

SUMMARY
The proper coordination of transcription with DNA replication and repair is central for genomic stability. We investigate how the INO80C chromatin remodeling enzyme might coordinate these genomic processes. We find that INO80C co-localizes with the origin recognition complex (ORC) at yeast replication origins and is bound to replication initiation sites in mouse embryonic stem cells (mESCs). In yeast· INO80C recruitment requires origin sequences but does not require ORC· suggesting that recruitment is independent of pre-replication complex assembly. In both yeast and ESCs· INO80C co-localizes at origins with Mot1 and NC2 transcription factors· and genetic studies suggest that they function together to promote genome stability. Interestingly· nascent transcript sequencing demonstrates that INO80C and Mot1 prevent pervasive transcription through origin sequences· and absence of these factors leads to formation of new DNA double-strand breaks. We propose that INO80C and Mot1/NC2 function through distinct pathways to limit origin transcription· maintaining genomic stability.
Graphical Abstract
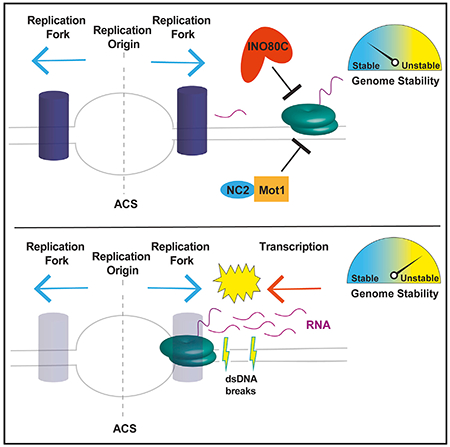
In Brief
The INO80C chromatin remodeler is known to regulate transcription and genomic stability. Topal et al. find that INO80C functions with Mot1 repressor to prevent pervasive transcription at replication origins in yeast and mESCs. Increased ncRNAs lead to new DNA double-strand breaks at origins, linking transcriptional regulation to genomic stability.
INTRODUCTION
Although many studies have focused on how chromatin contributes to regulation of gene transcription, nucleosome assembly affects all DNA-mediated processes within the cell nucleus, including DNA repair and replication. Indeed, recent in vitro studies have shown that chromatin regulates many steps of DNA replication, including binding of the origin recognition complex (ORC), origin licensing, origin activation, efficiency of fork progression, and lagging strand DNA synthesis (Eaton et al., 2010; Gros et al., 2014; Kurat et al., 2017). Proper coordination of gene transcription with DNA replication and repair is a central aspect of organismal survival because collisions between RNA and DNA polymerases can have deleterious effects on genomic stability (Helmrich et al., 2013). It is unclear how chromatin structure is regulated to coordinate these different genomic processes.
In budding yeast, replication origins are typically short (~150 bp) DNA sequence elements termed autonomous replication sequences (ARSs) that contain a consensus match to an 11 nt ARS consensus sequence (ACS) that is necessary but not sufficient for origin function (Bolon and Bielinsky, 2006; Newlon and Theis, 1993; Stinchcomb et al., 1979). The ACS is recognized by the multi-subunit ORC, which is bound to origins throughout the cell cycle and recruits several key replication factors in late G2/M and G1 phases to form a pre-replication complex (preRC) (Diffley et al., 1994, 1995; Hawkins et al., 2013). The preRC is activated at the G1/S boundary by cell cycle kinases, leading to origin activation and replication initiation (Aparicio, 2013; Sclafani and Holzen, 2007). Binding of ORC to origins is sufficient for preRC assembly in mammalian cells, though in this case, origins lack a specific DNA sequence (Takeda et al., 2005). Notably, yeast origins are largely nucleosome free even in the absence of ORC binding, and binding of ORC leads to further positioning of adjacent nucleosomes (Lipford and Bell, 2001). In vivo and in vitro studies have shown that alterations in nucleosome positioning can reduce origin efficiency by impinging on ORC binding and origin activation (Azmi et al., 2017; Lipford and Bell, 2001; Rodriguez et al., 2017). Yeast replication origins are defined as early or late origins on the basis of their respective replication timing during S phase, and it has been shown that efficiency of origins depends on their chromatin landscape (Heun et al., 2001; Raghuraman et al., 2001; Soriano et al., 2014). A key question is how nucleosome remodeling around the origin during replication is coordinated with transcription (Poli et al., 2017).
INO80C is an evolutionarily conserved, multi-subunit chromatin remodeling enzyme that plays roles in gene transcription, DNA double-strand break (DSB) repair, and DNA replication (Papamichos-Chronakis et al., 2006; Shimada et al., 2008; van Attikum et al., 2004; Xue et al., 2015). Early studies in yeast suggested that INO80C may be bound to many replication origins in G1 and that the enzyme is recruited to stalled replication forks (Papamichos-Chronakis and Peterson, 2008; Shimada et al., 2008). Furthermore, yeast that lacks an intact INO80C shows decreased replication fork elongation, instability of stalled forks, and poor ability of stalled forks to restart (Papamichos-Chronakis and Peterson, 2008; Shimada et al., 2008). Consequently, inactivation of INO80C causes sensitivity to replication stress agents (Papamichos-Chronakis and Peterson, 2008; Shen et al., 2000; van Attikum et al., 2004). How and why INO80C is recruited to replication origins is not clear, though recent studies suggest that INO80C may stabilize stalled replication forks in part by resolving encounters between elongating RNA polymerases and forks (Lafon et al., 2015; Poli et al., 2016). In vitro, INO80C has two known ATP-dependent activities: (1) mobilization and spacing of nucleosomes (Shen et al., 2003; Udugama et al., 2011) and (2) a nucleosome-editing activity whereby INO80C removes one or both variant H2A.Z/H2B dimers from a nucleosome and replaces them with a canonical H2A/H2B dimer (Papamichos-Chronakis et al., 2011). Accordingly, yeast INO80C is required for proper positioning of promoter proximal nucleosomes at many yeast genes (Tramantano et al., 2016; Yen et al., 2012), and loss of INO80C leads to aberrant, genome-wide accumulation of H2A.Z-containing nucleosomes (Chambers et al., 2012; Lademann et al., 2017; Papamichos-Chronakis et al., 2006).
In addition to its role in DNA replication, the ATPase subunit of INO80C was originally identified in a screen for mutants defective for activating genes in response to inositol depletion (Ebbert et al., 1999). Several further studies have shown that INO80C is required for activation of a subset of genes, including genes involved in metabolic pathways (Barbaric et al., 2007; Cai et al., 2007). INO80C occupies most transcription start sites (TSSs) of yeast promoters, and it is also present at transcription termination sites (TTSs) (Xue et al., 2015; Yen et al., 2012, 2013). Related to its proximity to promoter regions, INO80C prevents bidirectional transcription at functional promoters (Marquardt et al., 2014), and loss of INO80C leads to increases in noncoding transcription (Alcid and Tsukiyama, 2014; Xue et al., 2015, 2017). It is unclear why INO80C prevents transcription of noncoding RNAs (ncRNAs) near promoters and whether increased ncRNA caused by loss of INO80C affects DNA replication and repair.
Mot1 is a Snf2-like ATPase that removes and redistributes TATA binding protein (TBP) from DNA (Auble et al., 1994), while NC2 is a heterodimer that inhibits PIC formation (Cang and Prelich, 2002). Both Mot1 and NC2 have been shown to regulate antisense transcription by preventing PIC assembly near the 3′ end of genes (Koster and Timmers, 2015). Recently, we found that INO80C co-localizes with Mot1 and NC2 at intergenic regions in both yeast and mouse embryonic stem cells (ESCs) and that these factors function coordinately to suppress intergenic ncRNAs (Xue et al., 2017). How INO80C suppresses these ncRNAs is not yet known, though Mot1 and NC2 are likely to function by inhibiting binding of TBP to cryptic or low-affinity binding sites. Here we find that INO80C, Mot1, and NC2 co-localize with ORC at yeast replication origins as well as replication initiation sites (RISs) in ESCs, and loss of either INO80C or Mot1 leads to production of ncRNAs at origins. Yeast that lacks INO80C, Mot1, or NC2 is sensitive to replication stress agents, suggesting that suppression of ncRNAs is important for genome stability. Importantly, loss of INO80C and Mot1 causes an increase in DNA DSBs near origins, suggesting that the prevention of promiscuous transcription ensures genome stability.
RESULTS
INO80C Localization to Replication Origins Requires Origin DNA, but Not a preRC
Previous studies suggested that INO80C localizes to at least a subset of yeast replication origins, though how INO80C is recruited to origins is not known (Papamichos-Chronakis and Peterson, 2008; Shimada et al., 2008). First, we tested whether recruitment of INO80C to an origin requires cis-acting DNA sequences. Following arrest of cells in G1, samples were collected for chromatin immunoprecipitation (ChIP), monitoring enrichment of the Arp5 subunit of INO80C and the Orc2 subunit of ORC at an early-firing origin, ARS432.5, and at a late firing origin, ARS501. High levels of Orc2 were detected at both origins, whereas significant levels of Arp5 were detected only at the early origin, ARS432.5 (Figure S1A). Strikingly, a small, 100 bp deletion at ARS342.5 eliminated both Orc2 and Arp5 recruitment (Figure S1A). Thus, recruitment of INO80C to an early replication origin requires origin sequences.
We next investigated whether assembly of the preRC is required for INO80C recruitment to origins. To test this possibility, we used the anchor-away system to conditionally deplete Orc2 from the nucleus in G2/M-arrested cells (Haruki et al., 2008). Following Orc2 depletion, cells were released into media containing α-factor to arrest cells in the subsequent G1, followed by ChIP for Arp5 (INO80C) and Orc5 (preRC) (Figure 1A). As expected (Diffley et al., 1994, 1995), Orc5 was enriched at all annotated replication origins in the presence of Orc2, but depletion of Orc2 in the previous G2/M nearly eliminated Orc5 recruitment in G1 cells (Figure 1B). Strikingly, Arp5 was enriched at all replication origins, but recruitment of Arp5 was not affected by Orc2 depletion (Figure 1B). Thus, INO80C recruitment to origins does not require ORC, and consequently, recruitment does not require assembly of the pre-RC.
Figure 1. Ino80, Mot1, and NC2 Co-localize at Yeast Replication Origins.
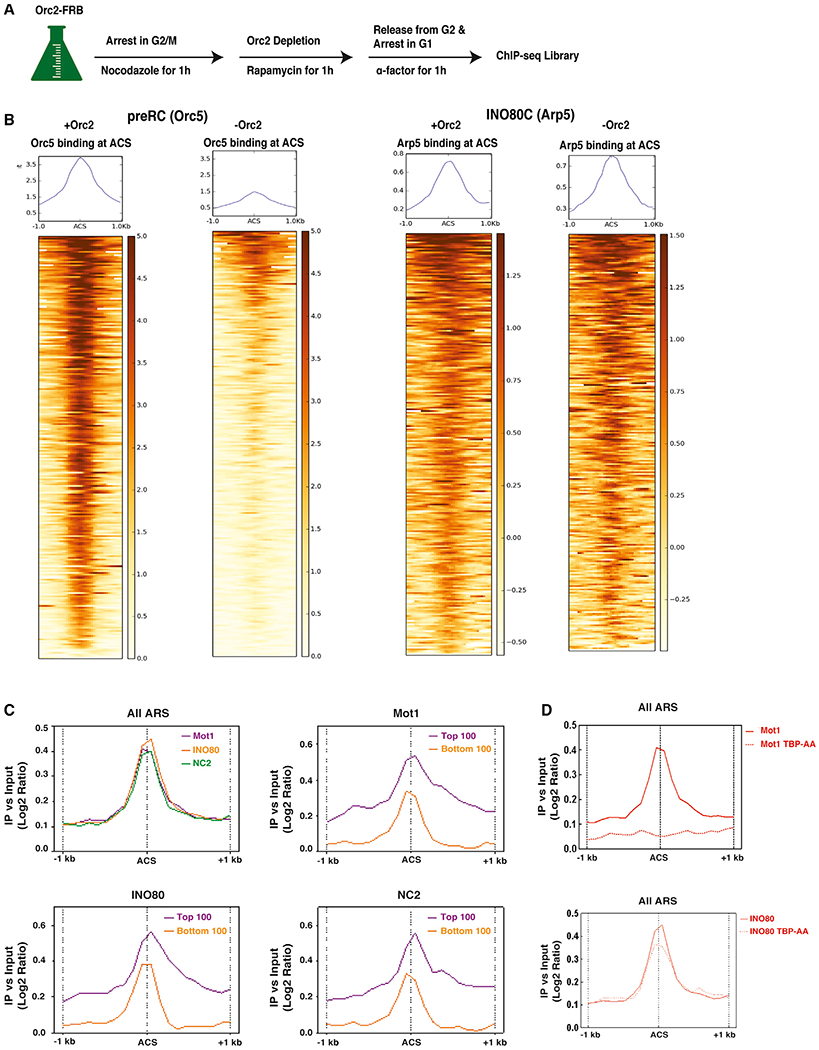
(A) Experimental design for ChIP-seq.
(B) Heatmaps showing log2 mean intensity values for an average of two biological replicates (n = 2) for all annotated replication origins (n = 253) for Orc5 (left two panels) or INO80C (right two panels) recruitment, comparing wild-type and Orc2-depleted cells.
(C) Average binding profiles of Ino80 (orange), Mot1 (purple), and NC2 (green) at all yeast replication origins (top left), Mot1 (top right), Ino80 (bottom left), and NC2 (bottom right) at top 100 (bound by Arp5; purple) and bottom 100 (bound by Arp5; orange) in wild-type by ChIP-seq.
(D) Average binding profile of Mot1 (top panel) or Ino80 (bottom panel) at all yeast replication origins in wild-type or after TBP depletion (TBP-AA cells). The moving averages of log2 Ino80, Mot1, or NC2 enrichment versus input were plotted by the distance from ARS, from 0 to 1 kb upstream and downstream (x axis).
There are 253 yeast replication origins with previously annotated ACSs (Nieduszynski et al., 2006; Soriano et al., 2014). We grouped origins by enrichment level for Arp5 as top 100 or bottom 100. Notably, top 100 origins showed 89% overlap with origins that fire early in S phase (Figure S1B). To ensure that these binding profiles were not unique to ChIP sequencing (ChIP-seq) analyses, we also analyzed a chromatin endogenous cleavage sequencing (ChEC-seq) dataset obtained from asynchronous yeast cells (Kubik et al., 2019). Although the peak of INO80C binding was more broad from this analysis, compared with ChIP-seq analysis in synchronized cells, binding was also enriched at replication origins, and the top 100 bound origins overlapped well between the two datasets (Figures S1C–S1E).
Recently, we found that INO80C co-localizes with Mot1 and NC2 at promoter proximal regions, and we suggested that these three factors may function together to limit ncRNAs (Xue et al., 2017). ChIP was used to investigate co-localization of these factors to replication origins. As shown in Figure 1C, all three factors co-localize at yeast origins, and increased levels of Mot1 and NC2 were also found at origins with higher levels of Arp5 (INO80C) (top 100; Figure 1C). Recruitment of Mot1 to origins was lost following nuclear depletion of TBP, consistent with the known role of Mot1 in targeting TBP displacement (Figure 1D). In contrast, recruitment of INO80C to origins was only partially disrupted by TBP depletion, indicating that binding of INO80C at origins is not strictly dependent on TBP or Mot1 (Figure 1D). Thus, recruitment of INO80C to origins appears to be distinct from binding of INO80C near gene TSSs where we previously found that depletion of TBP eliminated the majority of INO80C recruitment (Xue et al., 2017).
Roles of INO80C and Mot1 for Genic Transcription
Given that INO80C and Mot1 are enriched at early replication origins that are known to be adjacent to highly expressed genes, we tested whether their localization was linked to changes in gene transcription. We used the anchor-away strategy to induce the rapid depletion of either the Ino80 ATPase or Mot1 from the nucleus, followed after 1 h by analysis of nascent RNAPII transcripts by nascent elongating transcript sequencing (NET-seq) (Figure 2A). Fission yeast cells were used as “spike-in” controls for normalization of sequencing libraries. Surprisingly, scatter-plot analyses indicated that individual depletion of Ino80 or Mot1 had little impact on gene transcription compared with wild-type when a ≥ 1.5-fold change was used (false discovery rate [FDR] ≤ 0.05). Lowering the threshold to ≥ 1.25-fold change revealed that loss of either Ino80 or Mot1 led to decreased expression of ~1,200 genes (Figure 2B; Figure S2A). In the case of Ino80, this gene set showed a large overlap with genes that encode products involved with metabolism, similar to previously reported defects (Figure S2B; Yao et al., 2016). Furthermore, simultaneous depletion of both Ino80 and Mot1 led to a more global decrease in nascent transcripts, with 1,628 genes decreased by 1.5-fold or more (FDR ≤ 0.05) and 2,342 genes decreased by 1.25-fold or more (FDR ≤ 0.05) (Figure 2B; Figure S2A). Notably, transcription of genes located adjacent to replication origins with high occupancy levels of INO80C and Mot1 were not significantly decreased but rather showed slight increases in expression (Figure S2C).
Figure 2. Roles of INO80C and Mot1 for Genic Transcription.
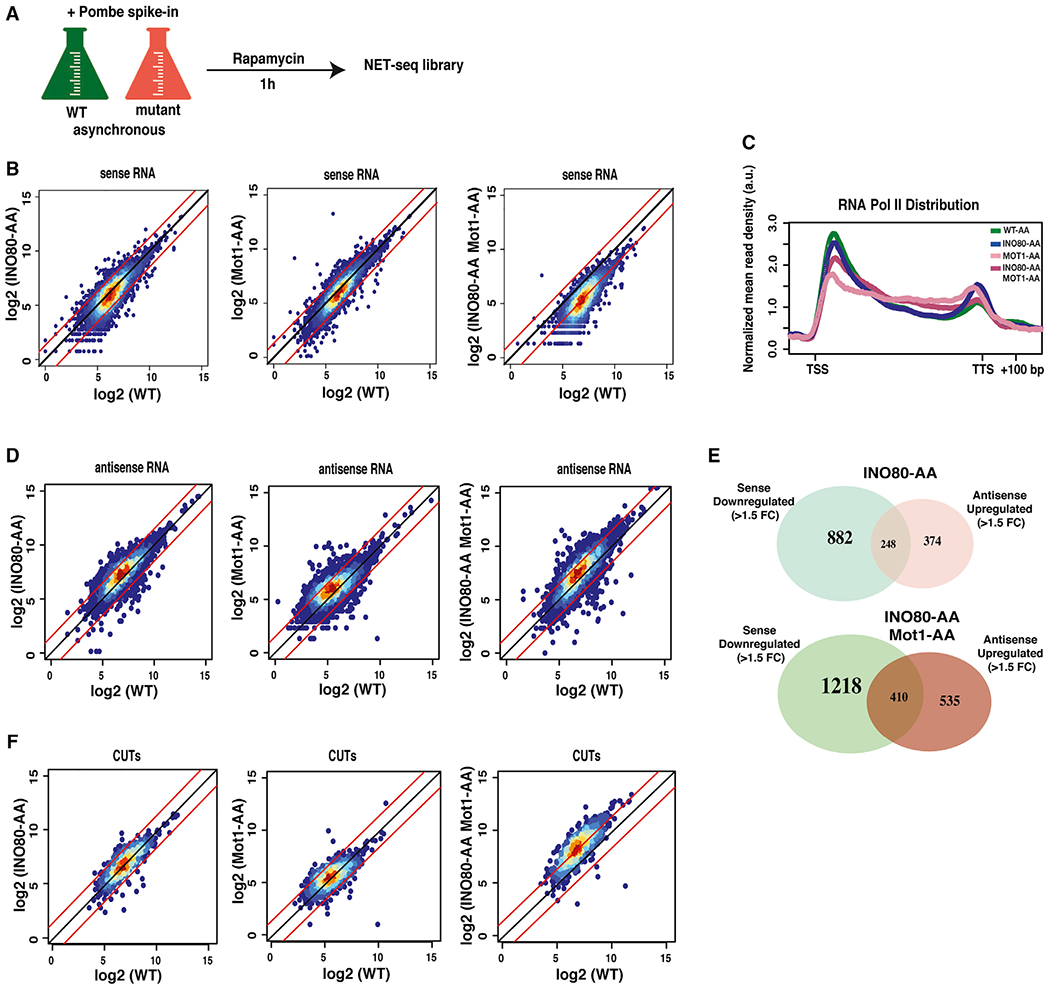
(A) Experimental design for NET-seq. Wild-type and mutant (AA [anchor-away]) cells were treated with rapamycin for 1 h. Following the addition of S. pombe cells, nascent RNAs associated with RNA polymerase (Pol) II were isolated and sequenced.
(B) NET-seq scatterplots showing log2 mean intensity values for an average of two biological replicates (n = 2) for all nascent coding transcripts (n = 5,302) for INO80-AA, Mot1-AA, and INO80-AA Mot1-AA.
(C) Metagene plot showing RNA Pol II distribution throughout the gene body from TSS to TTS (including 100 bp upstream of TSS and 200 bp downstream of TTS) fitted into a 500 bp window. WT-AA (green), INO80-AA (blue), Mot1-AA (pink), and INO80-AA Mot1-AA (fuchsia). The mean nascent transcript levels are normalized according to both spike-in numbers and each gene’s individual expression level. A.U., arbitrary unit.
(D) NET-seq scatterplots showing log2 mean intensity values for an average of two biological replicates (n = 2) for all antisense transcript levels for INO80-AA, Mot1-AA, and INO80-AA Mot1-AA. All reads are normalized according to S. pombe spike-in reads.
(E) Venn diagrams showing correlation in numbers of sense downregulated and antisense upregulated genes (≥ 1.5 fold change [FC], FDR ≤ 0.05) in INO80-AA (top) and INO80-AA Mot1-AA (bottom).
(F) NET-seq scatterplots showing log2 mean intensity values for an average of two biological replicates (n = 2) for nascent transcript levels of CUTs in INO80-AA, Mot1-AA, and INO80-AA Mot1-AA. Significance and p values were calculated by using the Mann-Whitney U test.
Next, we performed metagene analyses to analyze RNAPII distributions, normalizing RNAPII occupancy to the individual gene expression levels and plotting these values throughout all genes (~5,300 genes). Compared with wild-type, loss of Mot1 and Ino80 led to a global shift in the RNAPII distribution, with a large decrease in RNAPII at the 5′ end of genes and increased levels over the coding region (Figure 2C). This re-distribution of RNAPII could indicate a global decrease in transcriptional elongation or termination, or alternatively, the increased levels of RNAPII over coding regions might reflect increased use of cryptic promoters within genes. Consistent with the latter view, genic antisense transcripts were increased in the absence of either Mot1 or Ino80 (Figure 2D). Interestingly, a subset of these genic antisense transcripts overlapped with sense transcripts that decreased following Ino80 and Mot1 depletion, suggesting that some of the positive impact of Ino80 and Mot1 on the yeast transcriptome may be due to transcriptional interference (Figure 2E).
INO80C and Mot1 Prevent Cryptic Transcription around Yeast Replication Origins
The role of INO80C and Mot1 in suppressing antisense genic transcription is consistent with our previous findings that INO80C acts together with Mot1 and NC2 to prevent cryptic transcription in intergenic regions near a subset of yeast TSSs (Xue et al., 2017). Consistent with these previous studies, NET-seq analyses also detected increases in cryptic upstream transcripts (CUTs) in the absence of Ino80 or Mot1 (Figure 2F). We then exploited the high resolution of NET-seq to evaluate the impact of Ino80 and Mot1 on synthesis of nascent ncRNAs at replication origins. Whereas depletion of either Ino80 or Mot1 led to small increases in nascent RNA immediately adjacent to origins, the simultaneous depletion of both Ino80 and Mot1 led to large increases in ncRNA, consistent with complementary roles in preventing spurious ncRNA production (Figure 3). Furthermore, increased levels of ncRNAs were associated with early-firing origins that showed the highest enrichment for INO80C (Figures 3B–3D). Notably, nascent transcription levels were generally low at all origins in wild-type cells, with slightly higher levels observed at late firing origins with low INO80C occupancy (Figure 3). Increases in ncRNA levels adjacent to origins was also observed in RNA sequencing (RNA-seq) analyses (Figure S3). Strikingly, loss of INO80C and Mot1 led to production of ncRNAs primarily from the Crick DNA strand and downstream of the A-rich side of the ACS where initial DNA unwinding occurs (Figure 3; see also Figure S2D; Coster and Diffley, 2017). The A-rich side of origins also shows a higher frequency of matches to aTATA-box consensus sequence, suggesting the potential for a large number of cryptic promoters (Figure S2E).
Figure 3. INO80C and Mot1 Prevent Cryptic Transcription around Yeast Replication Origins.
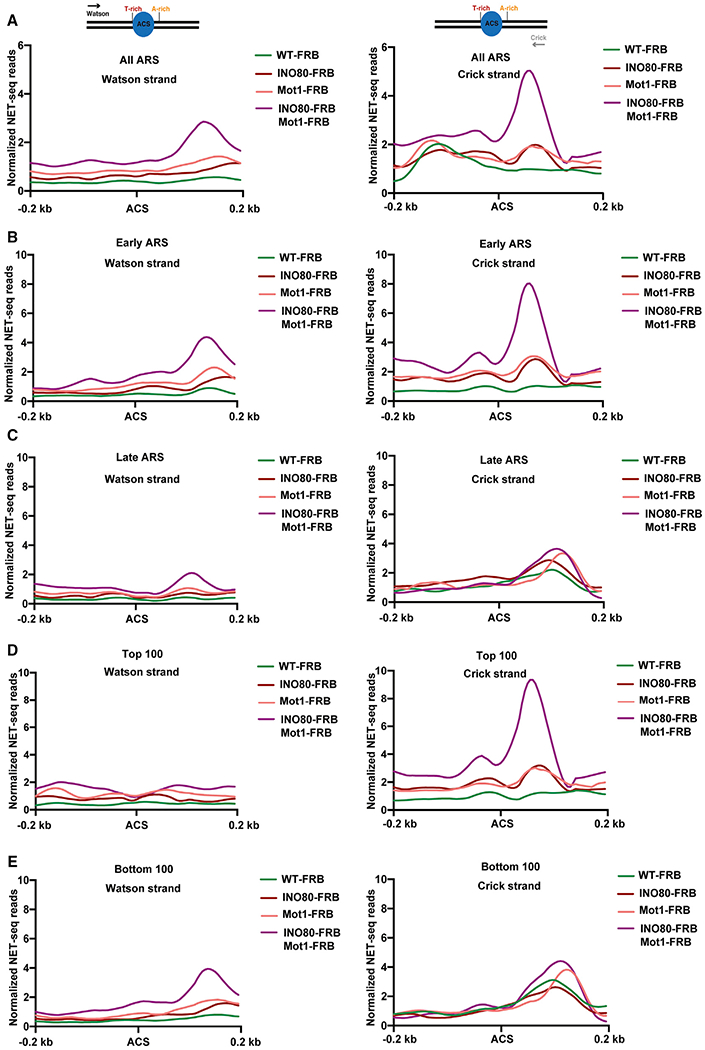
(A–E) Plots showing normalized NET-seq reads for an average of two biological replicates (n = 2) around ACS (flanking a region of 200 bp upstream and downstream of ACS) for transcriptional activity in Watson strand (left panels) and Crick strand (right panels) for WT-FRB, INO80-FRB, Mot1-FRB, and INO80-FRB Mot1-FRB for all yeast origins (A), for early-firing origins (B), for late-firing origins (C), for top 100 (D), and for bottom 100 (E). Schematics above each panel in (A) indicate the orientation of ACS and direction of corresponding strand.
Nucleosome Occupancy Is Disrupted in the Absence of Ino80
How might Ino80 prevent cryptic transcription around replication origins? Previous work has shown that INO80C is key for proper positioning of nucleosomes adjacent to the start site of genes transcribed by RNAPII (Tramantano et al., 2016). On the basis of these data, we asked whether INO80C regulates positioning or occupancy of nucleosomes surrounding yeast replication origins, as changes in nucleosome organization may uncover cryptic promoters and promote increased ncRNAs. We analyzed published Mnase-seq nucleosome mapping data and detected a small shift of nucleosomes in the absence of Ino80 on both sides of the ACS element, a shift similar in magnitude to that observed at promoter regions (Figure S4; Tramantano et al., 2016). Interestingly, we observed a more dramatic change in nucleosome occupancy surrounding origins following co-depletion of Ino80 and the Isw2 remodeler, or co-depletion of Ino80 and the Sth1 subunit of the RSC remodeler (Figure S4). Previous studies have indicated that INO80C and Isw2 function together during DNA replication (Vincent et al., 2008), and ChEC-seq analyses show strong enrichment of Isw2 at origins (Figure S1C). Furthermore, depletion of all three remodelers led to a more severe disruption of nucleosome architecture at origins (Figure S4). These results indicate that INO80C functions in concert with several other remodelers to enforce proper nucleosome organization around replication origins.
Increases in Cryptic Transcription Correlate with Increased DNA Breaks
Cells that lack Ino80 exhibit growth defects on media containing replication stress or DNA damaging agents, consistent with a role in genome stability pathways (Papamichos-Chronakis et al., 2006; Papamichos-Chronakis and Peterson, 2008; Shen et al., 2000; Shimada et al., 2008; van Attikum et al., 2004). We investigated whether loss of Mot1 or NC2 would also give rise to similar genome instability phenotypes. Consistent with previous work, Ino80 depletion led to slow growth on media containing genotoxic stress agents, such as hydroxyurea (HU), methylmethanesulfate (MMS), and camptothecin (CPT) (Papamichos-Chronakis and Peterson, 2008; Shen et al., 2000) (Figure S5). Interestingly, depletion of either Mot1 or NC2 did not lead to much sensitivity to genotoxic stress, but in every case, co-depletion of Mot1 or NC2 with Ino80 led to a synergistic sensitivity to genotoxic agents (Figure S5). These results are consistent with overlapping, partially redundant roles for INO80C, Mot1, and NC2 in genome stability pathways. We note, however, that anchor-away depletion is not equivalent to a null allele, and thus an epistasis analysis is less clear. It remains a possibility that these factors also function within the same pathway to promote genome stability.
INO80C and Mot1 Prevent DNA DSBs Near Yeast Replication Origins
We entertained the idea that sensitivity to genotoxic stress agents might be linked to increased cryptic transcription. For instance, increased transcription may lead to conflicts between the replication and transcription machineries, leading to DNA DSBs. To test this possibility, we used Break-seq to map DSBs genome-wide (Hoffman et al., 2015). We treated cells with rapamycin for 1 h to deplete proteins from the nucleus (Ino80, Mot1, or both), followed by treatment with or without 0.1 M HU for 1 h to create replication stress (Figure 4A). After cell lysis, DNA breaks were end-repaired with biotinylated ATP, and sequencing libraries were prepared as described previously (Hoffman et al., 2015). Initially, Genome Browser views indicated a greater number of DSBs in all mutants compared with wild-type (Figure 4B). Next, we used MACS2 to identify genomic regions enriched for end-labeled DSB signals, and bubble plots were used to illustrate both the number of peaks as well as their DSB density (e.g., break signal within each peak). In wild-type cells, the number of DSB peak regions increased following HU treatment, and there was also as a small increase in peak density (Figure 4C). In the absence of HU, depletion of Ino80 increased the number and intensity of peak regions, with more dramatic increases near origins (ARSs). Furthermore, HU treatment led to a large increase in DSB density near ARSs in the absence of Ino80 (Figure 4C). In contrast, loss of Mot1 had little impact on DSBs, though co-depletion of both Ino80 and Mot1 resulted in large increases in both the number and density of DSBs peaks, especially near replication origins (Figure 4C). Significantly, these increases were most prominent near ARS elements with high levels of Arp5 (top 100), correlating DSB formation to INO80C binding (Figure 4D). To test whether DSB formation also correlated with the level of promiscuous transcription, we grouped origins into quartiles on the basis of the level of ncRNAs in the absence of both INO80C and Mot1 and then used bubble plots to illustrate DSB formation. Importantly, this analysis showed a strong correlation between high levels of ncRNA near an origin and the formation of new DSBs (Figure 4E). These data support the view that INO80C and Mot1 prevent promiscuous transcription events that cause new DSBs.
Figure 4. INO80C and Mot1 Prevent Double-Strand Breaks Near Yeast Replication Origins.
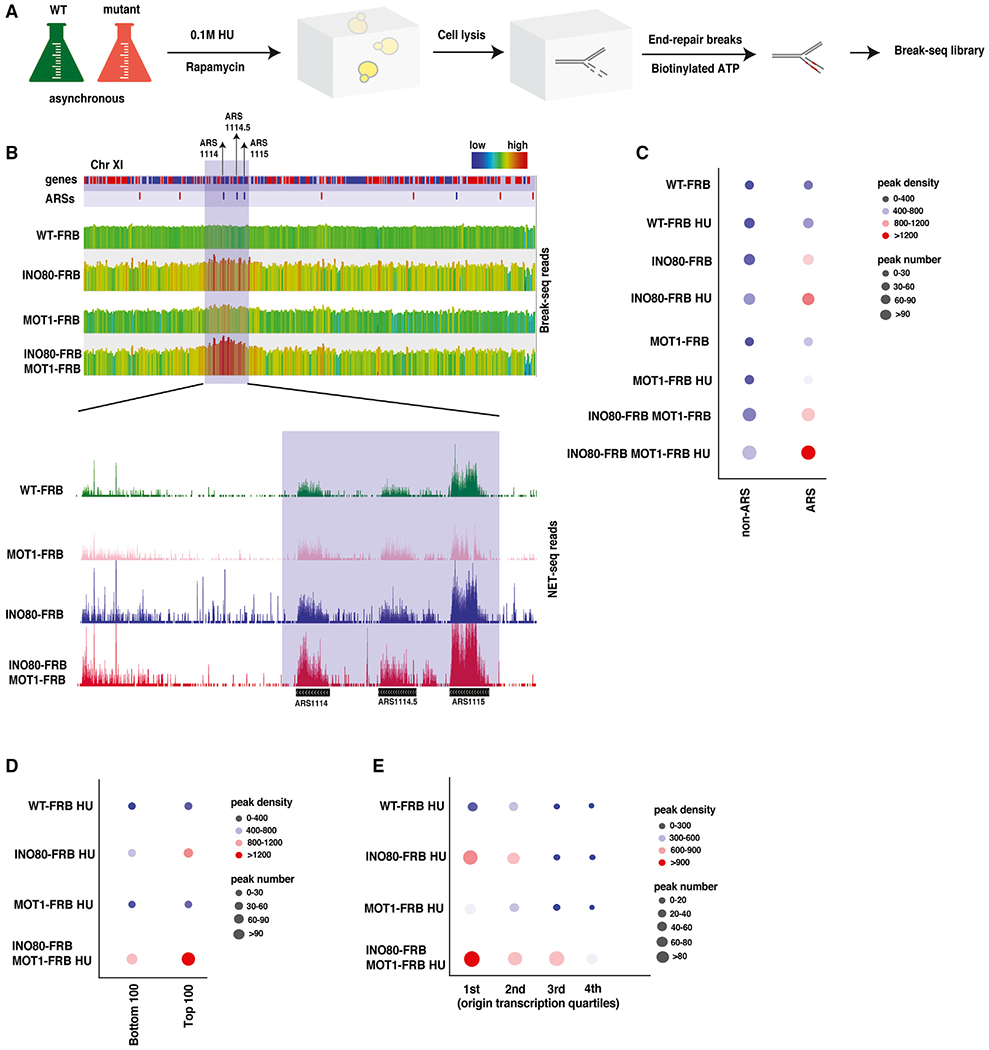
(A) Experimental design for Break-seq. Wild-type and mutant cells were treated with or without 0.1MHU and rapamycin for 1 h. Following cell lysis and end-repair for breaks, libraries were prepared.
(B) Representative Genome Browser views of Break-seq and NET-seq for WT-AA, INO80-AA, Mot1-AA, and INO80-AA Mot1-AA for a highlighted region in yeast chromosome (Chr) XI. Notice the increase in intensity of peaks over highlighted region for origins ARS1114, ARS1114.5, and ARS1115 in the Break-seq Genome Browser view (top panel). The highlighted region in Break-seq is zoomed in and represented in the NET-seq Genome Browser view to show nascent transcript levels for the same region (middle panel).
(C–E) Bubble plots representing both peak densities and peak numbers for WT-AA, INO80-AA, Mot1-AA, or INO80-AA Mot1-AA, with or without 0.1MHU for ARS or non-ARS regions in (C) and for all strains with 0.1MHU for bottom 100 or top 100 ARSs in (D), and for all yeast origins sorted from highest to lowest (first quartile being the highest and fourth quartile being the lowest) on the basis of cryptic transcription levels around corresponding yeast origins by NET-seq in (E). Number of yeast origins in each quartile is as following: first quartile (n = 63), second quartile (n = 63), third quartile (n = 63), and fourth quartile (n = 64).
Ino80 and Mot1 Regulate Nascent Transcription around Origins in mESCs
The existence of cryptic transcription in both yeast and mammals, combined with the evolutionary conservation of INO80C, Mot1, and NC2, led us to investigate whether these factors regulate cryptic transcription around origins in murine ESCs (mESCs). We compared ChIP-seq data for murine Mot1 (BTAF1), INO80C, and NC2 (termed MINC in our previous study; Xue et al., 2017) with the position of RISs in mESCs determined by sequencing of purified RNA-primed nascent DNA (Cayrou et al., 2015) (Figure 5). In metazoans, RIS lack a defined ARS and are typically mapped by a variety of ChIP and DNA sequencing (DNA-seq) techniques. We found that 40%–60% of MINC peaks overlap with RISs, while 40%–50% of RIS overlap with peaks of MINC (Figure 5A). We further compared the co-localization between MINC and RIS within a 3 kb region flanking either side of the RIS and divided these into two clusters (C1 and C2; Figure 5B). Note that these two clusters include all high-confidence peaks that were previously identified as RISs (Cayrou et al., 2015). MINC largely co-localized with strong RISs in cluster C1 but with weak RISs in cluster C2 (Figure 5C). Importantly, the increase in nascent RNA cryptic transcription, observed upon small interfering RNA (siRNA) knockdown of both Mot1 and Ino80, correlated closely with RISs in cluster C1 (Figure 5C). To identify the differences between cluster C1 and cluster C2, the genomic distribution of C1 and C2 was analyzed using the Cis-Regulatory Element Annotation System (CEAS) (Ji et al., 2006). CEAS analysis showed that cluster C1 comprises a large portion of promoter regions, while cluster C2 consists of introns and distal intergenic regions (Figure 5D). This observation was further confirmed using region-gene association plots, which showed that the majority of the positions (70%) in cluster C1 were localized within 5 kb of the TSS, while those in cluster C2 are >5 kb from TSS (Figure 5E). Collectively, the data suggest that the highly conserved Mot1, INO80C, and NC2 control cryptic transcription around yeast and mammalian origins. Thus, the regulation of chromatin remodeling and PIC assembly around origins is a conserved mechanistic principle to maintain genomic stability.
Figure 5. Ino80 and Mot1 Regulate Nascent Transcription around Origins in mESCs.
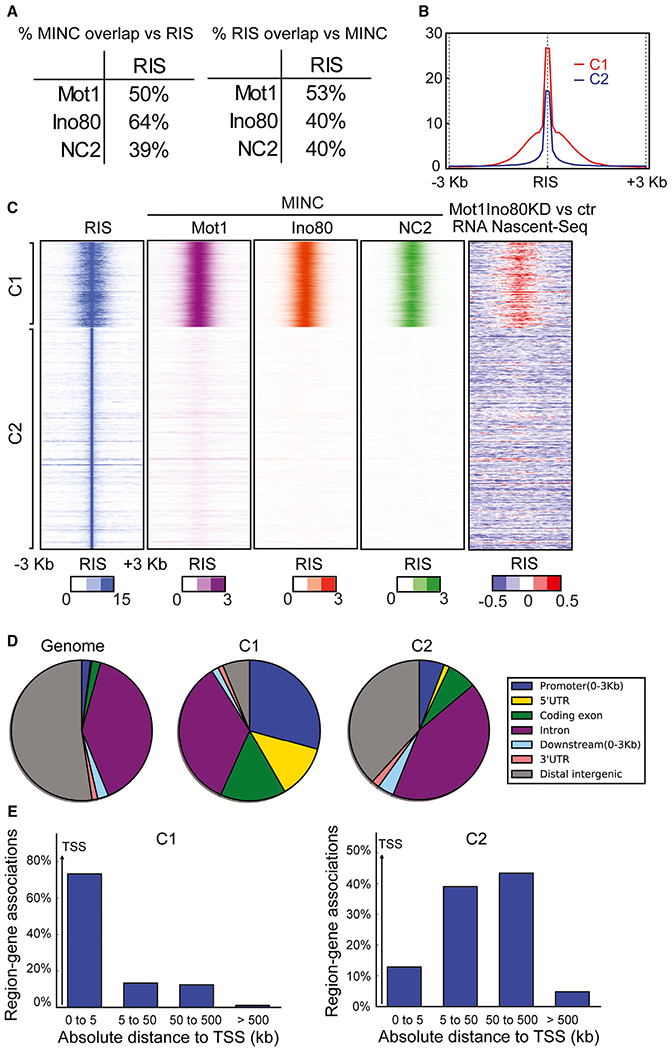
(A) Percentages of MINC overlap versus RIS.
(B) Plot showing average RIS signal intensities centered at RIS (and a region of 3 kb upstream and downstream of RIS) for two clusters C1 (red) and C2 (blue).
(C) Heatmaps of (RIS) densities, Mot1, INO80, and NC2 enrichment alongside changes in genic nascent RNA expression in INO80 Mot1 knockdown (log2 RPKM), separated in two clusters (C1 and C2) using the average enrichment of +3 or −3 kb around the RIS region.
(D) Pie charts showing percentages of different genomic regions represented in the two clusters, C1 and C2.
(E) Region-gene specific plot showing distribution of cluster C1 and C2 positions. The y axis represents percentage of peak positions found at a given genomic location from TSS (x axis) for two clusters C1 and C2.
DISCUSSION
Non-genic or pervasive transcription in yeast was initially discovered via mutations that eliminated transcription elongation factors controlling nucleosome assembly behind transcribing RNAPII (reviewed in Jensen et al., 2013). Further studies identified several classes of extragenic cryptic RNAs in yeast, including CUTs, SUTs, and XUTs, among others. Although the functionality of cryptic transcripts represents an active area of investigation, a less well studied but important question is how such transcripts affect genome stability. Collisions between RNA and DNA polymerases can generate DNA damage, leading to double-stranded DNA breaks (Helmrich et al., 2013), and cryptic transcription through an ARS has been suggested to decrease its efficiency of use during S phase (Candelli et al., 2018; Soudet et al., 2018). Collectively, nucleosome remodeling and PIC assembly must be tightly controlled to allow efficient origin firing and function. However, the proteins and mechanisms underlying such control are largely unknown.
Recently, we found that the INO80C remodeler localizes to gene boundaries, together with two transcription factors that negatively control PIC assembly, Mot1 and NC2. All three factors prevent the spreading of transcription into intergenic regions (Xue et al., 2017). Multiple studies have shown that INO80C regulates transcription in yeast and human cells (Cai et al., 2007; Shen et al., 2000; Yao et al., 2016). INO80C is also recruited to replication forks and helps maintain replisome stability and genomic integrity (Papamichos-Chronakis and Peterson, 2008; Shimada et al., 2008). However, whether INO80C’s role as a transcriptional regulator relates to its role in genome maintenance during replication has not been investigated. Here, we report that INO80C functions together with Mot1 at replication origins where they maintain genome stability by preventing cryptic transcription.
We have found that three components of what we previously named MINC—Mot1, INO80C, and NC2—bind to yeast and mammalian origin regions. Further characterization revealed that although the DNA sequence of a yeast origin is essential for binding of INO80C, the Orc2 subunit of the ORC is not required. This suggests that INO80C might use the nucleosome-depleted nature of ARS elements for recruitment, much in the same way that the related SWR1C complex is recruited to nucleosome-depleted regions (NDRs) at gene promoter regions (Ranjan et al., 2013). Alternatively, INO80C may be recruited by members of the forkhead family of transcription factors (e.g., Fkh1, Fkh2) which, like INO80C, bind primarily to early-firing yeast origins (Ostrow et al., 2014). Our data indicate that INO80C functions with other remodelers to reinforce proper positioning and occupancy of nucleosomes surrounding origins. Such positioning may facilitate more efficient origin firing and help occlude cryptic RNAPII promoters.
Together with the ability of Mot1 and NC2to block assembly of PICs, we envision that INO80C remodeling blocks spurious transcription from impinging on replication functions, such as limiting transcription-replication conflicts that can lead to DSBs.
Indeed, depletion of Ino80 and Mot1 leads to an increase in nascent transcripts from the Crick DNA strand on the A-rich side of ARS elements which supports the initial unwinding of origin DNA (Coster and Diffley, 2017). This model implies that INO80C and Mot1 may block spurious transcription during S phase, at the same time as replication initiation. Together with our observation that increased levels of ncRNAs correlates with greater numbers of DSBs near origins, these data strongly suggest that INO80C and Mot1 maintain genome stability by preventing promiscuous transcription events. Our results are also consistent with a recent study demonstrating that noncoding transcription correlates with low ARS efficiency and late replication timing (Soudet et al., 2018).
The conservation of INO80C and Mot1 (BTAF1) suggests the two proteins may play similar roles in in mammals. Indeed, depletion of both proteins individually or simultaneously in mESCs leads to increases in cryptic transcription around previously mapped RISs. Moreover, mining of The Cancer Genome Atlas (TCGA) has revealed that INO80C is widely amplified across cancer subtypes (Lee et al., 2017), and several cancers have demonstrated an inability to proliferate in the absence of INO80C (Zhang et al., 2017; Zhou et al., 2016). Although a role for INO80C in controlling cancer cell transcription has been proposed, the requirement of INO80C for tumor growth is unknown. On the basis of our findings in yeast, we speculate that INO80C may function as a genome protectant in fast-dividing tumor cells possibly through preventing transcription-replication conflicts.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Craig L. Peterson. (craig.peterson@umassmed.edu).
Materials Availability
Yeast strains generated in this study are available on request from the lead contact, Craig L. Peterson (craig.peterson@umassmed.edu).
Data and Code Availability
All data have been deposited in the Gene Expression Omnibus (GEO) under accession GSE144072 and GSE95633.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains used in this study are derivatives of either W303 (MATa his3-11, 15 leu2-3,112 trp1Δ ura3-1 ade2-1 can1-100) or BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0). Unless otherwise noted, cells were cultivated in YPD (10% yeast extract, 20% bacterial peptone, and 2% glucose) at 30°C. S. pombe cells (JY741, WT Flag-Rbp3) (courtesy of Dr. Makoto Kimura, Kyushu University) were cultivated in YES (yeast extract, 10Xaa supplement and 3% glucose). For α factor arrest, cells were grown to a density of 1.5-2.5 × 107 cells/ml in YPD and arrested by 5 μg/ml αF for 1 h. Arrest was confirmed by microscopic observation after 90 min. For nocodazole arrest, overnight cultured cells were diluted to OD600 = 0.1 and cells were grown to OD600 = 0.4 in YPD and arrested by 0.2M nocodazole for 1 h. The full strain list is shown in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT and RESOURCES | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG M2 Affinity Gel | Millipore Sigma | Cat#A2220 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080093 |
| Random Primers | Thermo Fisher Scientific | Cat#48190011 |
| Hydroxyurea (HU) | US Biological Life Sciences | Cat#127-07-1 |
| Methyl methanesulfonate | Millipore Sigma | Cat#129925 |
| Rapamycin | LC Laboratories | Cat#R-5000 |
| Camptothecin | Millipore Sigma | Cat#CP9911 |
| RNaseH | Thermo Fisher Scientific | Cat#18021071 |
| AmpureXP | Beckman Coulter | Cat#A63880 |
| RNase-Free DNase Set | QIAGEN | Cat#79254 |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher Scientific | Cat#65001 |
| Taq DNA Polymerase | New England Biolabs | Cat#M0273S |
| T4 DNA Ligase | New England Biolabs | Cat#M0202 |
| T4 DNA Polymerase | New England Biolabs | Cat#M0203 |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | Cat#M0201 |
| NEB Buffer 2 | New England Biolabs | Cat#B7002 |
| dATP Solution | New England Biolabs | Cat#N0440 |
| Klenow Fragment 3′ to 5′ Exo | New England Biolabs | Cat#M0212 |
| T4 DNA Ligase (Rapid) | Enzymatics | Cat#L6030-HC-L |
| Uracil-DNA Glycolylase (UDG) | New England Biolabs | Cat#M0280 |
| Phusion High Fidelity DNA Polymerase | New England Biolabs | Cat#M0530 |
| RQ1 RNase-Free DNase | Promega | Cat#M6101 |
| Manganese(II) Chloride Solution | Millipore Sigma | Cat#M1787 |
| 3x FLAG Peptide | Millipore Sigma | Cat#F4799 |
| SUPERase.In Rnase Inhibitor | Thermo Fisher Scientific | Cat#AM2694 |
| cOmplete, EDTA-Free Protease Inhibitor Cocktail | Millipore Sigma | Cat#11873580001 |
| Alpha-Factor Mating Pheromone | Zymo Research | Cat#Y1001 |
| T4 RNA Ligase 2, truncated | New England Biolabs | Cat#M0242 |
| Gel Loading Buffer II | Thermo Fisher Scientific | Cat#8546G |
| 10 bp DNA Ladder | Thermo Fisher Scientific | Cat#10821-015 |
| Novex TBE-Urea Gels 10% | Thermo Fisher Scientific | Cat#EC6875BOX |
| Novex TBE-Urea Gels 15% | Thermo Fisher Scientific | Cat#EC6885BOX |
| SYBR Gold Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat#S11494 |
| GlycoBlue Coprecipitant | Thermo Fisher Scientific | Cat#AM9515 |
| CircLigase ssDNA Ligase | Lucigen | Cat#CL4111K |
| Proteinase K | Millipore Sigma | Cat#P2308 |
| Biotin-14-dATP | Thermo Fisher Scientific | Cat#19524016 |
