

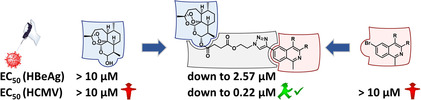
Abstract
Viral infections cause life‐threatening diseases in millions of people worldwide every year and there is an urgent need for new, effective antiviral drugs. Hybridization of two chemically diverse compounds into a new bioactive effector product is a successful concept to improve the properties of a hybrid drug relative to the parent compounds. In this study, (iso)quinoline–artemisinin hybrids, obtained through copper‐catalyzed azide–alkyne cycloaddition or metal‐free click reactions (in organic solvents or in the presence of water), were analyzed in vitro, for the first time, for their inhibitory activity against human cytomegalovirus (HCMV), relative to their parent compounds and the reference drug ganciclovir. EC50 (HCMV) values were obtained in a range 0.22–1.20 μm, which indicated highly potent antiviral properties in the absence of cytotoxic effects on normal cells (CC50>100 μm). The most active hybrid, 1 (EC50=0.22 μm), is 25 times more potent than its parent compound artesunic acid (EC50=5.41 μm) and 12 times more efficient than the standard drug ganciclovir (EC50=2.6 μm). Interestingly, hybrid 1 also shows inhibitory activity against hepatitis B virus in vitro (EC50 (HBeAg)=2.57 μm).
Keywords: antiviral agents, artemisinins, click chemistry, cycloaddition, quinolines
Combined strength: A series of (iso)quinoline–artemisinin hybrids are synthesized through click reactions and studied for their potency against human cytomegalovirus (HCMV). Seven hybrids show high in vitro activity and are more active than their parent compounds. Additionally, the most potent anti‐HCMV hybrid also shows inhibition activity against hepatitis B virus in vitro.
Introduction
Human cytomegalovirus (HCMV) is an opportunistic viral pathogen, which can take severe and sometimes life‐threatening courses in immunocompromised people, such as transplant recipients; cancer or AIDS patients; and, most importantly, unborn children and neonates.1 Notably, even after decades of intensive research, distinct medical questions concerning infection with HCMV remain highly relevant and have not yet been resolved. Ganciclovir (GCV; Figure 1 C) is a deoxyguanosine analogue and, in 1988, was the first drug to be approved for the treatment of HCMV. The emergence of HCMV resistance to currently available antiviral drugs, among them GCV, represents a constant limitation of therapy success.2 Another challenging virus, which leads to a wide spectrum of liver diseases, ranging from acute hepatitis to chronic (lifelong) hepatitis, cirrhosis, and hepatocellular carcinoma, is hepatitis B virus (HBV).3 The World Health Organization (WHO) estimates that, of the 2 billion people who have been infected with HBV, more than 350 million have chronic infection.4
Figure 1.
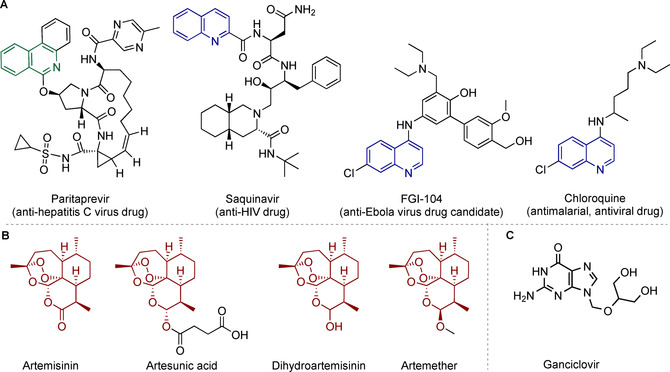
Structures of A) isoquinolines and quinolines with different bioactivities: antivirals Paritaprevir, saquinavir, FGI‐104, and antimalarial/antiviral chloroquine; B) artemisinin (ARN, naturally occurring) and its semisynthetic derivatives artesunic acid (ART), dihydroartemisinin (DHA), and artemether; C) the anti‐HCMV drug GCV, which is used as a reference compound in this study.
Hence, the development of new potent agents against HCMV and HBV are of high interest. For this task, hybridization can be used as a powerful concept to increase the biological potency or pharmacological efficacy (such as stability, distribution, or targeting) of the bioactive constituents of the hybrid molecule.5 One reason for the increased activity of hybrids versus the individual constituents could be given by the simultaneous cellular uptake of covalently linked pharmacophores in a way that the inhibitory kinetics of the two constituents may potentiate each other.6
Since their discovery in the 19th century, quinoline and isoquinoline heterocycles have been ubiquitous in pharmaceutical compounds and drugs.7 The class of quinolines shows a wide range of biological activities, such as anti‐inflammatory7a, 7e antifungal,7b and antibacterial.7c Furthermore, quinolines are also active against a wide range of viruses, such as coronaviruses,8 human immunodeficiency virus (HIV),9 respiratory syncytial virus,10 hepatitis C virus,11 West Nile virus,12 Zika virus,13 and Dengue virus.7e, 13 Known representatives in the field of antiviral agents containing an (iso)quinoline core structure are depicted in Figure 1 A, namely, the protease inhibitors Paritaprevir (a drug against hepatitis C virus),14 saquinavir (an anti‐HIV drug),15 FGI‐104 (a drug candidate against Ebola virus),16 and chloroquine (an antimalarial drug that is active against several viruses,17 including HIV; hepatitis C virus; Ebola virus; and, as recently reported, against coronaviruses, including newly emerged SARS‐CoV‐2).18
ARN is an enantiopure sesquiterpene lactone/trioxane (Figure 1 B), which is an approved antimalarial drug originally been isolated from the plant Artemisia annua L. The herb has been used since ancient times; however, structural identification of ARN was first possible in 1972 through X‐ray crystal structure analysis by Tu.19 Artesunate (the sodium salt of ART (Figure 1 B)), a semisynthetic derivative of ARN, has recently been characterized as an active compound against wild‐type, recombinant, GCV‐sensitive, and GCV‐resistant HCMVs, lacking cross‐resistance with HCMV drugs in vitro.20 In addition to artesunate, other derivatives, such as DHA and artemether (Figure 1 B), have been widely investigated as potential antiviral agents.
As alternatives to standard drugs, our ongoing search for highly active hybrid molecules,6 which exceed their parent compounds in activity against HCMV and malaria parasites, has resulted in the synthesis of novel (iso)quinoline–ARN hybrids (Figure 2). Notably, in our recent study, we reported that these hybrids were able to combat multidrug‐resistant malaria.6b Herein, we focus on their activities against HCMV. Twelve (iso)quinoline–ARN hybrids from our previous work were, therefore, analyzed, for the first time, for their potency as quinoline–ARN HCMV agents. The hybrid compounds exhibited high anti‐HCMV activities, without toxic effects on normal cells, and were more active than their parent compounds and more efficient than the standard drug GCV. In addition, the most potent anti‐HCMV hybrid also showed inhibition activity against HBV in vitro.
Figure 2.
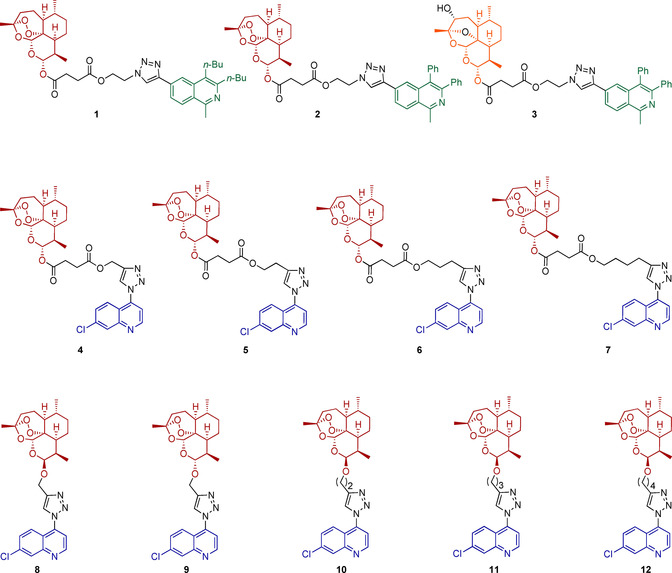
Structures of hybrids 1–12 applied for activity examination against HCMV. Red, blue, and green groups indicate parent pharmacophores of molecules. Black parts represent the varied linker groups of the molecules. The orange color indicates the 3‐hydroxydesoxydihydroartemisinin unit of hybrid 3.
Results and Discussion
Chemistry
The synthesis of hybrid compounds 1–12 (Figure 2) was described in our previous work (in which the compounds were studied against chloroquine‐resistant malaria parasites).6b Copper(I)‐catalyzed azide–alkyne cycloaddition (CuAAC) reactions were applied to couple the corresponding (iso)quinolines 15, 16, or 22 with ARN derivatives 17–21 and 23–27, giving hybrids 1–12 with different linkers, but all with a triazole moiety in common. The synthetic results are summarized in Scheme 1. The corresponding new isoquinolines 15 and 16, with an alkyne moiety, were prepared through recently developed, facile C−H cobaltation of O‐acetyl oximes with internal alkynes and a subsequent Sonogashira–Hagihara cross‐coupling with trimethylsilylacetylene.21 Additionally, hybrid 3 was isolated as a side product of hybrid 2 because the peroxide bridge of ARN was hydroxylated at the C3 position in the presence of copper(I) species, resulting in desoxydihydroartemisinin (Scheme 1 A).22 Hybrids 4–12 were obtained by coupling ART‐ (18–21) or ARN‐derived (23–27) alkynes with 4‐azido‐7‐chloroquinline (22) in the presence of the catalyst system CuSO4 ⋅5 H2O and sodium ascorbate (Scheme 1 B and C).
Scheme 1.
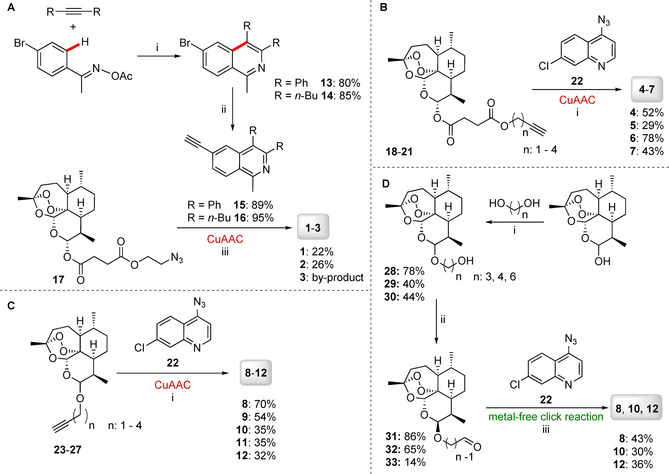
A) Synthesis of isoquinoline–ARN hybrids 1–3 through the CuAAC reaction. B) Synthesis of 7‐choloroquinoline–ART hybrids 4–7 through the CuAAC reaction. C) Synthesis of 7‐chloroquinoline–ARN hybrids 8–12 through the CuAAC reaction. D) Synthesis of hybrids 8, 10, and 12 through metal‐free click reactions. Reagents and conditions: A) i) [Co(CO)Cp*I2] (10 mol %; Cp*=1,2,3,4,5‐pentamethylcyclopentadiene), AgSbF6 (20 mol %), NaOAc (20 mol %), 1,2‐dichloroethane, 120 °C, 1 h; ii) 1) [PdCl2(PPh3)2] (1.0 mol %), trimethylsilylacetylene, triethylamine, 50 °C, 2 h; 2) K2CO3, MeOH, 25 °C, 4 h; iii) CuSO4 (5 mol %), sodium l‐ascorbate (10 mol %), CH2Cl2/H2O (1:1), RT, o/n; R=Ph or nBu. B) i) CuSO4 ⋅5 H2O (20 mol %), sodium ascorbate (40 mol %), CH2Cl2/H2O (1:1), RT, o/n; 18: n=1; 19: n=2; 20: n=3; 21: n=4. C) i) CuSO4 ⋅5 H2O (20 mol %), sodium ascorbate (40 mol %), CH2Cl2/H2O (1:1), RT, o/n; 23: n=1 (C‐10β); 24: n=1 (C‐10α); 25: n=2 (C‐10β); 26: n=3 (C‐10β); 27: n=4 (C‐10β). D) i) H3PW12O4 ⋅H2O (10 mol %), CH2Cl2/MeCN (1:1 (n=3), 5.5:4 (n=4), 8:2 (n=6)); ii) Dess–Martin periodinane (1.2 equiv), CH2Cl2; 28β: n=3 (C‐10β); 28α: n=3 (C‐10α); 29: n=4 (C‐10β); 30: n=6 (C‐10β); iii) 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU; 1 equiv), malononitrile (1 equiv), DMSO, RT, o/n; 31: n=3 (C‐10β); 32: n=4 (C‐10β); 33: n=6 (C‐10β).
To open up the opportunity to apply the formation of our hybrid compounds in future research in the field of bioorthogonal chemistry and, for example, enable the generation of hybrids in situ directly in living cells, we additionally applied an alternative metal‐free pathway, leading to the selected hybrid compounds 8, 10, and 12. The first metal‐free click reaction, which was reported independently in 2014 by the groups of Ramachary23 and Paixão,24 is a green method that might allow toxic copper(I) species25 to be replaced with DBU. The mechanism of regioselective synthesis of 1,4‐disubstituted‐1,2,3‐triazoles by applying a DBU/malononitrile co‐catalyzed 1,3‐dipolar cycloaddition strategy was proposed by Paixão et al.24 It starts with a Knoevenagel condensation of malononitrile with the aliphatic aldehyde, giving the alkylidene malononitrile A, which is subsequently deprotonated by DBU, forming vinylogous carbanion B (Scheme 2). Electron‐rich olefin B thereafter reacts with the aryl azide, proceeding via proposed transition‐state TS1, to give cycloaddition adduct D after protonation. Hypothetical transition‐states TS2, which could lead to isomer F (not observed), and TS3, which could give zwitterionic intermediate E, might be disfavored energetically. Finally, a syn‐elimination step results in the 1,4‐disubstituted‐1,2,3‐triazole products and recovers the malononitrile.24
Scheme 2.
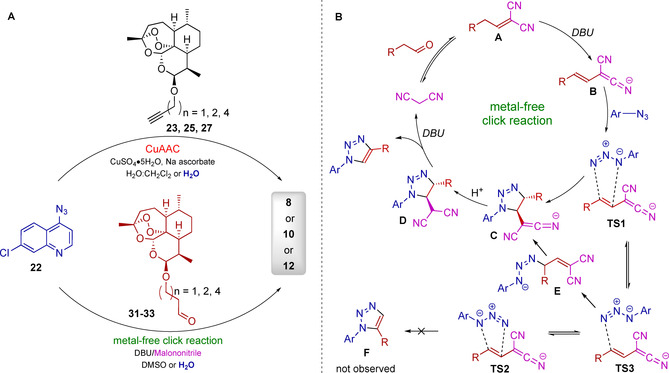
A) CuAAC and metal‐free click reactions, leading to the same hybrid compounds 8, 10, and 12. Detailed reaction conditions are described in Table 1. B) Proposed mechanism of metal‐free click reactions based on recent reports.24, 28
Aldehydes are used as substrates instead of alkynes, which are applied in CuAAC. Thus, for metal‐free click reaction, we used ARN‐derived aldehydes 31–33 and the previously applied azide 22 to form hybrids 8, 10, and 12. The aldehydes were obtained by etherification of DHA with the corresponding alcohols,26 catalyzed by H3PW12O4 ⋅H2O (Scheme 1 D), and subsequent Dess–Martin oxidation reaction to obtain aldehydes. Because a mixture of both DHA isomers was used, the alpha and beta isomers of the ARN‐derived alcohols were formed. In the case of compound 28, a mixture of both isomers (28β (C‐10β)/28α (C‐10α)=5:4) were isolated and used further. In the case of compounds 29 and 30, the β isomers were separately isolated in moderate yields (29: 40 %, 30: 44 %). The corresponding Dess–Martin oxidation27 gave the ARN‐derived aldehydes 31–33 (31: 86 %, 32: 65 %, 33: 14 %).
The DBU‐mediated 1,3‐dipolar cycloaddition was performed by stirring 22 with the corresponding ARN‐derived aldehydes 31–33 at RT in DMSO in the presence of malononitrile (1 equiv) and DBU (1 equiv). Hybrids 8, 10, and 12 were obtained in 43, 30, and 36 % yields, respectively (Scheme 1 D). Comparing the outcomes of the CuAAC and for metal‐free cycloaddition reactions in organic solvents (Scheme 2 and Table 1), the CuAAC catalysis gives hybrid 8, with the shortest linker, in a better yield (70 %, entry 1) than that through metal‐free click reaction (43 %, entry 5). If the linker contains more CH2 groups, the yields of hybrids 10 and 12 obtained in organic solvents through both synthetic pathways are almost identical: yields of CuAAC reactions were 35 (for hybrid 10, entry 2) and 32 % (for hybrid 12, entry 3), whereas yields of the corresponding metal‐free click reactions were 30 (hybrid 10, entry 6) and 36 % (hybrid 12; Table 1, entry 7), respectively.
Table 1.
A comparison of CuAAC and metal‐free cycloaddition reactions.
|
|
Entry |
Catalytic system[a] |
Solvent[a] |
Product |
Yield[a] [%] |
|---|---|---|---|---|---|
|
CuI‐catalyzed |
1 |
CuSO4 ⋅5 H2O (20 mol %), sodium ascorbate (40 mol %) |
CH2Cl2/H2O (1:1) |
8 |
70 |
|
2 |
CuSO4 ⋅5 H2O (20 mol %), sodium ascorbate (40 mol %) |
CH2Cl2/H2O (1:1) |
10 |
35 |
|
|
3 |
CuSO4 ⋅5 H2O (20 mol %), sodium ascorbate (40 mol %) |
CH2Cl2/H2O (1:1) |
12 |
32 |
|
|
4 |
CuSO4 ⋅5 H2O (1 equiv), sodium ascorbate (4 equiv) |
H2O |
8 |
9 |
|
|
metal‐free click reaction |
5 |
DBU (1 equiv), malononitrile (1 equiv) |
DMSO |
8 |
43 |
|
6 |
DBU (1 equiv), malononitrile (1 equiv) |
DMSO |
10 |
30 |
|
|
7 |
DBU (1 equiv), malononitrile (1 equiv) |
DMSO |
12 |
36 |
|
|
8 |
DBU (1 equiv), malononitrile (1 equiv) |
H2O |
8 |
30 |
[a] Reaction conditions and product yields correspond to the reaction mechanism depicted in Scheme 2.
As an initial test case for a metal‐free click reaction as a potential bioorthogonal reaction, we evaluated the tolerance of both catalytic systems to water as a solvent and studied how the yield of hybrid 8 was influenced by changing the solvent system from H2O/CH2Cl2 (CuAAC; Table 1, entry 4) and DMSO (metal‐free click reaction; Table 1, entry 8) to H2O. The experiments showed that, for the formation of quinoline–ARN hybrid 8 under aqueous conditions, the metal‐free system resulted in better yield (30 % yield, Table 1, entry 8) than that of the CuAAC reaction system (9 % yield, Table 1, entry 4). This higher yield of product 8, obtained through the metal‐free click reaction in the presence of water, could be explained by higher polarity of the aldehyde, relative to the alkyne, and thus, its higher solubility in water. In summary, such a metal‐free click reaction in water has the potential to be developed as a bioorthogonal metal‐free reaction under live‐cell conditions or in live cells.
Antiviral activity
In our previous study, the set of hybrids 1–12 enabled us to investigate variation of the antimalarial activities, depending on the type of linkage between ARN and (iso)quinolones.6b Herein, the extension of this study to the in vitro quinoline–ARN HCMV activity is presented (Table 2).
Table 2.
EC50 values for reference compounds GCV, ART, ARN, DHA, artemether, tenofovir alafenamide fumarate (TAF); parent compound 13; and hybrids 1–12, which were analyzed for anti‐HCMV and anti‐HBV activities.[a]
|
Compound |
HCMV EC50 [μm] |
LDH CC50 [μm] |
HepG2‐hNTCP CC50 [μm] |
HBeAg ELISA EC50 [μm] |
HBV DNA qPCR EC50 [μm] |
|---|---|---|---|---|---|
|
1 |
0.22±0.04 |
>100* |
29.9±1.1 |
2.57±1.51 |
≈10 |
|
2 |
0.67±0.03 |
>100 |
>50 |
>10 |
>10 |
|
3 |
none |
>100 |
n.d. |
n.d. |
n.d. |
|
4 |
none (strong cytotox. **) |
>100** |
n.d. |
n.d. |
n.d. |
|
5 |
none (strong cytotox. **) |
>100** |
>50 |
>10 |
>10 |
|
6 |
none (strong cytotox. **) |
>100** |
n.d. |
n.d. |
n.d. |
|
7 |
none (strong cytotox. **) |
>100** |
>50 |
>10 |
>10 |
|
8 |
0.71±0.03 |
n.d. |
n.d. |
n.d. |
n.d. |
|
9 |
1.20±0.11 |
>100 |
n.d. |
n.d. |
n.d. |
|
10 |
1.08±0.18 |
>100* |
n.d. |
n.d. |
n.d. |
|
11 |
0.30±0.02 |
>100 |
>50 |
>10 |
>10 |
|
12 |
0.38±0.03 |
>100 |
>50 |
>10 |
>10 |
|
13 |
>10 |
n.d. |
n.d. |
n.d. |
n.d. |
|
ARN[b] |
>10 |
>100 |
n.d. |
n.d. |
n.d. |
|
ART[c] |
5.41±0.61 |
n.d. |
>50 |
>10 |
>10 |
|
DHA[b] |
>10 |
n.d. |
>50 |
>10 |
>10 |
|
artemether |
>10 |
>100 |
n.d. |
n.d. |
n.d. |
|
GCV[d] |
2.60±0.5 |
>100 |
n.d. |
n.d. |
n.d. |
|
TAF |
– |
– |
27.2±0.7 |
3.93±0.8 |
0.00024±0.00004 |
[a] */** Microscopic inspection of cell morphology or cell lysis after 6–8 days, long‐term cytotoxicity (* moderate, ** strong). LDH: lactate dehydrogenase release assay, 24 h, acute cytotoxicity; “n.d.”—not determined. [b] EC50 values have been previously reported.6a, 29 [c] EC50 values have been previously reported.30 [d] EC50 values have been previously reported.31
Reference drug GCV; two quinoline compounds (34 and 35; see Table S1 in the Supporting Information); and the parent drugs ARN, ART, DHA, and artemether were used as comparative compounds in this analysis. Anti‐HCMV EC50 values of the hybrids and of precursor 13 were determined. GCV and ART showed EC50 values of (2.60±0.5) and (5.41±0.61) μm (Table 2), respectively, whereas ARN, DHA, and artemether were mostly inactive against HCMV. Similarly, quinoline compounds 34 and 35, as well as parent compound 13, only comprising the 7‐chloroquinoline unit, had no measurable effect on HCMV replication. Contrary to the antimalarial results, this antiviral analysis showed that dihydroartemisinin‐7‐chloroquinoline‐based hybrids 8–12 and isoquinoline–ARN hybrids 1 and 2 achieved the highest activity, with EC50 values ranging from (0.22±0.04) to (1.20±0.11) μm, even exerting higher in vitro activities than those of the parent and reference compounds. Isomers 8 (C‐10β‐isomer) and 9 (C‐10α‐isomer) exhibited a similar structure–activity relationship against HCMV to that observed for the antimalarial activity; the β‐isomer was more active than the α‐isomer. β‐Isomer 8 showed an EC50 value of (0.71±0.03) μm, whereas α‐isomer 9 showed (1.20±0.11) μm. Hybrids 10, 11, and 12 were particularly active, with EC50 values of (1.08±0.18), (0.30±0.02), and (0.38±0.03) μm (Table 2). The two longest linkers (hybrids 11 and 12), as spacers between pharmacophores, improved the activity; this was likely as a result of providing increased intramolecular flexibility.
Surprisingly, isoquinoline–ARN hybrids 1 and 2 showed 8‐ to 25‐fold higher activities than that of ART, with EC50 values of (0.22±0.04) and (0.67±0.03) μm, respectively, whereas none of the other ART‐based hybrids 4–7 were found to be active against HCMV (or the EC50 values could not be determined due to the induction of cytotoxicity). Of the three isoquinoline–ARN hybrids, only one, namely, hybrid 3, was inactive; this can be ascribed to hydroxylation of the peroxide bridge of the ARN unit. This finding was comparable to the antimalarial activity of 3, for which bioactivation was putatively based on the peroxide bridge. Notably, all active compounds were similar or even more active, in terms of in vitro anti‐HCMV efficacy, than that of the parent compounds and reference drugs. Moreover, none of these compounds exerted acute cytotoxicity, according to the LDH release assay, with CC50 values of >100 μm (Table 2).
Recently, the activities of ARN, artesunate, and whole extract of Chinese herb Artemisia capillaris were determined in HBV‐transfected HepG2.2.15 cells.32 Artesunate was shown to inhibit the HBV s antigen (HBsAg), with an IC50 value of 2.3 μm, and reduced the amount of HBV‐DNA secreted from HepG2.2.15 cells to the culture medium. On the other hand, HBsAg reduction by ARN was weaker, with an IC50 value of around 50 μm, and no reduction in HBV‐DNA was observed.32a Inspired by these results, we decided to assess the inhibitory potential of a selected subset of ARN‐derived hybrid compounds 1, 2, 5, 7, 11, and 12 against HBV. Inhibition of HBV was evaluated by measuring the extent to which the test compounds reduced the release of HBV e antigen (HBeAg) and HBV‐DNA after infection of HepG2‐hNTCP cells for 14 days.
The reference compounds TAF, DHA, and ART were used for comparison in the assays. Isoquinoline–ARN hybrid 1, containing n‐butyl moieties, inhibited HBeAg secretion in cell‐free supernatant, with EC50=(2.57±1.51) μm, and showed a reduction in HBV‐DNA, with an EC50 value of approximately 10 μm. This compound exhibited cytotoxicity in HepG2‐hNTCP cells, with CC50=(29.9±1.1) μm. The reference compound TAF showed similar cytotoxicity and reduction in HBeAg secretion, but, as a selective inhibitor of reverse transcriptase activity, it showed 10 000 times better inhibition of HBV‐DNA secretion in a medium than that of compound 1. The introduction of phenyl moieties in the place of n‐butyl in compound 2 abolished the anti‐HBV activity. Similarly, there was no anti‐HBV activity for dihydroartemisinin‐7‐chloroquinoline‐based hybrids 11 and 12.
Conclusion
Although HCMV infection is mostly inapparent in immunocompetent persons, it can be life‐threatening for immunonaïve or immunocompromised individuals, so that anti‐HCMV‐specific drug research is an ongoing issue investigated worldwide. To this end, the present study characterized new (iso)quinoline–ARN hybrid compounds for anti‐HCMV activity in vitro. Thus, three isoquinoline–ARN and nine quinoline–ARN hybrids, together with their precursors and reference drugs, were pharmacologically evaluated. Hybrids were obtained by facile C−H activation and CuAAC reactions.6b, 21 Additionally, hybrids 8, 10, and 12 were, for the first time, synthesized through a metal‐free cycloaddition reaction, with DBU and malononitrile as cocatalysts. Additionally, preliminary experiments of both cycloaddition reactions (CuAAC and metal‐free) were performed to analyze their suitability for future application in the context of living cells, by performing the selected reactions in the presence of water. Metal‐free click reaction proved to be more promising, since hybrid 8 could be isolated in 30 % yield, whereas CuAAC catalysis gave the hybrid product in only 9 % yield.
Remarkably, hybrids 1, 2, 8, 11, and 12 demonstrated pronounced activity against HCMV, that is, in vitro EC50 values in the sub‐micromolar range (down to 0.22 μm for hybrid 1). Notably, hybrid 1 also exhibited low micromolar activity against HBV (EC50 (HBeAg)=2.57 μm). Moreover, the cytotoxicity profiles of all hybrids were almost negative within the relevant range of antiviral concentrations in primary HFFs, that is, CC50 values were low or undetectable at concentrations up to 100 μm. Thus, these compounds, exhibiting high antiviral activities combined with a low toxicity/high selectivity profile, illustrate the potential of the hybridization concept as an alternative drug‐discovery approach, which can also be applied for current anti‐coronavirus drug development.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) through grant nos. TS 87/16‐3, MM 1289/7‐1/7‐3, and MM1289/11‐1. Generous support by the DFG (Gottfried Wilhelm Leibniz award to L.A.) is gratefully acknowledged. Financial support from the German Academic Exchange Service (DAAD) for a doctoral research fellowship to Aysun Çapcı and for the international research exchange program of M.M.’s team (DAAD‐Go8) is gratefully acknowledged. Generous support by the Alexander von Humboldt foundation (fellowship to M.M.L.) is gratefully acknowledged. We also thank, for support and funding, the Interdisciplinary Center for Molecular Materials (ICMM), DFG‐funded Graduate School GRK 2504 (A1/MM), the Graduate School Molecular Science (GSMS), and the Emerging Fields Initiative (EFI) “Chemistry in Live Cells” supported by Friedrich‐Alexander‐Universität Erlangen‐Nürnberg. Anti‐HBV activity determination was supported by the ERDF/ESF project ChemBioDrug CZ.02.1.01/0.0/0.0/16_019/0000729. Open access funding enabled and organized by Projekt DEAL.
A. Çapcı, M. M. Lorion, C. Mai, F. Hahn, J. Hodek, C. Wangen, J. Weber, M. Marschall, L. Ackermann, S. B. Tsogoeva, Chem. Eur. J. 2020, 26, 12019.
Contributor Information
Prof. Dr. Lutz Ackermann, Email: lutz.ackermann@chemie.uni-goettingen.de.
Prof. Dr. Svetlana B. Tsogoeva, Email: svetlana.tsogoeva@fau.de.
References
- 1. Dupont L., Reeves M. B., Rev. Med. Virol. 2016, 26, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gilbert C., Boivin G., Antimicrob. Agents Chemother. 2005, 49, 873–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liang T. J., Hepatology 2009, 49, S13-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nebbia G., Peppa D., Maini M. K., QJM 2012, 105, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Tietze L. F., Bell H. P., Chandrasekhar S., Angew. Chem. Int. Ed. 2003, 42, 3996–4028; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4128–4160; [Google Scholar]
- 5b. Gademann K., Chimia 2006, 60, 841–845; [Google Scholar]
- 5c. Tsogoeva S. B., Mini-Rev. Med. Chem. 2010, 10, 773–793; [DOI] [PubMed] [Google Scholar]
- 5d. Fröhlich T., Çapcı Karagöz A., Reiter C., Tsogoeva S. B., J. Med. Chem. 2016, 59, 7360–7388. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Held F. E., Guryev A. A., Fröhlich T., Hampel F., Kahnt A., Hutterer C., Steingruber M., Bahsi H., von Bojničić-Kninski C., Mattes D. S., Foertsch T. C., Nesterov-Mueller A., Marschall M., Tsogoeva S. B., Nat. Commun. 2017, 8, 15071; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Çapcı A., Lorion M. M., Wang H., Simon N., Leidenberger M., Borges Silva M. C., Moreira D. R. M., Zhu Y., Meng Y., Chen J. Y., Lee Y. M., Friedrich O., Kappes B., Wang J., Ackermann L., Tsogoeva S. B., Angew. Chem. Int. Ed. 2019, 58, 13066–13079; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13200–13213. [Google Scholar]
- 7.
- 7a. Ratheesh M., Sindhu G., Helen A., J. Inflamm. Res. 2013, 62, 367–376; [DOI] [PubMed] [Google Scholar]
- 7b. Vandekerckhove S., Tran H. G., Desmet T., D′hooghe M., Bioorg. Med. Chem. Lett. 2013, 23, 4641–4643; [DOI] [PubMed] [Google Scholar]
- 7c. Lam K.-H., Gambari R., Lee K. K.-H., Chen Y.-X., Kok S. H.-L., Wong R. S.-M., Lau F.-Y., Cheng C.-H., Wong W.-Y., Bian Z.-X., Chan A. S.-C., Tang J. C.-O., Chui C.-H., Bioorg. Med. Chem. Lett. 2014, 24, 367–370; [DOI] [PubMed] [Google Scholar]
- 7d.“Quinolines and Isoquinolines”: Finley K. T. in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2015; [Google Scholar]
- 7e. de la Guardia C., Stephens D. E., Dang H. T., Quijada M., Larionov O. V., Lleonart R., Molecules 2018, 23, 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vincent M. J., Bergeron E., Benjannet S., Erickson B. R., Rollin P. E., Ksiazek T. G., Seidah N. G., Nichol S. T., Virology 2005, 2, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rolain J.-M., Colson P., Raoult D., Int. J. Antimicrob. Agents 2007, 30, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng X., Wang L., Wang B., Miao K., Xiang K., Feng S., Gao L., Shen H. C., Yun H., ACS Med. Chem. Lett. 2016, 7, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Talamas F. X., Abbot S. C., Anand S., Brameld K. A., Carter D. S., Chen J., Davis D., de Vicente J., Fung A. D., Gong L., Harris S. F., Inbar P., Labadie S. S., Lee E. K., Lemoine R., Le Pogam S., Leveque V., Li J., McIntosh J., Nájera I., Park J., Railkar A., Rajyaguru S., Sangi M., Schoenfeld R. C., Staben L. R., Tan Y., Taygerly J. P., Villaseñor A. G., Weller P. E., J. Med. Chem. 2014, 57, 1914–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ezgimen M., Lai H., Mueller N. H., Lee K., Cuny G., Ostrov D. A., Padmanabhan R., Antiviral Res. 2012, 94, 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barbosa-Lima G., Moraes A. M., Araújo A. d. S., da Silva E. T., de Freitas C. S., Vieira Y. R., Marttorelli A., Neto J. C., Bozza P. T., de Souza M. V. N., Souza T. M. L., J. Med. Chem. 2017, 127, 334–340. [DOI] [PubMed] [Google Scholar]
- 14. De Clercq E., Li G., Clin. Microbiol. Rev. 2016, 29, 695–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vella S., Floridia M., Clin. Pharmacokinet. 1998, 34, 189–201. [DOI] [PubMed] [Google Scholar]
- 16.“A Focus on Ebola Virus Polymerase: Structure, Functions and Antiviral Therapies”: Pettini F., Trezza A., Spiga O. in Viral Polymerases (Ed.: Gupta), Academic Press, 2019, chap. 7. [Google Scholar]
- 17. D′Alessandro S., Scaccabarozzi D., Signorini L., Perego F., Ilboudo D. P., Ferrante P., Delbue S., Microorganisms 2020, 8, 85–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Keyaerts E., Li S., Vijgen L., Rysman E., Verbeeck J., Van Ranst M., Maes P., Antimicrob. Agents Chemother. 2009, 53, 3416–3421; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Cell Res. 2020, 30, 269–271; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18c. Devaux C. A., Rolain J.-M., Colson P., Raoult D., Int. J. Antimicrob. Agents 2020, 55, 105938–105943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Tu Y., Nat. Med. 2011, 17, 1217–1220; [DOI] [PubMed] [Google Scholar]
- 19b. Su X. Z., Miller L. H., Sci. China Life Sci. 2015, 58, 1175–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Efferth T., Marschall M., Wang X., Huong S.-M., Hauber I., Olbrich A., Kronschnabl M., Stamminger T., Huang E.-S., J. Mol. Med. 2002, 80, 233–242. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Ackermann L., Chem. Rev. 2011, 111, 1315–1345; [DOI] [PubMed] [Google Scholar]
- 21b. Wang H., Koeller J., Liu W., Ackermann L., Chem. Eur. J. 2015, 21, 15525–15528; [DOI] [PubMed] [Google Scholar]
- 21c. Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452. [DOI] [PubMed] [Google Scholar]
- 22. Kapkoti D. S., Singh S., Luqman S., Bhakuni R. S., New J. Chem. 2018, 42, 5978–5995. [Google Scholar]
- 23. Ramachary D. B., Shashank A. B., Karthik S., Angew. Chem. Int. Ed. 2014, 53, 10420–10424; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10588–10592. [Google Scholar]
- 24. Ali A., Corrêa A. G., Alves D., Zukerman-Schpector J., Westermann B., Ferreira M. A. B., Paixão M. W., Chem. Commun. 2014, 50, 11926–11929. [DOI] [PubMed] [Google Scholar]
- 25. Letelier M. E., Lepe A. M., Faúndez M., Salazar J., Marín R., Aracena P., Speisky H., Chem. Biol. Interact. 2005, 151, 71–82. [DOI] [PubMed] [Google Scholar]
- 26. Bora P. P., Baruah N., Bez G., Barua N. C., Synth. Commun. 2012, 42, 1218–1225. [Google Scholar]
- 27. Ji H., Li H., Martasek P., Roman L. J., Poulos T. L., Silverman R. B., J. Med. Chem. 2009, 52, 779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jalani H. B., Karagöz A. Ç., Tsogoeva S. B., Synthesis 2017, 49, 29–41. [Google Scholar]
- 29. Hutterer C., Niemann I., Milbradt J., Fröhlich T., Reiter C., Kadioglu O., Bahsi H., Zeitträger I., Wagner S., Einsiedel J., Gmeiner P., Vogel N., Wandinger S., Godl K., Stamminger T., Efferth T., Tsogoeva S. B., Marschall M., Antiviral Res. 2015, 124, 101–109. [DOI] [PubMed] [Google Scholar]
- 30. Chou S., Marousek G., Auerochs S., Stamminger T., Milbradt J., Marschall M., Antiviral Res. 2011, 92, 364–368. [DOI] [PubMed] [Google Scholar]
- 31. Kaptein S. J. F., Efferth T., Leis M., Rechter S., Auerochs S., Kalmer M., Bruggeman C. A., Vink C., Stamminger T., Marschall M., Antiviral Res. 2006, 69, 60–69. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Romero M. R., Efferth T., Serrano M. A., Castano B., Macias R. I., Briz O., Marin J. J., Antiviral Res. 2005, 68, 75–83; [DOI] [PubMed] [Google Scholar]
- 32b. Zhao Y., Geng C. A., Sun C. L., Ma Y. B., Huang X. Y., Cao T. W., He K., Wang H., Zhang X. M., Chen J. J., Fitoterapia 2014, 95, 187–193; [DOI] [PubMed] [Google Scholar]
- 32c. Geng C. A., Yang T. H., Huang X. Y., Yang J., Ma Y. B., Li T. Z., Zhang X. M., Chen J. J., J. Ethnopharmacol. 2018, 224, 283–289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary