

Abstract
Evidence for putative pathophysiological mechanisms of autism spectrum disorder (ASD), including peripheral inflammation, blood–brain barrier disruption, white matter alterations, and abnormal synaptic overgrowth, indicate a possible involvement of neuroinflammation in the disorder. Neuroinflammation plays a role in the development and maintenance of the dendritic spines involved in glutamatergic and GABAergic neurotransmission, and also influences blood–brain permeability. Cytokines released from microglia can impact the length, location or organization of dendritic spines on excitatory and inhibitory cells as well as recruit and impact glial cell function around the neurons. In this study, gene expression levels of anti‐ and pro‐inflammatory signaling molecules, as well as oligodendrocyte and astrocyte marker proteins, were measured in both gray and white matter tissue in the anterior cingulate cortex from ASD and age‐matched typically developing (TD) control brain donors, ranging from ages 4 to 37 years. Expression levels of the pro‐inflammatory gene, HLA‐DR, were significantly reduced in gray matter and expression levels of the anti‐inflammatory gene MRC1 were significantly elevated in white matter from ASD donors as compared to TD donors, but neither retained statistical significance after correction for multiple comparisons. Modest trends toward differences in expression levels were also observed for the pro‐inflammatory (CD68, IL1β) and anti‐inflammatory genes (IGF1, IGF1R) comparing ASD donors to TD donors. The direction of gene expression changes comparing ASD to TD donors did not reveal consistent findings implicating an elevated pro‐ or anti‐inflammatory state in ASD. However, altered expression of pro‐ and anti‐inflammatory gene expression indicates some involvement of neuroinflammation in ASD. Autism Res 2020, 13: 870‐884. © 2020 The Authors. Autism Research published by International Society for Autism Research and Wiley Periodicals LLC
Lay Summary
The anterior cingulate cortex is an integral brain region in modulating social behaviors including nonverbal communication. The study found that inflammatory gene expression levels were altered in this brain region. We hypothesize that the inflammatory changes in this area could impact neuronal function. The finding has future implications in using these molecular markers to identify potential environmental exposures and distinct cell differences in autism.
Keywords: autism, pathology, cytokines, neuroinflammation, white matter, postmortem
Introduction
Autism spectrum disorder (ASD) is a neurodevelopmental disorder that impacts social communication, emotional response and can result in repetitive or restrictive behaviors. Currently, ASD has a prevalence of one in 59 children with higher rates reported in males of one in 42 and is four times more likely to affect males than females (CDC surveillance data). Regardless of diagnostic consequences on prevalence, the financial impact of ASD is profound including increased patient office visits, pharmaceutical needs, and special education services across the lifespan [Buescher, Cidav, Knapp, & Mandell, 2014; Lavelle et al., 2014; Zerbo et al., 2019]. Early behavioral interventions are the only option for treating the core features of ASD and have been shown to affect behavioral outcomes [as reviewed by Zwaigenbaum et al., 2015]. The lack of specific biomarkers coupled with the benefits of early diagnosis sorely highlight the need to find a consistent, specific and reliable pathophysiology associated with the disorder that could lead to better diagnostic capability and possible preventative measures.
A core behavioral feature of the disorder is the display of atypical social actions that vary widely among those affected. Three important brain regions that contribute to the social brain network include the amygdala, anterior cingulate cortex (ACC), and the prefrontal cortical regions. Social encounters result in the activation of excitatory neurons in the amygdala to generate an action potential that will travel to the ACC where yet another action potential will elicit further downstream signaling in the prefrontal cortex where cognitive planning steps are executed. The series of action potentials are known to be part of the social brain loop and can exhibit dynamic plasticity during development [Blakemore, 2008]. Magnetic resonance imaging studies, both resting (MRI) and functional (fMRI), have been instrumental in determining that these areas are different between typically developing (TD) and ASD patients [Aylward et al., 1999; Gomot et al., 2006; Just, Cherkassky, Keller, Kana, & Minshew, 2007; Rausch et al., 2016]. Additionally, all three regions have demonstrated a unique neuropathology in ASD. In the amygdala, neuronal numbers are reduced overall but in particular in the lateral nucleus [Schumann & Amaral, 2006] while the microglia marker IBA‐1 was increased in the region [Morgan, Barger, Amaral, & Schumann, 2014]. Prefrontal cortical areas have been shown to have markers of neuronal overgrowth in young children diagnosed with ASD when compared to TD control brain tissue [Courchesne, Carper, & Akshoomoff, 2003; Hutsler & Zhang, 2010] while others have shown the number of interneurons were decreased [Ariza, Rogers, Hashemi, Noctor, & Martínez‐Cerdeño, 2018; Hashemi, Ariza, Rogers, Noctor, & Martínez‐Cerdeño, 2016]. In the ACC, decreased neuronal size and cell packing have been reported in layers I–III [Simms, Kemper, Timbie, Bauman, & Blatt, 2009] as well as a reduction in pyramidal cell size in layer V and a correlation between Von Economo neurons and ASD severity [Uppal et al., 2014]. Even when neuronal abnormalities have not been identified, there have been findings of columnar or laminar disorganization in cortical regions [Casanova, Buxhoeveden, Switala, & Roy, 2002; Trutzer, García‐Cabezas, & Zikopoulos, 2019]. It is possible that ASD neuronal pathology is the result of widely heterogeneous brain regional disconnections.
Concordance rates as high as 88% for males and 100% for females were found in monozygotic twin studies [Rosenberg et al., 2009] suggesting that ASD has a strong genetic contribution. Concordance rates of 30% have been reported in dizygotic twins [Hallmayer et al., 2011] and 11% in siblings with an additional one‐fifth of siblings displaying language delays [Constantino, Zhang, Frazier, Abbacchi, & Law, 2010]. Additionally, de novo copy number variations have been identified in ASD populations [Neale et al., 2012; Sanders et al., 2012; Sebat et al., 2007] and approximately 35% of children diagnosed with ASD have a recognizable genetic disorder or chromosomal deletion or duplication [Schaefer, 2016]. Genetic syndromes including Rett's syndrome, fragile X, and tuberous sclerosis complex (TSC 1/2) are referred to as syndromic ASD if behaviors are present. Interestingly, the syndromic disorders previously mentioned have a unique single gene mutation for each disorder that is linked to the synaptic protein, mammalian target of rapamycin or mTOR [Ricciardi et al., 2011; Sharma et al., 2010; Tee et al., 2002]. The mTOR protein is responsible for cell survival and dendritic plasticity and the association with syndromic ASD suggests that mTOR may be implicated in disrupted cell signaling in non‐syndromic or idiopathic ASD. While no single unique gene has been found to cause idiopathic autism, multiple autism susceptibility candidate genes have been identified [C Yuen et al., 2017; Glessner et al., 2009]. But while the evidence for a genetic etiology of ASD is strong, it has not yielded definitive results nor explains the heterogeneity of the spectrum disorder even though thousands of affected individuals and first‐order relatives have been sequenced. Environmental exposures or events may trigger neurodevelopmental alterations in utero or shortly thereafter making neuropathological studies of ASD essential in identifying definitive targets for etiological studies or therapeutic regimens.
A potential link between in utero or perinatal exposures to existing identifiable neuropathology may lie in the amount of inflammation that the brain is exposed to during development. Human epidemiology studies have suggested that maternal autoimmune disorders and situational environmental exposures are associated with the development of ASD [Atladottir, Henriksen, Schendel, & Parner, 2012; Atladottir et al., 2009; Keil et al., 2010; Stevens, Nash, Koren, & Rovet, 2013; Xiang et al., 2015]. Additionally, associations between exposures to anti‐seizure medications [Christensen et al., 2013] and antidepressants [Harrington, Lee, Crum, Zimmerman, & Hertz‐Picciotto, 2014] to a future ASD diagnosis have been found. A common thread in the aforementioned exposure studies is the ability to increase neuroinflammation in the fetal brain. The specific cytokines and degree of affect may contribute greatly to the wide heterogeneity of symptom presentation in the disorder. Interestingly, mothers with elevated IFNγ, IL‐4, and IL‐5 cytokine levels [Goines et al., 2011] and those with increased TNFɑ levels in amniotic fluid [Abdallah et al., 2013] had an increased risk for an ASD diagnosis in the child. These cytokines have the ability to cross the placental and blood–brain barrier to trigger cytokine expression in the brain. Microglia are the glial cell type responsible for regulating the majority of cytokine levels in extracellular areas of neighboring neurons and astrocytes; therefore, if microglia have been stimulated to release aberrant, localized levels of cytokines then astrocytes would be one of the first indicators by expressing the reactive protein GFAP that has been shown by multiple groups, including ours, in multiple brain regions in ASD [Crawford et al., 2015; Kotagiri, Chance, Szele, & Esiri, 2014; Laurence & Fatemi, 2005]. Low levels of extracellular cytokines work to provide the homeostatic balance during neuronal signaling, but pathophysiological levels of unknown amounts of cytokines identified in postmortem brain tissue may lead to inefficient synaptic development and function in ASD. For example, excessive levels of the cytokine, insulin‐like growth factor 1 (IGF1), demonstrated increased synaptic markers in both animal and cell culture studies [Carlson et al., 2014; Cheng et al., 2003; Corvin et al., 2012; Shcheglovitov et al., 2013] and prevented neurodegeneration in mouse models of brain injury [Madathil et al., 2013]. Conversely, increased levels of TGFβ reduce the number of neurons in the dentate gyrus of the hippocampus and result in social behavior deficits [Depino, Lucchina, & Pitossi, 2011]. Hence, abnormal gene expression levels associated with unique inflammatory phenotypes in postmortem brain tissue may offer clues regarding neuropathophysiology in ASD.
ASD is considered a spectrum disorder of atypical connections between brain areas, causing deficits in speech, motor skills, and social interaction. Most research to date has focused on the role of neurons in ASD pathology, while other major cell types in the brain have received far less attention. In the past, our lab has investigated the role of neurons and macroglia (astrocytes and oligodendrocytes) in ASD pathology, but not microglia. As the immune cells for the central nervous system (CNS), microglia can not only protect the brain from pathogenic factors and preserve homeostatic conditions via synaptic pruning and phagocytosis of apoptotic neurons but can also increase spine densities necessary for neurotransmission. Microglia are differentiated into distinct phenotypes that are associated with pro‐inflammatory and anti‐inflammatory responses. Pro‐inflammatory microglial responses produce cytokines and assume phagocytic roles that promote defense mechanisms, digest neurons and prune dendritic trees. In contrast, anti‐inflammatory microglial responses facilitate CNS healing by participating in phagocytosis, neuronal remodeling, and tissue regeneration. However, altered expression levels of either phenotype could have detrimental effects on neurotransmission. It is imperative to characterize and determine the potential contributory role of pro‐ and anti‐inflammatory mediators in the pathology of ASD.
Hypothesis
Given the growing evidence of a role of neuroinflammation in ASD, the present study was undertaken to examine the levels of gene expression of markers of inflammation in anterior cingulate cortical tissues from brain donors that had ASD and from typically developing, age‐matched donor tissue. The ACC was chosen because of the relationship to the amygdala and frontal cortex in the social behavior brain region network. Gene expression markers for two major glia cell types, astrocytes and oligodendrocytes, which respond to cytokine receptor activation, were included in this study.
Methods and Materials
Brain Tissue
Flash‐frozen postmortem BA24 tissue blocks from ASD donors and typically developed control donors were acquired from Autism BrainNet, a resource of the Simons Foundation Autism Research Initiative (additionally houses tissue from the Autism Tissue Program, Harvard Brain Tissue Resource Center, Belmont, MA) and Neurobiobank (formerly NICHD Brain and Tissue Bank for Developmental Disorders, Baltimore, MD). This study was reviewed by the Institutional Review Board of East Tennessee State University, who determined that it does not constitute human research under the Department of Health and Human Services exemption 45 CFR 46.101(b) relating to the use of publicly available unidentifiable pathology specimens. ASD (N = 17) and control donors (N = 17) were matched prior to experimentation by gender, age, and RNA quality. Control donors died by trauma (4), drowning (3), asphyxia (3), heart attack (2), respiratory issues (2), dilated cardiomyopathy (1), commotio cordis (1), and an unknown cause (1). ASD donors died by trauma (4), respiratory issues (3), asphyxia (2), cardiac arrhythmia (2), cardiopulmonary arrest (2), bowel obstruction (1), cancer (1), congestive heart failure (1), and diabetic ketoacidosis (1). Age and RNA quality were matched as closely as possible with a difference of no more than 3 years in age and one RNA integrity number for almost every ASD and control donor pair (Table 1). ASD donors were diagnosed using the Autism Diagnostic Interview‐Revised (ADI‐R) and met the diagnostic criteria outlined in the Diagnostic and Statistical Manual IV for autistic disorder. For the protection of the identity of the ASD and control subjects, causes of death were not incorporated in Table 1.
Table 1.
Subject Demographics Information for Postmortem Brain Tissue That was Used in the Study
| Pair | ID | Age | Gender | RIN | PMI (hours) | Toxicology | Tissue preservation | Matter type used for qPCR |
|---|---|---|---|---|---|---|---|---|
| Controls | ||||||||
| 1 | AN14757 | 24 | M | 7.8 | 21.33 | Frozen | WM | |
| 2 | AN07176 | 21 | M | 7.6 | 29.91 | Frozen | WM, GM | |
| 3 | AN07444 | 17 | M | 6.1 | 30.75 | Sertraline | Frozen | WM |
| 4 | 5408 | 6 | M | 5.8 | 16 | Frozen | WM | |
| 5 | 4848 | 16 | M | 7.5 | 15 | Frozen | WM, GM | |
| 6 | 5342 | 22 | M | 8.0 | 14 | Frozen | WM, GM | |
| 7 | 5079 | 33 | M | 5.3 | 16 | Ethanol | Frozen | WM, GM |
| 8 | M3231M | 37 | M | 4.9 | 24 | Frozen | WM, GM | |
| 9 | AN12137 | 31 | M | 4.5 | 32.92 | Frozen | WM, GM | |
| 10 | AN03217 | 19 | M | 5.3 | 18.58 | Frozen | WM, GM | |
| 11 | AN00544 | 17 | M | 5.8 | 28.92 | Frozen | WM, GM | |
| 12 | AN17425 | 16 | M | 6.8 | 26.16 | Frozen | WM, GM | |
| 13 | 4590 | 20 | M | 6.8 | 19 | Frozen | WM, GM | |
| 14 | 4670 | 4 | M | 6.2 | 17 | Frozen | WM, GM | |
| 17 | 4787 | 12 | M | 5.7 | 15 | Montelukast, Albuterol, Prednisone, Loratidine | Frozen | GM |
| 18 | 1105 | 16 | M | 7.8 | 17 | Frozen | GM | |
| 19 | 4334 | 12 | M | 5.7 | 15 | Frozen | GM | |
| Mean | 19.24 | 6.3 | 20.97 | |||||
| SEM | 2.12 | 0.3 | 1.56 | |||||
| ASD | ||||||||
| 1 | AN04166 | 24 | M | 8.1 | 18.51 | Frozen | WM | |
| 2 | AN03935 | 19 | M | 7.0 | 28 | Frozen | WM, GM | |
| 3 | AN02987 | 15 | M | 6.7 | 30.83 | Frozen | WM | |
| 4 | 5144 | 7 | M | 8.0 | 3 | Frozen | WM | |
| 5 | 5302 | 16 | M | 4.8 | 20 | Risperidone, Fluvoxamine, Clonidine, Insulin | Frozen | WM, GM |
| 6 | 5176 | 22 | M | 5.1 | 18 | Risperdal | Frozen | WM, GM |
| 7 | 5297 | 33 | M | 2.5 | 50 | Quetiapine, Fluoxetine, Divalproex Sodium, ziprasidone | Frozen | WM, GM |
| 8 | 5027 | 37 | M | 4.7 | 26 | Risperidone, Fluvoxamine | Frozen | WM, GM |
| 9 | AN11989 | 30 | M | 5.7 | 16.06 | Frozen | WM, GM | |
| 10 | AN07817 | 19 | M | 4.5 | 14.83 | Frozen | WM, GM | |
| 11 | AN00764 | 20 | M | 5.9 | 23.66 | Minocycline | Frozen | WM, GM |
| 12 | AN04682 | 15 | M | 5.6 | 23.23 | Frozen | WM, GM | |
| 13 | 4999 | 20 | M | 7.0 | 14 | Frozen | WM, GM | |
| 14 | 5308 | 4 | M | 7.0 | 21 | Frozen | WM, GM | |
| 17 | 5565 | 12 | M | 7.0 | 22 | Frozen | GM | |
| 18 | 5403 | 16 | M | 6.6 | 35 | Frozen | GM | |
| 19 | 5334 | 11 | M | 5.1 | 15 | Frozen | GM | |
| Mean | 18.88 | 6.0 | 22.30 | |||||
| SEM | 2.11 | 0.3 | 2.48 | |||||
| P‐valuea | 0.906 | 0.403 | 0.654 |
ID, identification number; RIN,, RNA integrity number; PMI,, postmortem interval; qPCR,, quantitative real‐time polymerase chain reaction; WM,, white matter; GM,, gray matter.
P‐value = results of an independent t‐test comparing control and ASD groups, statistically significant when P < 0.05.
Tissue Preparation and Sectioning
Frozen BA24 tissue was sectioned at thicknesses of 50 μm at −20°C using a cryostat microtome (Leica CM3050S) for differential gene expression analysis and immunohistochemical identification, respectively. To avoid cross‐contamination between subjects, the internal elements of the microtome were cleaned using 100% ethanol. Tissue sections from subject pairs were prepared on the same day to guarantee equal storage time. Gray matter punches were obtained by a placing the maximum diameter of a disposable 3.5 mm trephine tool at layer three of the neocortex. Superficial white matter was punch‐dissected at approximately 2 mm past the discernable neocortical layer VI. Dissected tissues were stored at −80°C and were later homogenized for RNA isolation.
RNA and cDNA Preparation
Total RNA was isolated from the laser capture microdissected (LCM) white matter using a Maxwell 16 LEV simplyRNA Tissue Kit (Promega, Madison, WI). To minimize tissue usage in future extractions, a Direct‐zol RNA MicroPrep Kit (Zymo Research, Irvine, CA) was used to extract total RNA from the gray matter and white matter punches for the analysis of the expression of the remaining genes. The RNA quality was assessed by measuring RNA integrity number (RIN) values with the Bioanalyzer RNA 6000 Nano chip (Agilent Technologies, Santa Carla, CA) and the Agilent 2200 TapeStation (Agilent Technologies), respectively. Double‐stranded cDNA was made by reverse transcription of the RNA samples using the Superscript III kit (Life Technologies, Grand Island, NY) that utilized both oligodTs and random hexamer primers during synthesis. Each cDNA reaction was standardized to 250 ng of input RNA.
Polymerase Chain Reactions
For primer temperature and cycle number optimization, end‐point polymerase chain reaction (PCR) was performed using a T100 Thermo Cycler (Bio‐Rad, Hercules, CA). Each reaction contained SYBR Green Master Mix (Qiagen, Valencia, CA), cDNA template, and gene‐specific primers (sequences shown in Table S1). Gene expression results from LCM white matter and white matter punches were compared by quantitative real‐time polymerase chain reaction (qPCR) to determine which tissue isolation technique ultimately yielded sufficient amounts of RNA for the gene expression studies. Once determined, qPCR was performed for genes. Information for genes used for normalization (GAPDH, TBP), glial activation (GFAP, MOG), pro‐inflammatory (CD68, HLADR, IL1B, NOS2), and anti‐inflammatory markers (ARG1, IGF1, IGF1R, MRC1, PPARγ, VDR) are shown in Table 2. Each PCR reaction was performed in triplicate with pairs of tissue represented on the plate if multiple plates were used to collect data for each gene.
Table 2.
Reference Genes, Glial Markers, Pro‐Inflammatory Microglial Markers, and Anti‐Inflammatory Microglial Markers Used For Gene Expression Studies
| Gene name | Alias | Protein name | Brief description of function |
|---|---|---|---|
| Reference genes | |||
| GAPDH | Glyceraldehyde‐3‐phosphate dehydrogenase | GAPDH | Catalyzes an energy‐yielding step in glycolysis |
| TBP | TATA‐box binding protein | TBP | Transcription factor |
| Glial marker genes | |||
| GFAP | Glial fibrillary astrocytic protein | GFAP | Intermediate cellular filament that can be used to identify astrocytes |
| MOG | Myelin oligodendrocyte protein | MOG | Component of the myelin sheath that encases neuronal axons; used to identify oligodendrocytes |
| Pro‐inflammatory microglial markers | |||
| CD68 | Cluster of differentiation 68 | CD68 | Cell surface protein that clears cellular debris, and promotes phagocytosis |
| HLA‐DRA (HLA‐DR) | Major histocompatibility complex, class II, DR alpha chain | HLA‐DRA | Presents peptide antigens that are able to create an immune response |
| IL1β | Interleukin 1 beta | IL1β | Cytokine mediator in inflammatory responses and involved in cell proliferation, differentiation, and apoptosis |
| NOS2 | Nitric oxide synthase 2 | iNOS | Enzyme that generates nitric oxide (reactive free radical) |
| Anti‐inflammatory microglial markers | |||
| ARG1 | Arginase 1 | ARG1 | Enzyme that converts arginine into compounds used for wound repair and downregulates nitric oxide |
| IGF1 | Insulin‐like growth factor 1 | IGF1 | Ligand that stimulates proliferation of oligodendrocytes (supports myelination of neuronal axons) |
| IGF1R | Insulin‐like growth factor 1 receptor | IGF1R | Tyrosine kinase cell surface receptor that binds with IGF1 |
| MRC1 | Mannose receptor, C type 1 | CD206 | Receptor that binds and internalizes mannosylated ligands on potentially pathogenic microorganisms so they can be neutralized by phagocytic engulfment |
| PPARγ | Peroxisome proliferative activated receptor gamma | PPARγ | Receptor that inhibits pro‐inflammatory gene expression |
| VDR | Vitamin D Receptor | VDR | Receptor found on the nucleus that binds to calcitriol, the active form of Vitamin D |
Statistical Analysis
Expression data for target genes were normalized to reference genes GAPDH and TBP. Fold changes in the expression of genes of interest comparing ASD to control subjects were obtained using the 2−ΔΔCt method [Livak & Schmittgen, 2001]. Median Ct values were used for the final calculations for all genes. For these calculations, the geometric means of Ct values of reference genes were used for normalizations. Outliers in data sets were identified and removed using robust regression and outlier removal (ROUT; GraphPad Prism v.8 for macOS). Gene expression levels from control and ASD donors were compared using independent t‐tests, with equality of variances analyzed using the Levene's test [SPSS (version 24, IBM, New York, NY)]. Potentially confounding effects of age, RIN values, and postmortem intervals (PMIs), on gene expression levels were considered by computing Pearson correlations (GraphPad Prism v.8; Tables S2 and S3). When correlations between potentially confounding factors and gene expression levels reached a level of significance of P < 0.01, then those data were reanalyzed using the significant confounder as a covariate in the analysis (as noted). Because multiple t‐tests were performed, statistics are reported before and after Holm‐Bonferroni correction [Gaetano, 2013; Holm, 1979] for the number of gene comparisons (Tables 3 and 4).
Table 3.
Results of Independent t‐Tests for Comparison of Gray Matter Gene Expressions in Controls and ASD Brain Donors
| Gene | Sample size (control, ASD) | t value (or F as noted) | df | P | P′ a |
|---|---|---|---|---|---|
| Reference genesb | 17, 17 | 0.886 | 32 | 0.382 | 1.000 |
| GFAP | 12, 10 | 0.391 | 20 | 0.700 | 1.000 |
| MOG | 13, 12 | −1.573 | 20 | 0.129 | 1.000 |
| HLDAR | 13, 11 | 3.312c | 1 | 0.040 | 0.520 |
| IL1β | 11, 11 | −1.581d | 10.2 | 0.144 | 1.000 |
| CD68 | 13, 10 | 1.951 | 21 | 0.065 | 0.756 |
| NOS2 | 11, 11 | 0.42 | 20 | 0.679 | 1.000 |
| IGF1 | 15, 15 | 1.356 | 28 | 0.186 | 1.000 |
| IGF1R | 14, 14 | 3.823c | 1 | 0.063 | 0.756 |
| VDR | 13, 14 | −1.073 | 25 | 0.293 | 1.000 |
| ARG1 | 12, 13 | −0.632 | 23 | 0.534 | 1.000 |
| MRC1 | 14, 15 | 0.093 | 27 | 0.926 | 1.000 |
| PPARγ | 13, 13 | −0.111 | 24 | 0.913 | 1.000 |
P‐value corrected by Holm–Bonferroni Sequential Correction for multiple comparisons.
Comparison of the deltaCt values of TBP and GAPDH gene expression for control and ASD donors.
Values of statistic shown reflect results of analysis of covariance based on significant correlations (P < 0.01) identified for age, PMI, or RINH (see Table S2).
Statistics shown were calculated assuming unequal variances base on results of the Levene's test for equality of variances.
Table 4.
Results of Independent t‐Tests for Comparison of White Matter Gene Expressions in Controls and ASD Brain Donors
| Gene | Sample size (control, ASD) | t value (or F as noted) | df | P | P′ a |
|---|---|---|---|---|---|
| Reference genesb | 15, 14 | 1.458 | 27 | 0.156 | 1.000 |
| GFAP | 10, 9 | 0.006 | 17 | 0.995 | 1.000 |
| MOG | 10, 9 | 1.526c | 1 | 0.235 | 1.000 |
| HLDAR | 10, 9 | −0.059 | 17 | 0.954 | 1.000 |
| IL1β | 12, 12 | −1.971d | 12.7 | 0.071 | 0.726 |
| NOS2 | 10, 9 | 0.218 | 17 | 0.830 | 1.000 |
| IGF1 | 13, 12 | −1.935d | 12.3 | 0.076 | 0.726 |
| IGF1R | 11, 11 | −0.156 | 20 | 0.878 | 1.000 |
| VDR | 11, 10 | 3.822c | 1 | 0.066 | 0.726 |
| ARG1 | 8, 9 | −0.667 | 15 | 0.515 | 1.000 |
| MRC1 | 12, 13 | −2.827 | 23 | 0.010 | 0.120 |
| PPARγ | 10, 9 | −0.990 | 17 | 0.336 | 1.000 |
P‐value corrected by Holm–Bonferroni Sequential Correction for multiple comparisons.
Comparison of the deltaCt values of TBP and GAPDH genes from control and ASD donors.
Values of statistics shown reflect results of analysis of covariance based on significant correlations identified for age, RIN, or PMI (see Table S3).
Statistics shown were calculated assuming unequal variances base on results of the Levene's test for equality of variances.
Results
Sample Characterization
There were no statistical differences between the ASD and TD group regarding age, RIN, and PMI (Table 1). Additionally, no significant differences in the expression levels of housekeeping genes GAPDH and TBP were observed comparing ASD to TD donors (Fig. 1). Levels of GFAP and MOG were measured to determine if glia activation could be identified by elevated gene expression in the ACC of the ASD group. Both MOG and GFAP were not significantly changed in ACC gray matter from ASD compared to TD donors (Fig. 2A; Table 3). Gene expression levels for both glia genes were unchanged in the white matter (Fig. 2B; Table 4).
Figure 1.
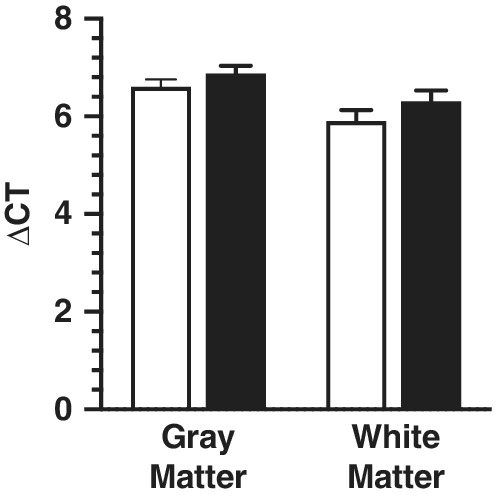
Difference between the Ct values of the two reference genes TBP and GAPDH in both gray (n = 17) and white matter (n = 14) using quantitative real‐time PCR. No significant differences were identified for either group in both brain areas.
Figure 2.
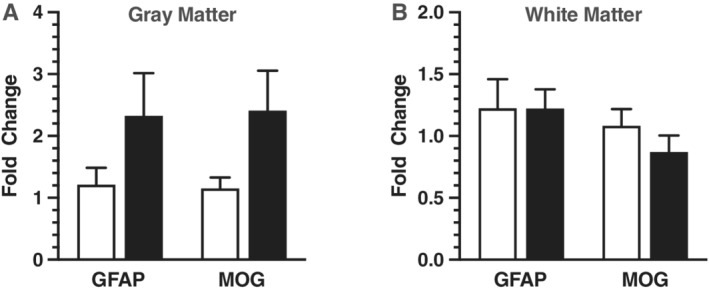
Panel A is real‐time quantitative PCR analysis of MOG (n = 13) and GFAP (n = 12) in gray matter using punch‐dissected anterior cingulate tissue centered at layer III from age‐matched pairs of male donor tissue from typically developing controls (unfilled bars) and males diagnosed with ASD (filled bars). Target gene expression was normalized to the geometric mean of TBP and GAPDH. Relative gene expression levels that were significant or close to significant are shown as fold changes using the Livak and Schmittgen method [Livak & Schmittgen, 2001]. Panel B is MOG (n = 10) and GFAP (n = 10) in white matter donor pairs.
Pro‐Inflammatory Related Cytokine Gene Expression
The expression levels of the pro‐inflammatory‐related genes HLA‐DR, IL1β, CD68 (gray only), and NOS2 were analyzed in gray and white matter from ASD and TD control donors (Fig. 3). Initial analysis of HLA‐DR expression levels in gray matter demonstrated significant correlations with age and PMI (Table S2). An analysis of variance with RIN and PMI as covariates indicated that HLA‐DR expression was significantly reduced in gray matter of ASD donors (P = 0.04; Fig. 3A); however, significance was lost after a Holm–Bonferroni correction for multiple comparisons was applied (Table 3). IL1β was modestly elevated in ACC white matter from ASD donors (P = 0.07; Fig. 3B; Table 4), while gray matter IL1β expression levels in gray matter showed the same trend (Table 4) with neither reaching statistical significance. CD68 expression levels in gray matter significantly correlated with RIN and PMI (Table S2) and covariate analysis of variance revealed a P‐value of 0.06 for the comparison of ASD to TD donors (Fig. 3A; Table 3). CD68 expression was not measured in white matter. No trending differences in NOS2 expression were found in either matter type comparing ASD to TD donors.
Figure 3.
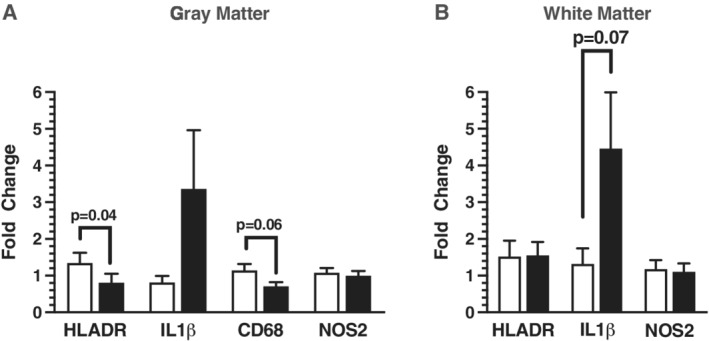
Real‐time quantitative polymerase chain reaction of pro‐inflammatory marker genes HLADR (n = 13), IL1β (n = 11), CD68 (n = 13), and NOS2 (n = 11) in gray matter shown in panel A with unfilled bars representing typically developing (TD) control donor tissue and filled bars representing ASD donor tissue punch‐dissected from layer III of the neocortex in gray matter. Panel B represents pro‐inflammatory related gene expression of HLADR (n = 10), IL1β (n = 12), and NOS2 (n = 10) in punch‐dissected superficial ACC white matter of TD and ASD donors. Relative gene expression was normalized to the geometric mean of TBP and GAPDH shown as fold changes using the Livak and Schmittgen method [Livak & Schmittgen, 2001].
Anti‐Inflammatory Related Cytokine Gene Expression
The level of expression of genes related to anti‐inflammatory mechanisms, including IGF1, IGF1R, VDR, ARG1, MRC1, and PPARγ were analyzed in white and gray matter from ASD and TD donors (Fig. 4). Expression levels for IGF1R demonstrated significant correlations with RIN and PMI (Table S2) in gray matter and covariate analysis of variance revealed modestly lower levels of IGF1R expression in gray matter from ASD donors as compared to TD donors (P = 0.06, Fig. 4A; Table 3). In white matter, both IGF1 (P = 0.08; Table 4) and VDR (P = 0.07; Table 4) demonstrated trends for elevated expression levels in ASD donors but did not make statistical significance. MRC1 expression levels were significantly elevated in white matter from ASD donors (P = 0.01; Fig. 4B; Table 4), but significance was lost upon application of the Holm–Bonferroni correction factor (Table 4). ARG1 and PPARγ did not demonstrate any differences in gene expression in gray or white matter comparing ASD to control donors.
Figure 4.
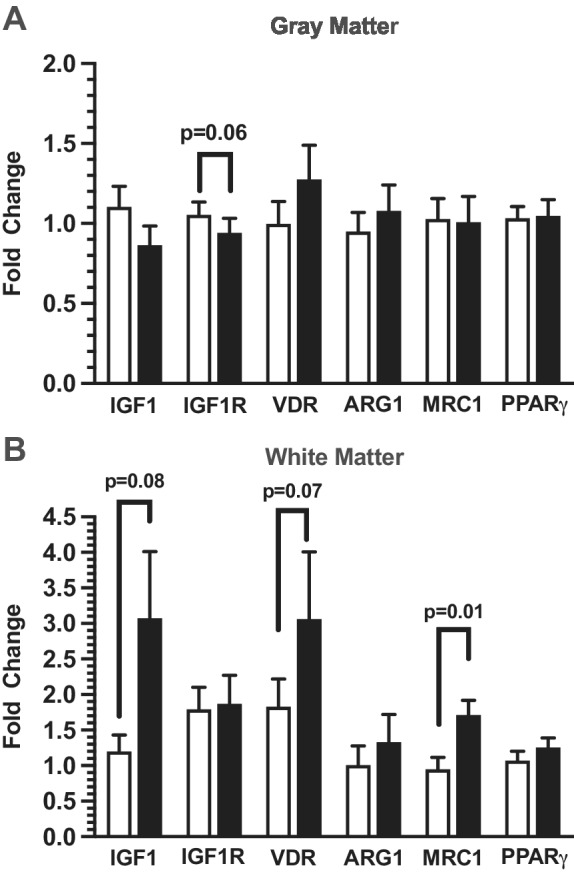
Real‐time polymerase chain analysis of anti‐inflammatory related genes IGF1 (n = 15), IGF1R (n = 14), VDR (n = 13), ARG1 (n = 12), MRC1 (n = 10), and PPARγ (n = 13) in ACC cortical gray matter punch‐dissected using layer three as the diameter in typically developing (TD) males (unfilled bars) and ASD donor tissue (filled bars) is shown in panel A. Panel B represents anti‐inflammatory related genes IGF1 (n = 13), IGF1R (n = 11), VDR (n = 11), ARG1 (n = 8), MRC1 (n = 12), and PPARγ (n = 10) in ACC superficial white matter. Relative gene expression was normalized to the geometric mean of TBP and GAPDH and shown as fold changes using the Livak and Schmittgen method [Livak & Schmittgen, 2001].
Discussion
Within the field of ASD research, pathology studies using postmortem brain tissues have been mainly limited to examining brain areas as a whole, without considering potential differences in white and gray matter. It is known that microglia reside in both white and gray matter; however, it is still unclear whether there is a difference in the functions of microglia that exist in white as compared to those that occur in gray matter [McKay, Brooks, Hu, & McLachlan, 2007]. The present research is one of the few studies that has characterized microglial phenotype‐specific gene expression levels in both white and gray matter brain tissue in ASD. Altered gene expression levels in both pro‐inflammatory and anti‐inflammatory markers in ACC from donors with ASD, when compared to TD controls, is the primary finding of this study.
Impaired neuronal communication likely through altered synaptic plasticity is considered a core neuropathophysiological feature of ASD. The pro‐inflammatory cytokine IL1β has a well‐documented history with neuronal synapse modulation. For example, early studies indicate that IL1β is necessary for long‐term potentiation in hippocampal CA1 as shown through IL1‐R (interleukin 1 receptor) knock‐out [Avital et al., 2003] and pharmacological methods [Coogan, O'Neill, & O'Connor, 1999; Schmid, Lynch, & Herron, 2009] in animals. Increases in IL1β gene expression follow long‐term potentiation (LTP) induction [Schneider et al., 1998] and ultimately inhibit excitatory synaptic transmission [Yang et al., 2005]. IL1β likely modulates LTP through changes in calcium conductance by way of the glutamate receptor family, NMDAR [Coogan & O'Connor, 1997; Gardoni et al., 2011; Viviani et al., 2003]. Very few findings have evaluated the impact of IL1β neuroinflammation on BDNF signaling in neurons. It has been shown using rat hippocampal slices that IL1β interferes with BDNF downstream signaling [Tong et al., 2012] and prevents the formation of dendritic spines in neurons [Tomasoni et al., 2017]. Additionally, IL1β interferes with BDNF‐induced cell survival specifically through mTOR signaling pathway [Smith et al., 2014]. It is possible that increases in IL1β impact synaptic signaling via BDNF in ASD. In human ASD, increased plasma levels of IL1β have been shown across the lifespan as reviewed by Saghazadeh et al. [2019]. Interestingly, our laboratory has shown changes in the gene expression levels of NMDAR receptors as well as downregulation of the BDNF receptor NTRK2 in ACC glutamatergic neurons in ASD as compared to cells captured in TD control ACC [Chandley, Crawford, Szebeni, Szebeni, & Ordway, 2015]. Although only trending toward statistical significance in this study for these subjects, altered levels of IL1β gene expression in human ASD brain tissue may indicate that immunity may play a direct role in etiology of the disorder.
Both CD‐68 and HLADR are membranous proteins that are associated with the pro‐inflammatory microglia phenotype. The findings in this study indicate that CD‐68 and HLA‐DR gene expression might be altered in the ACC (CD‐68 was only trending and statistical corrections eliminate the change in HLA‐DR). CD‐68 is associated with the innate immune response by signaling phagocytosis and debris clearance, while HLADR is associated with the complement immunity system and serves as a ligand for T‐cell binding. A previous study has shown that HLA‐DR protein levels is increased in ASD postmortem brain tissue [Vargas, Nascimbene, Krishnan, Zimmerman, & Pardo, 2005] and increases in serum complement cells were shown in blood from ASD patients [Ahmad et al., 2018]. The reduction in the mRNA of these molecules in this study would support that, at least at the gene level, alterations in the complement expression in the brain may result in synaptic hyperconnectivity as it has been shown in animals through reductions or knock‐out of pro‐inflammatory cytokines [Chu et al., 2010; Weinhard et al., 2018]. Excessive hyperconnectivity of synapses has been shown in mouse models of syndromic ASD including fragile X, tuberous sclerosis, and 15q duplication [Jawaid et al., 2018; Sato & Okabe, 2019; Tang et al., 2014].
Unlike IL1β, MRC1 expression does not have a well‐defined relationship with neurotransmission. Our findings indicate that MRC1 was elevated in ASD white matter, although significance was not retained after the correcting factor for multiple comparisons. Interestingly, MRC1 expression was originally identified on macrophages and represented a shift from anti‐inflammatory cells to active phagocytotic macrophages. The most prolific and well‐characterized function of MRC1 is to identify and facilitate ingestion of pathogens through binding to mannose found on bacteria. However, the receptor can bind a plethora of other molecules including oligosaccharides, glycoprotein hormones, myeloperoxidases, and lysosomal hydrolases. Binding to hydrolases and peroxidases indicates that MRC1 expressing cells are involved with the end stages of neuroinflammatory cellular events, that is, removal of apoptotic or necrotic cells. Mannose receptor activation can induce intracellular signaling events to trigger cytokine production of IL10 and other molecules that act to suppress the immune response [Chieppa et al., 2003]. Conversely, it has been shown through in vitro studies that IL10 can induce MRC1 receptor expression in cultured microglia [Liu et al., 2013; Makita, Hizukuri, Yamashiro, Murakawa, & Hayashi, 2015]. Evaluations using fluorescence activated cell sorting and other cell sorting methods have demonstrated that in human brain tissue there are subsets of microglia populations with detectable MRC1 protein expression [Böttcher et al., 2019; Galatro et al., 2017] and are considered representatives of the anti‐inflammatory microglial phenotype. Modulation of inflammatory levels by external environmental or therapeutic exposures can influence the shift in microglial phenotype. Dimethyl fumarate, used in the treatment of multiple sclerosis (MS), was found to reduce pro‐inflammatory levels [Spencer, Crabtree‐Hartman, Lehmann‐Horn, Cree, & Zamvil, 2015; Wilms et al., 2010] and in vitro studies with the drug led to increased gene expression of MRC1 [Han, Xiao, Zhai, & Hao, 2016; Kronenberg et al., 2019]. Overall, elevations of MRC1 in ASD tissue could indicate atypical levels of inflammation in white matter that would impact cell signaling between brain regions. Upregulation of MRC1 expression has been shown to be neuroprotective in animal models of spinal cord injuries [Ji et al., 2015; Stivers et al., 2017]. Interestingly, it was shown that rats exposed to lipopolysaccharide at postnatal day three demonstrated increased expression levels of MRC1 protein that had significant impairments in communication and cognitive functions [Pang et al., 2016]. While much information is lacking regarding the role of MRC1‐specific anti‐inflammatory microglia in neurotransmission, MRC1 could serve as a valuable target for future etiology or therapeutic studies in ASD.
Current clinical trials using treatments with IGF1 for idiopathic ASD, tuberous sclerosis, and Rett syndrome patients are underway. Our study did not identify significant gene expression changes for either the ligand (IGF1) or the receptor (IGF1R). However, it is worth mentioning that we did see trending elevations for IGF1 gene expression in ASD white matter brain tissue as well as trending reductions in the IGF1R gene expression in ASD gray matter when compared to TD control tissue. IGF1 is a mitogenic factor imperative for fetal brain development and growth. In the brain, IGF1 encourages the differentiation and maturation of oligodendrocytes, myelination, and neuronal survival. IGF1 Knock‐out mice display postnatal lethality, developmental retardation, defects in organ systems, and infertility [Liu, Yakar, & LeRoith, 2000] while overexpressing IGF1 protein in mice results in increased brain size and neuronal numbers [Ye et al., 2004]. IGF1 binds tightly with the receptor (IGF‐1R), triggering the autophosphorylation of the intracellular β‐subunit kinase domain of IGF‐1R to recruit adaptor proteins and activate several pathways including the MAP kinase and PI3‐kinase/Akt pathways. Both the MAP kinase and PI3‐kinase/Akt pathways activate mTOR, which through increasing the cap‐dependent translation initiation complex can increase mRNA translation to influence multiple developmental functions [Bondy & Cheng, 2004]. Interestingly, Faridar et al. [2014] reported increased activation of the MAP kinase pathway in mice with ASD‐like social and behavioral deficits. Increases in IGF1 expression in the white matter could promote elevated mTOR activation. An upregulation of mTOR and its activity in individuals with ASD would confirm a hypothesis made by other researchers that mTOR is in fact upregulated in ASD, causing an increase in unregulated protein synthesis [Chen, Alberts, & Li, 2014; Sawicka & Zukin, 2012; Wang & Doering, 2013]. Additionally, elevated IGF1 expression may reflect a compensatory mechanism of the brain in response to altered neurotransmission in the ACC leading to the decreased structural integrity of the ACC white matter that result in deficits in cognitive processing. Increased proliferation and differentiation of oligodendrocytes by IGF1 would in turn myelinate neuronal axons thereby increasing synchronization between brain areas and white matter structural integrity. A pilot study involving nine children with Phelan‐McDermid syndrome, a related syndromic ASD, reported an association between 3 months of IGF1 treatment and substantial improvement in social impairment and behavior [Kolevzon et al., 2014]. It is not possible at this point to know whether IGF1 gene expression changes are casual in the etiology of autism or a result of cellular processes that are activated to correct deficits that have their root causes in other pathophysiological mechanisms. With the prevalence of myelinating oligodendrocytes in the white matter, it is plausible that the demand for IGF1 in the white matter of ASD individuals may be higher than in the gray matter, consistent with our findings.
A most interesting finding of this study is the gene expression trends of specifically the anti‐inflammatory related gene expression changes in white matter. White matter has exhibited a unique pathology in ASD. For example, diffusor tensor imaging studies have shown that subjects with ASD exhibit reduced fractional anisotropy in white matter areas of brain areas associated with social behaviors, including the ACC, than in TD control subjects [Barnea‐Goraly et al., 2004] and can be correlated with diagnostic phenotype [Cheung et al., 2009; Noriuchi et al., 2010]. Our own laboratory found increased GFAP protein in white matter [Crawford et al., 2015] that could be related to elevated cytokine levels [Madathil et al., 2013]. Volumetric studies indicate that white matter volume is increased in ASD when compared to TD control brains using postmortem and imaging studies [as reviewed by Amaral, Schumann, & Nordahl, 2008]. Lastly, myelin thickness was altered in postmortem ASD donor tissue in white matter as compared to TD donor tissue, with no change in oligodendrocyte number and morphology [Zikopoulos & Barbas, 2010]. While white matter pathology exists in ASD, less clear is the relationship with gray matter neocortical pathology. In multiple sclerosis there is unique, discernable neuronal pathology in the neocortical gray matter [as reviewed by Lassmann, 2010], although the basis of the disease lies in demyelination of axons in the white matter. Interestingly, it was discovered in tumor metastatic studies that blood vessels travel through the brain tissue to coil in the gray‐white matter junction known as the superficial or radial white matter [Nonaka et al., 2003]. It is quite possible the inflammatory alterations are found in these areas of increased vasculature where they can alter myelination and ultimately exhibit a discernable neuronal pathology that occurred during critical developmental periods with a genetic susceptibility leading to a multifactorial etiology with a heterogeneous phenotypic presentation.
Limitations
The availability of postmortem ASD brain tissues is limited, preventing us from working with larger sample sizes. For all gene expression data, P‐values were corrected for the number of comparisons of dependent variables (gene expressions) using the Holm‐Bonferroni correction. The corrected P‐value indicated no statistically significant difference between ASD and control donors for gene expression in the ACC (Tables 3 and 4). We attribute a lack of Holm‐Bonferroni adjusted significance of the expression levels to our small sample size. Additionally, because of the limited availability of tissues, some ASD and control donors included in the study were exposed to medications (Table 1) that might potentially influence the outcomes of this study. Group donors were identified a priori by the brain banks and variation between groups was reduced as much as possible. Quantification of protein at a cellular level would give a more definitive result; however, ASD donor tissue is an extremely limited resource.
Conclusions
Genetic studies have introduced many candidate genes that have been identified in ASD and first‐order relatives. It has become apparent that one single gene is not responsible for ASD, but rather many closely related genes in conjunction with an environmental stimulus likely triggers alterations that are found in social behavior brain areas. Postmortem tissue is an important resource for the identification of those transcriptomic markers that are associated with symptom presentation or severity and will lead to mechanistic studies to find causes or treatments for ASD. Our study was able to identify alterations prior to statistical corrections of specific cytokines and cell markers for glial activation in a brain region associated with social behaviors. These markers may be a key part of the relationship between genome‐wide studies, environmental exposures, and the resulting neuropathological findings in ASD.
Conflict of Interest
No conflict of interest exists for any of the authors associated with the following manuscript.
Abbreviations
- ACC
Anterior cingulate cortex
- ASD
Autism spectrum disorder
- BA10
Brodmann Area 10
- BA24
Brodmann Area 24
- EEG
Electroencephalography
- GABA
gamma‐aminobutyric acid
- LCM
Laser capture microdissection
- NICHD
National Institutes for Child Health and Development
- NMDA
N‐methyl‐D‐aspartate
- PET
Positron emission tomography
- PMI
Postmortem interval
- RIN
RNA integrity number
- SPECT
single‐photon emission computed tomography
- SSRI
Selective serotonin reuptake inhibitors
Supporting information
Supplementary table 1: Primer sequences for PCR amplification reactions for each gene.
Supplementary Table 2: Pearson's correlation analyses for possible relationships between gene expression levels and age, RNA quality (RIN) and postmortem interval (PMI) for all genes in gray matter (GM).
Supplementary Table 3: Pearson's correlation analyses for possible relationships between gene expression levels and age, RNA quality (RIN) and postmortem interval (PMI) for all genes in white matter (WM).
Acknowledgments
We are extremely grateful to the families that participated in this study with the decision to donate brain tissue. This contribution is priceless and will support ASD research for many years. Tissues used in these studies were obtained from Autism BrainNet, a resource of the Simons Foundation Autism Research Initiative (SFARI) and manages the Autism Tissue Program, and NIH Neurobiobank (formerly the NICHD Brain and Tissue Bank for Developmental Disorders). The National Institutes of Health 1R15MH119628‐01 and the Department of Defense Grant AR110337 assisted this research by providing support for this study. Additional support was provided by the National Center for Research Resources grant number RR030651.
References
- Abdallah, M. W. , Larsen, N. , Grove, J. , Nørgaard‐Pedersen, B. , Thorsen, P. , Mortensen, E. L. , & Hougaard, D. M. (2013). Amniotic fluid inflammatory cytokines: Potential markers of immunologic dysfunction in autism spectrum disorders. The World Journal of Biological Psychiatry, 14(7), 528–538. 10.3109/15622975.2011.639803 [DOI] [PubMed] [Google Scholar]
- Ahmad, S. F. , Ansari, M. A. , Nadeem, A. , Bakheet, S. A. , Al‐Ayadhi, L. Y. , Alotaibi, M. R. , … Attia, S. M. (2018). Dysregulation of the expression of HLA‐DR, costimulatory molecule, and chemokine receptors on immune cells in children with autism. International Immunopharmacology, 65, 360–365. 10.1016/j.intimp.2018.10.027 [DOI] [PubMed] [Google Scholar]
- Amaral, D. G. , Schumann, C. M. , & Nordahl, C. W. (2008). Neuroanatomy of autism. Trends in Neurosciences, 31(3), 137–145. 10.1016/j.tins.2007.12.005 [DOI] [PubMed] [Google Scholar]
- Ariza, J. , Rogers, H. , Hashemi, E. , Noctor, S. C. , & Martínez‐Cerdeño, V. (2018). The number of chandelier and basket cells are differentially decreased in prefrontal cortex in autism. Cerebral Cortex, 28(2), 411–420. 10.1093/cercor/bhw349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atladottir, H. O. , Henriksen, T. B. , Schendel, D. E. , & Parner, E. T. (2012). Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics, 130(6), e1447–e1454. 10.1542/peds.2012-1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atladottir, H. O. , Pedersen, M. G. , Thorsen, P. , Mortensen, P. B. , Deleuran, B. , Eaton, W. W. , & Parner, E. T. (2009). Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics, 124(2), 687–694. 10.1542/peds.2008-2445 [DOI] [PubMed] [Google Scholar]
- Avital, A. , Goshen, I. , Kamsler, A. , Segal, M. , Iverfeldt, K. , Richter‐Levin, G. , & Yirmiya, R. (2003). Impaired interleukin‐1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus, 13(7), 826–834. 10.1002/hipo.10135 [DOI] [PubMed] [Google Scholar]
- Aylward, E. H. , Minshew, N. J. , Goldstein, G. , Honeycutt, N. A. , Augustine, A. M. , Yates, K. O. , … Pearlson, G. D. (1999). MRI volumes of amygdala and hippocampus in non‐mentally retarded autistic adolescents and adults. Neurology, 53(9), 2145–2150. 10.1212/wnl.53.9.2145 [DOI] [PubMed] [Google Scholar]
- Barnea‐Goraly, N. , Kwon, H. , Menon, V. , Eliez, S. , Lotspeich, L. , & Reiss, A. L. (2004). White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biological Psychiatry, 55(3), 323–326. [DOI] [PubMed] [Google Scholar]
- Blakemore, S.‐J. (2008). The social brain in adolescence. Nature Reviews Neuroscience, 9(4), 267–277. 10.1038/nrn2353 [DOI] [PubMed] [Google Scholar]
- Bondy, C. A. , & Cheng, C. M. (2004). Signaling by insulin‐like growth factor 1 in brain. European Journal of Pharmacology, 490(1–3), 25–31. 10.1016/j.ejphar.2004.02.042 [DOI] [PubMed] [Google Scholar]
- Böttcher, C. , Schlickeiser, S. , Sneeboer, M. A. M. , Kunkel, D. , Knop, A. , Paza, E. , … Priller, J. (2019). Human microglia regional heterogeneity and phenotypes determined by multiplexed single‐cell mass cytometry. Nature Neuroscience, 22(1), 78–90. 10.1038/s41593-018-0290-2 [DOI] [PubMed] [Google Scholar]
- Buescher, A. V. S. , Cidav, Z. , Knapp, M. , & Mandell, D. S. (2014). Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatrics, 168(8), 721–728. 10.1001/jamapediatrics.2014.210 [DOI] [PubMed] [Google Scholar]
- C Yuen, R. K. , Merico, D. , Bookman, M. , L Howe, J. , Thiruvahindrapuram, B. , Patel, R. V. , … Scherer, S. W. (2017). Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nature Neuroscience, 20(4), 602–611. 10.1038/nn.4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, S. W. , Madathil, S. K. , Sama, D. M. , Gao, X. , Chen, J. , & Saatman, K. E. (2014). Conditional overexpression of insulin‐like growth factor‐1 enhances hippocampal neurogenesis and restores immature neuron dendritic processes after traumatic brain injury. Journal of Neuropathology & Experimental Neurology, 73(8), 734–746. 10.1097/NEN.0000000000000092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova, M. F. , Buxhoeveden, D. P. , Switala, A. E. , & Roy, E. (2002). Minicolumnar pathology in autism. Neurology, 58(3), 428–432. 10.1212/wnl.58.3.428 [DOI] [PubMed] [Google Scholar]
- Chandley, M. J. , Crawford, J. D. , Szebeni, A. , Szebeni, K. , & Ordway, G. A. (2015). NTRK2 expression levels are reduced in laser captured pyramidal neurons from the anterior cingulate cortex in males with autism spectrum disorder. Molecular Autism, 6(1), 28 10.1186/s13229-015-0023-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Alberts, I. , & Li, X. (2014). Dysregulation of the IGF‐I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. International Journal of Developmental Neuroscience, 35, 35–41. 10.1016/j.ijdevneu.2014.03.006 [DOI] [PubMed] [Google Scholar]
- Cheng, C. M. , Mervis, R. F. , Niu, S.‐L. , Salem, N. , Witters, L. A. , Tseng, V. , … Bondy, C. A. (2003). Insulin‐like growth factor 1 is essential for normal dendritic growth. Journal of Neuroscience Research, 73(1), 1–9. 10.1002/jnr.10634 [DOI] [PubMed] [Google Scholar]
- Cheung, C. , Chua, S. E. , Cheung, V. , Khong, P. L. , Tai, K. S. , Wong, T. K. W. , … McAlonan, G. M. (2009). White matter fractional anisotrophy differences and correlates of diagnostic symptoms in autism. Journal of Child Psychology and Psychiatry, and Allied Disciplines, 50(9), 1102–1112. 10.1111/j.1469-7610.2009.02086.x [DOI] [PubMed] [Google Scholar]
- Chieppa, M. , Bianchi, G. , Doni, A. , Del Prete, A. , Sironi, M. , Laskarin, G. , … Allavena, P. (2003). Cross‐linking of the mannose receptor on monocyte‐derived dendritic cells activates an anti‐inflammatory immunosuppressive program. Journal of Immunology, 171(9), 4552–4560. 10.4049/jimmunol.171.9.4552 [DOI] [PubMed] [Google Scholar]
- Christensen, J. , Grønborg, T. K. , Sørensen, M. J. , Schendel, D. , Parner, E. T. , Pedersen, L. H. , & Vestergaard, M. (2013). Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA, 309(16), 1696–1703. 10.1001/jama.2013.2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, Y. , Jin, X. , Parada, I. , Pesic, A. , Stevens, B. , Barres, B. , & Prince, D. A. (2010). Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proceedings of the National Academy of Sciences of the United States of America, 107(17), 7975–7980. 10.1073/pnas.0913449107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantino, J. N. , Zhang, Y. , Frazier, T. , Abbacchi, A. M. , & Law, P. (2010). Sibling recurrence and the genetic epidemiology of autism. American Journal of Psychiatry, 167(11), 1349–1356. 10.1176/appi.ajp.2010.09101470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan, A. , & O'Connor, J. J. (1997). Inhibition of NMDA receptor‐mediated synaptic transmission in the rat dentate gyrus in vitro by IL‐1 beta. Neuroreport, 8(9–10), 2107–2110. 10.1097/00001756-199707070-00004 [DOI] [PubMed] [Google Scholar]
- Coogan, A. N. , O'Neill, L. A. , & O'Connor, J. J. (1999). The P38 mitogen‐activated protein kinase inhibitor SB203580 antagonizes the inhibitory effects of interleukin‐1beta on long‐term potentiation in the rat dentate gyrus in vitro. Neuroscience, 93(1), 57–69. 10.1016/s0306-4522(99)00100-1 [DOI] [PubMed] [Google Scholar]
- Corvin, A. P. , Molinos, I. , Little, G. , Donohoe, G. , Gill, M. , Morris, D. W. , & Tropea, D. (2012). Insulin‐like growth factor 1 (IGF1) and its active peptide (1–3)IGF1 enhance the expression of synaptic markers in neuronal circuits through different cellular mechanisms. Neuroscience Letters, 520(1), 51–56. 10.1016/j.neulet.2012.05.029 [DOI] [PubMed] [Google Scholar]
- Courchesne, E. , Carper, R. , & Akshoomoff, N. (2003). Evidence of brain overgrowth in the first year of life in autism. JAMA, 290(3), 337–344. 10.1001/jama.290.3.337 [DOI] [PubMed] [Google Scholar]
- Crawford, J. D. , Chandley, M. J. , Szebeni, K. , Szebeni, A. , Waters, B. , & Ordway, G. A. (2015). Elevated GFAP protein in anterior cingulate cortical white matter in males with autism spectrum disorder. Autism Research, 8(6), 649–657. 10.1002/aur.1480 [DOI] [PubMed] [Google Scholar]
- Depino, A. M. , Lucchina, L. , & Pitossi, F. (2011). Early and adult hippocampal TGF‐β1 overexpression have opposite effects on behavior. Brain, Behavior, and Immunity, 25(8), 1582–1591. 10.1016/j.bbi.2011.05.007 [DOI] [PubMed] [Google Scholar]
- Faridar, A. , Jones‐Davis, D. , Rider, E. , Li, J. , Gobius, I. , Morcom, L. , … Sherr, E. H. (2014). Mapk/Erk activation in an animal model of social deficits shows a possible link to autism. Molecular Autism, 5(1), 57 10.1186/2040-2392-5-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetano, J. (2013). Holm‐Bonferroni sequential correction: An EXCEL calculator. Retrieved from https://www.researchgate.net/publication/236969037_Holm‐Bonferroni_Sequential_Correction_An_EXCEL_Calculator.10.13140/RG.2.1.4466.9927 [Google Scholar]
- Galatro, T. F. , Holtman, I. R. , Lerario, A. M. , Vainchtein, I. D. , Brouwer, N. , Sola, P. R. , … Eggen, B. J. L. (2017). Transcriptomic analysis of purified human cortical microglia reveals age‐associated changes. Nature Neuroscience, 20(8), 1162–1171. 10.1038/nn.4597 [DOI] [PubMed] [Google Scholar]
- Gardoni, F. , Boraso, M. , Zianni, E. , Corsini, E. , Galli, C. L. , Cattabeni, F. , … Viviani, B. (2011). Distribution of interleukin‐1 receptor complex at the synaptic membrane driven by interleukin‐1β and NMDA stimulation. Journal of Neuroinflammation, 8(1), 14 10.1186/1742-2094-8-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner, J. T. , Wang, K. , Cai, G. , Korvatska, O. , Kim, C. E. , Wood, S. , … Hakonarson, H. (2009). Autism genome‐wide copy number variation reveals ubiquitin and neuronal genes. Nature, 459(7246), 569–573. 10.1038/nature07953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goines, P. E. , Croen, L. A. , Braunschweig, D. , Yoshida, C. K. , Grether, J. , Hansen, R. , … Van de Water, J. (2011). Increased midgestational IFN‐γ, IL‐4 and IL‐5 in women bearing a child with autism: A case‐control study. Molecular Autism, 2(1), 13 10.1186/2040-2392-2-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomot, M. , Bernard, F. A. , Davis, M. H. , Belmonte, M. K. , Ashwin, C. , Bullmore, E. T. , & Baron‐Cohen, S. (2006). Change detection in children with autism: An auditory event‐related fMRI study. NeuroImage, 29(2), 475–484. 10.1016/j.neuroimage.2005.07.027 [DOI] [PubMed] [Google Scholar]
- Hallmayer, J. , Cleveland, S. , Torres, A. , Phillips, J. , Cohen, B. , Torigoe, T. , … Risch, N. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Archives of General Psychiatry, 68(11), 1095–1102. 10.1001/archgenpsychiatry.2011.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, R. , Xiao, J. , Zhai, H. , & Hao, J. (2016). Dimethyl fumarate attenuates experimental autoimmune neuritis through the nuclear factor erythroid‐derived 2‐related factor 2/hemoxygenase‐1 pathway by altering the balance of M1/M2 macrophages. Journal of Neuroinflammation, 13(1), 97 10.1186/s12974-016-0559-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington, R. A. , Lee, L.‐C. , Crum, R. M. , Zimmerman, A. W. , & Hertz‐Picciotto, I. (2014). Prenatal SSRI use and offspring with autism spectrum disorder or developmental delay. Pediatrics, 133(5), e1241–e1248. 10.1542/peds.2013-3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashemi, E. , Ariza, J. , Rogers, H. , Noctor, S. C. , & Martínez‐Cerdeño, V. (2016). The number of parvalbumin‐expressing interneurons is decreased in the medial prefrontal cortex in autism. Cerebral Cortex, 27(3), bhw021 10.1093/cercor/bhw021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, S. (1979). A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics, 6, 65–70. 10.2307/4615733 [DOI] [Google Scholar]
- Hutsler, J. J. , & Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Research, 1309, 83–94. 10.1016/j.brainres.2009.09.120 [DOI] [PubMed] [Google Scholar]
- Jawaid, S. , Kidd, G. J. , Wang, J. , Swetlik, C. , Dutta, R. , & Trapp, B. D. (2018). Alterations in CA1 hippocampal synapses in a mouse model of fragile X syndrome. Glia, 66(4), 789–800. 10.1002/glia.23284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, X.‐C. , Dang, Y.‐Y. , Gao, H.‐Y. , Wang, Z.‐T. , Gao, M. , Yang, Y. , … Xu, R.‐X. (2015). Local injection of lenti–BDNF at the lesion site promotes M2 macrophage polarization and inhibits inflammatory response after spinal cord injury in mice. Cellular and Molecular Neurobiology, 35(6), 881–890. 10.1007/s10571-015-0182-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just, M. A. , Cherkassky, V. L. , Keller, T. A. , Kana, R. K. , & Minshew, N. J. (2007). Functional and anatomical cortical underconnectivity in autism: Evidence from an fMRI study of an executive function task and corpus callosum morphometry. Cerebral Cortex, 17(4), 951–961. 10.1093/cercor/bhl006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil, A. , Daniels, J. L. , Forssen, U. , Hultman, C. , Cnattingius, S. , Söderberg, K. C. , … Sparen, P. (2010). Parental autoimmune diseases associated with autism spectrum disorders in offspring. Epidemiology (Cambridge, Mass.), 21(6), 805–808. 10.1097/EDE.0b013e3181f26e3f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolevzon, A. , Bush, L. , Wang, A. , Halpern, D. , Frank, Y. , Grodberg, D. , … Buxbaum, J. D. (2014). A pilot controlled trial of insulin‐like growth factor‐1 in children with Phelan‐McDermid syndrome. Molecular Autism, 5(1), 54 10.1186/2040-2392-5-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotagiri, P. , Chance, S. A. , Szele, F. G. , & Esiri, M. M. (2014). Subventricular zone cytoarchitecture changes in Autism. Developmental Neurobiology, 74(1), 25–41. 10.1002/dneu.22127 [DOI] [PubMed] [Google Scholar]
- Kronenberg, J. , Pars, K. , Brieskorn, M. , Prajeeth, C. , Heckers, S. , Schwenkenbecher, P. , … Stangel, M. (2019). Fumaric acids directly influence gene expression of neuroprotective factors in rodent microglia. International Journal of Molecular Sciences, 20(2), 325 10.3390/ijms20020325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann, H. (2010). Axonal and neuronal pathology in multiple sclerosis: What have we learnt from animal models. Experimental Neurology, 225(1), 2–8. 10.1016/j.expneurol.2009.10.009 [DOI] [PubMed] [Google Scholar]
- Laurence, J. A. , & Fatemi, S. H. (2005). Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. The Cerebellum, 4(3), 206–210. 10.1080/14734220500208846 [DOI] [PubMed] [Google Scholar]
- Lavelle, T. A. , Weinstein, M. C. , Newhouse, J. P. , Munir, K. , Kuhlthau, K. A. , & Prosser, L. A. (2014). Economic burden of childhood autism spectrum disorders. Pediatrics, 133(3), e520–e529. 10.1542/peds.2013-0763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C.‐Y. , Xu, J.‐Y. , Shi, X.‐Y. , Huang, W. , Ruan, T.‐Y. , Xie, P. , & Ding, J.‐L. (2013). M2‐polarized tumor‐associated macrophages promoted epithelial‐mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL‐10 signaling pathway. Laboratory Investigation, 93(7), 844–854. 10.1038/labinvest.2013.69 [DOI] [PubMed] [Google Scholar]
- Liu, J. L. , Yakar, S. , & LeRoith, D. (2000). Conditional knockout of mouse insulin‐like growth factor‐1 gene using the Cre/loxP system. Proceedings of the Society for Experimental Biology and Medicine, 223(4), 344–351. 10.1046/j.1525-1373.2000.22349.x [DOI] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gne expression data using real‐time quantiative PCR and the 2(−delta delta C(t)) method. Methods, 25(4), 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Madathil, S. K. , Carlson, S. W. , Brelsfoard, J. M. , Ye, P. , D'Ercole, A. J. , & Saatman, K. E. (2013). Astrocyte‐specific overexpression of insulin‐like growth factor‐1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS One, 8(6), e67204 10.1371/journal.pone.0067204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita, N. , Hizukuri, Y. , Yamashiro, K. , Murakawa, M. , & Hayashi, Y. (2015). IL‐10 enhances the phenotype of M2 macrophages induced by IL‐4 and confers the ability to increase eosinophil migration. International Immunology, 27(3), 131–141. 10.1093/intimm/dxu090 [DOI] [PubMed] [Google Scholar]
- McKay, S. M. , Brooks, D. J. , Hu, P. , & McLachlan, E. M. (2007). Distinct types of microglial activation in white and grey matter of rat lumbosacral cord after mid‐thoracic spinal transection. Journal of Neuropathology and Experimental Neurology, 66(8), 698–710. 10.1097/nen.0b013e3181256b32 [DOI] [PubMed] [Google Scholar]
- Morgan, J. T. , Barger, N. , Amaral, D. G. , & Schumann, C. M. (2014). Stereological study of amygdala glial populations in adolescents and adults with autism spectrum disorder. PLoS One, 9(10), e110356 10.1371/journal.pone.0110356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, B. M. , Kou, Y. , Liu, L. , Ma'ayan, A. , Samocha, K. E. , Sabo, A. , … Daly, M. J. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485(7397), 242–245. 10.1038/nature11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka, H. , Akima, M. , Hatori, T. , Nagayama, T. , Zhang, Z. , & Ihara, F. (2003). The microvasculature of the cerebral white matter: Arteries of the subcortical white matter. Journal of Neuropathology & Experimental Neurology, 62(2), 154–161. 10.1093/jnen/62.2.154 [DOI] [PubMed] [Google Scholar]
- Noriuchi, M. , Kikuchi, Y. , Yoshiura, T. , Kira, R. , Shigeto, H. , Hara, T. , … Kamio, Y. (2010). Altered white matter fractional anisotropy and social impairment in children with autism spectrum disorder. Brain Research, 1362, 141–149. 10.1016/j.brainres.2010.09.051 [DOI] [PubMed] [Google Scholar]
- Pang, Y. , Dai, X. , Roller, A. , Carter, K. , Paul, I. , Bhatt, A. J. , … Fan, L. W. (2016). Early postnatal lipopolysaccharide exposure leads to enhanced neurogenesis and impaired communicative functions in rats. PloS one, 11(10), e0164403 10.1371/journal.pone.0164403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch, A. , Zhang, W. , Haak, K. V. , Mennes, M. , Hermans, E. J. , van Oort, E. , … Groen, W. B. (2016). Altered functional connectivity of the amygdaloid input nuclei in adolescents and young adults with autism spectrum disorder: A resting state fMRI study. Molecular Autism, 7(1), 13 10.1186/s13229-015-0060-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi, S. , Boggio, E. M. , Grosso, S. , Lonetti, G. , Forlani, G. , Stefanelli, G. , … Broccoli, V. (2011). Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Human Molecular Genetics, 20(6), 1182–1196. 10.1093/hmg/ddq563 [DOI] [PubMed] [Google Scholar]
- Rosenberg, R. E. , Law, J. K. , Yenokyan, G. , McGready, J. , Kaufmann, W. E. , & Law, P. A. (2009). Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Archives of Pediatrics & Adolescent Medicine, 163(10), 907–914. 10.1001/archpediatrics.2009.98 [DOI] [PubMed] [Google Scholar]
- Saghazadeh, A. , Ataeinia, B. , Keynejad, K. , Abdolalizadeh, A. , Hirbod‐Mobarakeh, A. , & Rezaei, N. (2019). A meta‐analysis of pro‐inflammatory cytokines in autism spectrum disorders: Effects of age, gender, and latitude. Journal of Psychiatric Research, 115, 90–102. 10.1016/j.jpsychires.2019.05.019 [DOI] [PubMed] [Google Scholar]
- Sanders, S. J. , Murtha, M. T. , Gupta, A. R. , Murdoch, J. D. , Raubeson, M. J. , Willsey, A. J. , … State, M. W. (2012). De novo mutations revealed by whole‐exome sequencing are strongly associated with autism. Nature, 485(7397), 237–241. 10.1038/nature10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, Y. , & Okabe, S. (2019). Nano‐scale analysis of synapse morphology in an autism mouse model with 15q11‐13 copy number variation using focused ion beam milling and scanning electron microscopy. Microscopy, 68(2), 122–132. 10.1093/jmicro/dfy128 [DOI] [PubMed] [Google Scholar]
- Sawicka, K. , & Zukin, R. S. (2012). Dysregulation of mTOR signaling in neuropsychiatric disorders: Therapeutic implications. Neuropsychopharmacology, 37(1), 305–306. 10.1038/npp.2011.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, G. (2016). clinical genetic aspects of autism spectrum disorders. International Journal of Molecular Sciences, 17(2), 180 10.3390/ijms17020180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, A. W. , Lynch, M. A. , & Herron, C. E. (2009). The effects of IL‐1 receptor antagonist on beta amyloid mediated depression of LTP in the rat CA1 in vivo. Hippocampus, 19(7), 670–676. 10.1002/hipo.20542 [DOI] [PubMed] [Google Scholar]
- Schneider, H. , Pitossi, F. , Balschun, D. , Wagner, A. , del Rey, A. , & Besedovsky, H. O. (1998). A neuromodulatory role of interleukin‐1 in the hippocampus. Proceedings of the National Academy of Sciences, 95(13), 7778–7783. 10.1073/pnas.95.13.7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann, C. M. , & Amaral, D. G. (2006). Stereological analysis of amygdala neuron number in autism. Journal of Neuroscience, 26(29), 7674–7679. 10.1523/JNEUROSCI.1285-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat, J. , Lakshmi, B. , Malhotra, D. , Troge, J. , Lese‐Martin, C. , Walsh, T. , … Wigler, M. (2007). Strong association of de novo copy number mutations with autism. Science, 316(5823), 445–449. 10.1126/science.1138659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A. , Hoeffer, C. A. , Takayasu, Y. , Miyawaki, T. , McBride, S. M. , Klann, E. , & Zukin, R. S. (2010). Dysregulation of mTOR signaling in fragile X syndrome. Journal of Neuroscience, 30(2), 694–702. 10.1523/JNEUROSCI.3696-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheglovitov, A. , Shcheglovitova, O. , Yazawa, M. , Portmann, T. , Shu, R. , Sebastiano, V. , … Dolmetsch, R. E. (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature, 503(7475), 267–271. 10.1038/nature12618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms, M. L. , Kemper, T. L. , Timbie, C. M. , Bauman, M. L. , & Blatt, G. J. (2009). The anterior cingulate cortex in autism: heterogeneity of qualitative and quantitative cytoarchitectonic features suggests possible subgroups. Acta Neuropathologica, 118(5), 673–684. 10.1007/s00401-009-0568-2 [DOI] [PubMed] [Google Scholar]
- Smith, E. D. , Prieto, G. A. , Tong, L. , Sears‐Kraxberger, I. , Rice, J. D. , Steward, O. , & Cotman, C. W. (2014). Rapamycin and interleukin‐1β impair brain‐derived neurotrophic factor‐dependent neuron survival by modulating autophagy. Journal of Biological Chemistry, 289(30), 20615–20629. 10.1074/jbc.M114.568659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, C. M. , Crabtree‐Hartman, E. C. , Lehmann‐Horn, K. , Cree, B. A. C. , & Zamvil, S. S. (2015). Reduction of CD8 + T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurology ‐ Neuroimmunology Neuroinflammation, 2(3), e76 10.1212/NXI.0000000000000076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens, S. A. , Nash, K. , Koren, G. , & Rovet, J. (2013). Autism characteristics in children with fetal alcohol spectrum disorders. Child Neuropsychology, 19(6), 579–587. 10.1080/09297049.2012.727791 [DOI] [PubMed] [Google Scholar]
- Stivers, N. S. , Pelisch, N. , Orem, B. C. , Williams, J. , Nally, J. M. , & Stirling, D. P. (2017). The toll‐like receptor 2 agonist Pam3CSK4 is neuroprotective after spinal cord injury. Experimental Neurology, 294, 1–11. 10.1016/j.expneurol.2017.04.012 [DOI] [PubMed] [Google Scholar]
- Tang, G. , Gudsnuk, K. , Kuo, S.‐H. , Cotrina, M. L. , Rosoklija, G. , Sosunov, A. , … Sulzer, D. (2014). Loss of mTOR‐dependent macroautophagy causes autistic‐like synaptic pruning deficits. Neuron, 83(5), 1131–1143. 10.1016/j.neuron.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee, A. R. , Fingar, D. C. , Manning, B. D. , Kwiatkowski, D. J. , Cantley, L. C. , & Blenis, J. (2002). Tuberous sclerosis complex‐1 and ‐2 gene products function together to inhibit mammalian target of rapamycin (mTOR)‐mediated downstream signaling. Proceedings of the National Academy of Sciences, 99(21), 13571–13576. 10.1073/pnas.202476899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasoni, R. , Morini, R. , Lopez‐Atalaya, J. P. , Corradini, I. , Canzi, A. , Rasile, M. , … Matteoli, M. (2017). Lack of IL‐1R8 in neurons causes hyperactivation of IL‐1 receptor pathway and induces MECP2‐dependent synaptic defects. eLife, 6, e21735 10.7554/eLife.21735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, L. , Prieto, G. A. , Kramar, E. A. , Smith, E. D. , Cribbs, D. H. , Lynch, G. , & Cotman, C. W. (2012). Brain‐derived neurotrophic factor‐dependent synaptic plasticity is suppressed by interleukin‐1 via p38 mitogen‐activated protein kinase. Journal of Neuroscience, 32(49), 17714–17724. 10.1523/JNEUROSCI.1253-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trutzer, I. M. , García‐Cabezas, M. Á. , & Zikopoulos, B. (2019). Postnatal development and maturation of layer 1 in the lateral prefrontal cortex and its disruption in autism. Acta Neuropathologica Communications, 7(1), 40 10.1186/s40478-019-0684-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppal, N. , Wicinski, B. , Buxbaum, J. D. , Heinsen, H. , Schmitz, C. , & Hof, P. R. (2014). Neuropathology of the anterior midcingulate cortex in young children with autism. Journal of Neuropathology & Experimental Neurology, 73(9), 891–902. 10.1097/NEN.0000000000000108 [DOI] [PubMed] [Google Scholar]
- Vargas, D. L. , Nascimbene, C. , Krishnan, C. , Zimmerman, A. W. , & Pardo, C. A. (2005). Neuroglial activation and neuroinflammation in the brain of patients with autism. Annals of Neurology, 57(1), 67–81. 10.1002/ana.20315 [DOI] [PubMed] [Google Scholar]
- Viviani, B. , Bartesaghi, S. , Gardoni, F. , Vezzani, A. , Behrens, M. M. , Bartfai, T. , … Marinovich, M. (2003). Interleukin‐1beta enhances NMDA receptor‐mediated intracellular calcium increase through activation of the Src family of kinases. The Journal of Neuroscience, 23(25), 8692–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , & Doering, L. C. (2013). Reversing autism by targeting downstream mTOR signaling. Frontiers in Cellular Neuroscience, 7, 28 10.3389/fncel.2013.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhard, L. , di Bartolomei, G. , Bolasco, G. , Machado, P. , Schieber, N. L. , Neniskyte, U. , … Gross, C. T. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature Communications, 9(1), 1228 10.1038/s41467-018-03566-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilms, H. , Sievers, J. , Rickert, U. , Rostami‐Yazdi, M. , Mrowietz, U. , & Lucius, R. (2010). Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL‐1beta, TNF‐alpha and IL‐6 in an in‐vitro model of brain inflammation. Journal of Neuroinflammation, 7(1), 30 10.1186/1742-2094-7-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang, A. H. , Wang, X. , Martinez, M. P. , Walthall, J. C. , Curry, E. S. , Page, K. , … Getahun, D. (2015). Association of maternal diabetes with autism in offspring. JAMA, 313(14), 1425–1434. 10.1001/jama.2015.2707 [DOI] [PubMed] [Google Scholar]
- Yang, S. , Liu, Z.‐W. , Wen, L. , Qiao, H.‐F. , Zhou, W.‐X. , & Zhang, Y.‐X. (2005). Interleukin‐1β enhances NMDA receptor‐mediated current but inhibits excitatory synaptic transmission. Brain Research, 1034((1–2)), 172–179. 10.1016/j.brainres.2004.11.018 [DOI] [PubMed] [Google Scholar]
- Ye, P. , Popken, G. J. , Kemper, A. , McCarthy, K. , Popko, B. , & D'Ercole, A. J. (2004). Astrocyte‐specific overexpression of insulin‐like growth factor‐I promotes brain overgrowth and glial fibrillary acidic protein expression. Journal of Neuroscience Research, 78(4), 472–484. 10.1002/jnr.20288 [DOI] [PubMed] [Google Scholar]
- Zerbo, O. , Qian, Y. , Ray, T. , Sidney, S. , Rich, S. , Massolo, M. , & Croen, L. A. (2019). Health care service utilization and cost among adults with autism spectrum disorders in a U.S. integrated health care system. Autism in Adulthood, 1(1),27–36. 10.1089/aut.2018.0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos, B. , & Barbas, H. (2010). Changes in prefrontal axons may disrupt the network in autism. The Journal of Neuroscience, 30(44), 14595–14609. 10.1523/JNEUROSCI.2257-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaigenbaum, L. , Bauman, M. L. , Choueiri, R. , Kasari, C. , Carter, A. , Granpeesheh, D. , … Natowicz, M. R. (2015). Early intervention for children with autism spectrum disorder under 3 years of age: Recommendations for practice and research. Pediatrics, 136(Suppl 1), S60–S81. 10.1542/peds.2014-3667E [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table 1: Primer sequences for PCR amplification reactions for each gene.
Supplementary Table 2: Pearson's correlation analyses for possible relationships between gene expression levels and age, RNA quality (RIN) and postmortem interval (PMI) for all genes in gray matter (GM).
Supplementary Table 3: Pearson's correlation analyses for possible relationships between gene expression levels and age, RNA quality (RIN) and postmortem interval (PMI) for all genes in white matter (WM).