| β-agarose | New England Biolabs | Cat#M0392L |
| HiFi HotStart Ready Mix | Kapa Biosystems | Cat#KK2601 |
| NuSieve GTG Agarose | Lonza | Cat#50081 |
| Critical Commercial Assays | ||
| QIAGEN RNeasy Mini Kit | QIAGEN | Cat#74104 |
| QIAGEN miRNeasy Mini Kit | QIAGEN | Cat#217004 |
| RNA Clean and Concentrator | Zymo Research | Cat#R1013 |
| NextSeq 500/550 High Output v2 Kit (75 cycles) | Illumina | Cat#FC-404-2005 |
| NextSeq 500/550 Mid Output v2 Kit (150 cycles) | Illumina | Cat#FC-404-2001 |
| Qubit 1X dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q33230 |
| End-It DNA End-Repair Kit | Thermo Fisher Scientific | Cat#ER81050 |
| miRNeasy Micro Kit | QIAGEN | Cat#217084 |
| Deposited Data | ||
| ChIP-seq, NET-seq, Break-seq | This study | GSE144072 |
| RNA-seq and ChIP-seq for mESCs | Xue et al., 2017 | GSE95633 |
| Experimental Models: Organisms/Strains | ||
| WT Rbp3-3xFLAG ((BY4741) MATa his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 rbp3::RBP3-3xFLAG::NAT) | Churchman and Weissman, 2011 | N/A |
| WT Rbp3-3xFLAG AA strain ((HHY221) MATa tor1-1 fpr1::loxP-LEU2-loxP RPL13A-2 x FKBP12:loxP BAR1Δ::HISG rbp3::RBP3-3xFLAG::NAT) | Topal et al., 2019 | N/A |
| WT-FRB, MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 tor1-1 fpr1::NAT RPL13A-2 x FKBP12::TRP1 Δade2-1::ADE2) | Marc Timmers | N/A |
| INO80-FRB, isogenic to WT-FRB except INO80-FRB::hyhMX6 | Marc Timmers | N/A |
| Mot1-FRB, isogenic to WT-FRB except Mot1-FRB::HIS3 | Marc Timmers | N/A |
| INO80-FRB Mot1-FRB, isogenic to WT-FRB except INO80-FRB::hyhMX6 Mot1-FRB::HIS3 | Marc Timmers | N/A |
| IN080-FRB Rpb3-3xFLAG, isogenic to WT-FRB Rpb3-3xFLAG strain except INO80-FRB::hyhMX6) | This study | N/A |
| Mot1-FRB Rpb3-3xFLAG, isogenic to WT-FRB Rpb3-3xFLAG strain except Mot1-FRB::HIS3) | This study | N/A |
| IN080-FRB Mot1-FRB Rpb3-3xFLAG, isogenic to WT-FRB Rpb3-3xFLAG strain except for INO80-FRB::hyhMX6 Mot1-FRB::HIS3)) | This study | N/A |
| Orc2-FRB, MATa tor1-1 fpr1::loxP-LEU2-loxP RPL13A-2x FKBP12:loxP Bar1Δ::HISG Orc2-FRB::HIS3MX6 ORC5-V5::hyhMX6 | This study | N/A |
| WT Rbp3-3xFLAG S. pombe strain (h-Flag-rbp3 ade6-M216ura4-D18leu1) | NBRP (Kimura et al., 2001) | FY17156 |
| Software and Algorithms | ||
| GraphPad Prism v 5.0 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| R | RStudio | https://www.r-project.org/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| TopHat2 | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/index.shtml. RRID: SCR_013035 |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/. RRID: SCR_002105 |
| Bedtools | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2. RRID: SCR_006646 |
| deepTools | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ RRID: SCR_016366 |
| PIVOT | Zhu et al., 2018 | https://kim.bio.upenn.edu/software/pivot.shtml |
| HTseq 0.9.1 | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.9.1/install.html RRID: SCR_005514 |
| Galaxy web platform | Afgan et al., 2018 | https://usegalaxy.org |
| MACS Peak Caller | Zhang et al., 2008 | https://github.com/macs3-project/MACS RRID: SCR_013291 |
| SeqMonk | N/A | http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/ RRID: SCR_001913 |
| Other | ||
| Whatman nitrocellulose membrane filters | Millipore Sigma | Cat#7184-009 |
| Mixer Mill MM 400 | Retsch | N/A |
| Qubit Assay tubes | Thermo Fisher Scientific | Cat#Q32856 |
| Bioruptor Standard | Diagenode | UCD-200 |
| 5′ PRIME Phase Lock Gel Heavy | VWR | Cat#10847-802 |
| Corning Costar SpinX Centrifuge Tube Filters | Millipore Sigma | Cat#CL8162 |
METHOD DETAILS
Serial Dilution Growth Assay
Cells were cultured to saturation in 5 mL YPD overnight. Cells were diluted to an OD600 = 0.1 and were grown until OD600 = 1. Cells were resuspended in sterile dH2O, serially diluted 10-fold four times, and 6 μL of each dilution was spotted onto plates of indicated media. Where used, DMSO was0.1% vol/vol, rapamycin was 8 μg/ml, methylmethanesulfonate (MMS) was 0.005% wt/vol, hydroxyurea (HU) was 0.1M and camptothecin was 10 μg/ml.
NET-seq
Library Construction
NET-seq conditions, immunoprecipitations, isolation of nascent RNA, and library construction were carried out as described (Churchman and Weissman, 2011) for 2 wild-type biological replicates and 2 mutant biological replicates (INO80-FRB, Mot1-FRB and INO80-FRB Mot1-FRB) with several modifications including addition of S. pombe cells as spike-in control. Overnight cultures from single yeast colonies were diluted to an OD600 = 0.05 in 1 L of YPD. Cells were grown at 30°C to OD600 = 0.8. Rapamycin was added to 8 μg/mL at OD600 = 0.25 for cells in the anchor-away background, and cells were grown for 1 h (OD600 = 0.7-0.8). To normalize sequencing libraries, S. pombe cells were mixed with S. cerevisiae cells at a 1:10 ratio, and the cells harvested by filtration and cryogenic lysis. 3xFLAG-tagged RNA Pol II was immunoprecipitated and nascent RNAs were purified using miRNAeasy mini kit (QIAGEN). Following ligation of pre-adenylated DNA linker onto purified nascent RNAs, RNAs were fragmented and reverse transcribed. Resulting cDNAs were circularized using DNA Circligase (Lucigen). Final PCR was performed to obtain double stranded product to sequence. Size of the library was determined by Fragment Analyzer and the concentrations were determined by Qubit 4.0 fluorometer (Invitrogen). 3′ end sequencing of all samples was carried out on an Illumina NextSeq 500 with a read length of 75 (single end).
Data Analysis
NET-seq reads were processed and aligned using the Galaxy web platform (Afgan et al., 2018). The adaptor sequence was (ATCTCGTATGCCGTCTTCTGCTTG) removed and the random hexamer sequence was removed from the 5′ end. The 3′ ends of the reads were then trimmed for quality using FASTQ Quality Timmer by sliding window (Blankenberg et al., 2010) with a window size of 10 and a step size of 5. The reads were trimmed until the aggregate score was ≥ 21. Reads were first aligned using Bowtie2 (Langmead and Salzberg, 2012; Langmead et al., 2009) to a combined FASTAfile of S. cerevisiae and S. pombe rRNA, tRNA, and RDN sequences to remove contaminating reads. Reads were then aligned to a combined version of the S. cerevisiae genome (SacCer3, SGD) and the S. pombe genome (ASM294v.2, PomBase) with TopHat2 (Kim et al., 2013), allowing up to three mismatches. The reads were separated by their respective genomes with SAMtools (Li et al., 2009), and only uniquely mapped reads were used for further analyses. Libraries were normalized by scaling the uniquely mapped S. pombe reads to 100,000 reads. This scaling factor was then used to scale the uniquely mapped S. cerevisiae reads. To account for differences between sequencing run depth for various NextSeq runs, the pombe-scaled WT S. cerevisiae read counts were then scaled to 1 M reads, and this additional scaling factor was included to scale the sample reads. Finally, only the 5′ end of the sequencing read, which corresponds to the 3′ end of the nascent RNA was recorded and used for downstream analyses. TSS and TTS annotation was obtained from (Xu et al., 2009). Read counts for genes and non-coding regions were obtained by summing normalized base pair reads over the region of interest. For average profiles, BAM files of biological replicates were merged and processed as above, and only genes longer than 500 bp were analyzed. Genes were scaled to 500 bp, and samples were scored in 1 bp bins using deepTools program (Ramírez et al., 2016). Reads were analyzed as described (Harlen et al., 2016). To calculate 5′ to 3′ ratios, the sum of reads from 1-250 bp from the TSS were divided by the sum of reads 250 bp upstream of the TTS to the TTS.
Break-seq
Library Construction
Break-seq libraries were prepared as described in Hoffman et al. (2015) for 2 biological replicates. Overnight cultures were diluted to an OD600 = 0.1 in 100 mLYPD. Cells were grown at 30C until OD600 = 0.45-50. Rapamycin was added at a final concentration of 8 μg/ml and cells were grown for 1 h (until OD600 = 1.0). Cells were harvested and resuspended in 1ml of 50 mM EDTA. 50 μL of cells were combined with 50 μL of 1% low melting temperature agarose (Lonza). Following in-gel labeling and sonication, libraries were prepared as described (Hoffman et al., 2015). Paired-end sequencing was performed on an Illumina NextSeq 500 with a read length of 75 bp.
Data Analysis
FASTQ files from paired end libraries were collapsed by barcode and the Illumina adaptor sequence was trimmed from the 3′ ends. Files were uploaded and analyzed using the Galaxy web platform (Afgan et al., 2018). Reads were aligned to S. cerevisiae genome (SacCer3, SGD) using Bowtie2 with a maximum fragment length for valid paired-end alignments set to 500 bp. Aligned reads were then filtered for quality and only uniquely mapped paired reads were used for future analyses. BAM files were uploaded to SeqMonk and normalized by scaling to 1 million reads. MACS Peak Caller was used to identify new peaks in INO80-FRB, Mot1-FRB, and INO80-FRB Mot1-FRB biological replicates using the mean WT-FRB file as the input (p value 1x10−5, 500 bp fragment size). Genome browser views were obtained by generating probes using a running window with a probe size of 1000 bp and a step size of 500 bp.
ChIP-seq
Library Construction for Yeast
Orc2-FRB cells were arrested in G2/M with 0.2M nocodazole for 1 h, and Orc2 was depleted by 8 μg/ml rapamycin for 1 h. Cells were washed with dH2O three times to release from G2 and arrested in G1 by 5 μg/ml α-factor for 1 h. After that, ChIP-seq libraries were prepared as described in Xue et al. (2017). Briefly, cells were collected and crosslinked with 1% formaldehyde. Samples were lysed with glass beads in lysis buffer (50 mM HEPES, 140 mM NaCl, 10 mM EDTA, 5 mM EGTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.5% N-Lauroyl Sarcosine) with protease inhibitors. Chromatin was collected by centrifugation, resuspended in lysis buffer and subjected to sonication. The supernatant from sonicated lysates were precleared with Protein A/G beads and ChIP was performed as described in Kitada et al. (2012) using commercial antibodies (α-V5 [Invitrogen]; α-Arp5 [Abcam]). Libraries were prepared with a KAPA LTP kit (Ilumina) and sequenced using the Illumina HiSeq 2000 for 50 bp single end reads.
Data Analysis
FASTQ files from paired end libraries were collapsed by barcode and the Illumina adaptor sequence was trimmed from the 3′ ends. Files were uploaded and analyzed using the Galaxy web platform (Afgan et al., 2018). Reads were aligned to S. cerevisiae genome (SacCer3, SGD) using Bowtie2. After filtering out the clonal reads, the IP and input samples were shuffled to similar reads, IP samples were normalized to input using a custom script. S. cerevisiae genomes were divided into 50-bp windows, and significant windows with a p value lower than 0.001 were selected as described in Ferrari et al. (2012). The log2 ratio of ChIP versus input at significant windows was used to generate profiles centered at the ACS. Yeast MINC binding sites were obtained from GSE95633 (Xue et al., 2017) for the replication origin analysis.
Yeast mRNA-Seq Replication Analysis
Yeast mRNA-seq in control, INO80, Mot1, Nc2α, Ino80&Nc2α and Mot1&INO80 anchor away were obtained from GSE95633 (Xue et al., 2017). The log2 ratio of RNA transcription level in mutant versus WT were calculated based on different sizes of region centered at ACS and plot over window size.
mESC Replication Analysis
MINC binding sites, nascent RNA-seq in control and Mot1&INO80 double siRNA knockdown, and Replication Initiation Sites (RIS) in mouse ES cells were obtained from GSE95633 (Xue et al., 2017) and GSE68347 (Cayrou et al., 2015) respectively. The overlap between MINC and RIS was conducted using BEDTools (Quinlan, 2014) and the MINC binding and RIS occupancy profiles centered at the RIS were separated into two groups using the k-means clustering algorithm in Cluster 3.0 (de Hoon et al., 2004). The positions in cluster C1 and C2 were further analyzed using CEAS (Ji et al., 2006) and GREAT (http://great.stanford.edu/public/html/) for the genomic distribution and distance to the TSS, respectively. The analysis of MINC binding, RIS occupancy, and relative nascent RNA change centered at either TSS or RIS were as in our previous study (Xue et al., 2017).
Nucleosome Occupancy Analysis
Nucleosome occupancy mapping data in Figure 4 was obtained from a previously published dataset under accession number GSE115412 (Kubik et al., 2019). Pre-processed and normalized bigwig files for WT and depletion of the chromatin remodelers INO80, Isw2 and Sth1 were reanalyzed in the Galaxy platform (Afgan et al., 2018). Briefly, bigwig files were used to generate normalized nucleosome occupancy values to plot over ARS region (and 500 bp upstream or downstream of ACS).
ChEC-seq Data Analysis
Heatmaps for INO80 and Isw2 enrichments over ACS regions and genome browser view for INO80 peaks in Figure S2 were obtained from a previously published dataset under accession number GSE115412 (Kubik et al., 2019). Pre-processed and normalized bigwig files for INO80 and Isw2 enrichments were reanalyzed in the Galaxy Platform (Afgan et al., 2018). Briefly, bigwig files were used to generate normalized ChEC-seq enrichment values to plot over ARS region (+/− 1 kb).
TATA-box Frequency Analysis
TATA-box frequency scores were obtained by searching the canonical TATAWAWR motif in genomic locations for yeast replication origins using FIMO from the MEME Suite with a threshold of p < 0.001. Then, the frequency scores were plotted over ACS (+/− 0.2 kb) for all annotated yeast replication origins.
Primers Used in This Study
DNA linker (Churchman and Weissman, 2011): /5rApp/(N1:25252525)(N1)(N1)(N1)(N1)(N1) CTGTAGGCACCATCAAT/3ddC/
RT primer oLSC007 (Churchman and Weissman, 2011): 5 phos/atctcgtatgccgtcttctgcttg/iSp18/cactca/iS p18/tccgacgatcatt gatggtgcctacag 3
Reverse primer oNTI231 (Churchman and Weissman, 2011): 5′ CAAGCAGAAGACGGCATACGA 3′
Custom primer for NET-seq oLSC006 (Churchman and Weissman, 2011): 5′ -TCCGACGATCATTGATGGTGCCTACAG 3′
Internal RNA control for NET-seq oGAB11 (Churchman and Weissman, 2011): 5′ agu cac uua gcg aug uac acu gac ugu g 3′
QUANTIFICATION AND STATISTICAL ANALYSIS
Software and statistical analysis details can be found in the Methods Details section of the STAR Methods, as well as the Key Resources table.
Supplementary Material
Highlights.
The INO80C remodeler is bound to replication origins in yeast and mESCs
INO80C and the Mot1 repressor prevent pervasive transcription at origins
INO80C works with other remodelers to organize nucleosomes at origins
Loss of INO80C and Mot1 leads to new DNA double-strand breaks near origins
ACKNOWLEDGMENTS
We thank members of the Peterson and Carey labs for helpful discussions and Mike Kronenberg for discussion comments. We also thank the Stirling Churchman (Harvard Medical School) and William Theurkauf (University of Massachusetts Medical School [UMMS]) labs for sharing sequencing protocols and helping with troubleshooting. We also thank Ruijia Wang (UMMS) for analysis of Break-seq data. This work was supported by grants from the National Institutes of Health (NIH) to C.L.P. (R35GM122519) and M.F.C. (R01GM074701). Y.X. was also supported by the National Natural Science Foundation of China (31670088).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108106.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. (2018). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46 (W1), W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcid EA, and Tsukiyama T (2014). ATP-dependent chromatin remodeling shapes the long noncoding RNA landscape. Genes Dev. 28, 2348–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl P, and Huber W (2015). HTSeq-a Python framework to work with high-throughput sequencing data. Bionformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio OM (2013). Location, location, location: it’s all in the timing for replication origins. Genes Dev. 27, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auble DT, Hansen KE, Mueller CG, Lane WS, Thorner J, and Hahn S (1994). Mot1, a global repressor of RNA polymerase II transcription, inhibits TBP binding to DNA by an ATP-dependent mechanism. Genes Dev. 8, 1920–1934. [DOI] [PubMed] [Google Scholar]
- Azmi IF, Watanabe S, Maloney MF, Kang S, Belsky JA, MacAlpine DM, Peterson CL, and Bell SP (2017). Nucleosomes influence multiple steps during replication initiation. eLife 6, e22512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaric S, Luckenbach T, Schmid A, Blaschke D, Horz W, and Korber P (2007). Redundancy of chromatin remodeling pathways for the induction of the yeast PHO5 promoter in vivo. J. Biol. Chem 282, 27610–27621. [DOI] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, and Nekrutenko A; Galaxy Team (2010). Manipulation of FASTQ data with Galaxy. Bioinformatics 26, 1783–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon YT, and Bielinsky AK (2006). The spatial arrangement of ORC binding modules determines the functionality of replication origins in budding yeast. Nucleic Acids Res. 34, 5069–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Jin J, Yao T, Gottschalk AJ, Swanson SK, Wu S, Shi Y, Washburn MP, Florens L, Conaway RC, and Conaway JW (2007). YY1 functions with INO80to activate transcription. Nat. Struct. Mol. Biol 14, 872–874. [DOI] [PubMed] [Google Scholar]
- Candelli T, Challal D, Briand JB, Boulay J, Porrua O, Colin J, and Libri D (2018). High-resolution transcription maps reveal the widespread impact of roadblock termination in yeast. EMBO J. 37, e97490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang Y, and Prelich G (2002). Direct stimulation of transcription by negative cofactor 2 (NC2) through TATA-binding protein (TBP). Proc. Natl. Acad. Sci. U S A 99, 12727–12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrou C, Ballester B, Peiffer I, Fenouil R, Coulombe P, Andrau JC, van Helden J, and Mechali M (2015). The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 25, 1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AL, Ormerod G, Durley SC, Sing TL, Brown GW, Kent NA, and Downs JA (2012). The INO80 chromatin remodeling complex prevents polyploidy and maintains normal chromatin structure at centromeres. Genes Dev. 26, 2590–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, and Weissman JS (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coster G, and Diffley JFX (2017). Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science 357, 314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, and Miyano S (2004). Open source clustering software. Bioinformatics 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Diffley JF, Cocker JH, Dowell SJ, and Rowley A (1994). Two steps in the assembly of complexes at yeast replication origins in vivo. Cell 78, 303–316. [DOI] [PubMed] [Google Scholar]
- Diffley JF, Cocker JH, Dowell SJ, Harwood J, and Rowley A (1995). Stepwise assembly of initiation complexes at budding yeast replication origins during the cell cycle. J. Cell Sci Suppl. 19, 67–72. [DOI] [PubMed] [Google Scholar]
- Eaton ML, Galani K, Kang S, Bell SP, and MacAlpine DM (2010). Conserved nucleosome positioning defines replication origins. Genes Dev. 24, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbert R, Birkmann A, and Schdller HJ (1999). The product of the SNF2/SWI2 paralogue INO80 of Saccharomyces cerevisiae required for efficient expression of various yeast structural genes is part of a high-molecular-weight protein complex. Mol. Microbiol 32, 741–751. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Su T, Li B, Bonora G, Oberai A, Chan Y, Sasidharan R, Berk AJ, Pellegrini M, and Kurdistani SK (2012). Reorganization of the host epigenome by a viral oncogene. Genome Res. 22, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros J, Devbhandari S, and Remus D (2014). Origin plasticity during budding yeast DNA replication in vitro. EMBO J. 33, 621–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Trotta KL, Smith EE, Mosaheb MM, Fuchs SM, and Churchman LS (2016). Comprehensive RNA polymerase II interactomes reveal distinct and varied roles for each phospho-CTD residue. Cell Rep. 15, 2147–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, and Laemmli UK (2008). The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell 31,925–932. [DOI] [PubMed] [Google Scholar]
- Hawkins M, Retkute R, Meller CA, Saner N, Tanaka TU, de Moura AP, and Nieduszynski CA (2013). High-resolution replication profiles define the stochastic nature of genome replication initiation and termination. Cell Rep. 5, 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Nudler E, and Tora L (2013). Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol 20, 412–418. [DOI] [PubMed] [Google Scholar]
- Heun P, Laroche T, Raghuraman MK, and Gasser SM (2001). The positioning and dynamics of origins of replication in the budding yeast nucleus. J. Cell Biol 152, 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EA, McCulley A, Haarer B, Arnak R, and Feng W (2015). Break-seq reveals hydroxyurea-induced chromosome fragility as a result of unscheduled conflict between DNA replication and transcription. Genome Res. 25, 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TH, Jacquier A, and Libri D (2013). Dealing with pervasive transcription. Mol. Cell 52, 473–484. [DOI] [PubMed] [Google Scholar]
- Ji X, Li W, Song J, Wei L, and Liu XS (2006). CEAS: cis-regulatory element annotation system. Nucleic Acids Res. 34, W551–W554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Sakurai H, and Ishihama A (2001). Intracellular contents and assembly states of all 12 subunits of the RNA polymerase II in the fission yeast Schizosaccharomyces pombe. European Journal of Biochemistry 268, 612–619. [DOI] [PubMed] [Google Scholar]
- Kitada T, Kuryan BG, Tran NN, Song C, Xue Y, Carey M, and Grunstein M (2012). Mechanism for epigenetic variegation of gene expression at yeast telomeric heterochromatin. Genes Dev. 26, 2443–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster MJ, and Timmers HT (2015). Regulation of anti-sense transcription by Mot1p and NC2 via removal of TATA-binding protein (TBP) from the 30-end of genes. Nucleic Acids Res. 43, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Challal D, Dreos R, Mattarocci S, Bucher P, Libri D, and Shore D (2019). Opposing chromatin remodelers control transcription initiation frequency and start site selection. Nat. Struct. Mol. Biol 26, 744–754. [DOI] [PubMed] [Google Scholar]
- Kurat CF, Yeeles JTP, Patel H, Early A, and Diffley JFX (2017). Chromatin controls DNA replication origin selection, lagging-strand synthesis, and replication fork rates. Mol. Cell 65, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lademann CA, Renkawitz J, Pfander B, and Jentsch S (2017). The INO80 complex removes H2A.Zto promote presynaptic filament formation during homologous recombination. Cell Rep. 19, 1294–1303. [DOI] [PubMed] [Google Scholar]
- Lafon A, Taranum S, Pietrocola F, Dingli F, Loew D, Brahma S, Bartholomew B, and Papamichos-Chronakis M (2015). INO80 Chromatin Remodeler Facilitates Release of RNA Polymerase II from Chromatin for Ubiquitin-Mediated Proteasomal Degradation. Molecular Cell 60, 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SA, Lee HS, Hur SK, Kang SW, Oh GT, Lee D, and Kwon J (2017). INO80 haploinsufficiency inhibits colon cancer tumorigenesis via replication stress-induced apoptosis. Oncotarget 8, 115041–115053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25,2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipford JR, and Bell SP (2001). Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol. Cell 7, 21–30. [DOI] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, and Buratowski S (2014). A chromatin-based mechanism for limiting divergent noncoding transcription. Cell 157, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon CS, and Theis JF (1993). The structure and function of yeast ARS elements. Curr. Opin. Genet. Dev 3, 752–758. [DOI] [PubMed] [Google Scholar]
- Nieduszynski CA, Knox Y, and Donaldson AD (2006). Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev. 20, 1874–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrow AZ, Nellimoottil T, Knott SRV, Fox CA, Tavare S, and Aparicio OM (2014). Fkh1 and Fkh2 bind multiple chromosomal elements in the S. cerevisiae genome with distinct specificities and cell cycle dynamics. PLoS ONE 9, e87647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, and Peterson CL (2008). The Ino80 chromatin-remodeling enzyme regulates replisome function and stability. Nat. Struct. Mol. Biol 15, 338–345. [DOI] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Krebs JE, and Peterson CL (2006). Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev. 20, 2437–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamichos-Chronakis M, Watanabe S, Rando OJ, and Peterson CL (2011). Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144, 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Gasser SM, and Papamichos-Chronakis M (2017). The INO80 remodeller in transcription, replication and repair. Philos. Trans. R. Soc. Lond. B Biol. Sci 372, 20160290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Gerhold C-B, Tosi A, Hustedt N, Seeber A, Sack R, Herzog F, Pasero P, Shimada K, Hopfner K-P, and Gasser S (2016). Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Genes and Development 30, 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014). BEDTools: the Swiss-Army tool for genome feature analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–11.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan Aaron, and Hall Ira (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, and Fangman WL (2001). Replication dynamics of the yeast genome. Science 294, 115–121. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Mizuguchi G, FitzGerald PC, Wei D, Wang F, Huang Y, Luk E, Woodcock CL, and Wu C (2013). Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 154, 1232–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Lee L, Lynch B, and Tsukiyama T (2017). Nucleosome occupancy as a novel chromatin parameter for replication origin functions. Genome Res. 27, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclafani RA, and Holzen TM (2007). Cell cycle regulation of DNA replication. Annu. Rev. Genet 41, 237–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Mizuguchi G, Hamiche A, and Wu C (2000). A chromatin remodelling complex involved in transcription and DNA processing. Nature 406, 541–544. [DOI] [PubMed] [Google Scholar]
- Shen X, Ranallo R, Choi E, and Wu C (2003). Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol. Cell 12, 147–155. [DOI] [PubMed] [Google Scholar]
- Shimada K, Oma Y, Schleker T, Kugou K, Ohta K, Harata M, and Gasser SM (2008). Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr. Biol 18, 566–575. [DOI] [PubMed] [Google Scholar]
- Soriano I, Morafraile EC, Vázquez E, Antequera F, and Segurado M (2014). Different nucleosomal architectures at early and late replicating origins in Saccharomyces cerevisiae. BMC Genomics 15, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudet J, Gill JK, and Stutz F (2018). Noncoding transcription influences the replication initiation program through chromatin regulation. Genome Res. 28,1882–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcomb DT, Struhl K, and Davis RW (1979). Isolation and characterisation of a yeast chromosomal replicator. Nature 282, 39–43. [DOI] [PubMed] [Google Scholar]
- Takeda DY, Shibata Y, Parvin JD, and Dutta A (2005). Recruitment of ORC or CDC6 to DNA is sufficient to create an artificial origin of replication in mammalian cells. Genes Dev. 19, 2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topal S, Vasseur P, Radman-Livaja M, and Peterson C (2019). Distinct transcriptional roles for Histone H3-K56 acetylation during the cell cycle in Yeast. Nature Communications 10, 4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramantano M, Sun L, Au C, Labuz D, Liu Z, Chou M, Shen C, and Luk E (2016). Constitutive turnover of histone H2A.Z at yeast promoters requires the preinitiation complex. eLife 5, e14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugama M, Sabri A, and Bartholomew B (2011). The INO80 ATP-dependent chromatin remodeling complexis a nucleosome spacing factor. Mol. Cell. Biol 31, 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Fritsch O, Hohn B, and Gasser SM (2004). Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 119, 777–788. [DOI] [PubMed] [Google Scholar]
- Vincent JA, Kwong TJ, and Tsukiyama T (2008). ATP-dependent chromatin remodeling shapes the DNA replication landscape. Nat. Struct. Mol. Biol 15, 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, and Steinmetz LM (2009). Bidirectional promoters generate pervasive transcription in yeast. Nature 457, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Van C, Pradhan SK, Su T, Gehrke J, Kuryan BG, Kitada T, Vashisht A,Tran N, Wohlschlegel J, et al. (2015). The Ino80 complex prevents invasion of euchromatin into silent chromatin. Genes Dev. 29, 350–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Pradhan SK, Sun F, Chronis C, Tran N, Su T, Van C, Vashisht A, Wohlschlegel J, Peterson CL, et al. (2017). Mot1, Ino80C, and NC2 function coordinately to regulate pervasive transcription in yeast and mammals. Mol. Cell 67, 594–607.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W, King DA, Beckwith SL, Gowans GJ, Yen K, Zhou C, and Morrison AJ (2016). The INO80 complex requires the Arp5-Ies6 subcomplex for chromatin remodeling and metabolic regulation. Mol. Cell. Biol 36, 979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, and Pugh BF (2012). Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, and Pugh BF (2013). SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell 154, 1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tao L, Meyer C, Eeckhoute J, Johnson D, Bernstein B, Nusbaum C, Meyers R, Brown M, Li W, and Liu X (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biology. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhou B, Wang L, Li P, Bennett BD, Snyder R, Garantziotis S, Fargo DC, Cox AD, Chen L, and Hu G (2017). INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene 36, 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Wang L, Zhang S, Bennett BD, He F, Zhang Y, Xiong C, Han L, Diao L, Li P, et al. (2016). INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 30, 1440–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Fisher S, Dueck H, Middleton S, Khaladkar M, and Kim J (2018). PIVOT: platform for interactive analysis and visualization of transcriptomics data. BMC Bioinformatics 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data have been deposited in the Gene Expression Omnibus (GEO) under accession GSE144072 and GSE95633.