

Abstract
Intestinal health relies on the immunosuppressive activity of CD4+ regulatory T (Treg) cells1. Foxp3 expression defines this lineage and can be induced extrathymically by dietary or commensal-derived antigens in a process assisted by the Foxp3 enhancer CNS1 (conserved non-coding sequence 1)2–4. Microbial fermentation products including butyrate facilitate the generation of peripherally-induced Treg (pTreg) cells5–7, indicating that metabolites shape colonic immune cell composition. In addition to dietary components, bacteria modify host-derived molecules, generating a number of bioactive substances. This is epitomized by transformation of bile acids (BAs), which creates a complex pool of steroids8 presenting a range of physiological functions9. Here, we screened the major species of deconjugated BAs for their ability to potentiate pTreg cell differentiation. We found that the secondary BA 3β-hydroxydeoxycholic acid (isoDCA) increased Foxp3 induction by acting on dendritic cells (DCs) to diminish their immunostimulatory properties. Farnesoid X receptor ablation in DCs enhanced Treg cell generation and imposed a transcriptional profile similar to isoDCA, suggesting interaction between this BA/nuclear receptor pair. To investigate isoDCA in vivo, we took a synthetic biology approach and designed minimal microbial consortia containing engineered Bacteroides strains. IsoDCA-producing consortia increased colonic RORγt+ Treg cells in a CNS1-dependent manner, indicative of enhanced extrathymic differentiation.
BAs are cholesterol-derived molecules involved in essential physiological processes including nutrient absorption, glucose homeostasis and regulation of energy expenditure10. Upon feeding, endocrine signals stimulate the emptying of the gallbladder into the duodenum, where BAs aid in the emulsification of dietary fats10. Primary or liver-derived BAs in mammals are mostly conjugated with taurine or glycine and undergo pervasive deconjugation by microbial bile salt hydrolases in the small intestine. Although the majority of BAs are transported back into the liver via enterohepatic circulation, a small fraction of this pool escapes reabsorption in the ileum and is subjected to further bacterial transformation in the colon, giving rise to secondary BAs10. Knowing that the generation of colonic pTreg cells is affected by microbial metabolites, we screened the major species of deconjugated BAs found in mice and humans for their ability to enhance Foxp3 induction in vitro (Fig.1a–c). Two secondary BAs, ω-muricholic acid (ω-MCA) and 3β-hydroxydeoxycholic acid (isoDCA) potently increased the frequency of Foxp3+ cells among naïve CD4+ T cells co-cultured with dendritic cells (DCs) under suboptimal Treg cell-polarizing conditions (Fig.1d). The differentiation of pro-inflammatory T helper 17 (Th17) cells was not affected by the presence of either BA (Extended Data Fig. 1a), suggesting a specific effect on the generation of suppressive Treg cells. Because both isoDCA and ω-MCA are isomers of molecules with no Treg cell-promoting activity, we hypothesized that the spatial orientation of specific hydroxyl (-OH) groups is required for their effect. The formation of isoDCA from DCA requires oxidation of the 3α-OH group to an -oxo intermediate and its subsequent reduction into a 3β-OH group11. Despite remaining poorly characterized, the conversion of β-MCA into ω-MCA was also reported to generate an -oxo intermediate12,13. We observed that the 3-oxo-derivative of DCA failed to potentiate Treg cell induction to the same extent as isoDCA (Fig.1e). Similarly, oxidation of the 6α-OH group abolished Treg cell induction by ω-MCA (Extended Data Fig.1b), indicating that microbial epimerization of BAs gives rise to metabolites with unique immunomodulatory properties. Unlike ω-MCA, isoDCA is found at significant levels in the intestinal contents of healthy adult humans (≈50 μM)14 and its biosynthesis is well-characterized11. Therefore, we focused on understanding the biological activity of isoDCA.
Fig. 1|. Bacterial epimerization of bile acids (BAs) generates molecules with Treg cell-inducing activity.
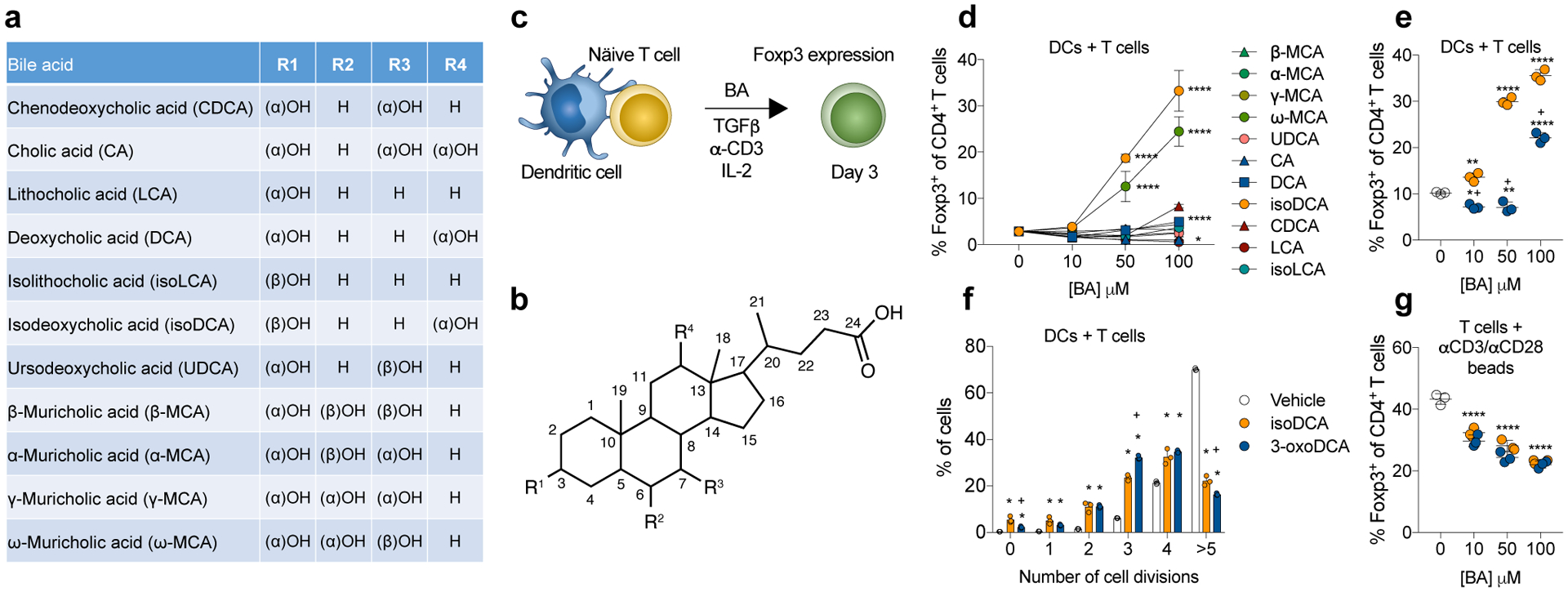
(a, b) Structure of BAs and summary of substitutions around the cholesterol backbone. (c) Screen setup: Naïve CD4+ T cells (5 × 104) were co-cultured with DCs (1 × 105) in suboptimal Treg-cell inducing conditions (1 ng/mL TGFβ, 1 μg/mL αCD3, 100 U/mL IL-2) and analyzed on day 3 by FACS. BAs listed in (a) were added at the indicated doses. (d) Frequencies of Foxp3+ CD4+ T cells after exposure to various concentrations of BA. (e) The 3β-OH of isoDCA is required for its Treg cell-inducing effects. Cells were co-cultured as described in (c) and incubated with 3-oxoDCA or isoDCA at the indicated concentrations. (f) Assessment of cell proliferation. Naïve CD4+ T cells were labeled with Cell Trace Violet (CTV) and cultured with DCs as described in (c) in the presence of isoDCA or 3-oxoDCA (100 μM). CTV dilution was assessed on day 3 by FACS. (g) Effect of BAs on Treg cell differentiation in the absence of DCs. Naïve CD4+ T cells were activated with αCD3/CD28 beads under suboptimal Treg cell-inducing conditions. IsoDCA and 3-oxoDCA were added at the indicated concentrations and cells were analyzed on day 3 by FACS. Showing mean ± SD of technical replicates (d, n=4; e-g, n=3). (*) p<0.05, (**) p<0.01, (****) p<0.0001 vs vehicle; (+) p<0.0001 vs isoDCA (paired concentration) by one-way (d,e,g) or two-way (f) ANOVA followed by a Dunnet (d) or Tukey (e-g) multiple comparison test. Data representative of at least three independent experiments.
The amphipathic nature of BAs makes these molecules natural detergents15 with potentially deleterious effects on cell viability and proliferation. Since conditions associated with reduced T cell proliferation are conducive to Foxp3 expression16,17, we assessed whether this mechanism can account for potentiation of BA-mediated Treg cell differentiation. IsoDCA significantly reduced T cell proliferation compared to vehicle (Fig.1f). We observed a similar effect in cells treated with 3-oxoDCA, which did not promote Treg generation in a dose-dependent manner, indicating that decreased proliferation alone cannot account for the increased frequency of Foxp3+ T cells. We next assessed whether DCs were required for isoDCA-mediated potentiation of Treg cell induction. Neither isoDCA nor 3-oxoDCA increased Treg cell frequencies when naïve T cells were activated by CD3/CD28 antibody-coated beads. Rather, both BAs caused a decrease in the percentage of Foxp3+ cells (Fig.1g), which was also accompanied by reduced proliferation (Extended Data Fig.1c). Consistent with these observations, isoDCA still enhanced Treg cell frequencies when naïve T cells lacking the Foxp3 enhancer CNS3 were co-cultured with DCs (data not shown). Together, these results suggested a requirement for antigen-presenting cells (APCs), i.e. DCs, in mediating the Treg cell-inducing effects of isoDCA.
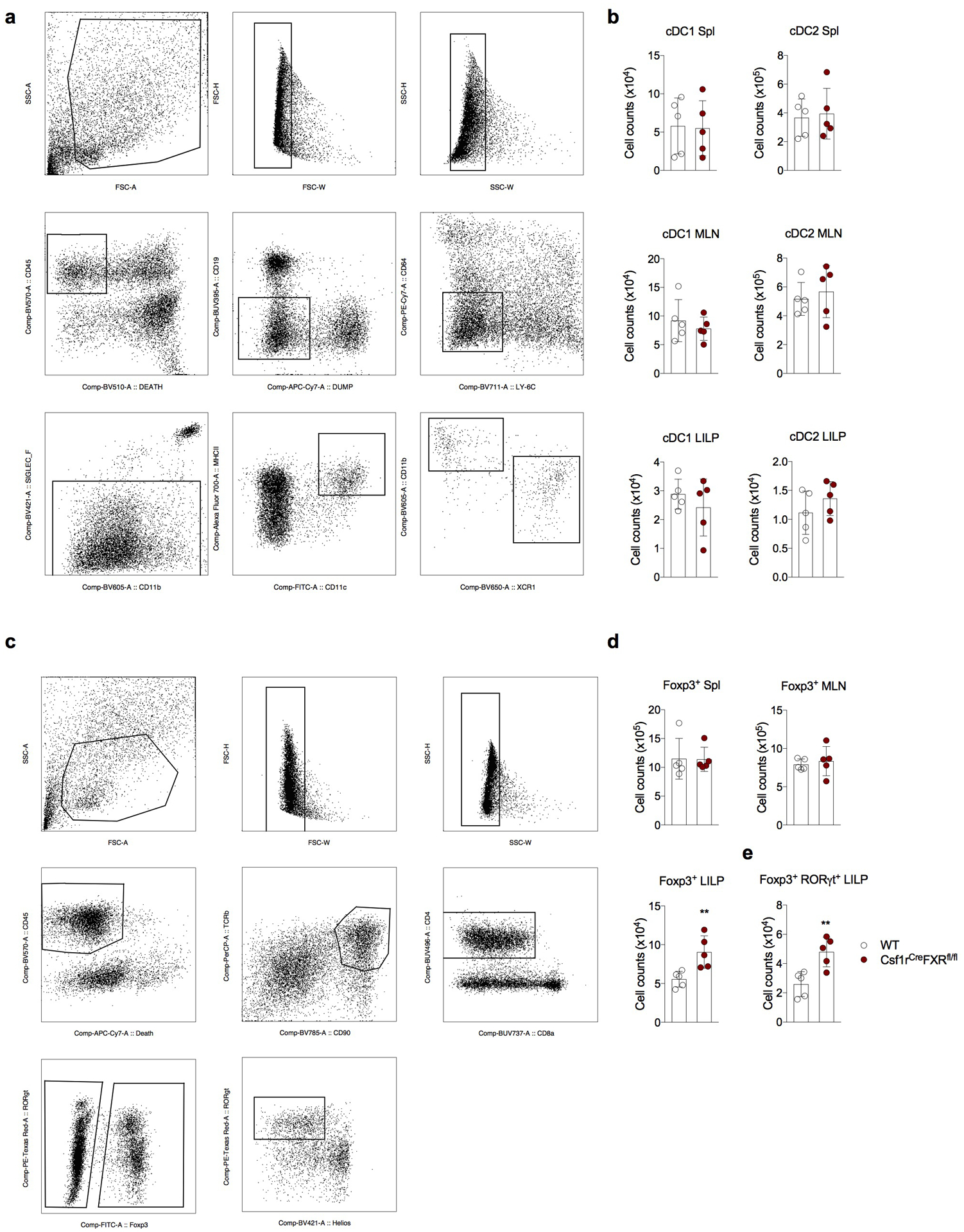
BAs affect mammalian physiology via interactions with multiple receptors, including the Farnesoid X Receptor (FXR)9. To assess a potential role for FXR in Treg cell induction by isoDCA, we tested the ability of FXR-deficient and sufficient naïve CD4+ T cells or DCs (isolated from CD4CreNr1h4fl/fl and Csf1rCreNr1h4fl/fl mice and their corresponding littermate controls, respectively) to respond to isoDCA by enhanced Foxp3 induction in an in vitro co-culture system. FXR deficiency in T cells did not change Foxp3 induction in response to isoDCA (Fig.2a, left panel). In contrast, DCs lacking FXR generated a higher frequency of Foxp3+ cells at baseline, and this increase could not be further enhanced by addition of isoDCA (Fig.2a, right panel). These results support our finding that isoDCA acts upon DCs to potentiate Treg cell induction and suggest that FXR is involved in this process. To rule out effects from potential differences in the splenic DC population used as APCs, we analyzed their composition in Csf1rCreNr1h4fl/fl mice and WT littermate controls. Although we failed to detect differences in the composition of the CD11c+ MHC class II+ cell population (mostly DCs) in the spleen and in other organs (Extended Data Fig.2b), we observed higher numbers of Foxp3+ cells in the large intestine lamina propria (LILP) of Csf1rCreNr1h4fl/fl mice (Extended Data Fig.2e), particularly within the Foxp3+RORγt+ subset (Extended Data Fig.2e). As pTreg cells arising in response to microbial antigens are predominantly RORγt+18,19, these data suggest that the absence of FXR in the myeloid compartment facilitates extrathymic generation of Treg cells in vivo.
Fig.2|. Potentiation of Treg cell generation by isoDCA acts requires FXR expression in DCs.
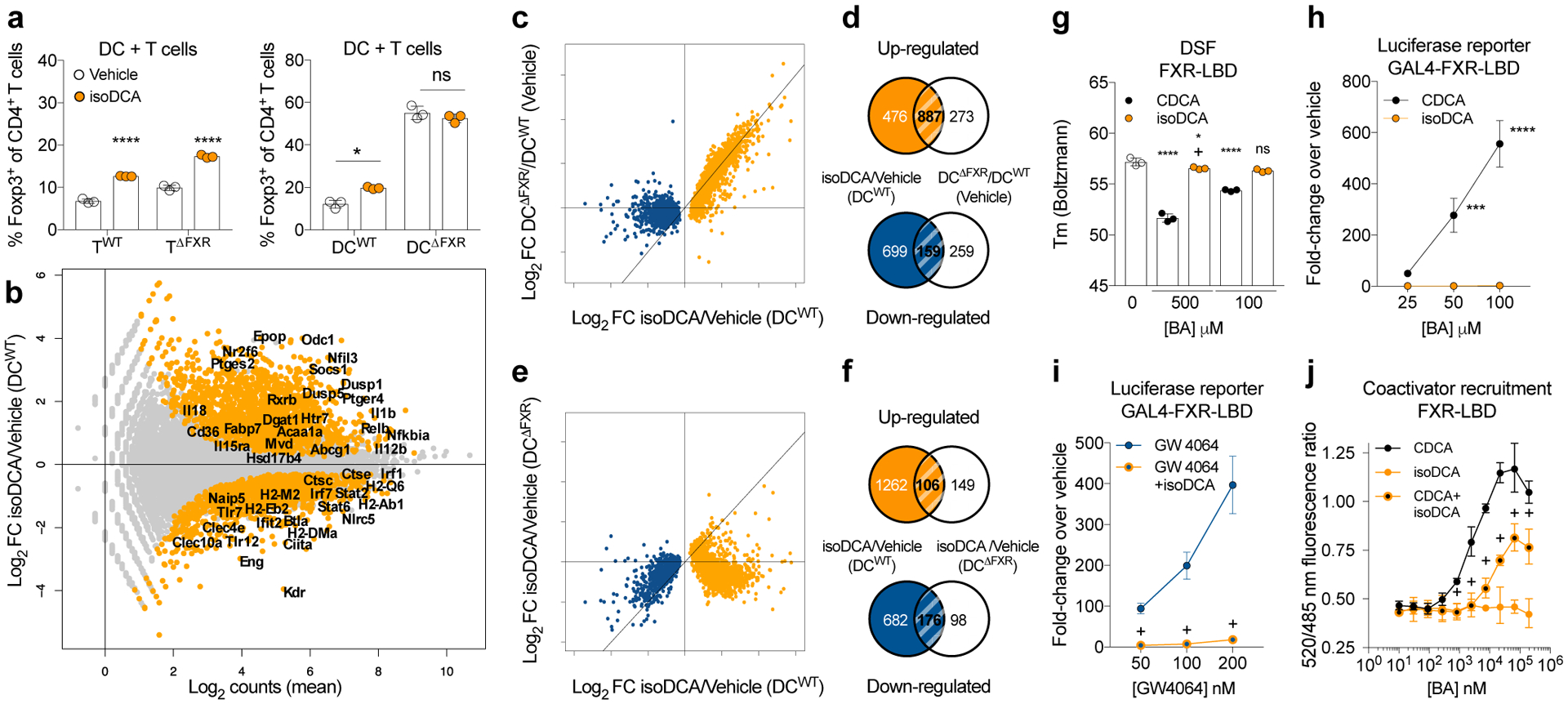
(a) Effects of FXR deficiency in T cells (TWT and TΔFXR) or DCs (DCWT and DCΔFXR) on Treg cell induction by isoDCA (50 μM). (b-f) RNA-seq analysis of FACS-purified DCs 24 hours after BA exposure. (b) Transcriptional profiling of WT DCs treated with isoDCA (50 μM). Differentially expressed genes in orange. (c) Fold-change (FC) vs. FC plot comparing the transcriptional changes induced by isoDCA treatment (x-axis) and FXR deficiency (y-axis). Genes reduced by isoDCA in blue; genes induced by isoDCA in orange. (d) Overlap between genes regulated by isoDCA treatment (orange) and FXR deficiency (blue). (e) FC vs. FC plot comparing the effects of isoDCA on WT (x-axis) and FXR-deficient (y-axis) DCs, color-coded as in (c). (f) Overlap between genes regulated by isoDCA in WT (orange) and FXR-deficient (blue) DCs. (g) Differential scanning fluorimetry (DSF) experiment with recombinant FXR-LBD and BAs at 1000-fold (500 μM) or 200-fold (100 μM) excess. Showing the Boltzmann melting temperature (Tm). (h-i) Luciferase reporter assays. Cells expressing a Gal4-FXR-LBD fusion protein were treated with vehicle, CDCA, GW4064 alone or combined with isoDCA (100 μM). (j) FRET-based co-activator recruitment assay. BAs were mixed with GST-tagged FXR-LBD (5 nM), FITC-SRC2–2 coactivator peptide (500 nM) and Tb α-GST antibody (5 nM). Showing ratio of fluorescence at 520/485 nm. Displaying mean ± SD of technical replicates (a, g, n=3; j, n=4), representative of three independent experiments. Data in (b-f, n=3) are from one experiment. Data in (h-i, n=3) shown as mean ± SD, pooled from three independent experiments. Statistical significance determined by a one- (g) or two-way (a, h-j) ANOVA followed by Tukey or Sidak’s test. (*) p<0.05, (***) p<0.001, (****) p<0.0001 vs vehicle; (+) p<0.05 vs paired concentration of agonist; (ns) not significant.
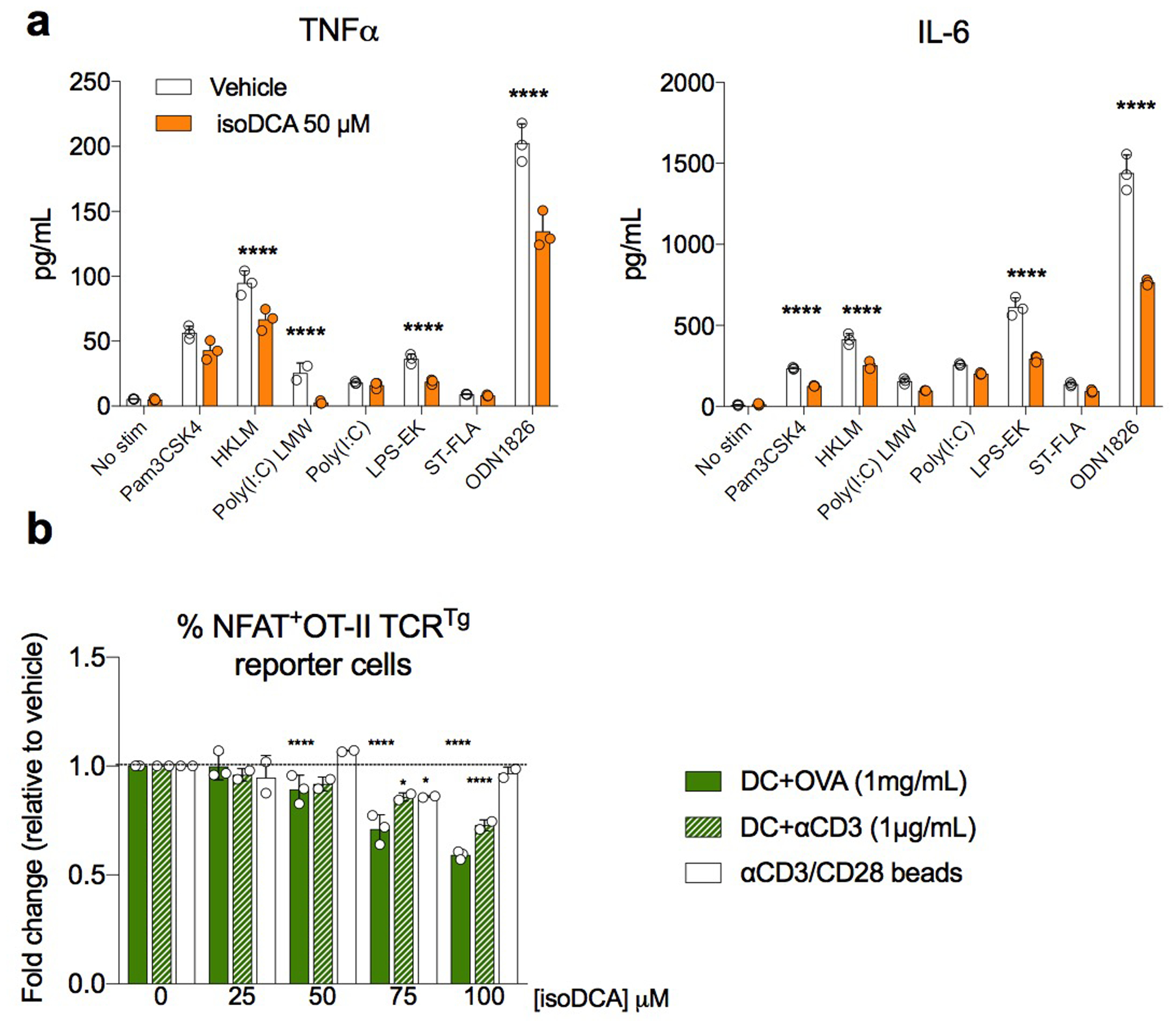
We next performed RNA-seq analysis to comprehensively assess the effects of isoDCA treatment and FXR ablation in antigen-presenting cells. DCs exposed to isoDCA showed reduced expression of several genes related to antigen processing and presentation, including Ciita, Ctse, H2ab, H2eb, and H2dma (Fig.2b). Genes involved in detecting and transducing pro-inflammatory cues such as Tlr7, Tlr12, Nlrc5, Stat2, Stat6, Irf1 and Irf7 were also decreased, as well as several genes downstream of interferon signaling. Among the genes induced by isoDCA treatment we identified negative regulators of NFκB, MAPK and cytokine receptor signaling, including Nfkbia, Dusp1, Dusp5 and Socs1. Since the transcriptional profile imposed by isoDCA suggested an overall anti-inflammatory state, we tested the effects of this bile acid on the ability of DCs to prime antigen specific T cells and to secrete cytokines in response to microbial cues. IsoDCA treatment decreased the production of the inflammatory cytokines TNFα and IL-6 upon TLR agonist stimulation (Extended Data Fig.3a). Furthermore, isoDCA reduced the activation of a NFAT-GFP reporter T cell line expressing the MHC II-restricted OT-II TCR by DCs pulsed for 4 hours with ovalbumin (OVA, Extended Data Fig.3b) indicating broad anti-inflammatory activities for this molecule.
Given the results implicating FXR in Treg cell induction by isoDCA, we compared the transcriptional changes elicited by FXR deficiency and bile acid exposure. Genes significantly modulated in response to isoDCA treatment were by and large differentially expressed between FXR-deficient and -sufficient cells (Fig.2c) indicating that these two perturbations lead to similar transcriptional changes in DCs. Notably, over 54% of genes induced by isoDCA treatment were also significantly upregulated in FXR-deficient DCs compared to WT (Fig.2c–d), implying that isoDCA may counteract FXR-mediated transcriptional repression. Although transcripts downregulated in response to isoDCA were generally present at lower levels in FXR-deficient DCs relative to WT (Fig.2c), their expression was further decreased when FXR-deficient DCs were treated with isoDCA (Fig.2e and f). Thus, while FXR may contribute to drive the expression of genes downregulated by isoDCA, these results suggest that additional, FXR-independent mechanisms regulate these targets. We next sought to characterize the molecular interaction between FXR and isoDCA. While the naturally occurring FXR agonist chenodeoxycholic acid (CDCA)20 induced a significant shift in the melting temperature of recombinant FXR ligand binding domain (LBD), isoDCA exerted a less pronounced change, only evident at a high ligand to protein ratio (Fig.2g). In line with this observation, CDCA elicited a robust signal in a FXR luciferase reporter assay, but isoDCA treatment had no effect (Fig.2h). These results suggested a distinct mode of interaction between FXR and isoDCA and raised the possibility of functional antagonism of this nuclear receptor. Indeed, we observed that isoDCA reduced the luciferase reporter signal in response to a synthetic FXR agonist, GW4064 (Fig.2i). Corroborating these data, we found that isoDCA limited CDCA-induced fluorescence on a FRET-based co-activator recruitment assay (Fig.2j). Altogether, these findings suggest that antagonizing the FXR-dependent transcriptional output of APCs may contribute to the pTreg cell-inducing effects of isoDCA.
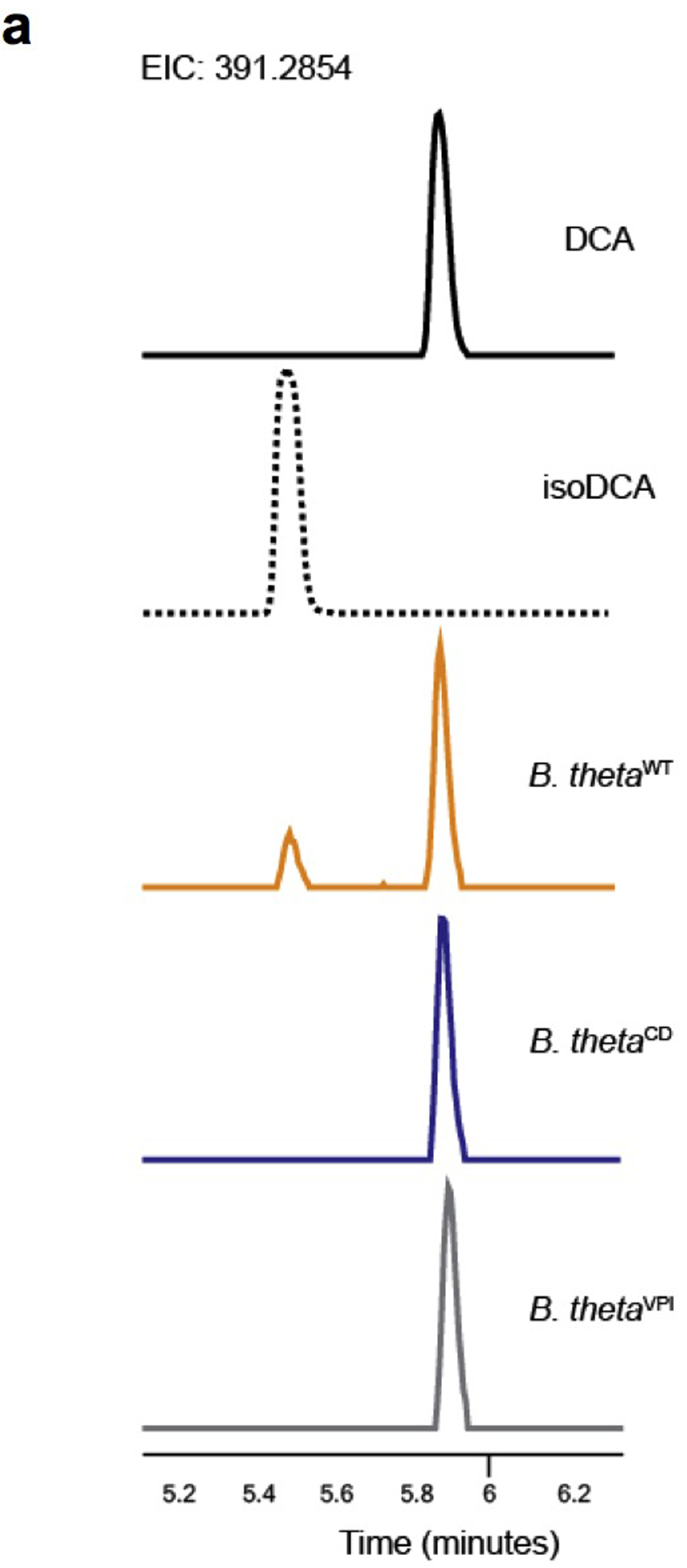
We then set out to explore the biological effects of isoDCA in vivo. Production of isoDCA from cholic acid (CA) involves chemical transformations performed by at least two different bacteria. The capacity for cleavage of the 7α-hydroxyl group from CA has been observed in Clostridium scindens8, while epimerization of the 3α-hydroxyl group of DCA was recently characterized in Ruminococcus gnavus11 (Fig.3a). To assess the effects of bacterial transformation of BAs in the colon, we reconstructed the isomerization pathway for isoDCA by inserting hydroxysteroid dehydrogenases from R. gnavus (Rumgna_02133 and Rumgna_00694) into Bacteroides thetaiotaomicron (B. theta), a genetically tractable commensal (Fig.3b). The active site of the enzyme encoded by Rumgna_00694 contains a tyrosine residue predicted to be conserved by homology modeling, which we changed to phenylalanine to create a catalytically dead (CD) mutant (Fig.3c). As expected, we observed robust production of isoDCA by the wild-type (WT) engineered B. theta strain assessed by thin layer chromatography, while conversion of DCA by the corresponding mutant strain was undetectable (Fig.3d and Extended Data Fig.4a). Because Bacteroides species lack 7α-dehydroxylation activity, we assembled an isoDCA-producing consortium by combining C. scindens with our engineered strains (Fig.4a–b). Both WT and CD consortia colonized mice to similar levels (Extended Data Fig.5a–b). All colonization conditions increased colonic Treg cells, including the RORγt+ cell subset (Fig.4c and Extended Data Fig.6a–b). Despite having similar frequencies of bulk Foxp3+ cells, recipients of the WT consortium showed a significant increase in the RORγt+ pTreg cell compartment compared to mice colonized with the control CD ensemble (Fig.4d). In agreement with the notion that Clostridium species and secondary BA are indigenous to the colon, we failed to detect differential levels of RORγt-expressing pTreg cells in the mesenteric lymph node or small intestine lamina propria (Extended Data Fig.6c–d). Although Foxp3−RORγt+ CD4+ T cells were robustly induced in the colon of mice conventionalized by fecal microbiota transplantation (FMT), this effector T cell population was comparable between recipients of the WT or CD consortia both at 10 days and 4 weeks after colonization (Extended Data Fig.6e–f), consistent with our in vitro finding that isoDCA had no substantial effect on the generation of Th17 cells (Extended Data Fig.1a). In contrast, the differences in colonic RORγt+ pTreg cell frequencies persisted at this later time point, albeit less pronounced (Extended Data Fig.6g).
Fig. 3|. Engineering an isoDCA producing strain of Bacteroides thetaiotaomicron (B. theta).
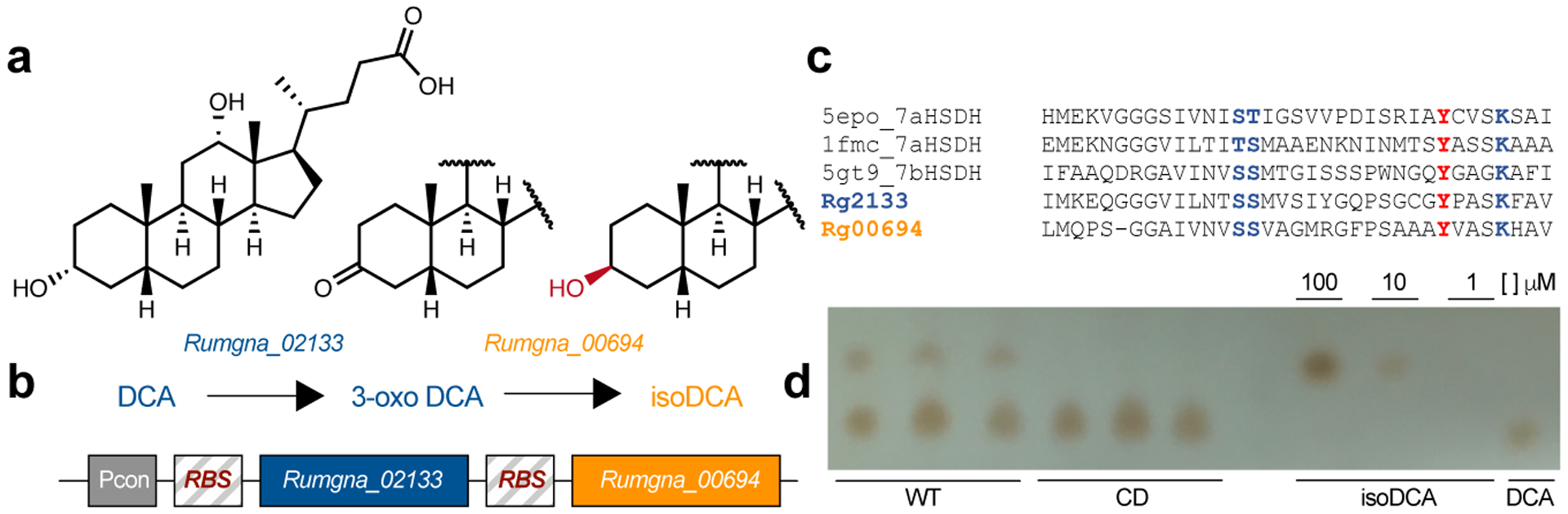
(a) Genes involved in isoDCA formation from DCA. Rumgna_02133 and Rumgna_00694, two hydroxysteroid dehydrogenases (HSDHs) present in Ruminococcus gnavus, were identified by Devlin et al. as key enzymes catalyzing epimerization of the 3-OH group of DCA by this bacterium. (b) Cloning strategy to reconstitute the pathway for isoDCA generation in B. theta: Constructs for Rumgna_02133 and Rumgna_00694 were codon-optimized, put under the control of a strong, constitutive promoter in B. theta and chromosomally integrated by conjugation. (c) Rationale for the design of catalytically-dead (CD) mutant of Rumgna_00694. A conserved tyrosine (red) in the putative active site of the enzyme was identified and mutated to phenylalanine (Y165F). (d) Characterization of the biochemical activity of WT and CD engineered strains of B. theta by thin layer chromatography (TLC). Bacteria were grown to exponential phase and transferred to media containing DCA. Following incubation for 24 hours, media was extracted in ethyl acetate and analyzed by TLC. DCA and isoDCA controls are shown on the 4 rightmost lanes. The WT strain (3 leftmost lanes) converts DCA into isoDCA, while the CD mutant (3 middle lanes) shows no activity. Data are representative of two experiments.
Fig. 4|. Defined bacterial consortia containing isoDCA-producing strains promote pTreg cell generation in vivo.
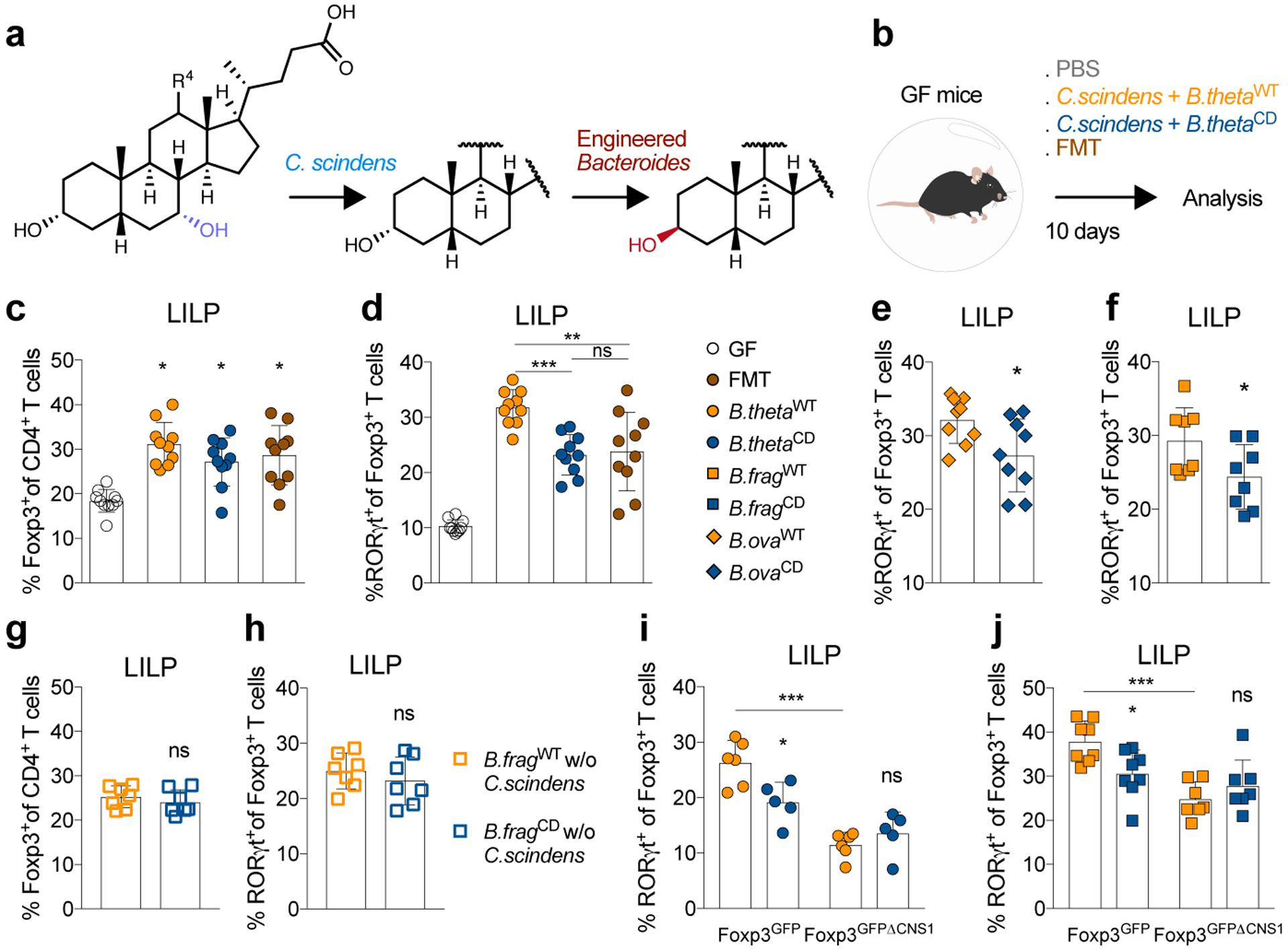
(a) Generation of isoDCA by a minimal microbial consortium. Enzymatic steps performed by C. scindens and the engineered Bacteroides sp. (B. thetaiotamicron, B. fragilis and B. ovatus). (b) Experimental setup. Germ-free (GF) mice were colonized with consortia containing C. scindens + B. thetaWT or C. scindens + B. thetaCD. Recipients of a fecal microbiota transplant (FMT) and non-colonized mice (PBS) served as references. Immune cell composition in the large intestine lamina propria (LILP) was analyzed on day 10 by FACS. Frequency of Foxp3+ (c) and RORγt+ (d) Treg cells. Frequency of RORγt+ Treg cells in GF mice colonized with C. scindens in combination with engineered B. ova (e) or B. frag (f). (g-h) B. fragWT or B. fragCD were administered to GF mice the absence of a 7-α dehydroxylating bacterium (w/o C. scindens). Showing frequencies of Foxp3+ (g) and RORγt+ (h) Treg cells. (i-j) GF Foxp3GFP and Foxp3GFPΔCNS1 littermate mice were gavaged with C. scindens + B. thetaWT (i) or C. scindens + B. fragWT (j) and compared to recipients of the respective CD consortia. Showing percentage of colonic RORγt+Foxp3+Treg cells. (c-j) Data combined from 2 independent experiments (except in i) and shown as mean ± SD. Statistical significance determined by two-tailed T-test (e, n=8; f, n=9; g-h, n=7), one-way (c-d, n=10) or two-way (i, n=5; j, n=7) ANOVA followed by Tukey’s multiple comparison’s test. (*) p<0.05; (**) p<0.01; (***) p<0.001; (ns) not significant.
To exclude potential effects of bacterial strain background and generalize these initial findings, we engineered two additional species of intestinal commensals to produce isoDCA. For this purpose, WT and CD strains of Bacteroides fragilis (B. frag) and Bacteroides ovatus (B. ova) were generated using the above strategy. All WT Bacteroides strains converted DCA into isoDCA, with B. frag and B. ova yielding similar levels to R. gnavus (R. gnavus ~B. fragWT ~B. ovaWT> B. thetaWT) as measured by liquid chromatography-mass spectrometry (Extended Data Fig.7a). No isoDCA was detected in the culture supernatants of CD strains. B. frag and B.ova WT strains also induced higher frequencies of colonic RORγt+ Treg cells compared to their respective CD counterparts (Fig. 4e–f and Extended Data Fig.7b), suggesting that this effect is not dependent on a particular strain background and unlikely to be caused by a potential “feedback” of isoDCA on the bacterium. Importantly, isoDCA levels in the cecal contents of animals colonized with a high-producing consortium (C. scindens + B. fragWT) did not surpass that of FMT recipient mice Extended Data Fig.7c), indicating that our reconstruction of this enzymatic pathway did not lead to supraphysiological BA levels. Production of short-chain fatty acids (SCFAs) was comparable between consortia (Extended Data Fig.8) indicating that BA transformation did not broadly impact bacterial metabolism and that isoDCA-producing bacteria were able to increase pTreg cells in the presence of other tolerogenic metabolites. Since the HSDH introduced into Bacteroides could potentially modify substrates other than DCA, we tested the effects of our engineered bacteria in the absence of the 7α-dehydroxylating commensal, i.e. without C. scindens. Mono-colonization of GF mice with B. fragWT or B. fragCD resulted in similar frequencies of colonic RORγt+ pTreg cells (Fig. 4g–h), demonstrating that the biological activity of our isoDCA-producing consortia depends on the presence of a DCA-generating bacterium. To confirm that our engineered strains promote bona fide pTreg cell generation in vivo, we colonized GF CNS1-sufficient (Foxp3GFP) and -deficient (Foxp3GFPΔCNS1) mice with WT or CD consortia. In comparison to CD groups, colonization with WT consortia led to significantly higher frequencies of Foxp3+RORγt+ pTreg cells in CNS1-sufficient animals (Fig. 4i–j). CNS1-deficient hosts with genetically impeded pTreg cell generation showed similarly low frequencies of Foxp3+RORγt+ CD4+ T cells when colonized with WT or CD consortia (Fig. 4i–j). These experiments demonstrate that colonization with isoDCA-producing microbial consortia promotes de novo generation of colonic pTreg cells.
Previously, pTreg cells were shown to dampen immune responses during colonization and to support the metabolic function of the gut microbiota21. The establishment of immunological tolerance to commensals and potentially other types of host-microbe interactions likely evolved around conserved features of microbial communities, such as their metabolic output. Supporting this notion, we found that, in addition to bacterial fermentation products5, the secondary BA isoDCA is also a potent inducer of pTreg cells. Using engineered Bacteroides strains as part of rationally designed, minimal microbial consortia, we showed that isoDCA-producing bacteria promote pTreg cell generation in vivo in a CNS1-dependent manner. IsoDCA limited FXR activity in DCs and conferred upon them an anti-inflammatory phenotype. Although our data support the involvement of myeloid cell-intrinsic FXR activity in pTreg cell induction, the relative contribution of this and other BA-sensing receptors in mediating the effects of isoDCA on the mucosal immune milieu remains to be investigated. In conclusion, our study suggests that microbial metabolism of endogenous steroids contributes to immunological balance in the colon.
Methods
Dendritic cell isolation
B16 melanoma cells secreting Flt3 ligand (kindly provided by Dr. Glenn Dranoff) were injected subcutaneously into the left flank of mice to expand splenic dendritic cells in vivo. Ten to twenty days after tumor injection, spleens were harvested and dissociated in RPMI 1640 medium containing 1.67 U/mL liberase TL (Roche) and 50 μg/mL DNAse I (Roche) for 20 min at 37 °C with vigorous shaking (250 rpm). Digested spleens were passed through a 100 μm strainer and washed in complete RPMI [RPMI 1640 with 10mM HEPES buffer (ThermoFisher), 1% penicillin/streptomycin (ThermoFisher), 1% L-glutamine (ThermoFisher) and 10% Fetal Bovine Serum (FBS, ThermoFisher). Single cell suspension was enriched for DCs using MACS CD11c Microbeads Ultrapure isolation kit (Miltenyi Biotec) according to manufacturer’s instructions. Purity of DC enrichment was assessed by FACS analysis (typical purity >92% CD11c+ MHCIIhi cells).
In vitro assays
T cell polarization
Naïve (CD44− CD62L+GFP−) CD4+T cells from Foxp3GFP mice were FACS-purified from spleen and peripheral (pooled inguinal, brachial, axial and submandibular) lymph nodes after a CD4-enrichment step (Dynabeads™11461D, Invitrogen™) performed as per manufacturer’s instructions. DC (1×105) plus naïve CD4+T cell (5×104) co-cultures were set up in the presence of 1 μg/mL of monoclonal anti-mouse CD3ε (InvivoMAb, BioXcel). Cholic acid (C19000–000), chenodeoxycholic acid (C0985–000), lithocholic acid (C1420–000), isolithocholic acid (C10475–00), deoxycholic acid (C1070–000), isodeoxycholic acid (C1165–000), 3-oxodeoxycholic acid (C1725–000), ursodeoxycholic (C1020–000), α-muricholic acid (C1890–00), β-muricholic acid (C1895–000), γ-muricholic acid (C1850–000) and ω-muricholic acid (C1888–000) were all purchased from Steraloids Inc. (Newport, RI). 6-oxo-muricholic acid (5β-cholanic acid-3α,7β-diol-6-one) was produced by the Organic Chemistry Synthesis core at MSKCC. 100 mM stocks of BAs in DMSO were stored at −80°C. For T cell activation in the absence of DCs, naïve CD4+ T cells (5×104) were incubated with Mouse T activator CD3/CD28 Dynabeads™ (Gibco) at 1:1 bead to cell ratio. For assessment of cell proliferation, naïve CD4+T cells were labeled with 5 μM Cell Trace Violet™ (Invitrogen) according to manufacturer’s instructions. Suboptimal Treg cell induction conditions consisted of 1 ng/mL of recombinant human TGFβ 1 (R&D) and 100 U/mL recombinant human IL-2 (Biological Resources Branch, NCI). For Th17 differentiation, cells were incubated with 2 ng/mL of recombinant human TGFβ 1 (R&D) and 20 ng/mL recombinant murine IL-6 (PeproTech). All in vitro polarization assays were carried in complete RPMI with 10% FBS (final volume of 200 μL) in flat-bottom 96 well plates (USA Scientific). Border wells were filled with media only (no cells) to minimize evaporation during incubation at 37°C/5% CO2. On D3 of culture, cells were transferred into V-bottom plates (Fisher), pelleted by centrifugation and incubated with antibody staining mix containing Ghost Dye™ Red 780 viability dye diluted in PBS for 15 minutes at 4°C. For cytokine production analyses, cells were incubated for 3.5 hours at 37°C/5% CO2 in re-stimulation media [complete RPMI 1640 with 5% FBS, 50 ng/mL PMA (Sigma), 500 ng/mL ionomycin (Sigma), 1 μg/mL brefeldin A (Sigma) and 2 μM monensin (Sigma)]. Extracellular antigens were stained for 15 minutes at 4°C with an antibody staining mix containing Ghost Dye™ Red 780 viability dye diluted in PBS. Cells were fixed and permeabilized with BD Cytofix/Cytoperm™ for 20 minutes at 4°C. Antibodies against intracellular antigens were diluted in 1x BD Perm/Wash™ buffer and cells were stained for 30 minutes at 4°C. Cytometry data were acquired on a LSRII (Becton Dickinson, NJ). Foxp3 induction was assessed by expression of GFP reporter protein. 123count eBeads™ (Invitrogen) were added at 5,000 beads/sample to quantify absolute cell numbers.
TLR agonist activation
DCs (1×105 per well, flat bottom 96-well plates) were pretreated with isoDCA or vehicle in complete RPMI 10% FBS for 5 hours before stimulation with various TLR ligands [Pam3CSK4 (0.5 μg/mL); HKLM (107 cells/mL); Poly(I:C) and Poly(I:C) LMW (5 μg/mL); LPS-EK (5 μg/mL); ST-FLA (1 μg/mL) and ODN1826 (2.5 μM)] from the Mouse TLR 1–9 agonist kit (Invivogen tlrl-kit1mw) for 18 hours at 37°C/5% CO2. TNFα and IL-6 levels were quantified in cell-free supernatants (~ 150 μL of 200 μL cell culture after centrifugation) by ELISA (eBioscience™, 5017331 and 5017218) according to the manufacturer’s instructions.
Antigen processing and presentation assays
DCs (1×104) were pulsed with ovalbumin (Sigma A5503, 1 mg/mL) or anti-mouse CD3ε (1 μg/mL) in the presence or absence of isoDCA for 1 hour at 37°C/5% CO2 in serum-free complete RPMI. Then, an equal volume of 10% FBS RPMI was added for a final serum concentration of 5%. After 5 hours, 1×104 reporter T cells (TCRα/β-null, BW5147 mouse thymoma cells22 with NFAT-controlled GFP expression23) carrying the OTII TCR were added, along with ovalbumin or anti-CD3 and isoDCA (to keep concentrations constant) in complete RPMI with 10% FBS. The frequency of GFP+ cells was determined 24 hours later by FACS.
RNA-sequencing of dendritic cells
In vitro DC-T cell co-culture assays in the presence of BAs were carried out as described above. Following 24 hours of incubation at 37°C/5% CO2, cells were transferred into V-bottom plates, pelleted by centrifugation and incubated with antibody staining mix containing Ghost Dye™ Red 780 viability dye diluted in PBS for 15 minutes at 4°C. Approximately 3×104 DCs were double-sorted on a FACSAria II (Becton Dickinson, NJ) based on viability and CD11c/MHC II expression. Purified cells were resuspended in Trizol™ and submitted for RNA-sequencing at the Integrated Genomics Core (iGO) at MSKCC. Samples underwent SMARTer amplification (Takara) and were sequenced on a Hiseq platform (Illumina) at a depth of 20–30 million paired-end 50bp reads per sample. RNA-sequencing reads were aligned to the reference mouse genome (Gencode m19) using STAR RNA-Seq aligner24, and read counts were obtained with HTSeq-count25. DESeq226 R package was used to perform differential gene expression analyses between groups. A cutoff of 0.05 was set on obtained p-values (adjusted using the Benjamini-Hochberg correction for multiple comparisons) to define statistically significant genes for each comparison.
Luciferase Assays
Reporter cells expressing GAL4-LBD fusions for human FXR (Indigo Biosciences, IB006001) were incubated for 22–24 hours in the presence of CDCA, the synthetic agonist GW4064 (provided with kit) and/or isoDCA. Cells were processed according to manufacturer’s instructions, including the recommended step to determine viability using the Live Cell Multiplex assay (LCM, Indigo Biosciences). Luminescence was read in a GloMax 96 Luminometer (Promega).
Differential scanning fluorimetry assay
IsoDCA and CDCA were tested for their ability to alter the melting temperature (Tm) of recombinant FXR-LBD (Invitrogen) using a modified version of the protocol established by Schmidt et al.27 Briefly, BAs and FXR-LBD (final concentration: 500 nM) were diluted in assay buffer [10 mM TRIS (pH 8.3), 0.5 mM EDTA, 100 mM NaCl and 5 mM DTT, added fresh before assay from a 100x frozen stock] and combined into transparent 384 well plates (Applied Biosystems). SYPRO Orange dye (Protein Thermal Shift Dye, Applied Biosystems) was added at a final concentration of 1: 2,000 in a reaction volume of 20 μL. Temperature-dependent changes in fluorescence were detected in a QuantStudio 6 Flex Real-time qPCR System (Applied Biosystems). The equipment was programmed with the following thermal profile: Step 1, Temp: 25 °C, Time: 2 minutes/ Step 2, Temp: 99 °C, Time: 2 minutes /Ramp mode: Continuous /Ramp rate: Step 1: 1.6°C /s, Step 2: 0.05°C /s. Data analysis was performed using the PTS software (Applied Biosystems).
FRET-based FXR co-activator recruitment assay
IsoDCA and CDCA were tested for their ability to activate FXR in a cell-free fluorescence resonance electron transfer (FRET) assay using LanthaScreen™ technology according to the manufacturer’s protocol (Thermo Fisher™, PV4833). Briefly, after combining diluted BAs with GST-tagged FXR-LBD, terbium anti-GST antibody and fluorescently-labeled SRC2 in white, flat-bottom 384 well plates (Greiner Bio-one), the reaction was incubated at RT in the dark under gentle shaking (60 rpm) for 1 hour before reading. Fluorescence detection was done in a Tecan Infinite® M100 Pro Microplate reader (Tecan Group, Switzerland) set up according to the LanthaScreen™ Terbium Assay Setup guide available at www.lifetechnologies.com/instrumentsetup. For the first Fluorescence Reading, settings were as follow: Wavelength > Excitation: 332 nm; Bandwith: 20.0 nm/ Emission: 485 nm; Bandwith: 20.0 nm/ Flashes > Mode 2 [100 Hz (20)]; Settle time: 0 ms/ Mode > Top/ Gain> Optimal/ Z-position > Calculated from well: (select well with appropriate substrate) / Integration > Lag time: 100 μs; Integration time: 200 μs. After a second Fluorescence Reading was added to the existing protocol, settings were adjusted to the same as described above, except Wavelength > Excitation: 332 nm; Bandwith: 20.0 nm/ Emission: 515 nm; Bandwith: 20.0 nm. Results were expressed as ratio of fluorescence at 520 nm/485 nm.
16s metagenomic sequencing of intestinal microbiota
DNA extraction from cecal contents was carried with Maxwell® RSC PureFood GMO and Authentication Kit (Promega, AS1600). Pre-weighted fecal material was deposited into a PowerBead glass 0.1 mm tube (Qiagen, 13118–50) and 1 mL of CTAB buffer + 20 μl of RNAse A solution were added. Samples were homogenized in buffer by gentle vortexing for 10 seconds and then incubated for 5 minutes in a ThermoMixer F2.0 (Eppendorf) shaking at 1,500 rpm. Homogenized samples were then vortexed horizontally at high-speed for 10 minutes. Samples were centrifuged at 12,700 rpm in an Eppendorf centrifuge for 10 minutes and extraction proceeded in an automated platform. Tubes were transferred to a MaxPrep Liquid Handler tube rack (Promega) and the Maxwell® RSC48 instrument (Promega, AS8500) was loaded with proteinase K tubes, lysis buffer, elution buffer, pipetting tips, 96-sample deep-well plate and Maxwell® RSC 48 sheath tips. The instrument was programed to use 300 μl of sample and transfer all sample lysate into the Maxwell® RSC 48 extraction cartridge for DNA extraction. Upon completion, the extraction cartridge was loaded into Maxwell® RSC 48 for DNA extraction and elution. DNA was eluted in 100 μl and transferred into 96-well plates. Quant-iT dsDNA High Sensitivity Assay (Thermo Fisher, Q33120) was used for DNA quantification. 16s libraries were generated according to the Earth Microbiome Project available at (http://press.igsb.anl.gov/earthmicrobiome/protocols-and-standards/16s/). Briefly, the V4 region was amplified using Hot Start PCR mix (ThermoFisher, 13000014). Primers were modified from Caporaso et al.28 (barcodes were moved to the 515F primer) and are listed below:
515F forward primer, barcoded (xrefxref) AATGATACGGCGACCACCGAGATCTACACGCT xrefxref TATGGTAATT GT GTGYCAGCMGCCGCGGTAA
806 R Reverse
CAAGCAGAAGACGGCATACGAGAT AGTCAGCCAG CC
Cycling conditions for 96 well plate thermocyclers:
94°C – 3 min
(94°C – 45s; 50°C – 60s; 72°C –90s) x35
72°C – 10 min
4°C – hold
PCR products were purified with MoBio UltraClean PCR Clean-Up Kit (12500) according to manufacturer’s instructions. Library concentrations were determined with Quant-iT dsDNA High Sensitivity Assay. Library quality and size verification were performed using PerkinElmer LabChip GXII instrument with DNA 1K Reagent Kit (CLS760673). Libraries were normalized and pooled at 2 nM before sequencing on a MiSeq instrument (Illumina) at a loading concentration of 5.5 pM with 10% PhiX, paired-end 250 using MiSeq Reagent Kit v2, 500 cycles (MS-102–2003). Custom sequencing primers were as follow:
Read 1: TATGGTAATT GT GTGYCAGCMGCCGCGGTAA
Index: AATGATACGGCGACCACCGAGATCTACACGCT
Read 2: AGTCAGCCAG CC GGACTACNVGGGTWTCTAAT
Note: The 5’ adapter sequence/index sequencing primer has an extra GCT at its 3’ end compared to Illumina’s usual index primer sequences. These bases were added to the 3’ end of the Illumina 5’ adapter sequence to increase the melting temperature for Read 1 during sequencing.
For analysis, demultiplexed raw reads were processed to generate an operational taxonomic unit (OTU) table using USEARCH version 11.0.66729. Specifically, forward and reverse reads were merged using a maximum of 5 mismatches in the overlap region, a minimum sequence identity in the overlap region of 90 percent, a minimum overlap length of 16 base pairs, and a minimum merged sequence length of 300 base pairs. PhiX contamination was then removed, followed by quality filtering based on FASTQ quality scores, with a maximum expected error number of 1.0. OTU clustering was performed using usearch -cluster_otus with default settings. Merged (pre-filter) reads were mapped to the OTU sequences to generate the OTU table. Taxonomic classification of OTU representative sequences was performed using usearch-sintax, an implementation of the SINTAX algorithm30 using version 16 of the Ribosomal Database Project (RDP) Training Set31.
Engineering recombinant Bacteroides strains
The 3α-HSDH Rg_2133 and the 3β-HSDH Rg_00694 genes were synthesized (GeneWiz) after codon-optimization for expression in B. theta. Golden Gate assembly kit (NEB) was used to assemble the following fragments and insert them into pNBU232 in 5’ to 3’ order: ppWW3806, ribosome binding site #8 (RBS), Rg_2133 open reading frame, RBS #8, Rg_00694, and pWW3810. The assembled construct was transformed into Stellar competent cells (Takara) and positive clones were selected and verified by Sanger sequencing. The plasmid was purified and re-transformed into E. coli S17–1 cells for conjugation into B. theta, B. frag and B. ova. Mid-log Bacteroides cultures and plasmid-containing E. coli were mixed 10:1, centrifuged and allowed to sit for 20 minutes before resuspending the pellet in PBS and spreading on Brain Heart Infusion (BHI) Agar with 10% Horse blood containing 25 μg/mL erythromycin and 200 μg/mL gentamycin, followed by incubation in an anaerobic chamber (Coy Labs) for 48 hours at 37°C. Individual colonies were screened by Sanger sequencing. For more information on bacterial strains and plasmids, please see Supplementary Information Table 1.
Thin Layer Chromatography
Strains were grown for 24 or 72 hours in YCFAC liquid medium (Anaerobe Systems) containing 100 μM DCA. 2g NaCl2 was added to 7 mL culture supernatant, followed by 700 μL 6M HCl. The supernatant was extracted with 5 mL ethyl acetate followed by dehydration in 2 g MgSO4, and a second extraction with ethyl acetate. The organic extract was filtered through 40 μm nylon filters (Falcon), then dried by vacuum centrifugation in glass tubes. The pellet was resuspended in methanol, tubes were washed in an equal volume of acetone and the solvent was again removed by vacuum centrifugation. The final dried pellet was resuspended in 100 μL acetone and was spotted onto glass-backed silica plates (Sigma), which were developed in 70:20:2 benzene/1,4-dioxane/acetic acid. Plates were stained in 10% w/v CuSO4 8% H3PO4 and dried over a hot plate.
Mass Spectrometry
IsoDCA quantification
Bacterial broth was mixed with an equal volume of methanol and centrifuged at 21,000 × g for 20 minutes. For in vivo quantification, about 10–30 mg of the dried fecal pellet was extracted in 150 μL of 50%MeOH in water and vortexed for 15 minutes. The mixture was then spun down at 21,000 × g for 15 minutes. The supernatant of bacterial broth or extracted fecal pellet was analyzed using an Agilent 1290 LC system coupled to an Agilent 6530 QTOF with a 2.1 μm, 2.1 × 50 mm ZORBAX Eclipse Plus C18 column (Agilent). Water with 0.05% formic acid (A) and acetone with 0.05% formic acid (B) was used as the mobile phase at a flow rate of 0.40 mL/min over a 11 min gradient: 0–1 min, 0% B; 1–3 min, 0–40% B; 3–10 min, 40–100% B; 10–11 min, 100–0% B. Reagents were MS grade purchased from Fisher Scientific. All data were collected in negative ion mode.
Short-Chain Fatty Acids (SCFAs) quantification
Cecal samples (~120 mg) were weighed into 2 mL microtubes containing 2.8 mm ceramic beads (Omni International) and resuspended to a final concentration of 100 mg/mL using 80:20 methanol:water containing acetate-d3, propionate-d5, butyrate-d7 and valerate-d9 as internal standards (Cambridge Isotope Laboratories). Homogenization was using a Bead Ruptor (Omni International) at 5.4 m/s for 3 minutes at 4°C. Samples were centrifuged for 20 minutes at 20,000 × g at 4°C and 100 μL of cecal extract was added to 100 μL of 100 mM borate buffer (pH 10). Subsequently, 400 μL of 100 mM pentafluorobenzyl bromide (Thermo Scientific) diluted in acetonitrile (Fisher) and 400 μL of cyclohexane (Acros Organics) were added and reaction vials were sealed. Samples were heated to 65°C for 1 hour with agitation and then cooled to room temperature and centrifuged to promote phase separation. 100 μL of the cyclohexane (upper) phase was transferred to a new autosampler vial and analyzed at 1:10 and 1:100 dilutions (made using cyclohexane). A calibration curve and quality control (QC) samples were prepared in borate buffer covering the range 0.1 – 50 mM. Analysis by GC-MS was using an Agilent 7890A GC and Agilent 5975C MS detector operating in negative chemical ionization (CI) mode. Methane was used as the CI reagent gas at 2 mL/min and a 1 μL splitless injection was made onto a HP-5MS column (30 m × 0.25 mm, 0.25 μm; Agilent Technologies). For SCFA quantification, the raw peak areas of acetate (m/z 59) and propionate (m/z 73) were normalized to acetate-d3 (m/z 62) and propionate-d5 (m/z 78) internal standards respectively; the C4 compounds butyrate and isobutyrate (m/z 87) were normalized to butyrate-d7 (m/z 94) and the C5 compounds 2-methylbutyrate, valerate and isovalerate (m/z 101) were normalized to valerate-d9 (m/z 110). Data analysis was performed with Agilent MassHunter Quantitative Analysis software (version 10.1, Agilent Technologies).
Colonization with engineered consortia
Frozen stocks of Clostridium scindens (ATCC 35704) or engineered Bacteroides strains were streaked on Columbia agar plates (BD) and grown at 37°C inside an anaerobic chamber. Bacteria were scraped from agar plates with bacteriological loops into sterile anaerobic PBS. Consortia were assembled and transported in airtight tubes to the animal facility and administered to experimental animals by oral gavage. Mice were housed in flexible PVC isolators (Park Bioservices) or Sentry Sealed positive pressure cages (SPP, Allentown Inc.) for the duration of the experiments. For experiments in SPP cages, animals were manipulated with sterile gloves using aseptic techniques inside biosafety cabinets.
Isolation of cells from the intestinal lamina propria
Approximately 12 cm of the distal small intestine and the combined cecum and colon were processed as the small intestine (SI) and large intestine (LI), respectively. After removal of adherent adipose tissue and resection of Peyer’s patches, intestines were opened longitudinally and shaken vigorously in PBS to release contents. Tissues were incubated in 25 mL IEL solution [1x PBS with 2% FBS (ThermoFisher), 10 mM HEPES buffer (ThermoFisher), 1% penicillin/streptomycin (ThermoFisher), 1% L-glutamine (ThermoFisher), plus 1 mM EDTA (Sigma) and 1 mM DTT (Sigma) added immediately before use] for 15 minutes at 37°C with vigorous shaking (250 rpm). Intestines were removed from IEL suspension, rinsed in PBS and transferred into 50 mL tubes containing 3x ¼inch ceramic beads (MP Biomedicals) and 25 mL collagenase solution [1x RPMI 1640 with 2% FBS (ThermoFisher), 10 mM HEPES buffer (ThermoFisher), 1% penicillin/streptomycin (ThermoFisher), 1% L-glutamine (ThermoFisher), 1 mg/mL collagenase A (Sigma) and 1 U/mL DNase I (Sigma)]. Following incubation for 30 minutes at 37°C with vigorous shaking (250 rpm), digested lamina propria (LP) samples were passed through a 100 μm strainer and centrifuged to remove collagenase solution. The LP fractions were washed by centrifugation (5 min at 450 × g) in 44% Percoll™ (ThermoFisher) in PBS to remove debris and excess epithelial cell contamination before ex-vivo re-stimulation for analysis of cytokine production. Foxp3 expression was assessed by detection of the fluorescent GFP reporter protein or intracellular staining using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience™). 123count eBeads™ (Invitrogen) were added at 5,000 beads/sample to determine cell numbers. Cytometry data were acquired on a LSRII (Becton Dickinson, NJ).
Mice
GF C57Bl/6 mice were purchased from Taconic and maintained in semi-rigid isolators (Park Bioservices) at Boehringer Ingelheim (Ridgefield, CT). Female mice were used in all experiments to facilitate distribution of animals into experimental groups. GF Foxp3GFPΔCNS1 and Foxp3GFP mice were re-derived as described previously21 and maintained in flexible isolators (Class Biologically Clean; USA) at Weill Cornell Medicine. Male and female littermate mice were used in all experiments. Animals were fed with autoclaved 5KA1 chow. GF status was routinely checked by aerobic and anaerobic cultures of fecal samples for bacteria and fungi and by PCR of fecal DNA samples for bacterial 16S and fungal/yeast 18S genes. GF were at least 8 weeks old at the initiation of experiments.
SPF mice were housed at the Research Animal Resource Center for Memorial Sloan Kettering Cancer Center and Weill Cornell Medicine with 12-hour light/dark cycles under ambient conditions and ad libitum access to food and water. Experimental mice were maintained in a standard rodent diet (5053, LabDiet). Dr. Frank Gonzalez (NIH, USA) kindly provided the Nr1h4fl mouse strain. Csf1rCre mice were provided by Dr. Frederic Geissman at MSKCC, USA. CD4Cre mice [Tg(Cd4-cre)1Cwi] were purchased from Jax laboratories and maintained in house. Experimental littermate animals were generated by mating mice homozygous for the Nr1h4fl allele, with one of the breeders (male of female) carrying one copy of the Cre-driver gene. Cells from male and female CD4CreNr1h4fl or Csf1rCreNr1h4fl were used for in vitro experiments. Male Csf1rCreNr1h4fl mice were analyzed at 6–8 weeks of age.
All studies were under protocol 08-10-023 and approved by the Sloan Kettering Institute Institutional Animal Care and Use Committee. GF mice housed at Boehringer Ingelheim were maintained under a protocol approved by the Institutional Animal Care and Use Committee of Boehringer Ingelheim Pharmaceuticals, Inc. All animals used in this study had no previous history of experimentation and were naïve at the time of analysis.
Statistical analyses
Statistical tests were performed with GraphPad Prism™ version 7.0.
Extended Data
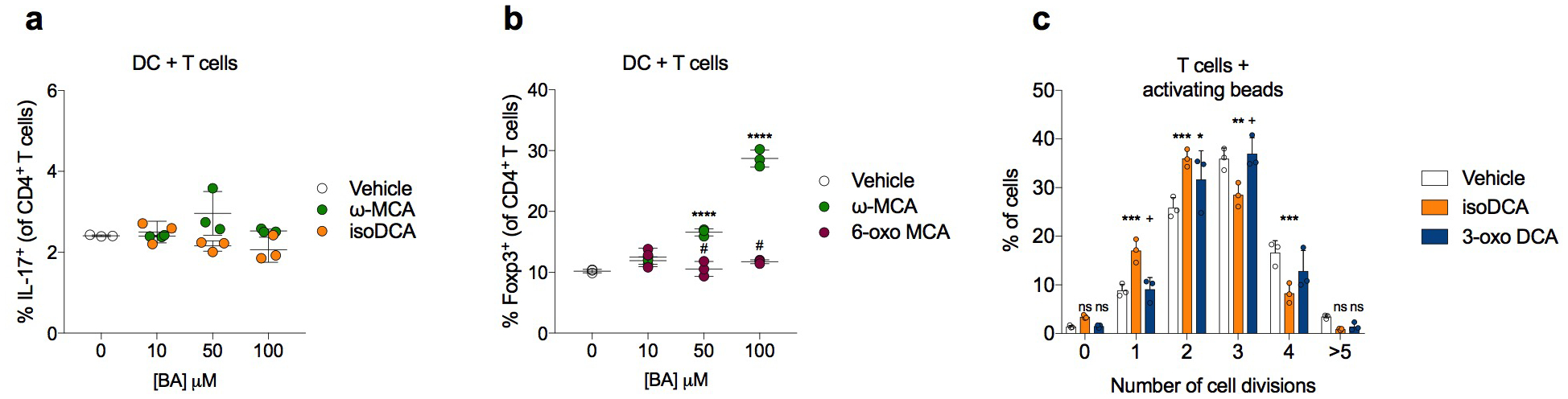
Extended Data Fig. 1|. Effects of iso- and oxo-bile acids on T cell differentiation and proliferation.
(a) Effects of Treg cell-inducing BAs on the in vitro generation of Th17 cells. Naïve CD4+ T cells were activated by DCs in Th17-polarizing conditions (2 ng/mL TGFβ, 1 μg/mL αCD3, and 20 ng/mL IL-6). On day 3, co-cultures were re-stimulated with PMA and ionomycin in the presence of brefeldin A and monensin for 3 hours before FACS analysis of IL-17A production. (b) The 6β-OH of ω−MCA is required for its Treg cell-inducing activity. Naïve CD4+ T cells were activated by DCs in suboptimal Treg-cell inducing conditions (1 ng/mL TGFβ, 1 μg/mL αCD3, 100 U/mL IL-2) and exposed to ω−MCA or 6-oxoMCA at the indicated concentrations. Foxp3 induction was assessed by FACS on day 3. (c) Assessment of cell division in the presence of isoDCA and 3-oxoDCA (100 μM). Naïve CD4+ T cells were labeled with Cell Trace Violet and activated with αCD3/αCD28 beads in the presence of TGFβ and IL-2 for 3 days before FACS analysis. Showing mean ± SD of replicates (a-c, n=3). Statistical significance determined by one-way (a,b) or two-way (c) ANOVA followed by a Dunnet (a) or Tukey (b, c) multiple comparison test. (*) p<0.05, (**) p<0.01, (***) p<0.001 vs vehicle; (#) p<0.05 vs ω−MCA (paired concentration); (+) p<0.05 vs isoDCA (paired concentration); (ns) not significant. Data representative of at least two independent experiments.
Extended Data Fig. 2|. Characterization of mice with FXR deficiency in the myeloid compartment.
(a-e) WT (Csf1rWT Nr1h4fl/fl) and DCΔFXR (Csf1rCreNr1h4fl/fl) littermate mice were analyzed between 6–8 weeks of age. Gating strategy (a) and quantification of cDC1 [Live CD45+ Lin− (dump: CD90, CD3; CD64−, Ly6C−, Siglec-F−) CD11c+ MHC class IIhi CD11b− XCR1+] and cDC2 [Live CD45+ Lin− (dump: CD90, CD3; CD64−, Ly6C−, Siglec-F−) CD11c+ MHC class IIhi CD11b+ XCR1−] in the spleen (Spl), mesenteric lymph node (MLN) and large intestine lamina propria (LILP, b). (c) Gating strategy for (d-e). (d) Number of total Foxp3+ Treg cells in the indicated organs. (e) Quantification of RORγt+ Foxp3+ Treg cells in the LILP. Data shown as mean ± SD (n=5), representative of two independent cohorts of mice. Statistical significance determined by a two-tailed T-test. (**) p<0.01.
Extended Data Fig. 3|. Anti-inflammatory effects of isoDCA treatment on DCs.
(a) DCs (1 × 105) were stimulated for 18 hours with various TLR agonists in the presence or absence of 50 μM isoDCA. Levels of the indicated cytokines were determined in the culture supernatant by ELISA. (b) DCs (1 × 104) were pulsed with ovalbumin (OVA, 1 mg/mL) in the presence of various concentrations of isoDCA for 1 hour in serum-free medium and allowed to process antigen for 4 hours in complete medium before addition of an NFAT-GFP reporter cell line expressing the MHC II-restricted OT-II TCR recognizing the ISQAVHAAHAEINEAGR peptide of OVA. The frequency of GFP+ cells was determined by FACS analysis after 24 hours. Co-cultures treated with αCD3 antibody (1 μg/mL) served as controls for DC-dependent, antigen-processing independent effects of isoDCA on the activation of reporter cells. Activation with αCD3/αCD28 beads in the presence of isoDCA served as control for DC-independent effects on reporter gene expression. Showing mean ± SD of replicates in (a) and fold-change relative to vehicle (0 μM isoDCA) within each condition (OVA, αCD3 or αCD3/CD28 beads) in (b). Statistical significance in (a) was determined by multiple T-tests using the Holm-Sidak correction method with alpha = 0.05. (****) p<0.001 vs vehicle. Statistical significance in (b) was determined by a two-way ANOVA followed by Dunnet’s multiple comparison’s test. (*) p<0.05; (****) p<0.001 vs vehicle in each condition. Data representative of 3 independent experiments.
Extended Data Fig.4|. LC-MS-based analysis of isoDCA production by engineered B. theta strains.
Bacteria were grown to exponential phase and transferred to media containing DCA. Following incubation for 24 hours, media was extracted with methanol and supernatants were analyzed by Liquid Chromatography-Mass Spectrometry (LC-MS). (a) Traces for spike-in controls with DCA and isoDCA standards (solid and dotted black lines, respectively), and media conditioned by B. thetaWT (yellow line), B. thetaCD (blue line) or the parental, un-manipulated B. thetaVPI strain VPI-5482 (gray line). Data are representative of two independent experiments carried in triplicates.
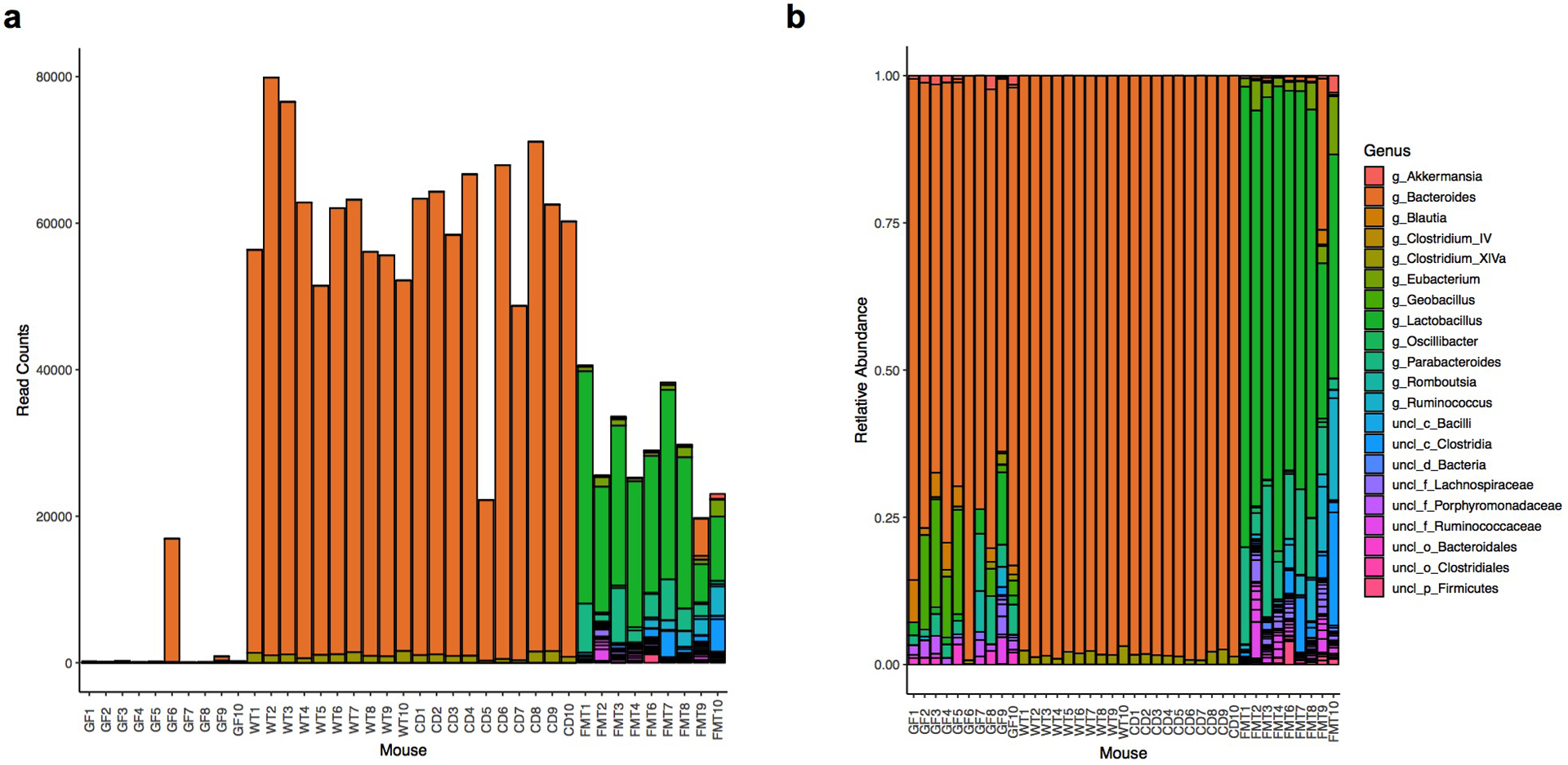
Extended Data Fig.5|. Analyses of microbial community composition in gnotobiotic and conventionalized mice.
Germ-free (GF) mice were gavaged with WT or CD engineered consortia (C. scindens + B. thetaWT or C. scindens + B. thetaCD, respectively). Recipients of a fecal microbiota transplant (FMT) and non-colonized mice (PBS) served as references. OTU composition of the cecal microbiota on day 10 post-colonization was determined by 16S sequencing. Total read counts (a) and relative abundances (b) of bacteria in individual experimental mice. Showing data pooled from two independent experiments (n=10).
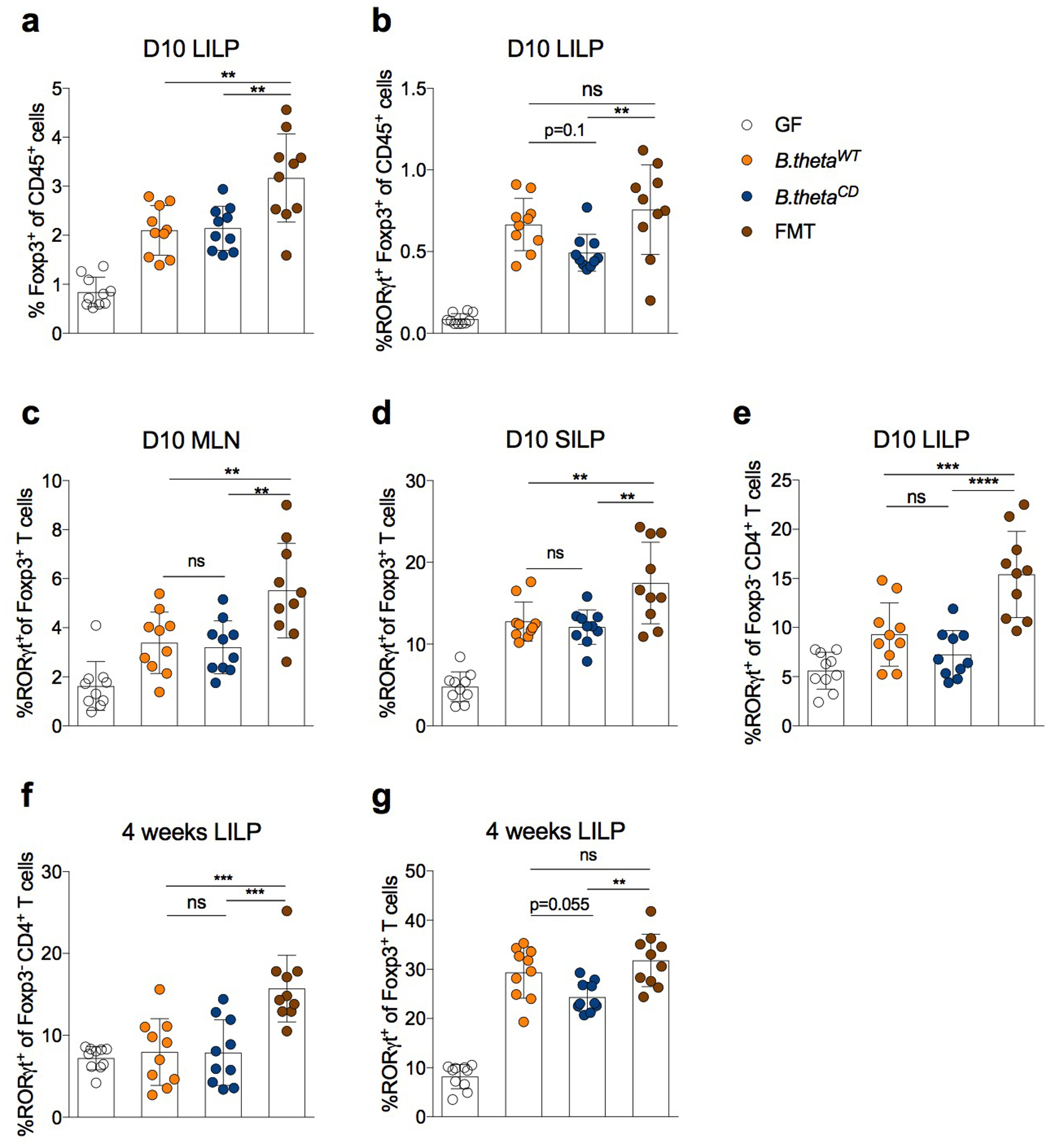
Extended Data Fig.6|. Effects of isoDCA-producing consortium on colonic lymphocytes.
Germ-free (GF) mice were gavaged with engineered consortia (C. scindens + B. thetaWT or C. scindens + B. thetaCD), PBS or a complex microbial community (FMT) as described in Fig. 4b. Immune cell composition in the LILP was analyzed by FACS on day 10 (a-e) or day 30 (f-g) post-colonization. Frequencies of total Foxp3+ (a) and RORγt+Foxp3+ (b) Treg cells among CD45+ cells. Frequency of RORγt+Foxp3+ cells in the mesenteric lymph node (MLN, c) and small intestine lamina propria (SILP, d). (e) Frequency of RORγt+ cells among Foxp3− (e, f) and Foxp3+ CD4+ T cells (g). Showing mean ± SD (n=10). Data are pooled from two independent experiments. Statistical significance determined by a one-way ANOVA followed by Tukey’s multiple comparison’s test. (**) p<0.01; (***) p<0.001; (****) p<0.0001; (ns) not significant.
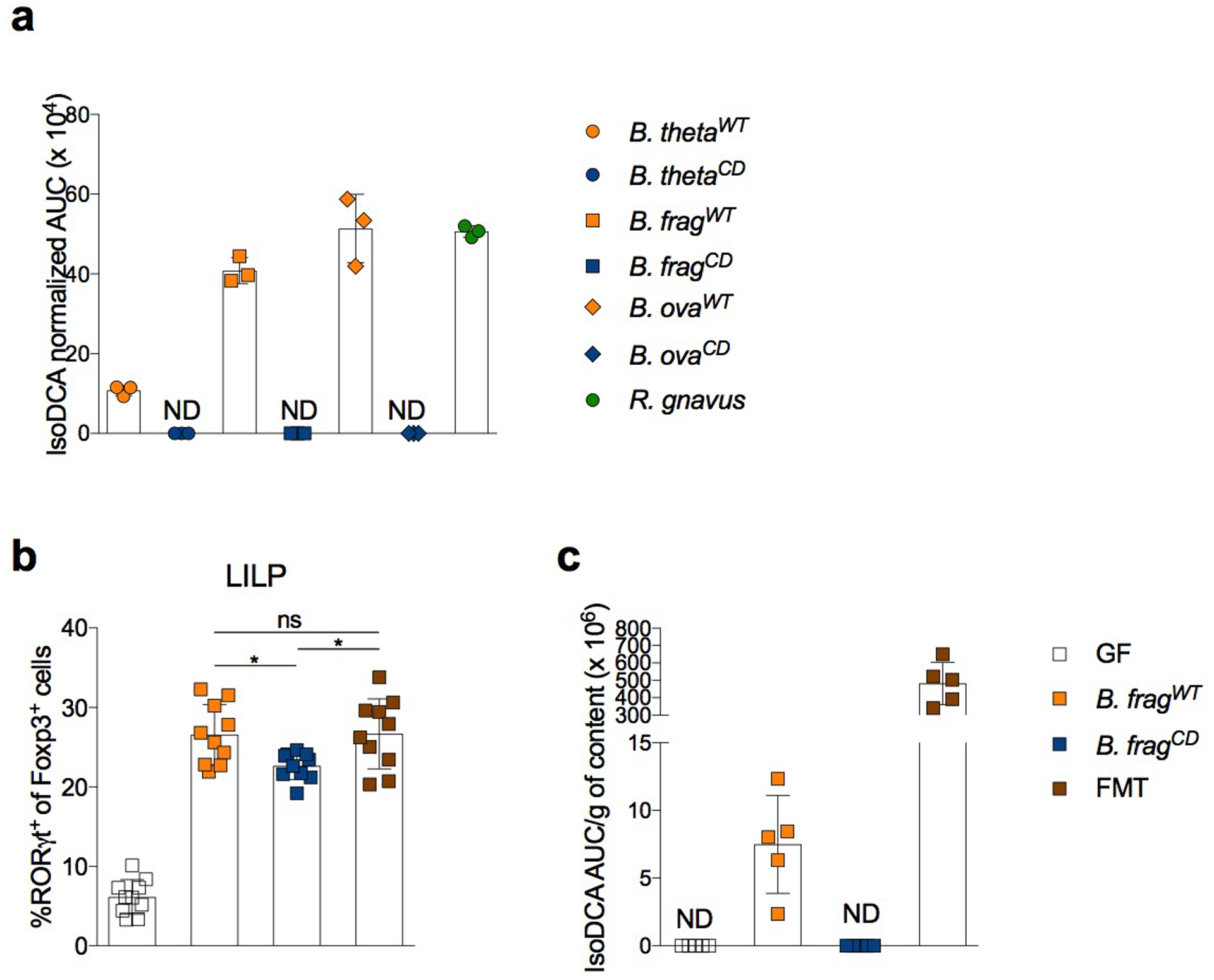
Extended Data Fig.7|. IsoDCA production by engineered Bacteroides sp. strains.
(a) Quantification of isoDCA production by engineered and reference strains in vitro. Bacteria were grown to exponential phase and transferred to media containing DCA. Following incubation for 24 hours, media was extracted with methanol and supernatants were analyzed by Liquid Chromatography-Mass Spectrometry (LC-MS). (b-c) Germ-free (GF) mice were colonized with consortia containing either the engineered strain of B. frag capable of producing isoDCA (WT) or the catalytically dead (CD) mutant in combination with C. scindens (C. scindens + B. fragWT and C. scindens + B. fragCD, respectively). Recipients of a fecal microbiota transplant (FMT) and non-colonized mice (PBS) served as references. Immune cell composition and isoDCA quantification were performed 10 days post-colonization. (b) FACS analysis of the frequency of RORγt+Foxp3+CD4+ T cells in the large intestine lamina propria (LILP). (c) Quantification of isoDCA in cecal contents. Fecal material was weighed, homogenized and extracted with methanol for LC-MS analysis. Reporting area under the curve (AUC) normalized by weight of input material. Showing mean ± SD (a, n=3; b, n=10; c, n=5). Data in (a, c) are representative of two independent experiments. Data in (b) are pooled from two independent experiments. Statistical significance determined by a one-way ANOVA followed by Tukey’s multiple comparison’s test. (*) p<0.05; (ns) not significant.
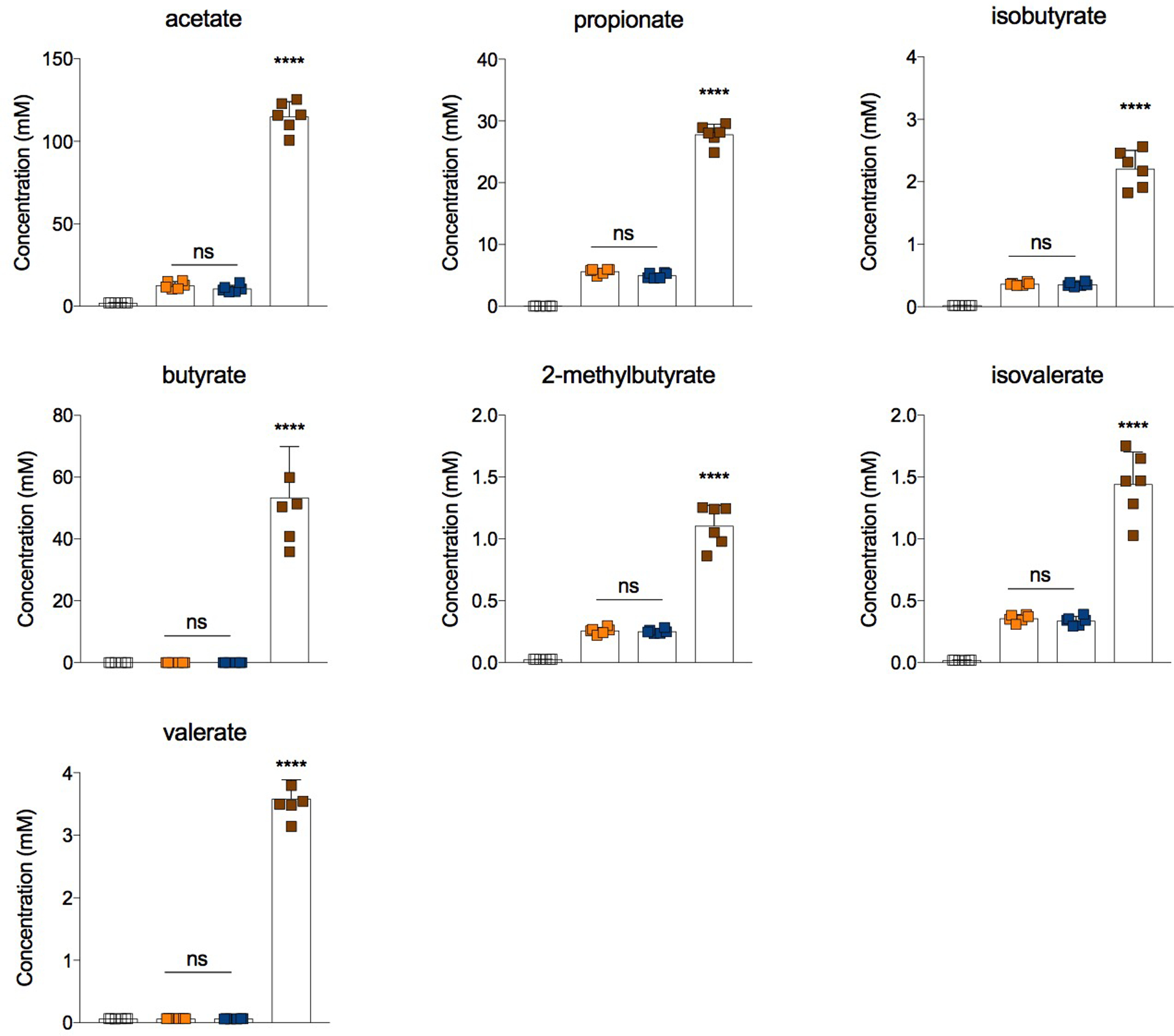
Extended Data Fig. 8|. Short-chain fatty acids (SCFAs) production by minimal, defined microbial consortia.
Germ-free (GF) mice were colonized with consortia containing either the engineered strain of B. frag capable of producing isoDCA (WT) or the catalytically dead (CD) mutant in combination with C. scindens (C. scindens + B. fragWT and C. scindens + B. fragCD, respectively). Recipients of a fecal microbiota transplant (FMT) and non-colonized mice (PBS) served as references. Cecal content material was weighed, homogenized and subjected to organic solvent extraction for GC-MS-based quantification of SCFAs levels. Showing mean ± SD (n=6). Data are pooled from two independent experiments. Statistical significance determined by a one-way ANOVA followed by Tukey’s multiple comparison’s test. (****) p<0.0001; (ns) not significant.
Supplementary Material
Acknowledgements:
We thank Dr. Frank Gonzalez (NIH) for kindly providing Nr1h4fl mice and Dr. Frederic Geissmann (MSKCC) for providing Csf1rCre mice. We thank Dr. Justin Sonnenburg (Stanford University) for providing genetic tools to engineer B. theta. We thank Su-Ellen Brown, Melissa Rosenthal, Thanh Nguyen, Paul Gonzales (BI) and Rielmer Pinedo (WCMC) for assistance with germ-free mice. We thank Dr. Ouathek Ouerfelli and the staff at the Organic Synthesis core at MSKCC for producing 6-oxoMCA. We thank Amanda Pickard (MKSCC) for assistance with metabolomic data analyses. We thank Dr. Everton D. D’Andrea (U. of Arizona) for assistance with DSF experiments. We thank Dr. Joris van der Veeken for discussion of gene expression data and all other members of the Rudensky lab for their suggestions and technical assistance. This study was supported by NIH grant R37, the Ludwig Institute for Cancer Research, the Hilton Foundation and Research Beyond Borders at Boehringer Ingelheim. W.G. and C.J.G were supported by 1DP2HD101401-01. A.Y.R is an investigator with the Howard Hughes Medical Institute.
An invention disclosure has been filed based on the data generated in this study. P.T.M. and A.Y.R. received funding from Boehringer Ingelheim, A.Y.R. is a co-founder and SAB member of and holds stock options in Vedanta Biosciences, P.T.M. receives licensing royalties from Seres Therapeutics and is a co-inventor on patent applications: US20170087196A1, US20180256653A1 and WO2018195467A1.
Footnotes
Data availability
RNA-sequencing and 16s metagenomics sequencing data are available under BioProject ID PRJNA600898 and PRJNA600979. Source data for Fig1–4 and Extended Data Fig.1–8 available as .xsl tables. Other relevant data are available from the corresponding authors upon reasonable request.
References
- 1.Josefowicz SZ et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399, doi: 10.1038/nature10772 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng Y et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812, doi: 10.1038/nature08750 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nutsch K et al. Rapid and Efficient Generation of Regulatory T Cells to Commensal Antigens in the Periphery. Cell Rep 17, 206–220, doi: 10.1016/j.celrep.2016.08.092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim KS et al. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863, doi: 10.1126/science.aac5560 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Arpaia N et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455, doi: 10.1038/nature12726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furusawa Y et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450, doi: 10.1038/nature12721 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573, doi: 10.1126/science.1241165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ridlon JM, Kang DJ & Hylemon PB Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259, doi: 10.1194/jlr.R500013-JLR200 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Ridlon JM, Harris SC, Bhowmik S, Kang DJ & Hylemon PB Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7, 22–39, doi: 10.1080/19490976.2015.1127483 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahlstrom A, Sayin SI, Marschall HU & Backhed F Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab 24, 41–50, doi: 10.1016/j.cmet.2016.05.005 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Devlin AS & Fischbach MA A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol 11, 685–690, doi: 10.1038/nchembio.1864 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eyssen H, De Pauw G, Stragier J & Verhulst A Cooperative formation of omega-muricholic acid by intestinal microorganisms. Appl Environ Microbiol 45, 141–147 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyssen HJ, De Pauw G & Van Eldere J Formation of hyodeoxycholic acid from muricholic acid and hyocholic acid by an unidentified gram-positive rod termed HDCA-1 isolated from rat intestinal microflora. Appl Environ Microbiol 65, 3158–3163 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton JP et al. Human cecal bile acids: concentration and spectrum. Am J Physiol Gastrointest Liver Physiol 293, G256–263, doi: 10.1152/ajpgi.00027.2007 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Hofmann AF & Small DM Detergent properties of bile salts: correlation with physiological function. Annu Rev Med 18, 333–376, doi: 10.1146/annurev.me.18.020167.002001 (1967). [DOI] [PubMed] [Google Scholar]
- 16.Kretschmer K et al. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol 6, 1219–1227, doi: 10.1038/ni1265 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Haxhinasto S, Mathis D & Benoist C The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 205, 565–574, doi: 10.1084/jem.20071477 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sefik E et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 349, 993–997, doi: 10.1126/science.aaa9420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohnmacht C et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science 349, 989–993, doi: 10.1126/science.aac4263 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Parks DJ et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Campbell C et al. Extrathymically Generated Regulatory T Cells Establish a Niche for Intestinal Border-Dwelling Bacteria and Affect Physiologic Metabolite Balance. Immunity 48, 1245–1257 e1249, doi: 10.1016/j.immuni.2018.04.013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White J et al. Two better cell lines for making hybridomas expressing specific T cell receptors. J Immunol 143, 1822–1825 (1989). [PubMed] [Google Scholar]
- 23.Hooijberg E, Bakker AQ, Ruizendaal JJ & Spits H NFAT-controlled expression of GFP permits visualization and isolation of antigen-stimulated primary human T cells. Blood 96, 459–466 (2000). [PubMed] [Google Scholar]
- 24.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, doi: 10.1093/bioinformatics/btu638 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt J et al. NSAIDs Ibuprofen, Indometacin, and Diclofenac do not interact with Farnesoid X Receptor. Sci Rep 5, 14782, doi: 10.1038/srep14782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caporaso JG et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108 Suppl 1, 4516–4522, doi: 10.1073/pnas.1000080107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, doi: 10.1093/bioinformatics/btq461 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Edgar RC SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. BioRxiv, doi: 10.1101/074161 (2016). [DOI] [Google Scholar]
- 31.Cole JR et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42, D633–642, doi: 10.1093/nar/gkt1244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitaker WR, Shepherd ES & Sonnenburg JL Tunable Expression Tools Enable Single-Cell Strain Distinction in the Gut Microbiome. Cell 169, 538–546.e512, doi: 10.1016/j.cell.2017.03.041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.