

Abstract
The versatile coordination behavior of the P4 butterfly complex [{Cp*Cr(CO)3}2(μ,η1:1‐P4)] (1) towards Lewis acidic pentacarbonyl compounds of Cr, Mo and W is reported. The reaction of 1 with [W(CO)4(nbd)] (nbd=norbornadiene) yields the complex [{Cp*Cr(CO)3}2(μ3,η1:1:1:1‐P4){W(CO)4}] (2) in which 1 serves as a chelating P4 butterfly ligand. In contrast, reactions of 1 with [M(CO)4(nbd)] (M=Cr (a), Mo (b)) result in the step‐wise formation of [{Cp*Cr(CO)2}2(μ3,η3:1:1‐P4){M(CO)5}] (3 a,b) and [{Cp*Cr(CO)2}2‐(μ4,η3:1:1:1‐P4){M(CO)5}2] (4 a,b) which contain a folded cyclo‐P4 unit. Complex 4 a undergoes an unprecedented P1/P3‐fragmentation yielding the cyclo‐P3 complex [Cp*Cr(CO)2(η3‐P3)] (5) and the as yet unknown phosphinidene complex [Cp*Cr(CO)2{Cr(CO)5}2(μ3‐P)] (6). The identity of 6 is confirmed by spectroscopic methods and by the in situ formation of [{Cp*Cr(CO)2(tBuNC)}P{Cr(CO)5}2(tBuNC)] (7). DFT calculations throw light on the bonding situation of the reported products.
Keywords: coordination, P1/P3-fragmentation, P4 activation, phosphinidene, polyphosphorus complexes
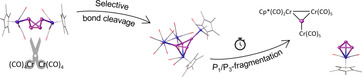
Unprecedented P1/P3‐fragmentation: The versatile P4 butterfly complex [{Cp*Cr(CO)3}2(μ,η1:1‐P4)] reacts with [Cr(CO)4] fragments to afford a folded deltoid cyclo‐P4 unit stabilized by [Cp*Cr(CO)2] and [Cr(CO)5] moieties. The new chromium compound undergoes a selective P1/P3‐fragmentation. Hereby, the cyclo‐P3 complex [Cp*Cr(CO)2(η3‐P3)] and the as yet unknown phosphinidene complex [Cp*Cr(CO)2{Cr(CO)5}2(μ3‐P)] are obtained (see scheme).
The research of the activation of small molecules is of great importance, as it can improve uneconomical industrial‐scale reactions by making them more atom‐efficient, clean, sustainable and inexpensive.1 In this field, investigations regarding the activation of P4 describe the subsequent P−P bond cleavage of the tetrahedral P4 molecule of white phosphorus.2 Ultimately, the goal of these studies is to provide insight into controlling the remarkable reactivity of P4 and to obtain organo‐phosphorus compounds in a more sustainable way. The first step of the selective degradation of the P4 tetrahedron is the formation of the tetraphospha‐bicyclo[1.1.0]butane moiety (often referred to as P4 butterfly due to its geometry). This moiety can be used as a ligand in coordination chemistry, typically displaying small bite angles, like common chelating diphosphine ligands.3 This was demonstrated by the synthesis of the transition‐metal‐stabilized bridging P4 butterfly complex [{Cp′′′Fe(CO)2}2(μ,η1:1‐P4)] (A, Cp′′′=η5‐C5H2 tBu3).4
The continuing degradation of white phosphorus via the P4 butterfly structure has been widely investigated and a plethora of polyphosphorus compounds could be isolated.2 However, only very few examples have been reported for a controlled fragmentation of the intact P4 butterfly moiety affording a P1 and a P3 fragment. One of the reported examples originates from the butterfly anion Li[Mes*P4 ⋅BR3].5 Lammertsma et al. studied its reactivity towards imidazolium salts and phenylisocyanate yielding stabilized phosphinidene adducts and [Mes*P3] fragments. They succeeded in isolating the P3 fragment as the respective dimer [Mes*P3]2 or as the Diels–Alder adduct [Mes*P3(C6H8)] after employing the trapping agent 1,3‐cyclohexadiene. Starting from elemental phosphorus, Zhang et al. used rare‐earth‐metal complexes to obtain bicyclo[4.1.0]triphospha‐heptanide ligands alongside with phospholyl lithium.6a, 6b, 6c Both of these fragmentation routes require additional reactants such as non‐innocent ligands in order to induce the P1/P3 fragmentation. Very recently, the group of Ghadwal reported on the P1/P3 fragmentation of P4 induced by mesoionic carbenes and anionic dicarbenes affording 1,2,3‐triphosphol‐2‐ides incorporating a cyclo‐C2P3 unit.6d, 6e A first step towards a more untouched P1/P3 fragmentation route was reported by reacting [Cp*Ni(μ‐CO)]2 with P4 in the presence of stabilizing [Cr(CO)5] fragments.7 Various steps of irradiation and thermolysis induce the fragmentation process affording a bent cyclo‐P4 complex. Finally, the formation of [Cp*Ni(η3‐P3){Cr(CO)5}3] and a [Cp*Ni≡P‐Cr(CO)5] intermediate that could be isolated as its corresponding dimer [{Cp*Ni}2(μ,η2‐P2){Cr(CO)5}2] was obtained.
DFT calculations predicted that a chelating coordination mode via the lone pairs of the two wing tip P atoms of A is energetically most favorable.8 Therefore, we studied the coordination behavior of A towards monovalent coinage metal salts and different FeII compounds.8, 9 As anticipated, chelating coordination products, for example, [{(Cp′′′Fe(CO)2)2(μ3,η1:1:1:1‐P4)}2FeBr2]9 (B, Scheme 1), in which A acts as a bidentate ligand could be obtained. With 70.27(3)°, the bite angle of B compares well to the bite angle reported for dppm (72(2)°, 1,2‐bis(diphenylphosphino)methane).3 In contrast, [Fe(MeCN)6]2+, a Lewis acid containing labile acetonitrile ligands, reacts with A by inducing an isomerization of the P4 butterfly unit forming the 6π‐aromatic cyclo‐P4 sandwiched dication [{(Cp’’’Fe(CO)2)2P4}2Fe]2+ (C, Scheme 1).9
Scheme 1.
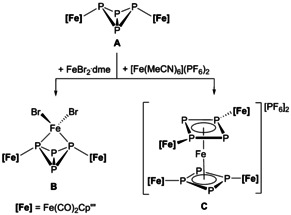
Coordination and isomerization obtained from the reaction of A with different FeII Lewis acids.
While the reactivity of A under photolytic10 and thermolytic4 conditions, its reactivity towards alkynes11 and its coordination chemistry8, 9 have been intensively studied, the reactivity of the isostructural chromium‐containing complex [{Cp*Cr(CO)3}2(μ,η1:1‐P4)] (1, Cp*=η5‐C5(CH3)5) has only been scarcely investigated.12 This encouraged us to further study the reactivity of 1 and the question arose whether a simple coordination chemistry as expected for a chelating polyphosphine would occur or a much more diverse reaction pathway would be unraveled. Herein, we report on the reaction of 1 with Lewis acidic group six carbonyl complexes [M(CO)4(nbd)] (M=Cr, Mo, W; nbd=norbornadiene). The weakly coordinating norbornadiene ligand is expected to be replaced by the more strongly donating P4 butterfly ligand affording new organometallic P4 coordination compounds. Unexpectedly, in the case of [Cr(CO)4(nbd)] an unprecedented reactivity occurs leding to a final P3/P1 fragmentation.
The reaction of 1 with 1.0 equiv. [W(CO)4(nbd)] selectively yields the chelating complex [{Cp*Cr(CO)3}2(μ3,η1:1:1:1‐P4){W(CO)4}] (2, Scheme 2). The formation of the tetraphosphatungsten‐tricyclo[1.1.1.02, 4]pentane compound could be verified by single crystal X‐ray diffraction analysis13 and only little influence of the bidentate coordination on the overall structure of the P4 scaffold could be detected in comparison to 1 (Figure 1). While for 2 bond lengths and angles similar to those for 1 are given, the central P4 unit in 2 is slightly distorted, whereas, in 1, this moiety is more symmetrical. Due to the distortion which probably perseveres in solution, 2 displays an AA′BB′ spin system with a relatively large δ AA’/δ BB’ separation affording two multiplets at δ=−168.8 ppm (bridgehead P atoms) and δ=−153.8 ppm (wing tip P atoms) in the 31P{1H} NMR spectrum. The outstanding feature of 2 is the extremely small P1‐W‐P2 bite angle of 64.21(11)° again highlighting the structural similarity with the dppm ligand.3
Scheme 2.
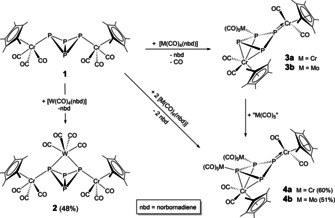
Reactions of 1 with Lewis acidic [M(CO)4(nbd)] (M=Cr, Mo, W). Yields are given in parentheses.
Figure 1.
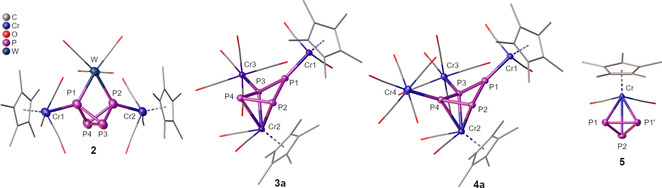
Molecular structures of 2, 3 a, 4 a and 5 in the solid state. H atoms and solvent molecules are omitted for clarity and CO as well as Cp* ligands are drawn in the wire frame model; thermal ellipsoids are drawn at 50 % probability level.
Surprisingly, the reactions of 1 with [M(CO)4(nbd)] (M=Cr (a), Mo (b)) do not result in P4 butterfly‐Lewis acid adducts, but afford the mono‐substituted [{Cp*Cr(CO)2}2(μ3,η3:1:1‐P4){M(CO)5}] (3) and the bis‐substituted derivative [{Cp*Cr(CO)2}2(μ4,η3:1:1:1‐P4){M(CO)5}2] (4) (Scheme 2). During the formation of 3 and 4, a cleavage of the P‐P bond between the former bridgehead P atoms of 1 is observed affording a folded deltoid cyclo‐P4 unit as the central structural moiety. This is a direct result of an initial CO shift from the [Cp*Cr(CO)3] substituents of 1 to the [M(CO)4] fragments yielding [Cp*Cr(CO)2] substituents and [M(CO)5] units. The consequential electron deficit on Cr2 is balanced by an additional coordination of the two former bridgehead phosphorus atoms towards Cr2, leading to the cleavage of the P−P bond (labeling according to Figure 1).
The electron deficit of the Cr1 fragment is balanced by the formation of a formal double bond between Cr1 and the adjacent P1 atom, and the former wing tip atom P1 reaches a planar coordination environment. The formal P=Cr double bond can be viewed as an additional coordination of the P lone pair to the Cr1 atom since the [Cp*Cr(CO)2] fragment requires three additional electrons according to the 18 VE rule. Various terminal or bridging, square planar or distorted cyclo‐P4 ligands have been reported either as bare polyphosphorus units or stabilized by different Lewis acids.2 However, the folded deltoid cyclo‐P4 unit of 3 and 4 represents a novel structural motif due to the adjacent P=Cr double bond that can be interpreted as the tail of the cyclo‐P4 kite. The closest related compound, from a structural perspective, to 3 and 4 is [{(CH3CN)2(CO)2WCl}(η3‐P3{W(CO)5}2P{(X)W(CO)5})] (X=Cl, OH) for which no phosphorus metal double bond but an additional stabilization with a chloride or hydroxy group, respectively, on the P1 atom is observed.14
According to DFT calculations, the reaction of 1 with [Cr(CO)4(nbd)] is slightly endothermic (1.34 kJ mol−1) while the reaction of 1 with [W(CO)4(nbd)] yielding 2 is exothermic (−6.49 kJ mol−1),which reinforces that a stable complex such as [{Cp*Cr(CO)3}2(μ3,η1:1:1:1‐P4){Cr(CO)4}] („2 a“) is not formed, but rather a CO shift from a [Cp*Cr(CO)3] unit to the [Cr(CO)4] moiety occurs.
When the reaction of 1 with [M(CO)4(nbd)] (M=Cr (a), Mo (b)) is performed in a 1:1 stoichiometry, the mono‐substituted compound 3 is the main product in the reaction solution alongside with traces of 4. However, the conversion of 1 is limited to 50 % due to the mismatched CO count. Two CO ligands are abstracted from each molecule of 1, but only one CO ligand is needed to obtain an [M(CO)5] fragment from [M(CO)4(nbd)] (M=Cr, Mo). In contrast, 4 is formed almost quantitatively (besides some impurities of 3), when the reaction of 1 with [M(CO)4(nbd)] (M=Cr, Mo) is performed in a 1:2 ratio. Attempts to isolate 3 from a 1:1 reaction by crystallization lead to a rearrangement to 4, which exclusively crystallizes from the solution. This process was monitored by 31P{1H} NMR spectroscopy (vide infra).
Therefore, reasonable amounts of pure 3 could not be isolated, although different isolation methods and alternative synthetic pathways were examined. Yet, a few single crystals of 3 a could be obtained from the reaction mixture after storage at −78 °C, whereas 4 a already crystallizes at −28 °C (Figure 1). Unfortunately, no single crystals could be obtained for 3 b, but a similar structure as compared to 3 a can be proposed based on NMR spectroscopic data.15 Compounds 3 a, 4 a and 4 b all crystallize readily in the form of stacked plates from saturated solutions in CH2Cl2 at −28 °C (3 a: P21/c; 4 a and 4 b: P ). The central deltoid cyclo‐P4 structural motif of 3 a, 4 a and 4 b is very similar. The distances for the P1−P2 bond (3 a: 2.2181(7) Å, 4 a: 2.2265(13) Å, 4 b: 2.2259(13) Å) and the P1−P3 bond (3 a: 2.1973(7) Å, 4 a: 2.2013(14) Å, 4 b: 2.1938(13) Å) comply well with the value for a P−P single bond (2.209(5) Å).16 In contrast, the P2−P4 bond (3 a: 2.1606(7) Å, 4 a: 2.1390(15) Å, 4 b: 2.1427 (13) Å) and P3−P4 bond (3 a: 2.1705(7) Å, 4 a: 2.1583(12) Å, 4 b: 2.1560(13) Å) are noticeably shortened indicating a delocalized electron system between P2, P3 and P4 (labeling according to Figure 1). The Cr1−P1 bond attached to the rearranged cyclo‐P4 unit (3 a: 2.1258(6) Å, 4 a: 2.1129(11) Å, 4 b: 2.1169(11) Å) is significantly shortened in comparison to the corresponding P−Cr bond length in 1 (2.529(2) Å) and the Cr3−P3 as well as Cr4−P4 distance in 3 a (Cr3‐P3 2.3664(5) Å) and 4 a (Cr3−P3 2.3564(12) Å, Cr4−P4 2.3517(11) Å), respectively, revealing a Cr1–P1 multiple bond character. Moreover, the degree of folding of the cyclo‐P4 unit is independent of the nature of the substituent pattern as 3 a, 4 a and 4 b display nearly identical folding angles (3 a: 135.50(8)°, 4 a: 135.92(6)°, 4 b: 136.18(8)°).
The 31P{1H} NMR spectra of 3 and 4 are very similar displaying an AMNX spin system.15 The chemical shift of the signal attributed to PA (δ=515.4 ppm (3 a), 515.4 (3 b), 489.1 ppm (4 a), 487.8 ppm (4 b); P1 in Figure 1) is in the typical range for a phosphorus atom in the planar environment that is part of a formal phosphorus metal multiple bond. In comparison to the formal [Cr=P(P)2] structural motif in 3 and 4, the 31P NMR chemical shift of the trigonal planar [Mn=P(Fe)2] moiety of [{(CpMn(CO)2)(μ3,η1:1:1‐P)}2{Fe2(CO)6}] is even more downfield shifted (δ(31P)=977 ppm).17
The 31P{1H} NMR spectrum of the reaction mixture after stirring 1 with 1.0 equiv. of [Cr(CO)4(nbd)] in thf for 3 days shows the signal set characteristic for 3 a (Figure S6a in SI) as the main product, as well as traces of 4 a. In contrast, the 31P{1H} NMR spectrum of the crystals obtained from storing the concentrated reaction mixture at −28 °C displays exclusively the signals of 4 a (Figure S6b in Supporting Information). Obviously, 3 a coordinates to an excess of [Cr(CO)5] units present in the solution during the crystallization process. Consequently, 4 a is formed, which crystallizes due to its lower solubility. The 31P{1H} NMR spectrum of the supernatant of the obtained crystals exhibits signals for both major compounds 3 a and 4 a. However, the intensity of the signals corresponding to 3 a decreased significantly in comparison to the signals corresponding to 4 a , now promoting 4 a to the primary component in solution after crystallization (Figure S6c in Supporting Information). In summary, storing the concentrated reaction solution at low temperatures leads to the formation of 4 a from 3 a and excess [Cr(CO)5] in the form of crystalline 4 a as well as in solution. The same observations can be reported for 3 b and 4 b, but in the 31P{1H} NMR spectrum of crystalline 4 b, minor amounts of 3 b can be detected indicating partial degradation after re‐dissolving.
In order to elucidate the electronic structure of 4 a, DFT calculations at the B3LYP/def2‐TZVP level were performed.18 The DFT optimized geometry of 4 a compares well with the experimental geometry. According to the NBO analysis, the Cr1−P1 bond is a double bond built from a σ‐type and a π‐type bond (Figure 2). The σ‐bond is realized over a sp0.9 hybrid orbital on phosphorus and a sd2.2 hybrid orbital on chromium, while the π‐orbital is realized over a pure p‐orbital on P and a pure d‐orbital on Cr. The partial double bond character is also reflected in the Wiberg Bond Index (WBI) of 1.16, while the WBI of the Cr3−P3 bond is 0.39.15 The WBIs of the P1−P2 and P1−P3 bonds are slightly lower (0.91 and 0.93) than the WBIs of the P2−P4 and P3−P4 bonds (1.06 and 0.99).
Figure 2.
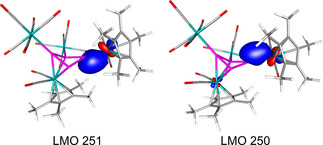
Localized molecular orbitals of 4 a representing the Cr1–P1 multiple bond.
A common observation for all experiments performed with 1 is the sensitivity of 1 towards temperature and light causing slow decomposition even at mild reaction conditions. In nearly all manipulations starting from 1, one characteristic singlet at approx. −270 ppm can be detected in the 31P{1H} NMR spectrum.
By comparison with literature data for similar compounds, Scherer et al. attributed this chemical shift to [Cp*Cr(CO)2(η3‐P3)] (5),19 and within this work, we were able to confirm this proposal by the first single crystal X‐ray diffraction of the isolated compound 5 (Figure 1).20 Alongside 5, an insoluble solid (probably a mixture of various polyphosphides) is obtained after the quantitative decomposition of 1. Remarkably, after stirring a solution of 4 a in thf for three days at 50 °C, a distinct second degradation product can be detected. In the 31P{1H} NMR spectrum, an additional singlet at δ=1123.7 ppm is recorded next to the characteristic singlet at δ=−273.3 ppm corresponding to 5. The drastic low field shift of this novel signal indicates a planar coordination sphere of the corresponding P atom, as it is typical for planar phosphinidene complexes as for instance the μ3‐bridging complex [{CpW(CO)2}(μ3‐P){Cr(CO)5}2] (B, δ(31P NMR)=945 ppm).21 Hence, we propose the structurally analog [{Cp*Cr(CO)2}(μ3‐P){Cr(CO)5}2] (6) as the second compound obtained from the degradation of 4 a attributable to the second signal at δ=1123.7 ppm) observed in the 31P{1H} NMR spectrum. Although 6 could not be isolated and characterized by single crystal X‐ray diffraction analysis, the mutual extreme low field 31P NMR chemical shifts of 6 and B validate the proposed structure of 6. Consequently, an unprecedented type of selective P1/P3‐fragmentation of 4 a yielding 5 and 6 can be proposed (Scheme 3). A closer look at the molecular structure of 4 a in the solid state further supports the proposed P1/P3‐fragmentation. As discussed above, two different P‐P bond lengths can be found in 4 a. The two longest and therefore comparatively weakest P‐P bonds (P1‐P2 and P1‐P3) appear to be the predetermined breaking points of 4 a finally affording 5 and 6.
Scheme 3.
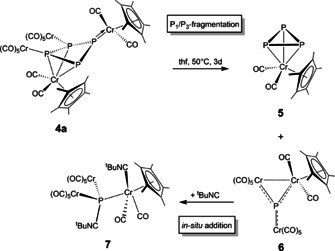
P1/P3‐fragmentation of 4 a and subsequent phosphinidene adduct formation of the obtained P1 fragment 6 yielding 7.
Since it was not possible to isolate 6 as a pure compound from the reaction mixture, we attempted to trap 6 with tBuNC, as this type of reaction is widely known for phosphinidene complexes. For instance, tBuNC reacts with [Cp*P{W(CO)5}2] affording the Lewis acid/base (LA/LB) adduct [Cp*P{W(CO)5}2(tBuNC)].22 Diagnostic is the extreme change in the 31P NMR chemical shift from δ=1076.5 ppm22a for the phosphinidene complex to δ=−73.1 ppm22b for the LA/LB adduct. Following this strategy, an excess of tBuNC was added to a solution of 6. The reaction was monitored by 31P{1H} NMR spectroscopy which shows that the characteristic signal for 6 at δ=1124 ppm disappears while a new signal at δ=−166 ppm appears indicating the full conversion of 6 into the proposed LA/LB adduct [{Cp*Cr(tBuNC)(CO)2}‐(μ3‐P){Cr(CO)5}2] (7, Scheme 3). In 7, the planar coordination geometry of the P atom is abrogated by the additional coordination of one tBuNC ligand yielding a pseudo‐tetrahedral phosphinidene adduct. Consequently, the deshielding of the P atom is strongly reduced leading to the drastic change in the chemical shift. A second tBuNC ligand additionally coordinates to the Cp*Cr(CO)2 fragment, compensating the electron deficit that occurred at the Cr atom accordingly. According to DFT calculations, the coordination of one tBuNC to the P atom in the center of 6 is exothermic with −48.5 kJ mol−1. The addition of the second tBuNC molecule to the Cp*Cr(CO)2 fragment is even more exothermic with −53.2 kJ mol−1, indicating that the coordination of two tBuNC ligands to 6 is to be expected. In order to prove the identity of 6 and 7, the 31P NMR chemical shifts of 5, B, 6 and 7 were calculated by DFT methods. To this effect, the geometry of the compounds was optimized in the gas phase at the BP86/Def2TZVP level of theory. For the calculation of the 31P NMR chemical shifts, using the GIAO method, the aug‐pcSseg‐2 basis set for phosphorus was utilized. The values of the calculated chemical shifts are in good agreement with the experimental values (Table 1), validating the proposed identity of 6 and 7.
Table 1.
Experimental (δ exp) and calculated (δ cal) 31P NMR chemical shifts of compounds 5, B, 6 and 7.
|
|
5 |
B |
6 |
7 |
|---|---|---|---|---|
|
δ exp/ppm |
−273[a] |
945[b] |
1124[a] |
−166[a] |
|
δ cal/ppm |
−242 |
1046 |
1221 |
−111 |
[a] Recorded in thf with C6D6 capillary at room temperature. [b] Recorded in CD2Cl2 at −20 °C.18
It has to be noted that the 31P chemical shifts are very sensitive to geometry changes. In order to evaluate the accuracy of the calculated chemical shifts, we also included the known phosphinidene complex [CpW(CO)2(Cr(CO)5)2(μ3‐P)] (B)21 in our calculations. The calculated 31P chemical shift of δ=1046 ppm is in good agrement with the experimental value of δ=954 ppm. This shows that the electronic structures of B and 6 are well described by the applied DFT methods and confirm the identity of 6.
In conclusion, we were able to illustrate the diverse coordination behavior of the P4 butterfly complex 1 towards Lewis acidic carbonyl compounds of Cr, Mo and W. On the one hand, a chelating coordination yielding 2, a complex with a P4 butterfly ligand that displays an exceedingly small bite angle, was achieved by implying [W(CO)4] fragments. On the other hand, 1 turned out to be a promising starting material for rearrangement processes, yielding new compounds with folded cyclo‐P4 units (3 and 4) when reacted with [M(CO)4] moieties (M=Cr (a), Mo (b)). Most importantly, an unprecedented P1/P3‐fragmentation route was observed starting from 4 a yielding the cyclo‐P3 complex 5 and the novel μ3‐bridging phosphinidene 6. The proposed structure of 6 could be verified by 31P{1H} NMR spectroscopy, DFT calculations and the in situ reaction with tBuNC, yielding the phosphinidene adduct 7. These results promote the ongoing implementation of P4 butterfly complexes as starting materials in the formation of unprecedented polyphosphorus compounds, which represent further steps is the P4 activation sequence.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/38‐1. RG is grateful to the Fonds der Chemischen Industrie for a PhD fellowship.
R. Grünbauer, G. Balázs, M. Scheer, Chem. Eur. J. 2020, 26, 11722.
Contributor Information
Dr. Rebecca Grünbauer, https://www.uni‐regensburg.de/chemie‐pharmazie/anorganische‐chemie‐scheer/startseite/index.html
Prof. Dr. Manfred Scheer, Email: Manfred.Scheer@ur.de.
References
- 1.
- 1a. Hazari N., Chem. Soc. Rev. 2010, 39, 4044–4056; [DOI] [PubMed] [Google Scholar]
- 1b. Mellone I., Bertini F., Gonsalvi L., Guerriero A., Peruzzini M., Chimia 2015, 69, 331–338; [DOI] [PubMed] [Google Scholar]
- 1c. Turner Z., Inorganics 2015, 3, 597; [Google Scholar]
- 1d. Henderson R. A., Trans. Met. Chem. 1990, 15, 330–336; [Google Scholar]
- 1e. Indrakanti V. P., Kubicki J. D., Schobert H. H., Energy Environ. Sci. 2009, 2, 745–758; [Google Scholar]
- 1f. Yin X., Moss J. R., Coord. Chem. Rev. 1999, 181, 27–59. [Google Scholar]
- 2.
- 2a. Cossairt B. M., Piro N. A., Cummins C. C., Chem. Rev. 2010, 110, 4164–4177; [DOI] [PubMed] [Google Scholar]
- 2b. Caporali M., Gonsalvi L., Rossin A., Peruzzini M., Chem. Rev. 2010, 110, 4178–4235; [DOI] [PubMed] [Google Scholar]
- 2c. Scheer M., Balázs G., Seitz A., Chem. Rev. 2010, 110, 4236–4256; [DOI] [PubMed] [Google Scholar]
- 2d. Giffin N. A., Masuda J. D., Coord. Chem. Rev. 2011, 255, 1342–1359. [Google Scholar]
- 3. van Leeuwen P. W. N. M., Kamer P. C. J., Reek J. N. H., Dierkes P., Chem. Rev. 2000, 100, 2741–2770. [DOI] [PubMed] [Google Scholar]
- 4. Scherer O. J., Hilt T., Wolmershäuser G., Organometallics 1998, 17, 4110–4112. [Google Scholar]
- 5. Borger J. E., Ehlers A. W., Lutz M., Slootweg J. C., Lammertsma K., Angew. Chem. Int. Ed. 2017, 56, 285–290; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 291–296. [Google Scholar]
- 6.
