

Abstract:
Myocardial infarction (MI) is an irreversible damage of the heart muscle, which often leads to adverse cardiac remodeling and progressive heart failure. After MI, immune cells play a vital role in the clearance of the dying tissue and cardiac remodeling. Post-MI events include the release of danger signals by necrotic cardiomyocytes and the migration of the inflammatory cells, such as dendritic cells, neutrophils, monocytes, and macrophages, into the site of the cardiac injury to digest the cell debris and secrete a variety of inflammatory factors activating the inflammatory response. In this review, we focus on the role of immune cells in the cardiac remodeling after MI and the novel immunotherapies targeting immune cells.
Key Words: macrophages, neutrophil, dendritic cells, lymphocytes, myocardial infarction, immunotherapy
INTRODUCTION
Myocardial infarction (MI), a permanent damage to the heart muscle, often leads to progressive heart failure (HF) due to the adverse cardiac remodeling (Table 1 for abbreviations). Following MI, the necrotized myocardium is eliminated by inflammatory cells and replaced by a granulation tissue, which eventually becomes collagen scars. Although the stable collagen scars, which replaced the necrotized myocardium, could maintain the cardiac function, a massive MI usually results in progressive adverse cardiac remodeling, which leads to HF.1 The inflammatory response and immune cells play a vital role in the cardiac remodeling following MI.2
TABLE 1.
Nonstandard Abbreviations and Acronym
| MI | Myocardial infarction |
| DCs | Dendritic cells |
| HF | Heart failure |
| DAMPs | Danger-associated molecular patterns |
| ROS | Reactive oxygen species |
| ECM | Extracellular matrix |
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| Th | Helper T cell |
| CD | Cluster of differentiation |
| CCR | C-C chemokine receptor |
| MHC | Major histocompatibility complex |
| Ly6C | Lymphocyte antigen 6 complex |
| LV | Left ventricular |
| IRF | Interferon regulatory factor |
| MR | Mineralocorticoid receptor |
| IFN | Interferon |
| IP | Interferon-inducible protein |
| BRG | Brahma-related gene |
| EF | Ejection fraction |
| I/R | Ischemia–reperfusion |
| XCR | Chemokine (C motif) receptor |
| Batf | Basic leucine zipper ATF-Like transcription factor |
| IRF | Interferon regulatory factor |
| BDCA | Blood dendritic cell antigen |
| APCs | Antigen-presenting cells |
| DTR | Diphtheria toxin receptor |
| DT | Diphtheria toxin |
| DEXs | Dendritic cell exosomes |
| cDCs | Conventional dendritic cells |
| pDCs | Plasmacytoid dendritic cells |
| MyHC | Myosin heavy chain |
| Foxp3 | Forkhead box P3 |
| ATR2 | Angiotensin II receptor 2 |
| Ig | Immunoglobulin |
| MPO | Myeloperoxidase |
| IRAK | Interleukin-1 receptor–associated kinase |
| tDCs | Tolerogenic dendritic cells |
| Tregs | Regulatory T cells |
The healing process of the heart following MI includes 3 overlapping phases: inflammatory, healing, and remodeling3 (Fig. 1). At the early stage after MI, the necrotic cardiomyocytes and cell debris release danger-associated molecular patterns (DAMPs) and cytokines; these attract the innate immune cells, including the neutrophils, monocyte/macrophages, and dendritic cells (DCs) into the infarct area. Neutrophils generate high levels of reactive oxygen species and proteases, which exacerbate the injury of local vessels and tissue.4 At the same time, neutrophils and proinflammatory monocytes promote clearing the cell debris and digesting the extracellular matrix (ECM),5 which are necessary for wound healing. Subsequently, monocyte/macrophages are recruited in the infarcted myocardium to remove the debris and apoptotic neutrophils; this leads to the activation of reparative pathways, which are necessary for the scar formation.2 Beside the unspecific inflammation, an emerging evidence indicates that lymphocyte-mediated immune responses also contribute to the cardiac remodeling after MI.3 After the early inflammatory phase, new ECM and blood vessels are rebuilt. At this stage, T and B lymphocytes massively infiltrate into the infarct area,6 and the local macrophages switch their phenotypic polarization to support the healing process rather than the inflammation reaction.7 Because the adult mammalian heart has a negligible regenerative capacity, MI often results in structural and functional changes in the heart, including scar formation, hypertrophy of the cardiomyocytes, and ventricular chamber dilation.8 Sufficient fibrous scar tissue after MI is important to maintain the cardiac function and avoid a cardiac rupture. However, the excessive fibrosis and remodeling often result in a progressive HF.9
FIGURE 1.
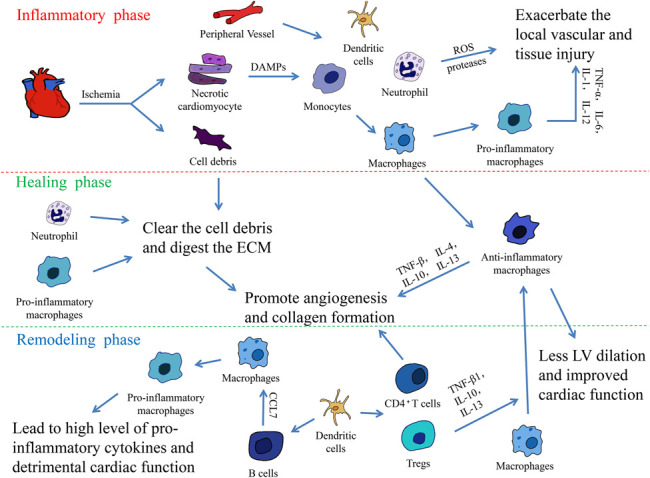
The 3 overlapping phases in the heart healing following MI. The inflammatory response following MI can be divided into 3 overlapping phases: The inflammatory, healing, and remodeling phases. In the inflammatory phase, the necrotic cardiomyocytes release DAMPs, HMGB1, extracellular RNA/eRNA, IL-1α, and other danger signals. These signals attract the innate immune cells, monocytes, neutrophils, and dendritic cells, which infiltrate into the infarct area from the peripheral vessels. At the early stage of MI, monocytes are quickly transformed into mature macrophages and they polarize into proinflammatory macrophages. The macrophages secrete various proinflammatory cytokines, including TNF-α, IL-1, IL-6, and IL-12, and contribute to the acute inflammatory response. Meanwhile, neutrophils generate high levels of reactive oxygen species and proteases, which exacerbates the injury of local vessels and tissue. After the inflammatory phase, neutrophils and proinflammatory monocytes promote the clearance of cell debris and digestion of ECM, preparing for wound healing. At the same time, local macrophages switch their phenotypic polarization to anti-inflammatory macrophages and promote angiogenesis and collagen formation via the secretion of anti-inflammatory cytokines, including TNF-β, IL-4, IL-10, and IL-13. At the terminal stage of MI, T and B lymphocytes massively infiltrate into the infarct area. Activated by DCs, CD4+ T cells facilitate wound healing of the myocardium, and regulatory CD4+ T cells (Tregs) improve healing after MI by producing TGF-β1, IL-13, and IL-10 to induce M2-like differentiation. The recruitment of B lymphocytes induces the mobilization of proinflammatory monocytes and leads to tissue injury and the deterioration of the myocardial function.
Therefore, it is urgent to investigate how to prevent adverse cardiac remodeling and HF, which requires a deep understanding of the immunological mechanisms following MI. In this review, we focus on the roles of immune cells, including the monocyte/macrophages, neutrophils, dendritic cells, and lymphocytes in the cardiac remodeling following MI.
The Role of Monocytes/Macrophages Following MI
Monocytes/macrophages play a central role in the innate immune response, inflammation, and host defense. After MI, monocytes massively infiltrate into the infarct area and differentiate into macrophages; these represent the major source of infarct macrophages during the first 7 days post-MI.10 The macrophages are generally divided into 2 subsets: activated macrophages (M1) and alternatively activated macrophages (M2).11 M1 macrophages express specific surface markers, such as CD68 and CD86. These cells are called proinflammatory cells because they secrete various proinflammatory cytokines, such as the tumor necrosis factor-α (TNF-α), IL-6, IL-1, and IL-12 and stimulate the activity of helper T cell-1 (Th1) during immune responses. On the other hand, M2 macrophages express different specific surface markers, such as CD206 and CD163. These cells are considered to be anti-inflammatory cells because they promote the Th2 activity during immune responses.12 Murine cardiac macrophages (CD45+CD11b+F4/80+CD64+ or CD68+) are further categorized by the expression of C-C chemokine receptor type 2 (CCR2), major histocompatibility complex II (MHC-II), and the lymphocyte antigen 6 complex (Ly6C).13 Ma et al14 reported that the M1 and M2 macrophages originate from the circulating Ly6Chigh and Ly6Clow monocytes, respectively, and the recruitment of Ly6Chigh monocytes depends on the CCR2 signaling, whereas the recruitment of Ly6Clow monocytes is CX3CR1 dependent. However, it must be acknowledged that so far, there is no consensus on the phenotypic classification of macrophages; thus, the respective function of the macrophages subsets is still controversial.13
Macrophages are critical players in the inflammation resolution/perpetuation and cardiac remodeling in the infarcted myocardium.14,15 A previous study reported that the macrophage depletion impaired wound healing and resulted in a high mortality rate accompanied by an increase in the left ventricular (LV) dilatation and wall thinning in murine.16 On the other hand, the adoptive transfer of activated macrophages improved the scar thickening and reduced the LV diastolic dilatation.17 These studies indicated the vital role of macrophages in the post-MI cardiac remodeling and the different functions of macrophages belonging to different subsets. Nahrendorf et al18 reported that the Ly6Chigh monocytes accumulated via CCR2 and dominated at the site of injury during the first 3 days, whereas the Ly6Clow monocytes preferentially accumulated via CX3CR1 from day 5 onward in MI mice models. Furthermore, they revealed that the Ly6Chigh monocytes removed the necrotic debris by the combination of the expression of inflammatory mediators, proteolysis, and phagocytosis, whereas the Ly6Clow monocytes promoted the reparative processes, such as the angiogenesis and ECM deposition.18 Overall, these observations are similar to the above-mentioned functions of M1 and M2 monocytes.
In addition, the macrophage polarization regulators, such as the interferon regulatory factor-5 (IRF5) and myeloid mineralocorticoid receptor, have been shown to be involved in the cardiac remodeling. Courties et al19 reported that in vivo silencing of the transcription factor IRF5 reduced the M1 macrophage subset polarization and diminished the post-MI increase in the LV volume. However, Hu et al20 reported that class A scavenger receptor played a protective role against MI by suppressing the polarization of the M1 macrophage subset.
These studies have shown that macrophages can be both protective and detrimental after MI. Although M1 macrophages are considered to be proinflammatory cells playing a detrimental role in the cardiac remodeling, M2 macrophages are considered to be antiinflammatory cells playing a protective role in the cardiac remodeling. Thus, macrophages could be a potential therapeutic target, which can influence the healing and ventricular remodeling after MI.
The Role of Neutrophils following MI
Neutrophils are commonly defined as CD45+CD11b+Ly6C+ cells.6 Similar to the M1 and M2 phenotypes of macrophages, neutrophils are divided into 2 subsets: N1 and N2. N1 neutrophils are defined as Ly6G+CD206− and express high levels of proinflammatory IL-12, whereas N2 neutrophils are defined as Ly6G+CD206+ and express high levels of anti-inflammatory IL-10.21 Ma et al21 reported the existence of N1 and N2 neutrophils in the infarct area. They found that N1 is the predominant neutrophil phenotype in the infarct area after MI (>80% of the total neutrophils), whereas the percentage of N2 neutrophils increased over time, which indicated that their role results in anti-inflammatory and wound repairing effects, similar to M2 macrophages. Neutrophils are vital for the clearance of pathogens or debris and also for the resolution of inflammation and returning to tissue homeostasis in acute inflammation.22
Previous studies reported the role of neutrophils after MI. Horckmans et al23 reported that the depletion of neutrophils worsened the cardiac function. The characteristic features of neutrophil depletion were larger end-systolic LV dimensions and a significant reduction of the LV ejection fraction and cardiac output. Furthermore, they found that the number of Ly6Chigh monocytes was decreased in neutrophil-depleted mice, whereas the proliferation of the cardiac macrophages was enhanced. Meanwhile, the macrophage subtype analysis revealed that the cardiac expression of M1 markers (IL-12, TNF-α, IFN-γ, IP-10, IL-1β) was reduced, whereas the M2 markers (CX3CR1, arginase, YM1, IL-4) were increased in neutrophil-depleted mice; they then further confirmed that the neutrophil secretome promoted the macrophage polarization toward M2.23
In addition, it was recently reported by Zhang et al24 that the deletion of the endothelial brahma-related gene 1 (BRG1), which could mediate the neutrophil endothelium adhesion, reduces the infarct size and LV fibrosis and improves the ejection fraction following ischemia–reperfusion (I/R) in mice, which was attributed to the reduction of neutrophil infiltration in the infarct area. Overall, neutrophils play a detrimental role after MI. However, the function of the different subsets of neutrophils needs to be further explored.
The Role of DCs in the Regulation of Post-MI Inflammation and Ventricular Remodeling
Dendritic cells are generally classified into 2 subsets: conventional dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs). cDCs could be further divided into cDC1 and cDC2,25–27 whereas CD11c is expressed on all the DC subsets. cDC1 cells express CD103 and XCR-1 and depend on the Batf3 and IRF8 transcription factors; cDC2 cells express CD11b and CD172α and depend on the IRF4 transcription factor,28 and all the cDCs subsets depend on the Zbtb46 transcription factor.29 In pDCs, BDCA2 and CD123 are highly expressed, and these cells depend on the E2-2 transcription factor and others.30 As the most potent antigen-presenting cells (APCs), DCs are regarded as the bridge between innate and adaptive immunity and are thus termed as professional APCs.31
Nagai et al32 investigated 24 autopsy subjects after an ST-elevation MI, and they found that the decrease in the number of DCs in the human infarcted myocardial tissue was associated with the increased macrophage infiltration, impaired reparative fibrosis, and development of cardiac rupture after MI. These findings suggest that DCs have a protective role in post-MI inflammation and the subsequent healing process.32 In addition, Anzai et al33 transferred bone marrow cells from CD11c diphtheria toxin receptor (CD11c-DTR) transgenic mice into lethally irradiated wild-type recipient mice and used the diphtheria toxin (DT) to eliminate the CD11c+ DCs in the recipient mice before the ligation of the left coronary artery. They found that the DC deletion group demonstrated an enhanced and sustained expression of the inflammatory cytokines, such as IL-1β, IL-18, and TNF-α, compared with the control group. The mice with DC deletion showed an acceleration in the cardiac dilatation and deterioration of the LV function.33 Meanwhile, they revealed that the DC deletion enhanced the infiltration of proinflammatory Ly6Chigh monocytes and impaired the recruitment of anti-inflammatory Ly6Clow monocytes in the infarct area. These results suggest that DCs play a protective role during the healing process after MI via its control of the monocyte/macrophage homeostasis.33 In addition, our previous study has showed that the injection of MI-DEXs, which are derived from the dendritic cells, improved the post-MI cardiac function in mice via mediating the activation of CD4+ T cells through an endocrine mechanism.34 Our findings also support the protective role of DCs after MI, but different results were reported when investigating the functions of the different DCs subsets.
Lee et al35 used the DT treatment to eliminate the CD103+ and CD11b+ cDCs in MI mice that were reconstituted with bone marrow cells from Zbtb46-DTR mice. Then, they found that these DT-treated Zbtb46-DTR mice showed a dramatic reduction in the infarct size, via preventing the ventricular remodeling, and a significantly improved LV fractional shortening compared with the phosphate-buffered saline–treated control group.35 This result suggests that cDCs play a detrimental role in wound healing and may even expand the infarct size. However, another study crossed mice expressing CRE recombinase under the control of the CD11c promoter (Cd11cCre mice) with Irf8flox or Irf4flox mice to generate mice that are lacking IRF8 or IRF4 expression to selectively eliminate the cDC1 or cDC2. They found that cDC1s generate myosin-specific regulatory T cells (Tregs) in the heart-draining lymph node, whereas cDC2s are the main presenters of αMyHC to CD4+ T cells.28 The activation of Treg has been proven to improve the heart healing after MI by triggering the differentiation of M2-like macrophages. Treg-cell ablation in Foxp3-DTR mice resulted in an increased infarct size, LV dilation, and impaired cardiac function.36 These findings suggested that cDCs play a protective role in the cardiac remodeling.
To assess the role of pDCs after MI, Lee et al35 gave the DT treatment to selectively eliminate the heart pDCs in MI mice models that were reconstituted with bone marrow cells from BDCA2-DTR mice. They found that the left ventricle function was not affected in the mice without pDCs. The function of cardiac pDCs may lie in their capacity to produce type I IFN and protect the tissue against viral infections.35
The Role of T-Lymphocytes in Ventricular Remodeling After MI
T lymphocytes are generally divided into cytotoxic (CD8+) and helper (CD4+) T-cells. T-helper cells could be further subdivided into 2 subsets: Th1 and Th2.37 Besides the CD4+ and CD8+ T cell subsets, regulatory CD4+ T cells (Tregs) have also been characterized based on their ability to counterbalance the classical immune responses.38 Tregs, which express the Forkhead box P3 (Foxp3) transcription factor, are important components to maintain the immune system homeostasis and suppress the pro-inflammatory immune responses.38
Maisel et al39 demonstrated that the adoptive transfer of splenocytes from rats after MI produced myocarditis in naïve recipient rats, which was the first experimental evidence that myocardial autoantigens could activate T cells. Then, Hofmann et al40 showed that both conventional effector CD4+ T cells and Foxp3+ Tregs have infiltrated into the myocardium within a few days after MI, they found that the absence of CD4+ T cells in CD4-deficient or MHC II-deficient mice was associated with a worse outcome. They speculated that the activation of CD4+ T cells is driven by the recognition of cardiac autoantigens via MHC II+ cells, and it facilitates the myocardium wound healing.40 Anzai et al33 provided another important evidence regarding the interaction between CD4+ T cells and DCs after MI. They removed CD11c+ DCs from a transgenic mouse MI model and found similar results to those in the CD4+ T cells-deficient mice model. The absence of CD11c+ DCs resulted in a deterioration in the LV function and remodeling after MI.33 However, the mechanism of CD11c+ DCs in the activation of CD4+ T cells was not further explored in this study.
To further explore the role of CD4+ T cells after MI, Hofmann et al40 performed a permanent coronary artery ligation in CD4 knockout mice, which lack the CD4+ T cells, and found that the absence of CD4+ T cells increased the number of Ly6Chigh monocytes in the infarct area and promoted the inflammatory response. Meanwhile, they also found that the CD4+ T cells–deficient mice showed disturbed scar formation, increased LV dilation, and higher mortality. These results suggest that the CD4+ T cells play a protective role after MI.
Saxena et al41 reported that Tregs were found in the infarcted myocardium after 24 hours of reperfusion. Additionally, they found that Treg-depleted mice showed accentuation of the postinfarction inflammatory response, increased collagen deposition, and significant ventricular dilation after MI. Similar results were reported in another study indicating that the adoptive transfer of Tregs in MI mice attenuated both the postinfarction inflammatory response and adverse remodeling.42 To further explore the mechanism behind these results, Saxena et al41 found that Tregs may modulate the fibroblast phenotype and function in the infarcted myocardium. Weirather et al36 proved that Foxp3+ Tregs improve the healing after MI by modulating the differentiation of monocytes/macrophages. They showed that the depletion of Tregs induced the M1-like differentiation, whereas their activation produced TGF-β1, IL-13, and IL-10 to induce M2-like differentiation. Nevertheless, these findings are not sufficient to fully explain the protective role of Tregs, and further exploration is still needed.
As for CD8+ T cells, some recent studies reported their role in the cardiac remodeling after MI. Curato et al43 reported that a subset of CD8+ T cells expressing the angiotensin II receptor 2 (ATR2) accumulated in the infarct area of rats at day 7 after MI, expanding the response to myocardial ischemic injury and producing IL-10 in response to angiotensin II stimulation, which differed from the classical CD8+AT2R− CD8+ T cells. Further examination revealed that the intramyocardial transplantation of these CD8+AT2R+ T cells lead to a significant reduction in the infarct volume.43 Although these results indicated the protective role of CD8+AT2R+ T cells after MI, further studies need to be done in the future.
The Role of B Lymphocytes in Ventricular Remodeling After MI
B lymphocytes are generally classified into 2 subsets: B1 and B2 cells. The long-lived and self-renewing B1 cells produce antibodies, which are crucial for defending against encapsulated bacteria, whereas B2 cells are activated upon encountering an antigen, and they expand and generate short-lived plasma cells. Some activated B2 cells will generate memory cells and long-lived plasma cells in the germinal center. Long-lived plasma cells populate the bone marrow.44 In the course of maturation, B cells express different sets of surface molecules, such as CD19, CD20, CD52, and CD22, which help to distinguish between different developmental stages.45 Furthermore, CD19 and CD22 are highly expressed on B1 cells, whereas CD20 are highly expressed on B2 cells.46
Previous studies showed that CD19+ IgD+ IgMlow B cells are recruited to the murine infarcted myocardium after MI, whereas the CD20+ B cells have been found in the human heart biopsy from MI patients at day 1 and day 6.6,47 To assess the role of B lymphocytes in the cardiac remodeling after MI, Zouggari et al depleted B lymphocytes using a CD20-specific monoclonal antibody (CD20 mAb). They found that compared with the control mice, the deletion of B lymphocytes led to smaller infarct size, lower levels of proinflammatory cytokines, and significant improvement in the cardiac function after MI. Furthermore, they described that the CCl2 levels were reduced by anti-CD20. Previous studies described that CCL2, a major CCR2 ligand together with CCL7, is a chemokine that plays a pivotal role in the MI by inducing monocyte mobilization and ventricular and vascular remodeling.48–50 Therefore, we could infer that B lymphocytes induce the mobilization and recruitment of Ly6Chigh monocytes to the infarcted myocardium, which leads to tissue injury and the deterioration of the myocardial function.47
However, another study demonstrated that the rats receiving intramyocardial injections of B cells showed an improved LV function compared with the sham-injected control animals.51 These conflicting results indicated that further investigation is required to get more insights into the role and mechanism of B cell recruitment to the infarct region after MI.
Immunotherapy for MI
The rising use of immunotherapy has revolutionized the treatment of cancer and autoimmune diseases,52,53 and targeting inflammation has also become a realistic goal in the treatment of cardiovascular disease. Immune cells play a vital role in the cardiac remodeling; thus, immunotherapies targeting different immune cells have attracted more attention.
Approaches Targeting Macrophages and Neutrophils
Duerr et al54 have proved that the endocannabinoid-CB2 receptor axis protected the ischemic heart via modulating the macrophage polarization (M1 to M2) and attenuated the inflammatory response and adverse cardiac remodeling after myocardial ischemia and reperfusion. Vasilyev et al55 reported that the deletion of myeloperoxidase (MPO), which is predominantly produced by neutrophils and Ly6Chigh monocytes when they infiltrate into the infarct area, could reduce the LV dilatation and dysfunction, but it has no effect on the infarct size. Ali et al56 found that PF-1355, an MPO inhibitor, improves the ventricular function and remodeling after MI, and a longer treatment period could even restore the cardiac function.
Approaches Targeting Dendritic Cells and T Lymphocytes
Maekawa et al57 reported that the deletion of the interleukin-1 receptor–associated kinase 4 (IRAK-4) has favorable effects on the survival and LV remodeling after MI. These effects are achieved via the modification of the host inflammatory process by blunting the mobilization of the detrimental bone marrow dendritic cells after myocardial ischemia. Choo et al58 injected tolerogenic dendritic cells, which could modulate the immune responses and induce Tregs, to MI mice and found that the tolerogenic dendritic cells treatment could activate Tregs and elicit an inflammatory-to-reparative macrophage shift. In addition, our previous study has showed that the injection of MI-DEXs improved the cardiac function in mice after MI via mediating the activation of CD4+ T cells through an endocrine mechanism.34 Matsumoto et al42 reported that the adoptive transfer of Tregs in MI mice attenuated both the postinfarction inflammatory response and adverse remodeling. Similar results were reported from another study, which showed that promoting the Treg expansion using an anti-CD28 superagonist or performing Treg adoptive cell transfer improved the myocardial repair.59
Approaches Targeting B Lymphocytes
Although several approaches targeting B lymphocytes have been reported, they showed contradicting results. Goodchild et al51 found that the intramyocardial injection of B lymphocytes into early postischemic myocardium preserved the cardiac function. However, the anti-CD20mAb (Rituximab) that was used by Zouggari et al47 to deplete the mature B lymphocytes led to smaller infarct size, lower levels proinflammatory cytokines, and significant improvement in the cardiac function.
However, it should be noticed that this field is still in its infancy, and such novel therapeutic approaches are still in a premature stage.
CONCLUSIONS
Due to the poor regenerative capacity of the adult mammals' heart, the injured myocardium is replaced with a fibrous tissue. Although this reaction maintains and restores the integrity of the heart, it often leads to adverse cardiac remodeling and progressive HF. The inflammatory response plays a vital role in this process, but it is a double-edged sword.60 After the MI, large numbers of immune cells are recruited to the heart to remove the dying tissue and promote the healing process. However, this is done under some circumstances because the immune cells can cause an irreversible damage, contributing to the HF.61 Therefore, it is crucial to deeply understand the cellular and molecular mechanisms behind the post-MI inflammatory response. Many recent studies indicated some answers but also raised more questions on the role of immune cells in the inflammatory response after MI.
As for the immunotherapies, it is widely accepted that MI patients receiving immunosuppressive therapies with corticosteroids exhibit a worse outcome due to the impaired healing response62; thus, novel immunotherapies should be more precise and focus on the immune cells rather than simply silencing the inflammatory response. Although we have reviewed many novel immunotherapies in this review, this field is still in its infancy.
Overall, it needs to be acknowledged that although the role of immune cells in the post-MI inflammatory response is promising, there is still a long way to fully explore the molecular mechanisms behind it, and the novel therapeutic approaches are still in a premature stage.
Footnotes
Supported by a project of the National Natural Science Foundation of China (Grant No. 81770350).
The authors report no conflicts of interest.
Y. Zhang and W. Wen contribute equally to this paper.
REFERENCES
- 1.McMurray JJ, Adamopoulos S, Anker SD, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European society of cardiology. Developed in collaboration with the heart failure association (HFA) of the ESC. Eur Heart J. 2012;33:1787–1847. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nunes-Silva V, Frantz S, Ramos GC. Lymphocytes at the heart of wound healing. Adv Exp Med Biol. 2017;1003:225–250. [DOI] [PubMed] [Google Scholar]
- 4.Ma Y, Yabluchanskiy A, Lindsey ML. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair. 2013;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan X, Anzai A, Katsumata Y, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. [DOI] [PubMed] [Google Scholar]
- 7.Harel-Adar T, Ben Mordechai T, Amsalem Y, et al. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci U S A. 2011;108:1827–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang B, Liao R. The paradoxical role of inflammation in cardiac repair and regeneration. J Cardiovasc Transl Res. 2010;3:410–416. [DOI] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. Inflammation in cardiac injury, repair and regeneration. Curr Opin Cardiol. 2015;30:240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heidt T, Courties G, Dutta P, et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res. 2014;115:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang YH, He M, Wang Y, et al. Modulators of the balance between M1 and M2 macrophages during pregnancy. Front Immunol. 2017;8:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nasser MI, Zhu S, Huang H, et al. Macrophages: first guards in the prevention of cardiovascular diseases. Life Sci. 2020;250:117559. [DOI] [PubMed] [Google Scholar]
- 13.Gomez I, Duval V, Silvestre JS. Cardiomyocytes and macrophages discourse on the method to govern cardiac repair. Front Cardiovasc Med. 2018;5:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vagnozzi RJ, Maillet M, Sargent MA, et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature. 2020;577:405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Amerongen MJ, Harmsen MC, van Rooijen N, et al. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leor J, Rozen L, Zuloff-Shani A, et al. Ex vivo activated human macrophages improve healing, remodeling, and function of the infarcted heart. Circulation. 2006;114(1 suppl):I94–I100. [DOI] [PubMed] [Google Scholar]
- 18.Nahrendorf M, Swirski FK, Aikawa E, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Courties G, Heidt T, Sebas M, et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2014;63:1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Zhang H, Lu Y, et al. Class A scavenger receptor attenuates myocardial infarction-induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res Cardiol. 2011;106:1311–1328. [DOI] [PubMed] [Google Scholar]
- 21.Ma Y, Yabluchanskiy A, Iyer RP, et al. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 23.Horckmans M, Ring L, Duchene J, et al. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–197. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Liu S, Weng X, et al. Brg1 deficiency in vascular endothelial cells blocks neutrophil recruitment and ameliorates cardiac ischemia-reperfusion injury in mice. Int J Cardiol. 2018;269:250–258. [DOI] [PubMed] [Google Scholar]
- 25.Guilliams M, van de Laar L. A hitchhiker's guide to myeloid cell subsets: practical implementation of a novel mononuclear phagocyte classification system. Front Immunol. 2015;6:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. [DOI] [PubMed] [Google Scholar]
- 27.Ismahil T, Hamid S, Bansal B, et al. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic Axis. Circ Res. 2014;114:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van der Borght K, Scott CL, Nindl V, et al. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep. 2017;18:3005–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sichien D, Lambrecht BN, Guilliams M, et al. Development of conventional dendritic cells: from common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. 2017;10:831–844. [DOI] [PubMed] [Google Scholar]
- 30.Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15:471–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon KB, Davies SS, Kirabo A. Dendritic cells and isolevuglandins in immunity, inflammation, and hypertension. Am J Physiol Heart Circ Physiol. 2017;312:H368–H374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagai T, Honda S, Sugano Y, et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J Am Heart Assoc. 2014;3:e000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anzai A, Anzai T, Nagai S, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125:1234–1245. [DOI] [PubMed] [Google Scholar]
- 34.Liu H, Gao W, Yuan J, et al. Exosomes derived from dendritic cells improve cardiac function via activation of CD4(+) T lymphocytes after myocardial infarction. J Mol Cell Cardiol. 2016;91:123–133. [DOI] [PubMed] [Google Scholar]
- 35.Lee JS, Jeong SJ, Kim S, et al. Conventional dendritic cells impair recovery after myocardial infarction. J Immunol. 2018;201:1784–1798. [DOI] [PubMed] [Google Scholar]
- 36.Weirather J, Hofmann UD, Beyersdorf N, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. [DOI] [PubMed] [Google Scholar]
- 37.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liston A, Nutsch KM, Farr AG, et al. Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc Natl Acad Sci. 2008;105:11903–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maisel A, Cesario D, Baird S, et al. Experimental autoimmune myocarditis produced by adoptive transfer of splenocytes after myocardial infarction. Circ Res. 1998;82:458–463. [DOI] [PubMed] [Google Scholar]
- 40.Hofmann U, Beyersdorf N, Weirather J, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652–1663. [DOI] [PubMed] [Google Scholar]
- 41.Saxena A, Dobaczewski M, Rai V, et al. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circul Physiol. 2014;307:H1233–H1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsumoto K, Ogawa M, Suzuki J, et al. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int Heart J. 2011;52:382–387. [DOI] [PubMed] [Google Scholar]
- 43.Curato C, Slavic S, Dong J, et al. Identification of noncytotoxic and IL-10-producing CD8+AT2R+ T cell population in response to ischemic heart injury. J Immunol. 2010;185:6286–6293. [DOI] [PubMed] [Google Scholar]
- 44.Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006;5:564–576. [DOI] [PubMed] [Google Scholar]
- 45.Rahmanzadeh R, Weber MS, Bruck W, et al. B cells in multiple sclerosis therapy-A comprehensive review. Acta Neurol Scand. 2018;137:544–556. [DOI] [PubMed] [Google Scholar]
- 46.Porsch F, Binder CJ. Impact of B-Cell-Targeted therapies on cardiovascular disease. Arterioscler Thromb Vasc Biol. 2019;39:1705–1714. [DOI] [PubMed] [Google Scholar]
- 47.Zouggari Y, Ait-Oufella H, Bonnin P, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maddaluno M, Grassia G, Di Lauro MV, et al. Bindarit inhibits human coronary artery smooth muscle cell proliferation, migration and phenotypic switching. PLoS One. 2012;7:e47464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dobaczewski M, Frangogiannis NG. Chemokines and cardiac fibrosis. Front Biosci. 2009;1:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodchild TT, Robinson KA, Pang W, et al. Bone marrow-derived B cells preserve ventricular function after acute myocardial infarction. JACC Cardiovasc Interv. 2009;2:1005–1016. [DOI] [PubMed] [Google Scholar]
- 52.Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and prevention of pancreatic cancer. Trends Cancer. 2018;4:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Young A, Quandt Z, Bluestone JA. The balancing act between cancer immunity and autoimmunity in response to immunotherapy. Cancer Immunol Res. 2018;6:1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duerr GD, Heinemann JC, Suchan G, et al. The endocannabinoid-CB2 receptor axis protects the ischemic heart at the early stage of cardiomyopathy. Basic Res Cardiol. 2014;109:425. [DOI] [PubMed] [Google Scholar]
- 55.Vasilyev N, Williams T, Brennan ML, et al. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation. 2005;112:2812–2820. [DOI] [PubMed] [Google Scholar]
- 56.Ali M, Pulli B, Courties G, et al. Myeloperoxidase inhibition improves ventricular function and remodeling after experimental myocardial infarction. JACC Basic Transl Sci. 2016;1:633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maekawa Y, Mizue N, Chan A, et al. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin-1 receptor-associated kinase-4 signaling: a regulator of bone marrow-derived dendritic cells. Circulation. 2009;120:1401–1414. [DOI] [PubMed] [Google Scholar]
- 58.Choo EH, Lee JH, Park EH, et al. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation. 2017;135:1444–1457. [DOI] [PubMed] [Google Scholar]
- 59.Wang YP, Xie Y, Ma H, et al. Regulatory T lymphocytes in myocardial infarction: a promising new therapeutic target. Int J Cardiol. 2016;203:923–928. [DOI] [PubMed] [Google Scholar]
- 60.Anzai T. Post-infarction inflammation and left ventricular remodeling: a double-edged sword. Circ J. 2013;77:580–587. [DOI] [PubMed] [Google Scholar]
- 61.Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18:733–744. [DOI] [PubMed] [Google Scholar]
- 62.Kloner RA, Fishbein MC, Lew H, et al. Mummification of the infarcted myocardium by high dose corticosteroids. Circulation. 1978;57:56–63. [DOI] [PubMed] [Google Scholar]