adolescents, the median ABR in the Q4D prophylaxis arm, at 1.19 (IQR: 0.40; 3.63), was slightly higher than in adults, who had a median ABR of 0.75 (IQR: 0.00; 2.29). A similar trend was observed in the Q7D prophylaxis arm, in which adolescents had a median ABR of 5.23 (IQR: 0.50; 9.91) and adults a median ABR of 1.64 (IQR: 0.40; 3.53).
3.5. Bleeding episodes
A total of 2758 bleeds in 152 patients (81.7%) were treated with N8‐GP during the trial (Table 2). Almost half (46%) of all bleeding episodes were reported by patients being treated on‐demand. Overall, 28.8% of patients in the Q4D prophylaxis arm reported no bleeds during the entire trial. Furthermore, 38.4% of patients in the Q4D prophylaxis arm reported no spontaneous bleeds during the entire trial. A greater proportion of the bleeds in the on‐demand group were spontaneous (76.5%) compared with the Q4D (55.9%) or the Q7D (58.5%) prophylaxis arms.
Table 2.
Details of bleeding episodes for patients receiving prophylactic or on‐demand treatment with N8‐GP during the pathfinder2 completed trial
|
Prophylaxis 50 IU/kg Q4D (N = 177) |
Prophylaxis 75 IU/kg Q7D (N = 61) |
On‐demand 20‐75 IU/kg (n = 12) |
Total (N = 186) | |
|---|---|---|---|---|
| Patients with bleeds, N (%) | 126 (71.2) | 53 (86.9) | 12 (100.0) | 152 (81.7) |
| Bleeding episodes, N | 1312 | 176 | 1270 | 2758 |
| Cause of bleed, N (%) | ||||
| Spontaneous | 733 (55.9) | 103 (58.5) | 971 (76.5) | 1807 (65.5) |
| Traumatic | 572 (43.6) | 71 (40.3) | 288 (22.7) | 931 (33.8) |
| Minor surgery | 7 (0.5) | 2 (1.1) | 11 (0.9) | 20 (0.7) |
| Re‐bleed a , N (%) | ||||
| No | 1291 (98.4) | 176 (100.0) | 1252 (98.6) | 2719 (98.6) |
| Yes | 21 (1.6) | — | 18 (1.4) | 39 (1.4) |
| Site of bleed, N (%) | ||||
| Joint | 989 (75.4) | 119 (67.6) | 627 (49.4) | 1735 (62.9) |
| Mucosal | 47 (3.6) | 7 (4.0) | 58 (4.6) | 112 (4.1) |
| Muscular | 147 (11.2) | 19 (10.8) | 170 (13.4) | 336 (12.2) |
| Subcutaneous | 41 (3.1) | 11 (6.3) | 359 (28.3) | 411 (14.9) |
| Gastrointestinal | 11 (0.8) | 4 (2.3) | 6 (0.5) | 21 (0.8) |
| Other | 77 (5.9) | 16 (9.1) | 50 (3.9) | 143 (5.2) |
| Classification of bleed, N (%) | ||||
| Mild/moderate | 1286 (98.0) | 169 (96.0) | 1262 (99.4) | 2717 (98.5) |
| Severe | 22 (1.7) | 7 (4.0) | 6 (0.5) | 35 (1.3) |
| Unknown b | 4 (0.3) | — | 2 (0.2) | 6 (0.2) |
Abbreviations: Q4D, every fourth day treatment; Q7D, weekly treatment.
Re‐bleed defined as a worsening of symptoms in the same location after an initial period of improvement, either on treatment or within 72 hours of stopping treatment.
“Not done,” “not known,” and “missing” were considered unknown. Several patients changed treatment regimen during the trial; therefore, a patient may be included in more than one treatment arm, but only counted once in the total.
An exploratory, post‐hoc analysis of the patients who completed this entire trial (n = 110) showed that the proportion of patients with no bleeding episodes in a given year increased with time on regimen. During the first year of treatment with Q4D prophylaxis, 39.1% of patients experienced no bleeds compared with 56.5% and 64.4% of patients during the fifth and sixth years of treatment, respectively (Figure 2), with little change in patient numbers in years 3‐6. Similar trends were observed for patients on Q7D treatment (Figure 2).
Figure 2.
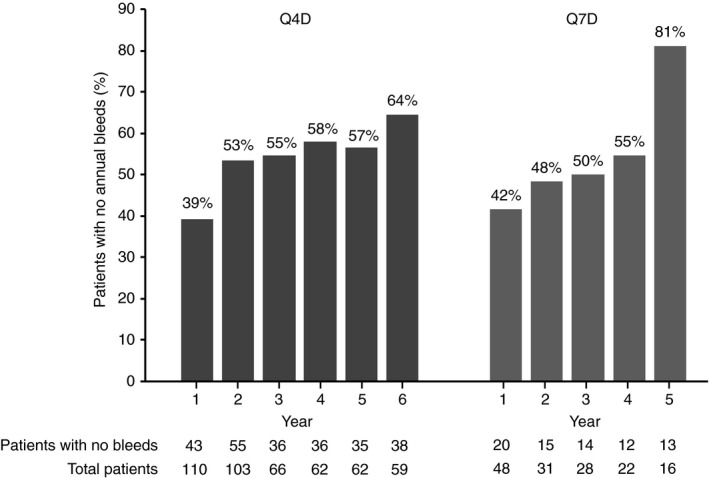
Proportion of patients in pathfinder2 who experienced no bleeds per year. Results from a post‐hoc analysis of patients who completed the entire trial (n = 110) are shown. Only the initial treatment regimen and first consecutive regimen were included in calculations of the proportion of patients who experienced no yearly bleeds. Q4D, every fourth day treatment; Q7D, weekly treatment
In total, 62.9% of all bleeds occurred in joints, with most of the patients with joint bleeds experiencing one or two bleeds during the trial. Nearly all bleeding episodes (98.5%) were classified as mild/moderate. There were no apparent differences between adolescents and adults in terms of bleeds, except for a higher frequency of traumatic bleeds among adolescents (51.4%) compared with adults (32.5%), and a longer duration of bleeds among the adolescents (mean 43.9 hours, standard deviation [SD] 56.7 hours) compared with adults (mean 23.9 hours, SD 42.2 hours).
3.6. Hemostatic response
The success rate for the treatment of all bleeding episodes (with missing responses counted as failure) was 83.2% (95% CI: 79.7; 86.2; Table 3), confirming the hemostatic effect of N8‐GP, as the lower limit of the 95% CI was above 65%. In total, 80.9% of bleeds were treated with 1 injection and 94.9% were treated with ≤2 injections. The success rate for the treatment of bleeds was higher in patients receiving on‐demand treatment than those receiving prophylactic treatment (Table 3), higher for adults (84.0%) than adolescents (77.2%), and similar for spontaneous (82.4%) and traumatic bleeds (84.2%).
Table 3.
Hemostatic response during the pathfinder2 completed trial
|
Prophylaxis 50 IU/kg Q4D (N = 177) |
Prophylaxis 75 IU/kg Q7D (N = 61) |
On‐demand 20‐75 IU/kg (n = 12) |
Total (N = 186) | |
|---|---|---|---|---|
| Bleeding episodes | 1312 | 176 | 1270 | 2758 |
| Hemostatic response, N (%) | ||||
| Excellent | 600 (45.7) | 75 (42.6) | 859 (67.6) | 1534 (55.6) |
| Good | 532 (40.5) | 65 (36.9) | 339 (26.7) | 936 (33.9) |
| Moderate | 153 (11.7) | 29 (16.5) | 71 (5.6) | 253 (9.2) |
| None | 6 (0.5) | 2 (1.1) | 1 (0.1) | 9 (0.3) |
| Missing | 21 (1.6) | 5 (2.8) | 0 (0.0) | 26 (0.9) |
| Success rate a | ||||
| Rate, % (95% CI) | 83.7 (79.8; 87.0) | 80.0 (71.8; 86.3) | 88.6 (81.1; 93.4) | 83.2 (79.7; 86.2) |
| Number of injections to treat bleed, N (%) | ||||
| ≤2 | 1226 (93.4) | 159 (90.3) | 1232 (97.0) | 2617 (94.9) |
| >2 | 86 (6.6) | 17 (9.7) | 38 (3.0) | 141 (5.1) |
Abbreviations: CI, confidence interval; Q4D, every fourth day treatment; Q7D, weekly treatment.
“Missing” included as failure. Several patients changed treatment regimen during the trial; therefore, a patient may be included in more than one treatment arm, but only counted once in the total. Success rate analyzed using logistic regression accounting for repeated measures within subject assuming compound symmetry working correlation.
The mean N8‐GP dose administered from start to stop of a bleed was 55.3 IU/kg; 68.1 IU/kg in the Q4D prophylaxis arm, 88.7 IU/kg in the Q7D prophylaxis arm, and 37.5 IU/kg in the on‐demand arm. The mean N8‐GP dose used from start to stop of a bleed was 90.2 IU/kg in adolescents and 52.9 IU/kg in adults.
3.7. Safety
Among 186 patients exposed to N8‐GP for a total of 785.4 years, one adolescent patient developed a FVIII inhibitor (≥0.6 BU) during the main phase of the trial. Following withdrawal from the trial, the patient returned to his previous FVIII product (see Giangrande et al 8 for full details). Transient N8‐GP‐binding antibodies were detected at a single visit (visit 5) in one patient during the main phase of the trial (see Giangrande et al 8 for full details). A further three patients had pre‐existing non‐neutralizing anti‐N8‐GP antibodies at baseline in the main phase and continued in the trial (see Giangrande et al 8 for full details).
A total of 23 patients tested positive for anti‐PEG antibodies at some point during the trial, with the highest reported anti‐PEG antibody titer being 4. Twelve (6.5%) patients had anti‐PEG antibodies prior to N8‐GP exposure; five of these were still positive at the last visit and the other seven became negative during the course of this trial. Overall, 174 (93.5%) patients had negative measurements for anti‐PEG antibodies at baseline, and of these 163 (87.6%) patients had negative measurements throughout the trial. Of the 11 patients who developed anti‐PEG antibodies during the trial, 8 were negative at the last visit. There was no discernable pattern to the incidence, which occurred at isolated visits. No cases of anti‐PEG antibodies were associated with AEs.
A total of 1827 AEs were reported in 170 patients (91.4%), resulting in a rate of 2.33 AEs per patient‐year of exposure (Table 4). There were no apparent differences in the safety profiles between adolescents and adults or between the different N8‐GP treatment regimens.
Table 4.
Summary of AEs during the pathfinder2 completed trial
|
Prophylaxis 50 IU/kg Q4D (N = 177) |
Prophylaxis 75 IU/kg Q7D (N = 61) |
On‐demand 20‐75 IU/kg (N = 12) |
Total (N = 186) | |
|---|---|---|---|---|
| Exposure days | 57,723 | 7,255 | 1,599 | 66,577 |
| All AEs, N (%) [E] | 158 (89.3) [1,326] | 53 (86.9) [369] | 10 (83.3) [132] | 170 (91.4) [1827] |
| Probably or possibly related | 24 (13.6) [59] | 6 (9.8) [7] | 4 (33.3) [11] | 30 (16.1) [77] |
| SAEs, N (%) [E] | 24 (13.6) [39] | 7 (11.5) [16] | 3 (25.0) [8] | 33 (17.7) [63] |
| Probably or possibly related | 2 (1.1) [2] | — | — | 2 (1.1) [2] |
| AEs by severity, N (%) [E] | ||||
| Mild | 147 (83.1) [1012] | 50 (82.0) [279] | 9 (75.0) [82] | 161 (86.6) [1373] |
| Moderate | 98 (55.4) [273] | 27 (44.3) [71] | 9 (75.0) [40] | 115 (61.8) [384] |
| Severe | 28 (15.8) [41] | 8 (13.1) [19] | 2 (16.7) [9] | 36 (19.4) [69] |
| Not known | — | — | 1 (8.3) [1] | 1 (0.5) [1] |
| MESIs, N (%) [E] | 16 (9.0) [20] | 3 (4.9) [3] | 1 (8.3) [1] | 19 (10.2) [24] |
Abbreviations: AE, adverse event; E, number of events; MESI, medical event of special interest; N, Number of patients with adverse event; Q4D, every fourth day treatment; Q7D, weekly treatment; SAE, serious AE.
Notes: Patients who switched arms are represented in multiple columns. Numbers based on the number of patients treated on each treatment arm at any time during the trial. Exposure days and events are based on the treatment arm at the time of each assessment.
Most AEs were of mild or moderate severity. AEs classified as severe were reported in 19.4% of patients. The most common AEs were nasopharyngitis (incidence 32.3%), headache (23.1%), upper respiratory tract infections (22.6%), arthralgia (22.6%), and diarrhea (13.4%).
In total, 77 AEs in 30 patients (16.1%) were evaluated by the investigator to be possibly or probably related to trial product (0.10 events per patient‐year of exposure), the most common being “investigations” (such as aberrant laboratory values; 36 AEs in 14 patients). Only three of these events were classified as severe: one event of intervertebral discitis, one event of gastritis, and one event of muscle hemorrhage.
A total of 63 SAEs were reported in 33 patients (17.7%). Two of these events in two patients (FVIII inhibitor and intervertebral discitis, reported in the main phase 8 ) were evaluated by the investigator as possibly or probably related to trial product. Six patients were withdrawn from the trial due to SAEs. The one event of FVIII inhibitors in main phase (see Giangrande et al 8 for full details) was considered related to the trial product. The other five SAEs were considered unlikely to be related to N8‐GP treatment (fatal pancreatic metastatic carcinoma in a 67‐year‐old patient, hepatocellular carcinoma, duodenal ulcer, road traffic accident, and ankle fracture).
With regard to MESIs, one suspected thromboembolic SAE (suspicion of cranial micro‐infarction) was reported during the trial; however, there was no definitive diagnosis and a relationship to N8‐GP was reported as unlikely. The final diagnosis was atypical hypertrophic cardiomyopathy and aortic valve stenosis, with both events reported as having an unlikely relationship to N8‐GP. Allergic reactions were reported as MESIs for 14 patients. None of these events required systemic treatment. For two patients, the allergic reactions (rash and mild erythema) were evaluated as possibly or probably related to N8‐GP treatment.
Results of laboratory parameters, vital signs, and other safety‐related examinations did not indicate any clinically relevant changes as a result of N8‐GP treatment.
3.8. Patient‐reported outcomes
Reductions in HAEM‐A‐QOL and Hemo‐Sat score represent improvements in quality of life. Among patients aged ≥17 years old on N8‐GP prophylaxis who participated in extension phase part 2 and completed a questionnaire in the given year of treatment, the mean change in HAEM‐A‐QOL (119 completed questionnaires) from baseline was −3.1 (SD 11.2; 157 questionnaires) after 5–<6 years treatment and −3.5 (SD 9.0) after 6–<7 years treatment (22 questionnaires). Changes in Hemo‐Sat scores for patients who participated in extension phase part 2 and completed a questionnaire in the given year of treatment suggested slight reduction of score in ease and convenience, efficacy, and general satisfaction with both prophylaxis and on‐demand treatment.
Increases in the EQ‐5D‐VAS score indicate a positive change in the patients’ state of health. For prophylaxis patients participating in extension phase part 2 and who completed a questionnaire in the given year of treatment, the mean change in EQ‐5D‐VAS score from baseline was 2.2 (SD 13.2; 167 questionnaires) after 5–<6 years of treatment and 7.5 (SD 13.6; 23 questionnaires) after 6–<7 years of treatment. Mean changes in EQ‐5D utility index score for prophylaxis patients who participated in extension phase part 2 and completed a questionnaire in the given year of treatment were small over the years, indicating that quality of life was maintained during the trial.
4. DISCUSSION
This multi‐national, multi‐center trial was a key element of the pathfinder clinical development program and is one of the largest and most comprehensive clinical trials in hemophilia A treatment. The trial design involved two extension phases, following patients treated with N8‐GP for up to approximately 6.6 years (median 5.4 years) of exposure. The first extension phase provided the opportunity to investigate the safety and efficacy of weekly (Q7D) N8‐GP prophylaxis by randomizing a subset of patients to either Q7D or Q4D treatment. The second extension phase, during which some patients could switch between Q4D and Q7D prophylaxis, aimed to collect long‐term safety and efficacy data.
Overall, almost 30% of the patients on N8‐GP Q4D prophylaxis experienced no bleeds and nearly 40% experienced no spontaneous bleeds over the duration of the entire pathfinder2 trial—a median of approximately 4.0 years on regimen. Due to the duration of this trial, inter‐trial comparisons regarding the proportion of patients with no bleeds in trial are not appropriate. More specifically, the time per patient on Q4D prophylaxis in the present study was approximately five times longer than (and included) the main phase, 8 and seven times longer than other studies that reported proportions of patients with no bleeds in trial. 16 Importantly, the proportion of patients with no bleeds in a given year increased with time on regimen in the Q4D treatment group with relatively little change in overall patient numbers in groups the last 4 years of treatment.
Even though patients spent almost double the amount of time on the Q4D regimen, fewer Q4D patients experienced bleeding episodes compared with Q7D dosing (71.2% versus 86.9%, respectively). Nevertheless, a proportion of the Q7D subgroup (13.1% of patients) experienced no bleeds over a median time of more than 2 years on regimen. The extension phase part 2 of the pathfinder2 trial was non‐randomized, and the treatment‐group size imbalances, as well as pre‐selection of patients with a low bleeding rate for Q7D dosing preclude a direct comparison of the two prophylaxis regimens similar to that performed in extension phase part 1. 9 Despite this limitation, these end‐of‐trial results support the previous finding that the Q7D N8‐GP prophylaxis regimen may be appropriate for patients who bleed at a lower rate.
As in the main phase and extension phase part 1, 8 , 9 the prophylactic effect of both Q4D and Q7D N8‐GP dosing was demonstrated in the analysis of data from the completed trial. As expected, the median ABR among patients who received on‐demand N8‐GP treatment (30.98) was higher than among the patients treated prophylactically (Q4D, 0.84; Q7D, 1.64), although, as previously noted, there were imbalances in treatment‐group sizes and the extension phase part 2 of the trial was non‐randomized.
The observed ABRs (Q4D, 2.14; Q7D, 1.31) in the completed study comprising all phases were comparable to those reported in the 24‐week randomized extension phase part 1 (Q4D, 1.66; Q7D, 1.65), 9 and complete trial ABRs for Q4D prophylaxis were lower than those reported for the main phase (3.04). 8 Other studies of FVIII replacement products often only report median observed ABRs. 17 , 18 , 19 Thus, the median observed ABR is the most reliable value for inter‐trial comparison. The Q4D prophylaxis arm in the completed study showed a slightly lower median observed ABR compared with similar regimens reported for other EHL FVIII replacement products. 17 , 18 , 19
In total, 80.9% of bleeds were treated with 1 injection of N8‐GP and 94.9% were treated with ≤2 injections, a rate similar to the main phase results 8 and to that reported for another EHL FVIII product. 17 The incidence of severe bleeds in the completed study was just 1.3% on any regimen, and the success rate for treatment of all bleeds (83.2% [95% CI: 79.7; 86.2]) demonstrated the hemostatic effect of N8‐GP according to the predefined criteria and was consistent with that reported for other commercially available FVIII products. 17 , 20 , 21 , 22 , 23 , 24
A greater proportion of patients in the on‐demand group experienced spontaneous bleeds than in the prophylaxis groups. This observation may involve multiple contributing factors. Patients receiving prophylactic treatment may exhibit fewer spontaneous bleeds due to better treatment coverage and a possible improvement in joint status due to fewer bleeds with greater time on regimen. Furthermore, patients may feel more protected with prophylaxis and have more active lifestyles, thus leading to a shift in the proportions of spontaneous and traumatic bleeds.
Trends possibly attributable to lifestyle choices might also be reflected in the results obtained for the adolescent and adult subgroups. Adolescents, who may typically have more active and higher‐risk lifestyles, had slightly higher ABRs than adults in both the Q4D and Q7D prophylaxis arms. Adolescents also reported a higher frequency of traumatic bleeds (51.4%) compared with adults (32.5%) and had a longer duration of bleeds.
Overall, for patients on Q4D prophylaxis, the mean FVIII trough level across all patients was 3.1 IU/dL, with only 17.8% of 3012 measurements being below the lower level of quantitation, meaning that most of the patients were in the range of moderate/mild hemophilia A most of the time on this regimen. Furthermore, incremental recovery was stable during extension phase part 2, with mean values between 2.0 and 3.0 (IU/dL)/(IU/kg), indicating sustained efficacy. This, combined with the efficacy results of the Q4D prophylaxis regimen, indicates that a simple, fixed‐dose N8‐GP regimen without the need for FVIII activity monitoring is possible for the management of most patients with hemophilia A.
PROs, quality of life, and treatment satisfaction scores showed minor changes with long‐term N8‐GP treatment. For adults on prophylaxis, there were small improvements in HAEM‐A‐QOL, Hemo‐Sat, and EQ‐5D‐VAS scores. The EQ‐5D utility index score was maintained over the course of the trial.
Overall, in the pathfinder2 trial, 186 patients were exposed to N8‐GP for a total of 785.4 years. No safety concerns were detected from the assessment of antibodies, AEs, vital signs, physical examinations, or clinical laboratory parameters. As previously reported, a single patient developed a FVIII inhibitor (≥0.6 BU) after 93 exposure days. As this incidence was below the pre‐specified limit of 6.8%, this primary test demonstrated adequate safety with regard to inhibitor development. 8 Of the 23 patients who tested positive for anti‐PEG antibodies, only 11 patients developed anti‐PEG antibodies during the trial, and there was no discernable pattern to the incidence, which occurred at isolated visits, while none of these antibodies were associated with a clinical effect.
In conclusion, with 186 patients exposed to N8‐GP for a median time on regimen of approximately 5.4 years, pathfinder2 was one of the largest and longest‐running clinical trials on the treatment of severe hemophilia A. N8‐GP allowed for effective, well‐tolerated, fixed‐dose, prophylactic treatment every 4 days without the need to monitor FVIII activity levels for dose adjustment. Data from the complete pathfinder2 trial confirm the long‐term efficacy and safety of N8‐GP treatment among previously treated adolescent and adult patients with severe hemophilia A.
CONFLICTS OF INTEREST
PG has received speaker and consultancy fees from Bayer, CSL Behring, Novo Nordisk, and Pfizer. FAK reports nothing to disclose. LN has received speaker or consultancy fees from Bayer, Baxalta, CSL Behring, Novo Nordisk, Pfizer, and Octapharma. CWY reports nothing to disclose. MSG and AL are employees and stockholders of Novo Nordisk A/S. NC has led research for CSL Behring and received speaker or consultancy fees from LFB, Bayer, Sobi, Novo Nordisk, and Pfizer.
AUTHOR CONTRIBUTIONS
P. Giangrande, F. Abdul Karim, L. Nemes, C. W. You, M. S. Geybels, A. Landorph, and N. Curry contributed to the writing and review of the manuscript and approved the final version.
Supporting information
Table S1
ACKNOWLEDGMENTS
The authors thank the patients who participated in the trial and their families as well as all investigators, co‐investigators, and trial nurses who were involved in pathfinder2. This trial was sponsored by Novo Nordisk A/S. The authors would also like to thank Soraya Benchikh el Fegoun and Ekaterina Gresko from Novo Nordisk for their review and input to the manuscript. This research was supported by Novo Nordisk. Medical writing support was provided by Physicians World Europe GmbH, Mannheim, Germany and was financially supported by Novo Nordisk A/S. Please find a full list of investigators and countries in the Table S1.
Giangrande P, Abdul Karim F, Nemes L, et al. Long-term safety and efficacy of N8-GP in previously treated adults and adolescents with hemophilia A: Final results from pathfinder2. J Thromb Haemost. 2020;18(Suppl. 1):5–14. 10.1111/jth.14959
Trial registration: NCT01480180
Manuscript handled by: David Lillicrap
Final decision: David Lillicrap, 4 June 2020
REFERENCES
- 1. Khawaji M, Astermark J, Berntorp E. Lifelong prophylaxis in a large cohort of adult patients with severe haemophilia: a beneficial effect on orthopaedic outcome and quality of life. Eur J Haematol. 2012;88:329‐335. [DOI] [PubMed] [Google Scholar]
- 2. Fischer K, Steen Carlsson K, Petrini P, et al. Intermediate‐dose versus high‐dose prophylaxis for severe hemophilia: comparing outcome and costs since the 1970s. Blood. 2013;122:1129‐1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1‐e47. [DOI] [PubMed] [Google Scholar]
- 4. Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first‐in‐human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11:670‐678. [DOI] [PubMed] [Google Scholar]
- 5. Medical and Scientific Advisory Council . MASAC recommendation #241 concerning prophylaxis 2016; Available from: https://www.hemophilia.org/Researchers‐Healthcare‐Providers/Medical‐and‐Scientific‐Advisory‐Council‐MASAC/MASAC‐Recommendations/MASAC‐Recommendation‐Concerning‐Prophylaxis
- 6. Oldenburg J, Albert T. Novel products for haemostasis ‐ current status. Haemophilia. 2014;20(Suppl 4):23‐28. [DOI] [PubMed] [Google Scholar]
- 7. Meunier S, Alamelu J, Ehrenforth S, et al. Safety and efficacy of a glycoPEGylated rFVIII (turoctocog alpha pegol, N8‐GP) in paediatric patients with severe haemophilia A. Thromb Haemost. 2017;117:1705‐1713. [DOI] [PubMed] [Google Scholar]
- 8. Giangrande P, Andreeva T, Chowdary P, et al. Clinical evaluation of glycoPEGylated recombinant FVIII: Efficacy and safety in severe haemophilia A. Thromb Haemost. 2017;117:252‐261. [DOI] [PubMed] [Google Scholar]
- 9. Curry N, Albayrak C, Escobar M, et al. Once‐weekly prophylaxis with glycoPEGylated recombinant factor VIII (N8‐GP) in severe haemophilia A: Safety and efficacy results from pathfinder 2 (randomized phase III trial). Haemophilia. 2019;25:373‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Curry N, Chowdary P, Clausen WH, et al. Long‐ term Safety of N8‐ GP (turoctocog alfa pegol) Prophylaxis in Patients with Severe Haemophilia A. Res Pract Thromb Haemost. 2019;3:420.31294330 [Google Scholar]
- 11. Turoctocog alfa pegol summary of product characteristics: European Medicines Agency; Available from: https://www.ema.europa.eu/en/documents/product‐information/esperoct‐epar‐product‐information_en.pdf
- 12. Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser‐Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII: C inhibitors: improved specificity and reliability. Thromb Haemost. 1995;73:247‐251. [PubMed] [Google Scholar]
- 13. Giles AR, Verbruggen B, Rivard GE, Teitel J, Walker I. A detailed comparison of the performance of the standard versus the Nijmegen modification of the Bethesda assay in detecting factor VIII: C inhibitors in the haemophilia A population of Canada. Association of Hemophilia Centre Directors of Canada. Factor VIII/IX Subcommittee of Scientific and Standardization Committee of International Society on Thrombosis and Haemostasis. Thromb Haemost. 1998;79:872‐875. [PubMed] [Google Scholar]
- 14. Kearney S, Raffini LJ, Pham TP, et al. Health‐related quality‐of‐life and treatment satisfaction of individuals with hemophilia A treated with turoctocog alfa pegol (N8‐GP): a new recombinant extended half‐life FVIII. Patient Prefer Adherence. 2019;13:497‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hampton K, Chowdary P, Dunkley S, et al. First report on the safety and efficacy of an extended half‐life glycoPEGylated recombinant FVIII for major surgery in severe haemophilia A. Haemophilia. 2017;23:689‐696. [DOI] [PubMed] [Google Scholar]
- 16. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377:809‐818. [DOI] [PubMed] [Google Scholar]
- 17. Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reding MT, Ng HJ, Poulsen LH, et al. Safety and efficacy of BAY 94–9027, a prolonged‐half‐life factor VIII. J Thromb Haemost. 2017;15:411‐419. [DOI] [PubMed] [Google Scholar]
- 19. Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full‐length, recombinant factor VIII for prophylactic and on‐demand treatment of severe hemophilia A. Blood. 2015;126:1078‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abshire TC, Brackmann HH, Scharrer I, et al. Sucrose formulated recombinant human antihemophilic factor VIII is safe and efficacious for treatment of hemophilia A in home therapy–International Kogenate‐FS Study Group. Thromb Haemost. 2000;83:811‐816. [PubMed] [Google Scholar]
- 21. Lusher JM, Lee CA, Kessler CM, Bedrosian CL, ReFacto Phase 3 Study G . The safety and efficacy of B‐domain deleted recombinant factor VIII concentrate in patients with severe haemophilia A. Haemophilia. 2003;9:38‐49. [DOI] [PubMed] [Google Scholar]
- 22. Tarantino MD, Collins PW, Hay CR, et al. Clinical evaluation of an advanced category antihaemophilic factor prepared using a plasma/albumin‐free method: pharmacokinetics, efficacy, and safety in previously treated patients with haemophilia A. Haemophilia. 2004;10:428‐437. [DOI] [PubMed] [Google Scholar]
- 23. Valentino LA, Mamonov V, Hellmann A, et al. A randomized comparison of two prophylaxis regimens and a paired comparison of on‐demand and prophylaxis treatments in hemophilia A management. J Thromb Haemost. 2012;10:359‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lentz SR, Misgav M, Ozelo M, et al. Results from a large multinational clinical trial (guardian1) using prophylactic treatment with turoctocog alfa in adolescent and adult patients with severe haemophilia A: safety and efficacy. Haemophilia. 2013;19:691‐697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1