- 6a. Du S., Yin J., Chi Y., Xu L., Zang W.-X., Angew. Chem. Int. Ed. 2017, 56, 15886–15890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16102–16106; [Google Scholar]
- 6b. Xu L., Chi Y., Du S., Zhang W.-X., Xi Z., Angew. Chem. Int. Ed. 2016, 55, 9187–9190; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9333–9336; [Google Scholar]
- 6c. Du S., Chai Z., Hu J., Zhang W.-X., Xi Z., Chin. J. Org. Chem. 2019, 39, 2338–2342; [Google Scholar]
- 6d. Rottschäfer D., Blomeyer S., Neumann B., Stammler H.-G., Ghadwal R. S., Chem. Sci. 2019, 10, 11078–11085; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Rottschäfer D., Glodde T., Neumann B., Stammler H.-G., Ghadwal R. S., Chem. Commun. 2020, 56, 2027–2030. [DOI] [PubMed] [Google Scholar]
- 7. Scheer M., Becker U., Chem. Ber. 1996, 129, 1307–1310. [Google Scholar]
- 8. Schwarzmaier C., Heinl S., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2015, 54, 13116–13121; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13309–13314. [Google Scholar]
- 9. Müller J., Heinl S., Schwarzmaier C., Balázs G., Keilwerth M., Meyer K., Scheer M., Angew. Chem. Int. Ed. 2017, 56, 7312–7317; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7418–7423. [Google Scholar]
- 10. Scherer O. J., Schwarz G., Wolmershäuser G., Z. Anorg. Allg. Chem. 1996, 622, 951–957. [Google Scholar]
- 11. Scherer O. J., Hilt T., Wolmershäuser G., Angew. Chem. Int. Ed. 2000, 39, 1425–1427; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 1483–1485. [Google Scholar]
- 12. Schwarzmaier C., Timoshkin A. Y., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2014, 53, 9077–9081; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9223–9227. [Google Scholar]
- 13. Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scheer M., Dargatz M., Jones P. G., J. Organomet. Chem. 1993, 447, 259–264. [Google Scholar]
- 15.See the Supporting Information for further details.
- 16.
- 16a. Simon A., Borrmann H., Craubner H., Phosphorus Sulfur Silicon Relat. Elem. 1987, 30, 507–710; [Google Scholar]
- 16b. Okudera H., Dinnebier Robert E., Simon A., Z. Kristallogr. 2005, 220, 259. [Google Scholar]
- 17. Lang H., Zsolnai L., Huttner G., Angew. Chem. Int. Ed. Engl. 1983, 22, 976; [Google Scholar]; Angew. Chem. 1983, 95, 1017. [Google Scholar]
- 18.The electronic structures of 3 a, 3 b, 4 a and 4 b are very similar, therefore only 4 a will be discussed. For details on the electronic structure of 4 a cf. Supporting Information.
- 19. Scherer O. J., Schwalb J., Wolmershäuser G., Kaim W., Groß R., Angew. Chem. 1986, 98, 349–350. [Google Scholar]
- 20.The structure of 5 in the solid state complies well with the known Cp analog [CpCr(CO)2(η3-P3)]; Goh L. Y., Chu C. K., Wong R. C. S., J. Chem. Soc. Dalton Trans. 1989, 1, 1951–1956. [Google Scholar]
- 21. Huttner G., Weber U., Sigwarth B., Scheidsteger O., Lang H., Zsolnai L., J. Organomet. Chem. 1985, 282, 331–348. [Google Scholar]
- 22.
- 22a. Scheer M., Leiner E., Kramkowski P., Schiffer M., Baum G., Chem. Eur. J. 1998, 4, 1917–1923; [Google Scholar]
- 22b. Seidl M., Schiffer M., Bodensteiner M., Timoshkin A. Y., Scheer M., Chem. Eur. J. 2013, 19, 13783–13791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary