

Abstract
Defined sialoglycoconjugates are important molecular probes for studying the role of sialylated glycans in biological systems. We show that the α2,3‐sialyltransferase from Photobacterium phosphoreum JT‐ISH‐467 (2,3SiaTpph) tolerates a very broad substrate scope for modifications in the sialic acid part, including bulky amide variation, C5/C9 substitution, and C5 stereoinversion. To reduce the enzyme's hydrolytic activity, which erodes the product yield, an extensive structure‐guided mutagenesis study identified three variants that show up to five times higher catalytic efficiency for sialyltransfer, up to ten times lower efficiency for substrate hydrolysis, and drastically reduced product hydrolysis. Variant 2,3SiaTpph (A151D) displayed the best performance overall in the synthesis of the GM3 trisaccharide (α2,3‐Neu5Ac‐Lac) from lactose in a one‐pot, two‐enzyme cascade. Our study demonstrates that several complementary solutions can be found to suppress the common problem of undesired hydrolysis activity of microbial GT80 sialyltransferases. The new enzymes are powerful catalysts for the synthesis of a wide variety of complex natural and new‐to‐nature sialoconjugates for biological studies.
Keywords: biocatalysis, carbohydrates, enzyme promiscuity, protein engineering, sialoconjugates
Be tolerant: An engineered sialyltransferase has exceptional tolerance for a wide variety of substrate modifications, both in glycosyl donor and acceptor moieties. Its undesired promiscuous hydrolytic activity was significantly reduced through deep mutational scanning around the active site. The resultant enzyme is a powerful catalyst for the one‐pot cascade synthesis of novel molecular probes for biological studies.
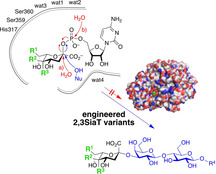
Introduction
Glycoconjugates containing terminal sialic acids are omnipresent glycan structures in animal glycoproteins and glycolipids, which are typically positioned at the outermost cell surface.1, 2 N‐Acetylneuraminic acid (Neu5Ac) is the most common form of sialic acid, which owing to its exposed capping position at the nonreducing ends or branches of glycans is of outstanding importance in the regulation of many biological processes, including cell–cell adhesion, organ development, intercellular communication, inflammation, and immunological responses. However, it is also associated with many cancers, autoimmune disorders, and diabetes as well as infections by pathogenic organisms such as trypanosomes, bacteria, and viruses.1, 3, 4, 5, 6, 7, 8 To further develop insights into the critical role of sialylated glycans in biological systems, an efficient synthesis of defined sialoglycoconjugates as individual molecular probes is crucial.9, 10, 11 Likewise, the modification of biopharmaceuticals requires efficient tools for specific glycoconjugation. Sialylation of glycans can be achieved chemically,12, 13, 14 but direct enzymatic sialylation by using enzymes of the Leloir biosynthesis pathway can avoid multistep synthetic operations.15
Sialyltransferases (SiaTs) catalyze the regio‐ and stereospecific transfer of cytidine monophosphate (CMP)‐activated sialic acid, typically CMP‐Neu5Ac, to a suitable glycan acceptor moiety with inversion of configuration.16, 17 For synthetic applications, bacterial SiaTs are preferred over their mammalian orthologues, because the latter are glycosylated proteins that are difficult to express and less stable to process conditions.18 Previous work from our group has demonstrated that the 2,6SiaT from Photobacterium leiognathi JT‐SHIZ‐145 is a widely useful catalyst for the preparative transfer of native and synthetic sialic acid derivatives to furnish the corresponding α2,6‐linked sialoconjugates in high yield.19, 20 In continuation of these studies, we were interested in a suitable bacterial 2,3SiaT catalyst for an effective approach toward the regioisomeric α2,3‐linked sialoconjugates.
The 2,3SiaT from the luminous marine bacterium Photobacterium phosphoreum JT‐ISH‐467 (2,3SiaTpph) was reported to belong to the GT80 family and to form α2,3‐sialyl linkages exclusively.21 The enzyme has been deduced from radiolabeling assays to offer a remarkably wide acceptor substrate scope that not only includes various galactosylated oligosaccharides, but also methyl‐α/β‐glycosides of d‐galactose, d‐glucose, d‐mannose, and d‐fucose.21 Its X‐ray crystal structure has been solved with bound CMP to assist in substrate binding studies.22 However, no investigation of its potential synthetic utility has been conducted so far. Thus, we selected this enzyme to study complementary tolerance to structural modifications in the sialic acid moiety and its efficiency in the synthesis of α2,3‐linked sialosides. Herein, we report on studies for the synthesis of novel sialoconjugates and a structure‐guided protein‐engineering approach to improve the enzyme's reaction selectivity, in particular to suppress promiscuous hydrolytic activities.
Results and Discussion
Substrate scope of native 2,3SiaTpph
The native 2,3SiaTpph contains an N‐terminal lipophilic signal sequence that makes handling difficult but is dispensable for catalysis.21 Therefore, we used a truncated synthetic gene that codes for a protein lacking 24 amino acid residues at its N terminus (Δ24‐α2,3SiaTpph) but carries an N‐terminal His6 tag instead. This recombinant enzyme was readily expressed from Escherichia coli BL21(DE3) and purified by Ni‐affinity column chromatography.
For synthetic studies, the purified 2,3SiaTpph was studied for its transfer activity with a test set of three natural (N‐acetyl‐neuraminic acid, 1, N‐glycoloyl‐neuraminic acid, 2, and KDN, 3) and three new‐to‐nature sialic acid derivatives (4–6, Scheme 1) that were available from previous studies.19, 20 Sialic acids were activated in situ by incubation with CSS from Neisseria meningitidis 23 in the presence of cytidine triphosphate (CTP). Transfer to a fluorophore‐labeled lactoside 7 19 was then monitored by HPLC, which showed the smooth conversion of all sialic acid compounds with generation of the expected labelled GM3 (Neu5Acα2‐3Galβ1‐4Glc) trisaccharide 8 and the corresponding analogues 9–13 (Scheme 1). From preparative studies, products were isolated in good yields by solid‐phase extraction on reversed‐phase silica, making use of the hydrophobic labeling tag. Exhaustive NMR analysis confirmed their structure and unambiguously verified that the 2,3SiaTpph exclusively forms a Sia‐Gal α2,3‐linkage as indicated by low‐field shift of the 3′‐H signal (=4.0–4.2 ppm vs. =3.5 ppm for 2,6‐linkage) and a HMBC crosspeak between 3′‐H and 2′′‐C. This confirms that 2,3SiaTpph is indeed a highly useful catalyst for sialoconjugation that shows broad substrate tolerance for various structural modifications in the sialic acid moiety to be transferred, including bulky variation of the acylamide group, as well as substitutions at C5 and C9 and stereoinversion at C5. Together with the previously demonstrated wide acceptor substrate scope,21 2,3SiaTpph is a promising candidate for the preparation of an extensive array of sialoconjugates for studies in glycobiology.
Scheme 1.
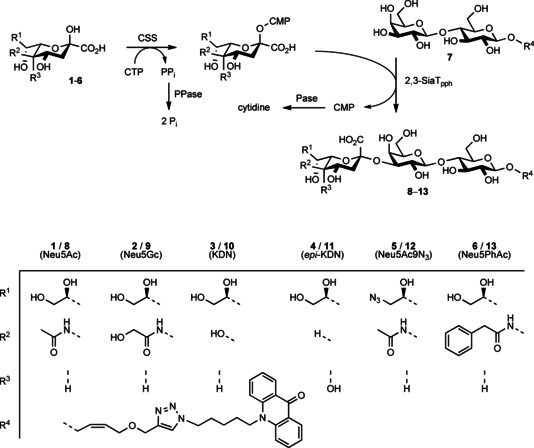
One‐pot synthesis of fluorophore‐labeled α2,3‐Neu5Ac‐Lac derivatives using Δ24‐α2,3SiaTpph.
However, we noted that isolated product yields were somewhat lower than anticipated when compared to the complementary reactions using the P. leiognathi 2,6SiaT,19, 20 particularly when using only stoichiometric quantities of CTP. For example, upon transfer of the parent Neu5Ac a limiting maximum yield of 84 % resulted for the labelled GM3, whereas the corresponding α2,6‐sialylated trisaccharide had been obtained in 93 % yield.20 Likewise, the synthesis of unlabeled GM3 trisaccharide from lactose by 2,3SiaTpph catalysis proceeded with an incomplete yield of 86 % only. This indicated that the 2,3‐sialyltransfer was suffering from a hydrolytic uncoupling that was partially causing an unproductive consumption of the limiting CTP quantity. Indeed, many other 2,3SiaT and 2,6SiaT enzymes from the GT80 family have been shown to display multifunctionality: Besides the desired sialyltransfer activity (A), promiscuous activities such as CMP‐Neu5Ac hydrolase (B; sialyltransfer to water or phosphodiester hydrolysis) and sialidase (C, hydrolytic cleavage of sialoside linkages) have been recorded (Scheme 2).21, 24, 25, 26, 27, 28, 29 Reversibility of sialyltransfer (A) was recognized as the cause of a purported trans‐sialidase activity, which arises from transient CMP‐Neu5Ac formation.30 In fact, the reverse sialyltransfer/ CMP‐NeuAc hydrolysis activity (i.e., retro‐A+B) of related bacterial 2,6SiaTs was recently employed for regiospecific removal of α2,6‐linked sialoconjugates31 and could be optimized by protein engineering.32 In synthetic direction, however, such side activity directly degrades the CMP‐activated starting material and erodes the yield of the transfer product by subsequent cleavage, respectively. Increasing the relative equivalents of CTP correspondingly can compensate for an undesired hydrolytic activity but renders preparative synthesis less economical. Clearly, the relative selectivity factor of transfer vs. hydrolysis from the activated CMP‐Sia conjugate requires optimization. Therefore, we decided to investigate the origin of incomplete transfer selectivity of the 2,3SiaTpph by structure‐guided protein engineering.
Scheme 2.
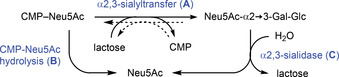
Reactions catalyzed by multifunctional α2,3‐sialyltransferases. A) sialyltransfer, B) substrate hydrolysis, C) product hydrolysis.
Protein engineering of 2,3SiaTpph
There have been various attempts to control hydrolytic side reactivity of the 2,3SiaT and 2,6SiaT from Pasteurella multocida,33 Photobacterium sp.34, 35 and P. phosphoreum,36 Pasteurella dagmatis,37, 38 or Photobacterium damselae,25 by simple addition of organic solvents, pH adjustment, or rational active‐site‐directed mutagenesis. Unfortunately, most efforts in suppressing side reactions by site‐directed mutagenesis were accompanied by drastically decreased transfer rates.33, 38 The highly polar nature and large molecular sizes of both the CMP‐Sia‐ and the Sia‐accepting glycan substrates, which have to be juxtaposed in the transition state for regiospecific sialyl transfer, require an intricate hydrogen‐bonding network to protein residues in the active site, as evident from several X‐ray crystal structures of GT80 enzymes with bound substrate analogues. In order to address this specific situation for the 2,3SiaTpph, we decided to embark on a directed evolution study involving an extended set of 13 amino acid residues in the active site that were either known or presumed to be in close contact with substrates during the catalytic transfer event. In particular, the published structure of the truncated 2,3SiaTpph with bound CMP was useful to select residues that are making up the phosphate binding site, involved in contact to bridging water molecules, or presumed to contact the acceptor substrate lactose. For screening and kinetic data analysis of enzyme activity we used a continuous colorimetric assay method previously published by our group, which is based on the pH‐shift occurring upon liberation of CMP.19 Because the competing enzymatic hydrolysis rates were comparatively slow and assay results less accurate, sialidase activity (C) further was analyzed directly by in situ NMR studies.
The major reactivity to be suppressed is that of CMP‐Neu5Ac hydrolysis (B), which is equivalent to a sialyltransfer to water instead of the 3‐OH group of a galactosyl moiety. However, water as an alternative nucleophile is unavoidable for enzymatic reactions being present in large excess as the solvent. Rather, a tighter substrate binding is desirable for a better discrimination of the sugar hydroxy group as a nucleophile that is slightly more basic and a somewhat softer nucleophile than water. To restrict space for water and suppress its unspecific activation, mutagenesis of the binding residues in direct contact is required. Subtle effects have to be expected not only due to the complex hydrogen binding network around the highly polar substrates but also due to the charge re‐distribution upon nucleophilic attack at C2 of the sialyl moiety. This involves dissociation of an anionically charged phosphoryl leaving group and simultaneous inversion of the sialyl configuration with consequent re‐positioning of the C1 carboxyl group (Scheme 3), which requires extensive mobility of the active site arrangement.30, 37, 39
Scheme 3.
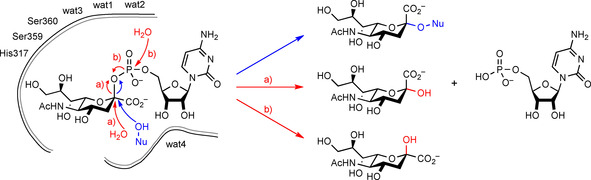
Mechanism of sialyltransfer (blue) and possible paths for hydrolysis (red).
For an identification of substrate binding residues the 3D crystal structure of the 2,3SiaTpph with bound CMP (PDB ID: 2ZWI) offers orientation but is insufficient because the critical sugar moieties of both the sialyl electrophile and galactoside nucleophile are missing.22 Iwatani et al. already argued that this rather “open” protein structure is unlikely to be the active conformation.22 Therefore, a superimposed 3D model of the original 2,3SiaTpph structure was constructed by alignment with that of the closed structure of the 2,3SiaT from P. multocida using the alignment tool of PyMOL. This model allowed the likely orientation of bound CMP‐3F‐Neu5Ac (PDB ID: 2IHZ)40 and α‐lactose (PDB ID: 2ILV)40 in the 2,3SiaTpph active site to be imitated. The 2ILV lactose structure was chosen in our aligned model, because the required 3′‐OH nucleophile is correctly positioned for a correct nucleophilic trajectory against the sialyl moiety towards the transition state, whereas lactose is bound in an unproductive orientation in the 2IHZ structure. Because the nucleotide‐binding pocket is highly conserved in this protein family,21, 41 a particular weight was placed on alignment of the corresponding CMP structural entities to achieve an optimum orientation. This arrangement revealed the positioning of CMP‐3F‐Neu5Ac and α‐lactose substrates in their respective reactive conformation relative to the active‐site protein residues of 2,3‐SiaTpph and was used as the basis for analyzing the direct protein–substrate interactions (Figure 1). The model also allowed the individual distances between substrate and corresponding amino acid residues in particular to be evaluated (Table S6 in the Supporting Information) so as to gain a coarse indication of the extent of conformational mobility required to close the active site for binding of substrate parts more distant from the nucleotide moiety, such as the sialic acid and lactose portions. Interestingly enough, in the crystal structure of the 2,3‐SiaTpph several ordered water molecules (wat 1–4) form a compact hydrogen‐bonding network around the glycoside phosphate unit, which probably facilitates the large structural and conformational changes happening during the catalytic event.
Figure 1.
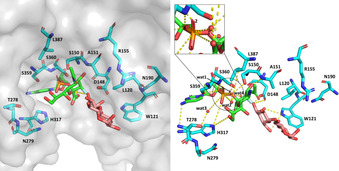
Homology 3D model constructed by aligning SiaT protein from P. phosphoreum JT‐ISH‐467 (PDB ID: 2ZWI) with CMP‐3F‐Neu5Ac (PDB ID: 2IHZ) and α‐lactose (PDB ID: 2ILV) as bound to SiaT from P. multocida. Possible polar contacts, also involving substrate and bound crystallographic water, are indicated by broken yellow lines.
Site‐directed saturation mutagenesis
In the judicious selection of active‐site residues, a sequence alignment of various sialyltransferases (Table 1) was further used to correlate highly conserved sequence motifs with rational criteria based on the 3D model to identify appropriate candidates to be considered in a site‐directed mutagenesis approach. Yamamoto et al. identified three highly conserved sequence motifs for the GT80 family.41 They assumed the so‐called YDDGS, FKGHP and SS motifs to play an essential mechanistic role in the transfer reaction, proton provision and binding conditions.
Table 1.
Partial protein sequence alignment around residues in substrate contact for sialyltransferases from the GT80 family; arrows identify positions selected for mutagenesis.
|
|
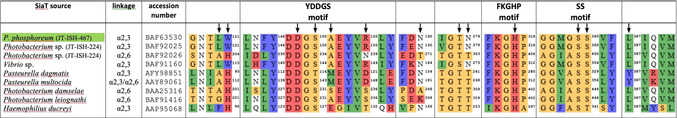
From the sequence alignment, positions S150, A151 and R155 within or adjacent to the YDDGS motif seemed interesting to be included because these three positions show some variability despite the high conservation of the YDDGS motif in the GT80 family. For example, S150 in P. dagmatis naturally is replaced by T116 and was shown to be involved in control of sialyltransfer stereoselectivity and the level of sialidase activity.27 Position A151 is replaced by M144/117 in P. multocida and P. dagmatis or by S232 in P. damselae. The contact of S150 to water molecule 1 (Figure 1) as well as the close distances to sialic acid (1.7–4.3 Å) might potentially impact the reaction course. The FKGHP and SS motifs are highly conserved on the other hand, with little variation only in the sequence of the Haemophilius ducreyi enzyme. The SS motif was included in our set due to its very close distance to phosphate (1.3–3.9 Å, Table S6) and water molecules 1–4 (3.6–5.6 Å), and its possible involvement in activating a water nucleophile in competition to lactose. Also included were the completely conserved opposing residues D148 and H317, which were predicted by Iwatani et al.to act as catalytic base and acid, respectively.22 With closest ostensible distances of 1.9–2.4 Å to the galactose moiety and 3.4–4.0 Å to the sialic acid moiety for D148 on one side, and 4.1–5.6 Å to the sialyl phosphate moiety of CMP‐Neu5Ac for H317, these are both essential in binding and coordinating interactions to and among the substrates. Somewhat farther away but still in contact to the sialic acid moiety are T278 and N279. Also, T278 makes contact to water molecule 3. With a distance of 8.7 Å to phosphate L387 is relatively distant to the reaction center of transfer/hydrolysis but with 4.1–5.2 Å to the Neu5Ac acyl group still in substrate contact, and its side chain directly oriented towards water molecules 1 and 2. Residues L120 and W121 were considered because they are located at the entrance to the active site with several contacts to lactose in distances of 3.4–5.0 Å, and because of their likely influence on the correct positioning of the galactoside moiety receiving the transferred sialyl group.
Having selected the 13 candidate positions (Table S6), it was necessary to define the mutagenesis strategy for site‐specific randomization and restrict the number of codons to keep library sizes manageable. Adjacent positions (278/279 and 359/360, respectively) were jointly mutated for efficiency. For residue types S, T, and N279, the WVC codon (coding for N, T, S, Y) was chosen, while others were saturated individually using NNK codons (coding for all 20 AA). All 11 plasmid libraries were used for transformation of BL21 Tuner cells for enzyme expression, in which galactosidase activity is practically absent so that crude lysates would not interfere with the assay components. For each single site library 96 colonies were picked, while for double site WVC mutagenesis 192 colonies were picked to secure ≥95 % coverage in all cases.
Activity screening and hit characterization
Most methods for assaying sialyltransferase activity reported so far are discontinuous and involve laborious quenching of the catalytic reaction by acid or heating followed by product analysis.36, 38, 42 HPLC analysis of fluorescently labeled substrates33 is indirect, and surrogate substrates may show a kinetic behavior different from the native substrates.43 The only continuous assay for SiaT activity is based on the indirect measurement of CMP release by an enzymatic link to NADPH consumption, which is inappropriate for discrimination of sialidase activity.44
In contrast to HPLC, radiolabeling, capillary electrophoresis or ESI‐MS assays, which require expensive dedicated instrumentation, assays based on monitoring a pH shift as a consequence of reaction progress are simple to operate in continuous mode,45 and also universal in that they are independent of substrate variations. Upon transfer of the sialyl group from CMP‐Neu5Ac, a proton is released as a consequence of CMP formation causing the reaction medium to turn acidic. Thus, proton release can be detected via color change of an appropriate pH indicator and quantified under suitably controlled conditions. Therefore, by adapting conditions of our analysis of CMP‐sialate synthase activity published previously,19 we developed a simple colorimetric pH assay for the measurement of sialyl transfer activity by using phenol red as the pH indicator under minimum buffering conditions. Phenol red has a pK a of 7.4 and a high absorption coefficient (Δϵ=56 000 m −1 cm−1 at 557 nm) imparting high assay sensitivity.46 The assay is started at pH 8.0 in 2 mm Tris buffer (pK a 8.06), so that the corresponding pH monitoring range (6.8–8.4) coincides with the most appropriate alkaline stability range of sialyl conjugates and the very broad activity range of α2,3‐SiaTpph catalysis between pH 5–11.21 Due to the buffered conditions and determination of an initial velocity within a short timeframe, only a minimum pH change is observed, which allows for a robust analysis. When measuring a calibration curve in the presence of all assay components, corresponding enzyme activity can be measured for both screening purposes as well as determination of kinetic constants. However, for practical reasons CMP‐Neu5Ac concentration was used at subsaturating concentration to avoid potential interference with the assay principle and to minimize background stemming from hydrolytic instability.
To identify variants that display the desired high transfer activity and reduced CMP‐Neu5Ac hydrolysis rates, cell lysates of all variants were tested in a duplicate activity screening. Each plate was tested for sialyltransfer activity in the presence of lactose as sialyl acceptor, and for CMP‐Neu5Ac hydrolysis activity in the absence of lactose by following the OD change at 560 nm. All variants with a transfer versus hydrolysis ratio better than that of the native enzyme of approximately 2:1 were chosen for rescreening, which was repeated twice by the same procedure to validate true positives and eliminate false positives. Variants that showed matching results from both runs were chosen for sequencing. This procedure identified nine unique variants showing either reduced hydrolysis activity, or improved transfer rate, or both, and including mutations at six different positions: S150T, A151D, A151L, A151M, A151N, N279S, S359T, S359T/S360T, and L387A. Not quite unexpectedly, hot spot positions S150 and A151 coincided with allowed variations that occur naturally in orthologous enzymes (Table 1).
For a detailed kinetic data analysis the nine variants were expressed, purified by Ni‐affinity chromatography and analyzed by SDS‐PAGE. Protein concentration was determined using the BCA assay.47 By keeping CMP‐Neu5Ac and lactose concentrations constant at 1 and 5 mm, respectively, apparent conditional kinetic data were reproducibly obtained that facilitated the comparison of catalysts (Table 2). The analysis revealed three interesting variants A151D, S359T/S360T and L387A that showed significantly improved properties as compared to the native enzyme. For all three variants the conditional K M values for CMP‐Neu5Ac indicate an improved binding affinity, combined with a faster transfer reaction rate for variants A151D and L387A. Under the specific assay conditions chosen the catalytic transfer efficiency for all three variants is 2 to 5 times higher. At the same time a much slower hydrolysis rate (all three variants), combined with a weaker binding of water, causes a catalytic efficiency for hydrolysis that is 4.3 to ten times worse (A151D, L387A, respectively). The reduced binding affinity for lactose (A151D, L387A) is compensated with a higher reaction rate than that of the native enzyme, and for preparative applications can be balanced such as with corresponding higher concentrations of lactose (or related sialyl acceptor). Overall, A151D shows the best improvement from a higher binding affinity towards CMP‐Neu5Ac, better conditional k cat values for CMP‐Neu5Ac and lactose and a hydrolase efficiency reduced by 4.3 times. Interestingly, the A151D hit in 2,3SiaTpph corresponds to the beneficial M144D mutation reported for the P. multocida enzyme. This change of methionine to aspartate caused a 20‐fold reduction in hydrolysis activity, but at the cost of an 18‐fold decrease in efficiency for α2,3‐sialyltransfer to lactose with ninefold lower binding affinity.33 In contrast, our 2,3SiaTpph (A151D) variant has a significantly reduced hydrolysis activity combined with a better conditional k cat /K M for CMP‐Neu5Ac and only 3.7‐fold lower k cat /K M for lactose. For preparative‐scale experiments the lower binding affinity for lactose can be easily compensated with a higher concentration of the inexpensive disaccharide. Obviously, the change of position 151 from a hydrophobic alanine to a charged aspartate has the strongest impact on reducing hydrolysis activity. This may possibly be explained by D151 blocking extra space that in the native enzyme is occupied by the water molecule, which is acting as the hydrolysis nucleophile, or by modifying the water molecule's nucleophilic trajectory, or more generally by shifting the pK a of the catalytically active D148.
Table 2.
Apparent conditional steady‐state kinetic parameters for variants of α2,3‐SiaTpph.[a]
|
|
|
Native SiaT |
S150T |
A151D |
A151L |
A151M |
A151N |
N279S |
S359T |
S359T/S360T |
L387A |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
CMP‐Neu5Ac[b,c] |
K M [mm] |
1.1±0.1 |
1.9±0.8 |
0.8±0.3 |
0.4±0.1 |
0.3±0.03 |
0.7±0.4 |
1.0±0.1 |
1.0±0.5 |
0.05±0.01 |
0.7±0.2 |
|
k cat [s−1] |
1.4±0.05 |
0.5±0.04 |
2.0±0.1 |
0.7±0.05 |
0.8±0.0 |
0.7±0.02 |
1.2±0.03 |
0.2±0.03 |
0.3±0.01 |
1.8±0.1 |
|
|
k cat/K M [mm −1 s−1] |
1.3 |
0.3 |
2.5 |
1.7 |
2.9 |
1.0 |
1.1 |
0.2 |
6.3 |
2.4 |
|
|
lactose[c,e] |
K M [mm] |
0.5±0.2 |
11.0±1.3 |
9.1±1.2 |
0.9±0.3 |
1.4±0.1 |
1.5±0.3 |
8.3±0.6 |
0.9±0.9 |
3.4±1.3 |
19±4 |
|
k cat [s−1] |
0.5±0.05 |
1.4±0.07 |
2.4±0.3 |
0.5±0.04 |
0.7±0.01 |
0.6±0.03 |
1.5±0.05 |
0.5±1.0 |
0.2±0.02 |
1.3±0.1 |
|
|
k cat/K M [mm −1 s−1] |
1.0 |
0.1 |
0.3 |
0.5 |
0.5 |
0.4 |
0.2 |
0.5 |
0.1 |
0.1 |
|
|
hydrolysis[d,f] |
K M [mm] |
0.14±0.03 |
0.02±0.01 |
0.38±0.11 |
0.05±0.00 |
0.08±0.02 |
0.08±0.01 |
0.11±0.01 |
0.21±0.09 |
0.07±0.03 |
0.42±0.08 |
|
k cat [s−1] |
0.40±0.02 |
0.03±0.00 |
0.25±0.02 |
0.11±0.00 |
0.11±0.00 |
0.06±0.00 |
0.14±0.00 |
0.19±0.01 |
0.04±0.00 |
0.12±0.01 |
|
|
k cat/K M [mm −1 s−1] |
2.9 |
1.7 |
0.7 |
2.2 |
1.3 |
0.8 |
1.3 |
0.9 |
0.6 |
0.3 |
[a] Assays were performed under subsaturating conditions, and relative kinetic data were obtained by fitting to Michaelis‐Menten equation using Origin software. [b] Lactose concentration was constant at 5.0 mm. [c] CMP‐Neu5Ac concentration was held constant at 1.0 mm. [d] Lactose was omitted and CMP‐Neu5Ac concentration was varied. [e] Enzyme quantities used for the measurements varied between 2.6 to 15.9 μg per assay. [f] Enzyme quantities used for the measurements varied between 13.2 to 38.8 μg per assay.
Second‐generation combination variants
Mutagenesis of proteins often leads to reduced thermodynamic stability against unfolding, rendering them unstable upon further mutagenesis and/or against conditions required for synthetic applications. Therefore, the thermal stability of the native enzyme and the most promising new variants was analyzed by nanoscale differential scanning fluorimetry (nanoDSF), which is a valuable tool to rapidly and reliably determine protein melting points by monitoring autofluorescence changes upon thermal activation in solution under variable conditions.48 The results for the native SiaT (Table S7) are congruent with the fact that it is derived from a mesophilic organism (T m=47.1 °C), and that single point mutagenesis causes only a small (ca. 2°) decrease in T m, while the double exchange of serines to threonines seems to be even slightly stabilizing. All three variants deemed suitable for further mutagenesis.
For a second round of mutagenesis, a combinatorial library of the most effective hot spots at positions 150 and 151 with those at 359 and 360 was created to sample potential cooperative effects. Combinations were based on the starting gene as well as on that containing the S359T/S360T double substitution (Table S3). Mutagenesis of the spatially close catalytic D148 to residues of similar size but different polarity profiles (serine, lysine) was included as a control. However, second‐generation library screening and re‐screening did not reveal any significant further improvement in transfer activity or reduction of hydrolysis activity. As was to be expected, variants at position 148 were catalytically ineffective and also showed dominant hydrolysis over transfer activities (lower transfer/hydrolysis ratio of only 0.5–1 vs. native SiaT ca. 2). Therefore, in order to gain a better understanding of the specific effect of combined mutations, specific individual combination variants had to be created individually from the three best hits (A151D, S359T/S360T, L387A) for a comparative kinetic analysis (Table 3). In addition, we also constructed the variant E342A, which is located in the CMP binding pocket, for a direct comparison to our findings. It was claimed that residue E342 in 2,3SiaTpph is important for sialidase activity (path C), because the mutation E342A abolished α2,3‐sialyllactose hydrolysis.36
Table 3.
Apparent conditional steady‐state kinetic parameters for combined α2,3‐SiaTpph variants.[a]
|
|
|
Native SiaT |
A151D/L387A |
A151D/S359T/S360T |
A151D/S359T/S360T/L387A |
E342A |
A151D/E342A |
|---|---|---|---|---|---|---|---|
|
CMP‐Neu5Ac[b,e] |
K M [mm] |
1.1±0.1 |
1.0±0.5 |
4.4±3.4 |
1.1±0.5 |
45±17 |
18±8 |
|
k cat [s−1] |
1.4±0.05 |
0.2±0.03 |
0.5±0.1 |
0.3±0.03 |
0.9±0.2 |
0.4±0.1 |
|
|
k cat/K M [mm −1 s−1] |
1.3 |
0.2 |
1.1 |
0.2 |
0.0 |
0.0 |
|
|
lactose[c,e] |
K M [mm] |
0.5±0.2 |
1.8±1.2 |
1.0±0.5 |
0.09±0.03 |
16.5±9.4 |
5.7±2.6 |
|
k cat [s−1] |
0.5±0.1 |
0.2±0.04 |
0.5±0.07 |
0.26±0.02 |
0.3±0.1 |
0.1±0.01 |
|
|
k cat/K M [mm −1 s−1] |
1.0 |
0.1 |
0.5 |
3.0 |
0.0 |
0.0 |
|
|
hydrolysis[d,e] |
K M [mm] |
0.14±0.03 |
0.64±0.30 |
0.73±0.33 |
0.19±0.13 |
0.07±0.03 |
0.13±0.09 |
|
k cat [s−1] |
0.40±0.02 |
0.06±0.01 |
0.34±0.04 |
0.34±0.04 |
0.02±0.00 |
0.04±0.00 |
|
|
k cat/K M [mm −1 s−1] |
2.9 |
0.1 |
0.5 |
1.8 |
0.2 |
0.3 |
[a] Assays were performed under subsaturating conditions, and relative kinetic data were obtained by fitting to Michaelis‐Menten equation using Origin software. [b] Lactose concentration was constant at 5.0 mm. [c] CMP‐Neu5Ac concentration was constant at 1.0 mm. [d] Lactose was omitted and CMP‐Neu5Ac concentration was varied. [e] Enzyme quantities used for the measurements varied between 1.9 to 12.3 μg per assay.
Kinetic data for the selected combination variants indeed did not show further improvement compared to the parent A151D, L387A, or S350T/S360T variants. The main similarity of the combined variants is the significant reduction of k cat values for sialyltransfer activity. Even a major reduction in hydrolytic efficiency for the combined A151D/L387A mutation is not useful for practical applications because the low sialyltransfer activity would cause a high stationary concentration of CMP‐Neu5Ac over a prolonged reaction period, which favors chemical background hydrolysis as proven by semipreparative‐scale synthesis (Figure 3). To our dismay, we could not confirm the literature data reported for variant E342A, which was claimed to have approximately five times higher k cat for CMP‐Neu5Ac and nine times higher k cat for lactose.36 Conversely, under our assay conditions we detected about 40 % reduced k cat values and a 40‐fold lower binding affinity towards CMP‐Neu5Ac (Table S5). The latter is plausible because the mutation concerns a highly conserved residue of the CMP‐binding motif in GT80 sialyltransferases.21, 41 According to the crystal structure with bound CMP (2ZWI) the carboxyl group of E342 makes parallel bidentate hydrogen bonds to both oxygen atoms O2 and O3 of the ribose unit.22 Thus, E342 cannot be involved in contacts to sialic acid or lactose substrate structures due to its location in the nucleotide‐binding pocket but may reduce apparent sialidase activity (via reverse sialyltransfer and CMP‐Sia hydrolysis; retro‐A+B) by weakening CMP affinity. Therefore this variant, as well as a variant carrying this mutation combined with our first generation best hit (A151D/E342A), were discarded from further analysis due to their dramatic losses of sialyltransfer efficiency.
Figure 3.
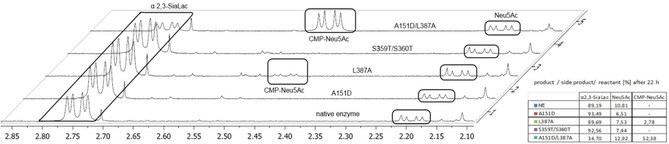
1H NMR (500 MHz) analysis of the formation of main product α2,3‐SiaLac, side product Neu5Ac, and remaining substrate CMP‐Neu5Ac after 22 h of reaction. Full conversion was observed for parent 2,3SiaTpph, as well as A151D and S359T/S360T variants; for variants L387A and (A151D/L387A) 2.8 and 52.4 % of CMP‐Neu5Ac remained, respectively, due to incomplete conversion.
Quantification of hydrolytic activity by preparative synthesis
To verify the consequences of the kinetic data gained from the mutagenesis study, the new variants were challenged by a preparative‐scale sialylation of lactose to evaluate their potential synthetic benefit. Thus, 15 mm CMP‐Neu5Ac, 15 mm lactose and 10 μg purified enzyme in Tris buffer (20 mm; 560 μL scale) were reacted for 22 h at pH 8.0 and 30 °C. At discrete intervals 10 μL samples of the reaction mixture were analyzed by a high‐performance thin‐layer chromatography (HPTLC). Quantitative monitoring of product (SiaLac) and side product (Neu5Ac) formation was performed after staining by comparison against authentic standards of SiaLac and Neu5Ac that were used as positive controls (Figure 2). The remaining reaction mixture was diluted with methanol to stop the reaction, and enzyme removed. The supernatant was lyophilized, and the residue was analyzed for endpoint quantification by 1H NMR analysis (Figure 3). The distinct chemical shifts and non‐overlapping C3 proton signals in free sialic acid and in conjugates α2,3‐SiaLac and CMP‐Neu5Ac, respectively, render a relative quantification highly accurate and allow simultaneous determination of conversion level as well as product distribution.
Figure 2.
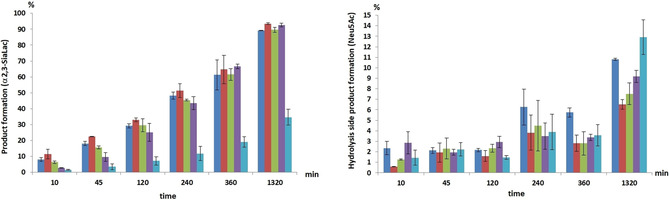
HPTLC analysis of the formation of sialyltransfer product α2,3‐SiaLac and hydrolysis side product Neu5Ac by using ▪: native 2,3SiaTpph and variants ▪: A151D, ▪: L387A, ▪: S359T/S360T, ▪: A151D/L387A. Data from duplicate experiments (error bars depicted as standard error of the mean (SEM)) were averaged and corrected with a scaling factor for absolute percentages determined by NMR analysis of endpoint product [%] and side product [%] after 22 h.
From an overall appraisal of results from the kinetic analysis and synthetic evaluation of the studied variants, 2,3SiaTpph (A151D) turns out to be the best catalyst. The formation of side product from hydrolysis was reduced by 40 % while product formation proceeded at similar rate but with consistently 5 % higher product yield relative to the native enzyme. In comparison, the S359T/S360T variant showed a slower sialyltransfer rate but after 22 h the reaction also reached complete conversion with competing hydrolysis decreased by 31 %. Reaction catalyzed by variant L387A showed 30 % reduced hydrolysis but was still incomplete after 22 h. The combined variant (A151D/L387A) is significantly less active, resulting in a 20 % relative higher fraction of hydrolysis product compared to the native enzyme arising from joint chemical and enzymatic loss of unstable CMP‐Neu5Ac.
Assessment of product hydrolysis activity
Incomplete formation of the desired sialyltransfer product can arise from CMP‐Neu5Ac hydrolysis along path B (Scheme 2) resulting from sialyltransfer to water or phosphodiester hydrolysis, which becomes particularly aggravated if sialyltransfer is rapidly reversible (retro‐A). However, the same net outcome (formation of free Neu5Ac) also can arise as a consequence of product hydrolysis along path C, if the 2,3SiaT enzyme shows direct sialidase activity.27, 33, 34, 36 The latter is more difficult to distinguish because catalytic quantities of CMP are sufficient for back transfer of the sialyl moiety onto CMP followed by CMP‐Neu5Ac hydrolysis.30 For the quantification of a potential sialidase activity we performed a series of NMR experiments with selected enzyme variants to determine the phenomenon and compare the initial velocities of the cleavage rates for liberation of Neu5Ac from α2,3‐SiaLac. To this end, 14.9 mm α2,3‐SiaLac was incubated in 520 μL Tris‐buffered (20 mm) D2O in a NMR tube with 10 μg samples of native enzyme, A151D, L387A and S359T/S360T variants, respectively, for 48 hours at 30 °C, and the reactions monitored after 18, 20, 22, 24, 42 and 48 hours by in situ 500 MHz 1H NMR spectroscopy using the 3‐H key signals of released Neu5Ac for quantification (Figure 4). Due to practical limitations the experiment was run only for a single α2,3‐SiaLac concentration, which displays the results as a comparison of initial velocities of sialidase reaction at 14.9 mm of α2,3‐SiaLac.
Figure 4.
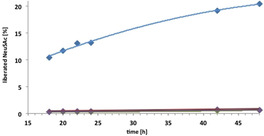
Quantification of sialidase activity of 2,3SiaTpph and variants by 1H NMR (500 MHz) in situ monitoring of Neu5Ac liberation from α2,3‐SiaLac. ▪: native enzyme, ▪: A151D, ▪: L387A, ▪: S359T/S360T.
Quantification of free Neu5Ac over the course of 2 days clearly revealed a considerable sialidase activity for the native 2,3SiaTpph enzyme. Interestingly enough, the level of sialidase activity determined for the native 2,3SiaTpph enzyme is similar to the catalytic rate of substrate hydrolysis determined in the kinetic evaluations above, explaining the lower synthetic yields due to parallel substrate and product eroding pathways. To discriminate direct sialidase from an indirect CMP catalyzed pathway, control experiments were performed in the presence of alkaline phosphatase to cleave any traces of CMP (Figure S7), which showed practically identical amounts of liberated Neu5Ac. This seems to be in favor of a direct hydrolysis along path C and incompatible with an indirect CMP dependent hydrolysis mechanism. Indeed, a previous complementary study of 2,3SiaTpph concluded that sialidase activity was not enhanced in the presence of added CMP.22 However, we cannot rule out an extremely tight binding of catalytic CMP that might be inaccessible to phosphatase decomposition.30 Quite satisfactorily, a dramatic reduction in initial velocities of sialidase activity was determined relative to the parent (1.3×10−3 mmol min−1) for the newly found 2,3SiaTpph variants (A151D: 4.9×10−5 mmol min−1, L387A: 1.9×10−5 mmol min−1, and S359T/S360T: 7.1×10−5 mmol min−1), which are reduction by factors of 26, 68 and 18, respectively. Amounts of liberated Neu5Ac within 48 h remained below 1 % for all variants (0.92 % for A151D, 0.65 % for L387A and 0.66 % for S359T/S360T), while the native enzyme produced 20.45 % Neu5Ac. This means that for all three variants, sialidase activity (path C) could be almost completely eliminated, whereas substrate hydrolysis (path B) was significantly reduced and transfer rates even somewhat increased relative to the parent enzyme.
Thus, a final experiment was performed for synthesis of Sia‐Lac (GM3, 14) as a one‐pot, two‐enzyme cascade reaction, similar to the conversion of labeled lactose described above. Activation of Neu5Ac by the CSS from N. meningitidis was followed by sialyltransfer using engineered variant 2,3SiaTpph (A151D) under standard conditions (3 equiv lactose). Product isolation by gel permeation chromatography furnished a total yield of 93.4 mg α2,3‐SiaLac 14 (91 %), which is a 5 % improvement compared to the corresponding experiment with wild‐type enzyme (yield 86 %), reflecting the gain to be expected from reduced hydrolysis. NMR spectroscopic analysis proved that like the parent enzyme the mutated enzyme variant exclusively generated the desired α2,3‐coupled GM3 trisaccharide (2D NMR; HMBC signal between 3′‐H and 2“‐C and proton signal H‐3′ at 4.18 ppm). From the accumulated data, the engineered 2,3‐SiaTpph (A151D) catalyst compares quite favorably to other 2,3SiaT variants that previously had been engineered for higher reaction specificity.33
Conclusions
We have shown that the truncated α2,3‐sialyltransferase from P. phosphoreum is a useful catalyst for the regio‐ and stereospecific synthesis of sialoconjugates. The enzyme offers a very broad substrate scope for structural variation in its substrates, covering both the sialyl donor and acceptor moieties. In particular, the enzyme tolerates substantial structural modifications in the sialic acid moiety, including bulky variation of the amide group at C5, replacement of the amide for a hydroxy group (viz., KDN), substitution at C9, and stereoinversion of KDN at C5. With all substrate analogues tested, regio‐ and stereospecific α2,3‐sialyltransfer occurred to lactose and a fluorescence‐tagged lactoside.
We also confirmed that the native 2,3SiaTpph enzyme displays a promiscuous multispecificity for sialyltransfer, CMP‐Neu5Ac substrate hydrolysis and sialidase activities. Whereas a larger quantity of CTP can be applied to compensate for the competitive loss of CMP‐Neu5Ac by hydrolysis in one‐pot operations, this results in a significant cost factor upon scale‐up. Therefore, we attempted to evolve the 2,3SiaTpph by site‐directed mutagenesis to minimize its hydrolytic activities. For this purpose, we developed a novel continuous colorimetric assay method for measuring SiaT activity by pH shift‐based determination, which in principle is universal in that activity determination can be conducted practically independent of substrate modifications for both sialic donor and acceptor structures, thus covering the entire substrate scope. Structure‐based saturation mutagenesis of 2,3SiaTpph at 13 amino acid positions around its reaction center in single‐site and combined double‐site fashion, followed by screening for transfer from CMP‐Neu5Ac to lactose versus CMP‐Neu5Ac hydrolysis revealed hot spots at six positions. Kinetic characterization identified three variants having up to five times higher catalytic efficiency for sialyltransfer (S359T/S360T) and up to ten times reduced efficiency for hydrolysis (L387A). An NMR study finally confirmed that these variants have an up to 68‐fold reduced sialidase activity (L387A). On a preparative scale, the isolated yield of the GM3 trisaccharide (α2,3‐Neu5Ac‐Lac, 14) from lactose in a one‐pot, two‐enzyme cascade could be significantly improved from 86 % (native enzyme) to 91 % when using the engineered 2,3‐SiaTpph (A151D) variant. Our study demonstrates that these powerful catalysts could prove extremely useful for synthetic access to a wide variety of complex natural and new‐to‐nature sialoconjugates for biological studies.49, 50, 51, 52
Experimental Section
Cloning, expression, and purification of 2,3SiaTpph: The codon‐optimized truncated gene coding for Δ24‐2,3SiaTpph was obtained commercially with NdeI and BamHI restriction sites at the 5′ and 3′ termini, respectively, for further cloning work. The gene was ligated into the pET19b vector (Figure S1) and the construct pET19b‐SiaTpph used for transformation of BL21(DE3) host cells for over‐expression. A positive colony was picked, cultured in 5 mL LB‐Amp medium at 37 °C and stored in 16 % glycerol at −80 °C. SDS‐PAGE analysis verified that the recombinant truncated 2,3SiaTpph is well expressed in soluble form (Figure S2). Expression was performed in baffled 2 L bottles with 0.5 L auto induction medium containing 100 μg mL−1 ampicillin in an Ecotron shaker (Infors AG, Bottmingen, CH) for 6 h at 37 °C, followed by 18 h at 30 °C and 220 rpm. Cells were lysed at 37 °C for 90 min in 20 mm potassium phosphate buffer (pH 7.4) supplemented with 0.5 mg mL−1 lysozyme and 5 U mL−1 DNase I. The lysate was adjusted to 0.5 m NaCl and centrifuged at 12 000 g for 30 min for clarification.
Site‐directed mutagenesis: Mutagenic PCR was performed on plasmid pET19b‐SiaTpph using the Q5 Site‐Directed Mutagenesis Kit (New England Biolabs). For a list of primers used for mutagenesis see Tables S1–S5. The linear, unmethylated PCR product was phosphorylated by polynucleotide kinase and ligated by T4 DNA ligase. Prior to transformation of chemically competent NEB5α cells the methylated template was degraded by Dpn1 restriction. The PCR product was analyzed by agarose gel electrophoresis to confirm successful amplification and complete DpnI digestion. For libraries a portion of the transformation mixture was incubated overnight in liquid culture for plasmid preparation, and the plasmid mixture was used for sequencing to confirm the poly mutation site. The plasmids were used to transform BL21(DE3)pLacI Tuner expression cells by electroporation. Cells were grown on agar plates at 37 °C for 16–18 h, then individual clones were picked for inoculation in 96‐well plates with 130 μL LB‐Amp. These master plates were incubated for 20 h at 37 °C, supplemented with 50 μL 50 % glycerol and sealed with acetate foil for storage at −80 °C.
Library screening: BL21 TunerTM cells (Novagen) transformed with the corresponding plasmids were induced to produce the variant enzymes and incubated with 0.1 mm IPTG at 37 °C in deep‐well plates for 20 h. After centrifugation, removal of the supernatant and cell lysis by addition of lysozyme (0.15 mg/well) and Cyanase nuclease (0.6 U/well) (Roth), the plates were again centrifuged and the enzyme containing supernatant was transferred to assay plates. For the screening 40 μL clarified lysate was used. Every plate was tested in parallel for transfer activity in the presence of lactose (5 mm) and for hydrolysis activity without lactose. Each well contained final concentrations of Tris buffer (2 mm, pH 8.0), phenol red (0.028 mm), lactose (5 mm/ 0 mm) and enzyme solution (40 μL lysate). The assay was started by addition of CMP‐Neu5Ac solution (0.5 mm) and the reaction rate was obtained by following the OD change at 560 nm. The linear slope was calculated using Microsoft Excel for comparison of rates for transfer (presence of lactose) and hydrolysis (absence of lactose).
Enzyme kinetic parameters: Components were pipetted consecutively to give a total assay volume of 200 μL per well with following end concentration: Tris buffer (2 mm, pH 8.0; 90–170 μL), phenol red (2 μL 10 mg L−1, 0.028 mm), variable substrate (lactose or CMP‐Neu5Ac, respectively, 0–80 μL) and enzyme (4–10 μL; for exact substrate and enzyme concentrations see Table 3 and Table S5). All solutions were carefully adjusted to pH 8.0 and tempered to 30 °C. The plate was shaken for 45 s at 30 °C. The assay was started by addition of the constant substrate (20 μL freshly prepared CMP‐Neu5Ac or lactose solution), the plate was immediately shaken again for 5 s and then directly measured by plate reader (SpectraMAX 190; Molecular Devices, Software: SoftMax Pro 6.5.1) at 560 nm. Experimental data were fitted to the Michaelis–Menten equation by nonlinear regression using Origin software (v9.1G, OriginLab) to obtain apparent kinetic parameters. Protein concentration was measured by BCA assay (bicinchoninic acid assay, Sigma–Aldrich).
HPTLC analysis of transfer and hydrolysis: 15 mm CMP‐Neu5Ac, 15 mm lactose and 10 μg purified enzyme variants were reacted in 560 μL Tris buffer (20 mm, pH 8.0) at 30 °C (preliminary test runs indicated complete conversion for native 2,3SiaTpph and the A151D variant after 22 h). At time points of 10 min, 45 min, 2, 4, 6, and 22 h 10 μL reaction mixture were withdrawn, respectively, diluted with 90 μL methanol to stop the reaction and stored in the freezer at −20 °C. After centrifugation the supernatant was analyzed by high performance thin layer chromatography (HPTLC; CAMAG Automatic TLC Sampler 4) on Merck silica gel plates 60 F254. For each time point measurement two identical 10 μL‐samples were sprayed and developed with different solvent mixtures to discriminate partially overlapping bands and quantify the amount of product (α2,3‐SiaLac) and hydrolysis side product (Neu5Ac). The basic solvent mix consisted of n‐propanol/H2O/NH3(20 %) (7:3:1), and acidic solvent mix of n‐butanol/acetone/AcOH/H2O (35:35:7:23). After anisaldehyde staining a quantitative analysis was performed with the CAMAG TLC Scanner 4 device against standards for SiaLac and Neu5Ac as positive controls. Scan data were normalized against the end point analysis data obtained from quantitative NMR analysis.
In situ NMR experiments: Reaction mixtures were prepared in D2O solvent. NMR spectra were recorded on a Bruker DRX500 spectrometer; chemical shifts are referenced to HOD, δ=4.79 ppm. For quantitative NMR determinations accurate manual phase correction and baseline correction with Whittaker Smoother method were performed using MestReNova 11.0.3. For sialidase measurements by in situ NMR analysis the samples were tempered to 30 °C for 48 h. Initial velocities were calculated from percentage of free Neu5Ac formed between 18–24 h. In parallel control experiments 20 U of alkaline phosphatase were added to eliminate potentially catalytic traces CMP.
Synthesis of sialylated lactosides by native 2,3SiaTpph: CTP (25.2 mg; 2 equiv) and sialic acid (Neu5Ac 1 or corresponding analogues 2–6; 1.5 equiv) were dissolved in 5 mL Tris⋅HCl buffer (50 mm, pH 8.6) containing 0.5 m NaCl, 0.1 % Triton×100 and 20 mm MgCl2. Reaction was started by addition of 5 mg CSS from N. meningitis and inorganic pyrophosphatase (2 U). When CMP activation was completed, lactoside 7 (20 mg; 1 equiv), 0.5 mg recombinant 2,3SiaTpph and alkaline phosphatase (160 U) were added and the pH was readjusted. When the reaction was finished, 30 mL cold methanol (−20 °C) was added to stop the reaction. Protein precipitate was removed by filtration. The filtrate was passed over a plug of reversed‐phase C18 silica, and the product was eluted with a 20–50 % gradient of aqueous methanol. The solution was concentrated to 10 mL and separated by Biogel P‐2 (Bio‐Rad, Germany) column chromatography (3×100 cm) using 5 mm Tris buffer (pH 8.5). The product fractions were collected and lyophilized for characterization.
Synthesis of α2,3‐SiaLac (GM3 trisaccharide, 14) by native enzyme and engineered 2,3SiaTpph (A151D): Neu5Ac (50 mg, 1 equiv) and CTP (117.2 mg, 1.5 equiv) were dissolved in 12 mL Tris⋅HCl buffer (50 mm, pH 8.5) containing 20 mm MgCl2. After addition of inorganic pyrophosphatase (10 U) the reaction mixture was re‐adjusted to pH 8.5 and started by adding 10 mg CSS, while keeping the pH constant by autotitration. After 2 h at 37 °C TLC analysis indicated the disappearance of Neu5Ac, then lactose (174.7 mg, 3 equiv), native 2,3SiaTpph or 2,3SiaTpph (A151D) (0.5 mg) and alkaline phosphatase (319 U) were added and incubation continued overnight. When conversion was completed, an equivalent volume of cold methanol (−20 °C) was added to stop the reaction. Protein precipitate was removed by centrifugation, methanol evaporated under reduced pressure, and the remaining solution directly purified over Biogel P‐2 (Bio‐Rad, Germany; 3×100 cm column). The product fractions were collected and lyophilized for NMR analysis. Product 14 was obtained as colorless solid (88.2 mg, 86 % for native 2,3SiaTpph/ 93.4 mg, 91 % for 2,3SiaTpph (A151D)).
Supporting Information: 2,3SiaTpph cloning and expression; primers used for mutagenesis; HPTLC documentation, characterization data of sialoconjugates; plasmid library sequencing.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by a PhD scholarship from the Chinese Research Council (to D.Y.) and by funds from the State of Hessia. We thank Iska Steffens and Juliane Joswig for assistance with the mutagenesis. Open access funding enabled and organized by Projekt DEAL.
A. Mertsch, N. He, D. Yi, M. Kickstein, W.-D. Fessner, Chem. Eur. J. 2020, 26, 11614.
References
- 1. Varki A., Nature 2007, 446, 1023–1029. [DOI] [PubMed] [Google Scholar]
- 2. Schauer R., Glycoconjugate J. 2000, 17, 485–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Varki A., Gagneux P., Ann. N. Y. Acad. Sci. 2012, 1253, 16–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schauer R., Curr. Opin. Struct. Biol. 2009, 19, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vimr E. R., ISRN Microbiology 2013, 816713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pearce O. M. T., Laeubli H., Glycobiology 2016, 26, 111–128. [DOI] [PubMed] [Google Scholar]
- 7. Bhide G. P., Colley K. J., Histochem. Cell Biol. 2017, 147, 149–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li F., Ding J., Protein Cell 2019, 10, 550–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fessner W.-D., He N., Yi D., Unruh P., Knorst M. in Cascade Biocatalysis (Eds.: S. Riva, W.-D. Fessner), Wiley-VCH, Weinheim, 2014, pp. 361–392. [Google Scholar]
- 10. Bayón C., He N., Deir-Kaspar M., Blasco P., Andre S., Gabius H.-J., Rumbero A., Jimenez-Barbero J., Fessner W.-D., Hernaiz M. J., Chem. Eur. J. 2017, 23, 1623–1633. [DOI] [PubMed] [Google Scholar]
- 11. Yu C.-C., Withers S. G., Adv. Synth. Catal. 2015, 357, 1633–1654. [Google Scholar]
- 12. Boons G.-J., Demchenko A. V., Chem. Rev. 2000, 100, 4539–4565. [DOI] [PubMed] [Google Scholar]
- 13. Izumi M., Shen G.-J., Wacowich-Sgarbi S., Nakatani T., Plettenburg O., Wong C.-H., J. Am. Chem. Soc. 2001, 123, 10909–10918. [DOI] [PubMed] [Google Scholar]
- 14. Pazynina G., Nasonov V., Belyanchikov I., Brossmer R., Maisel M., Tuzikov A., Bovin N., Int. J. Carbohydr. Chem. 2010, 594247. [Google Scholar]
- 15. Chen X., Varki A., ACS Chem. Biol. 2010, 5, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Audry M., Jeanneau C., Imberty A., Harduin-Lepers A., Delannoy P., Breton C., Glycobiology 2011, 21, 716–726. [DOI] [PubMed] [Google Scholar]
- 17. Kajiwara H., Mine T., Yamamoto T., J. Appl. Glycosci. 2009, 56, 77–82. [Google Scholar]
- 18. Yamamoto T., Mar. Drugs 2010, 8, 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He N., Yi D., Fessner W.-D., Adv. Synth. Catal. 2011, 353, 2384–2398. [Google Scholar]
- 20. Yi D., He N., Kickstein M., Metzner J., Weiss M., Berry A., Fessner W.-D., Adv. Synth. Catal. 2013, 355, 3597–3612. [Google Scholar]
- 21. Tsukamoto H., Takakura Y., Yamamoto T., J. Biol. Chem. 2007, 282, 29794–29802. [DOI] [PubMed] [Google Scholar]
- 22. Iwatani T., Okino N., Sakakura M., Kajiwara H., Takakura Y., Kimura M., Ito M., Yamamoto T., Kakuta Y., FEBS Lett. 2009, 583, 2083–2087. [DOI] [PubMed] [Google Scholar]
- 23. Knorst M., Fessner W.-D., Adv. Synth. Catal. 2001, 343, 698–710. [Google Scholar]
- 24. Yu H., Chokhawala H., Karpel R., Yu H., Wu B., Zhang J., Zhang Y., Jia Q., Chen X., J. Am. Chem. Soc. 2005, 127, 17618–17619. [DOI] [PubMed] [Google Scholar]
- 25. Cheng J., Huang S., Yu H., Li Y., Lau K., Chen X., Glycobiology 2010, 20, 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mine T., Katayama S., Kajiwara H., Tsunashima M., Tsukamoto H., Takakura Y., Yamamoto T., Glycobiology 2010, 20, 158–165. [DOI] [PubMed] [Google Scholar]
- 27. Schmölzer K., Ribitsch D., Czabany T., Luley-Goedl C., Kokot D., Lyskowski A., Zitzenbacher S., Schwab H., Nidetzky B., Glycobiology 2013, 23, 1293–1304. [DOI] [PubMed] [Google Scholar]
- 28. Guo Y., Jers C., Meyer A. S., Arnous A., Li H., Kirpekar F., Mikkelsen J. D., J. Biotechnol. 2014, 170, 60–67. [DOI] [PubMed] [Google Scholar]
- 29. Talafová K., Hrabarova E., Nahalka J., J. Biotechnol. 2015, 216, 116–124. [DOI] [PubMed] [Google Scholar]
- 30. Mehr K., Withers S. G., Glycobiology 2016, 26, 353–359. [DOI] [PubMed] [Google Scholar]
- 31. Both P., Riese M., Gray C. J., Huang K., Pallister E. G., Kosov I., Conway L. P., Voglmeir J., Flitsch S. L., Glycobiology 2018, 28, 261–268. [DOI] [PubMed] [Google Scholar]
- 32. McArthur J. B., Yu H., Tasnima N., Lee C. M., Fisher A. J., Chen X., ACS Chem. Biol. 2018, 13, 1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sugiarto G., Lau K., Qu J., Li Y., Lim S., Mu S., Ames J. B., Fisher A. J., Chen X., ACS Chem. Biol. 2012, 7, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagashima I., Mine T., Yamamoto T., Shimizu H., Carbohydr. Res. 2012, 358, 31–36. [DOI] [PubMed] [Google Scholar]
- 35. Kang J.-Y., Lim S.-J., Kwon O., Lee S.-G., Kim H. H., Oh D.-B., PLoS One 2015, 10, e0133739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kajiwara H., Katayama S., Kakuta Y., Okino N., Ito M., Mine T., Yamamoto T., Biosci. Biotechnol. Biochem. 2012, 76, 1639–1644. [DOI] [PubMed] [Google Scholar]
- 37. Schmölzer K., Luley-Goedl C., Czabany T., Ribitsch D., Schwab H., Weber H., Nidetzky B., FEBS Lett. 2014, 588, 2978–2984. [DOI] [PubMed] [Google Scholar]
- 38. Schmölzer K., Eibinger M., Nidetzky B., ChemBioChem 2017, 18, 1544–1550. [DOI] [PubMed] [Google Scholar]
- 39. Hamada Y., Kanematsu Y., Tachikawa M., Biochemistry 2016, 55, 5764–5771. [DOI] [PubMed] [Google Scholar]
- 40. Ni L., Chokhawala H. A., Cao H., Henning R., Ng L., Huang S., Yu H., Chen X., Fisher A. J., Biochemistry 2007, 46, 6288–6298. [DOI] [PubMed] [Google Scholar]
- 41. Yamamoto T., Ichikawa M., Takakura Y., Biochem. Biophys. Res. Commun. 2008, 365, 340–343. [DOI] [PubMed] [Google Scholar]
- 42. Czabany T., Schmoelzer K., Luley-Goedl C., Ribitsch D., Nidetzky B., Anal. Biochem. 2015, 483, 47–53. [DOI] [PubMed] [Google Scholar]
- 43. Noel M., Gilormini P.-A., Cogez V., Lion C., Biot C., Harduin-Lepers A., Guerardel Y., Bioconjugate Chem. 2018, 29, 3377–3384. [DOI] [PubMed] [Google Scholar]
- 44. Gosselin S., Alhussaini M., Streiff M. B., Takabayashi K., Palcic M. M., Anal. Biochem. 1994, 220, 92–97. [DOI] [PubMed] [Google Scholar]
- 45. Wagner G. K., Pesnot T., ChemBioChem 2010, 11, 1939–1949. [DOI] [PubMed] [Google Scholar]
- 46. Chapman E., Wong C.-H., Bioorg. Med. Chem. 2002, 10, 551–555. [DOI] [PubMed] [Google Scholar]
- 47. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C., Anal. Biochem. 1985, 150, 76–85. [DOI] [PubMed] [Google Scholar]
- 48. Magnusson A. O., Szekrenyi A., Joosten H. J., Finnigan J., Charnock S., Fessner W. D., FEBS J. 2019, 286, 184–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rillahan C. D., Schwartz E., McBride R., Fokin V. V., Paulson J. C., Angew. Chem. Int. Ed. 2012, 51, 11014–11018; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11176–11180. [Google Scholar]
- 50. Zhang J., Chen C., Gadi M. R., Gibbons C., Guo Y., Cao X., Edmunds G., Wang S., Liu D., Yu J., Wen L., Wang P. G., Angew. Chem. Int. Ed. 2018, 57, 16638–16642; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16880–16884. [Google Scholar]
- 51. Yamakawa N., Vanbeselaere J., Chang L.-Y., Yu S.-Y., Ducrocq L., Harduin-Lepers A., Kurata J., Aoki-Kinoshita K. F., Sato C., Khoo K.-H., Kitajima K., Guerardel Y., Nat. Commun. 2018, 9, 4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lopez Aguilar A., Meng L., Hou X., Li W., Moremen K. W., Wu P., Bioconjugate Chem. 2018, 29, 1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary