

SUMMARY
Neuroscience relies on techniques for imaging the structure and dynamics of neural circuits, but the cell bodies of individual neurons are often obscured by overlapping fluorescence from axons and dendrites in surrounding neuropil. Here, we describe two strategies for using the ribosome to restrict the expression of fluorescent proteins to the neuronal soma. We show first that a ribosome-tethered nanobody can be used to trap GFP in the cell body, thereby enabling direct visualization of previously undetectable GFP fluorescence. We then design a ribosome-tethered GCaMP for imaging calcium dynamics. We show that this reporter faithfully tracks somatic calcium dynamics in the mouse brain while eliminating cross-talk between neurons caused by contaminating neuropil. In worms, this reporter enables whole-brain imaging with faster kinetics and brighter fluorescence than commonly used nuclear GCaMPs. These two approaches provide a general way to enhance the specificity of imaging in neurobiology.
INTRODUCTION
Neural computations can only be understood by studying populations of neurons. The most common way to visualize populations of neurons is by imaging fluorescent proteins, such as GFP or GCaMPs (Chen et al., 2013; Nakai et al., 2001; Tsien, 1998). Rapid improvements in these fluorescent reporters has driven their widespread use in many contexts (Tian et al., 2012), but most of the applications in systems neuroscience require isolating the fluorescence signal from individual, intermingled neurons. This task is often complicated by the unique structure of neural tissue, in which the cell bodies of neurons are enmeshed within the axons and dendrites of surrounding cells.
Neuropil fluorescence causes two fundamental problems for imaging. The first challenge is that overlapping neuropil fluorescence can contaminate the somatic fluorescence signal being measured. This issue is particularly critical in calcium imaging experiments, in which contaminating neuropil fluorescence can result in the appearance of spurious correlations between the activity of neurons and external stimuli or each other (Harris et al., 2016). A second challenge is that neuropil fluorescence blurs the boundaries between neurons, making it difficult to define the underlying cells being imaged. This problem is particularly acute when fluorescent proteins are expressed at high cell densities or resolution is limited by extensive light scattering.
These problems have been addressed in several ways. One approach is to increase the resolution of imaging so that overlapping fluorescence is limited. However, this improvement comes at the expense of speed, sensitivity, and throughput, and even two-photon (2P) imaging cannot completely separate intermingled somatic and neuropil fluorescence signals (Keemink et al., 2018). An alternative strategy is to use post hoc computational approaches to deconvolve the signals that originate from different neural elements. For calcium imaging data, this is commonly performed by using algorithms, such as constrained non-negative matrix factorization (CNMF), that model the fluorescence signal from a region of space as the cumulative fluorescence of overlapping cells with different kinetics (Keemink et al., 2018; Pnevmatikakis et al., 2016; Zhou et al., 2018). While these tools are powerful, they also have important limitations, including the fact that they can over- and undercompensate for neuropil fluorescence, thereby creating the spurious appearance of activity. This distortion is challenging to detect and correct because the algorithms rely on assumptions that cannot be easily tested experimentally (Harris et al., 2016).
An alternative solution is to restrict fluorescent proteins to the cell body, thereby eliminating neuropil fluorescence directly. One way this has been achieved is through nuclear-localized (nls)-GCaMPs, which are widely used in worms and zebrafish. However, this nuclear localization comes at the expense of speed due to the slower kinetics of nuclear calcium (Kim et al., 2014). Here, we describe an alternative approach that uses the ribosome as a docking site for targeting fluorescent proteins to the neuronal soma. We describe first a method for inducible ribosome tethering that enables the ultrasensitive detection of GFP expression in brain slices. We then develop an analogous strategy for targeting GCaMPs to the neuronal soma in worms and mice, showing that this enables the crisp segmentation of intermingled cells and the elimination of neuropil contamination from somatic calcium signals. These two approaches provide a general way to enhance the subcellular specificity of neurobiological imaging.
RESULTS
Ribosome Tethering Dramatically Enhances Fluorescence from GFP Reporter Mice
We previously generated transgenic mice that express the GFP nanobody displayed on the surface of the ribosome (Nano-L10 mice; Figure 1A) (Ekstrand et al., 2014). The GFP nanobody is a small, high-affinity GFP binding protein (Rothbauer et al., 2006). We linked this GFP nanobody to ribosome protein L10, which is a component of the 60S ribosome and therefore localized to the cytoplasm of all cells. In the Nano-L10 mouse, the GFP nanobody is presented on the surface of the ribosome, such that intracellular GFP becomes tethered to the ribosome via the high-affinity nanobody-GFP interaction (Figure 1B). This property has been exploited to develop a ribosome profiling strategy known as retroTRAP (Ekstrand et al., 2014).
Figure 1. Ribosome Tethering Enables Ultrasensitive Visualization of GFP Reporters.
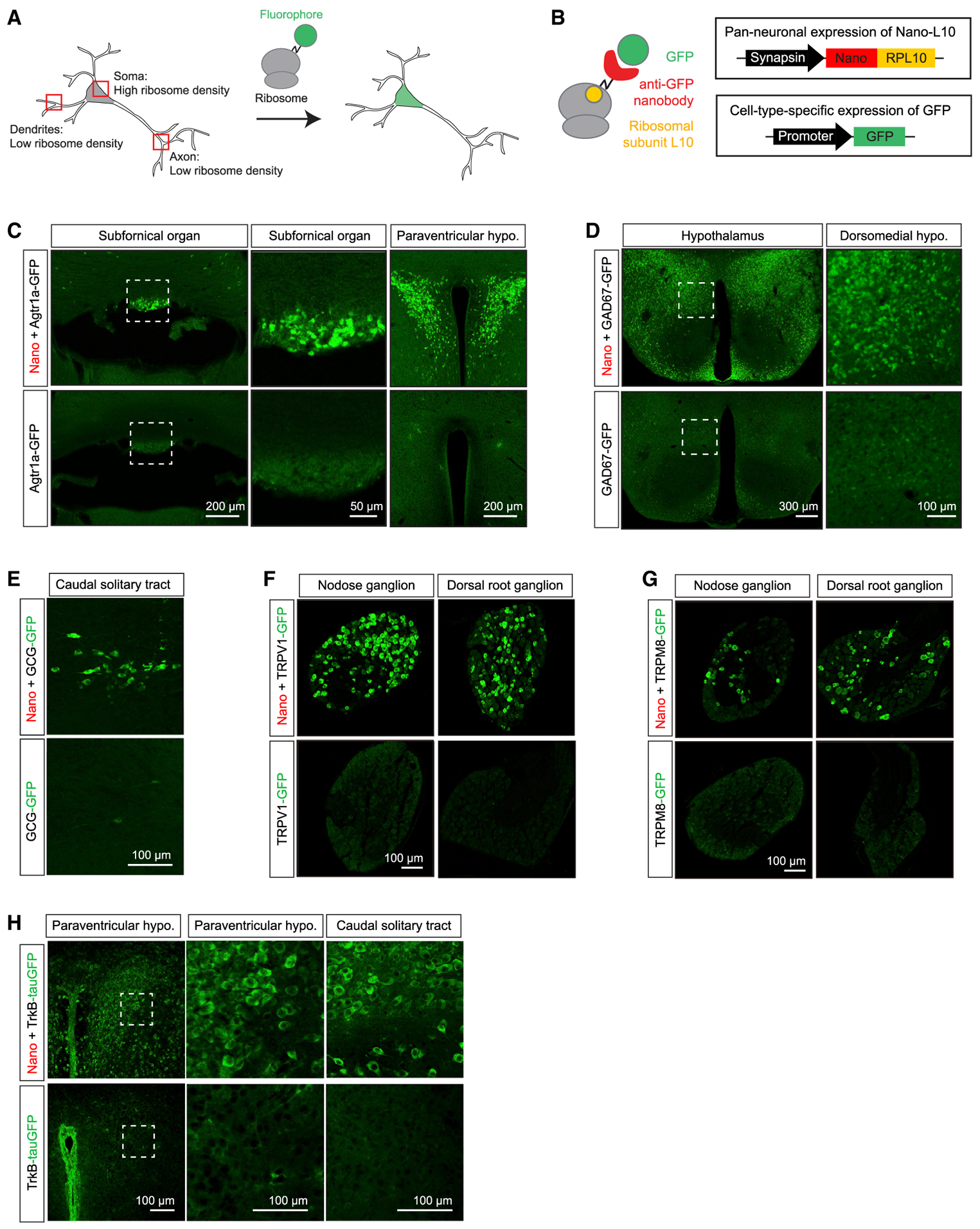
(A) Synthetic proteins tagged with ribosomal unit L10a become tethered to ribosomes and are enriched in the soma.
(B) The Nano-L10 transgenic mouse expresses a GFP-binding nanobody fused to ribosomal protein L10 in all neurons. When crossed to a second transgenic line expressing GFP from a cell-type-specific promoter, the GFP is captured by the nanobody, anchoring it in the soma.
(C–H) Images from the brains and peripheral ganglia of GFP reporter mice, either alone (bottom row) or after crossing to the Nano-L10 mouse (top row). All of the images show direct GFP fluorescence, except for (D), which is immunostained for GFP. Panels show Agtr1a-GFP (C), GAD67-GFP (D), GCG-GFP (E), TRPV1-GFP
(F), TRPM8-GFP (G), and TrkB-tauGFP (H). Scale bar sizes are indicated in each panel. See also Figure S1.
During the course of unrelated studies, we noticed that co-expression of the Nano-L10 transgene with GFP reporters caused a striking enhancement in GFP fluorescence. For example, transgenic mice expressing GFP from the Agtr1a gene promoter (Agtr1a-GFP mice) have been generated to enable the visualization of Agtr1a-expressing cells (Gonzalez et al., 2012), but we could detect only dim and diffuse GFP fluorescence in brain slices from these mice, even within brain regions where Agtr1a is highly expressed, such as the subfornical organ (SFO) and paraventricular hypothalamus (PVH) (Figure 1C, bottom). In contrast, after crossing Agtr1a-GFP mice into the Nano-L10 background, we observed bright GFP fluorescence in each of these structures (Figure 1C, top). This fluorescence was highly localized to discrete neuronal cell bodies, such that individual neurons could be easily identified by native fluorescence for applications such as patch clamping.
We observed a similar effect when we crossed the Nano-L10 mouse to other lines of GFP-expressing mice. For example, in brain slices from GAD67-GFP knockin mice (Tamamaki et al., 2003), we observed diffuse GFP fluorescence in most of the brain regions expressing GAD67 (Figure 1D, bottom). In the presence of the Nano-L10 transgene, this diffuse fluorescence was sharpened and amplified, such that individual neuronal cell bodies became readily apparent (Figure 1D, top). Likewise, after crossing the Nano-L10 line to GCG-GFP knockin mice, GCG neurons in the solitary tract became brightly visible by direct fluorescence (Figures 1E and S1A), whereas these same cells were largely undetectable in the parent GCG-GFP mouse line (Hayashi et al., 2009).
To explore whether this approach would work in peripheral neurons, we tested two lines of transgenic mice that express GFP from the TRPV1 and TRPM8 promoters (Pogorzala et al., 2013). TRPV1 and TRPM8 are expressed in subsets of sensory neurons in nodose and dorsal root ganglia, but we were unable to detect GFP in these structures by direct fluorescence (Figures 1F and 1G) or GFP immunostaining (Figure S1B) of tissue samples from either TRPV1-GFP or TRPM8-GFP animals. After crossing these two lines to the Nano-L10 mouse, we observed bright, native fluorescence in both nodose and dorsal root ganglia (Figures 1F and 1G). In every case we examined, the native GFP fluorescence in the presence of Nano-L10 was brighter and clearer than the indirect fluorescence we could observe after immunostaining tissue samples from the parent GFP mouse lines. This suggests that the Nano-L10 transgene should be broadly useful as a general enhancer for the visualization of neurons expressing GFP.
Nano-L10 expression results in the tight restriction of GFP fluorescence to the soma (Figures 1C–1G). This is consistent with the somatic localization of the ribosome and suggests that Nano-L10 improves the visualization of neurons in part by trapping and concentrating GFP within the cell body (Figure 1B). To test this, we used a TrkB-tauGFP knockin mouse (Li et al., 2011) in which GFP is targeted to axons via fusion to the micro-tubule binding protein Tau. In this mouse line, the extensive axonal localization of GFP renders the fluorescence from cell bodies indistinguishable from surrounding neuropil (Figure 1H, bottom). After crossing this TrkB-tauGFP reporter into the Nano-L10 background, we observed the extensive redistribution of fluorescence from axons to the soma, such that individual cell bodies became clearly distinct (Figure 1H, top). This confirms that the Nano-L10 transgene acts by enforcing the somatic localization of GFP, even in the presence of competing targeting domains, and suggests that ribosome tethering of fluorophores may be a general way to improve the visualization of neuronal cell bodies in the brain.
Ribosome Tethering Enables Somatic Targeting of GCaMP6
We reasoned that tethering GCaMPs to the ribosome may enable a similar improvement in calcium imaging. Because the GFP nanobody is not predicted to bind to GCaMPs (Kubala et al., 2010), we instead tethered GCaMP6 directly to the ribosome via fusion to the N terminus of ribosomal protein L10, analogous to the tagging approach that has been used for ribosome profiling (Heiman et al., 2008) (Figures 2A and 2B). We refer to this construct as a ribo-tagged GCaMP (ribo-GCaMP).
Figure 2. Ribo-tagging Restricts GCaMP to the Neuronal Soma.
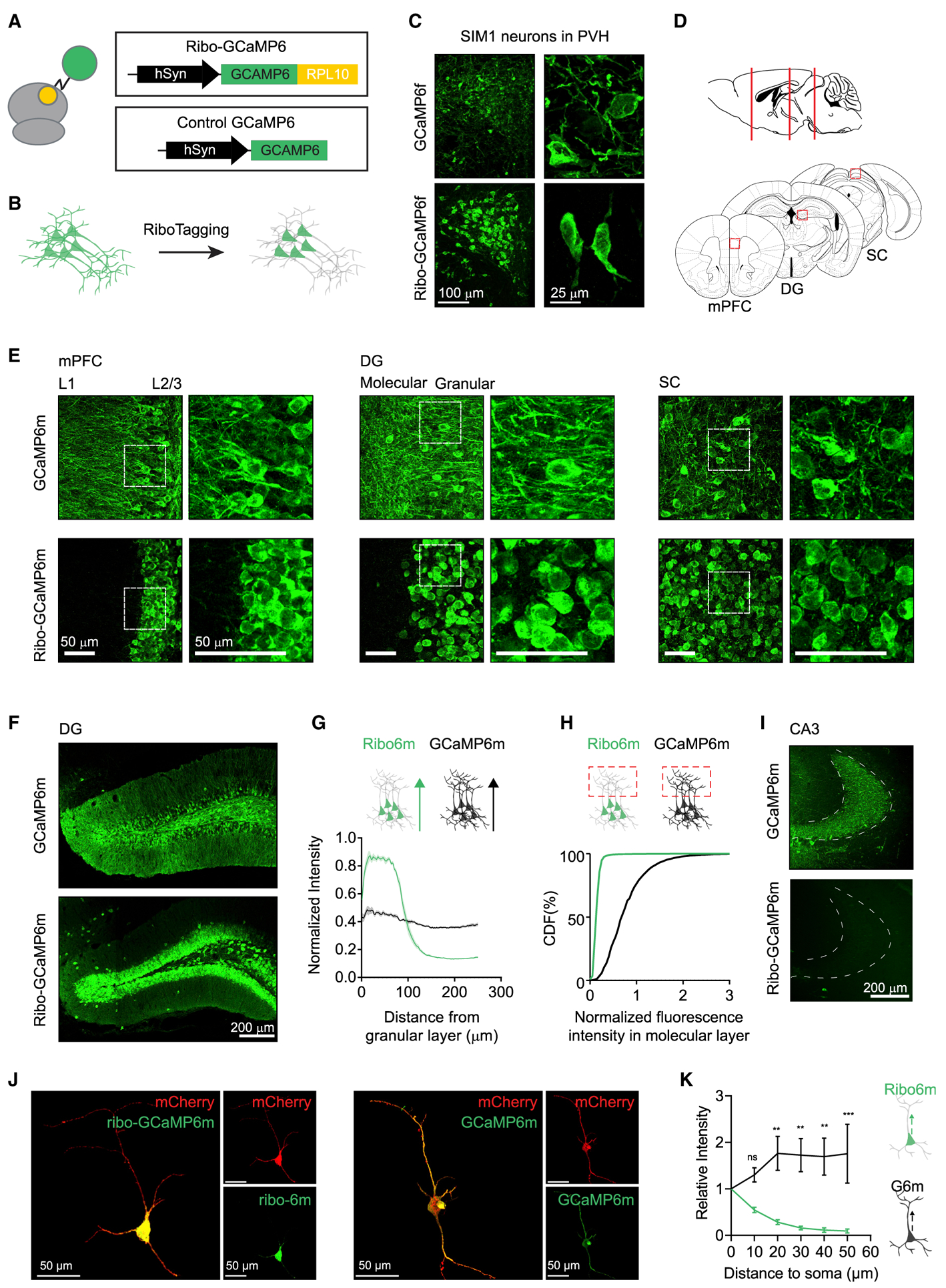
(A and B) Linking GCaMP to ribosomal protein L10a (A) enables soma targeting of GCaMP fluorescence (B).
(C) Ribo-tagging restricts GCaMP6f to the soma of PVH-SIM1 neurons.
(D and E) Ribo-GCaMP6m, when expressed in medial prefrontal cortex (mPFC), dentate gyrus (DG), and superior colliculus (SC) (D), is soma restricted compared to regular GCaMP6m (E) (white box indicates magnified region).
(F) Comparison of ribo-GCaMP6m and regular GCaMP6m when expressed in DG.
(G) Fluorescence intensity from the start of the granular layer to the molecular layer (250 μm length) of DG in samples expressing ribo-GAMP6m (n = 11) or regular GCaMP6m (n = 9). Sample size indicates number of brain slices used in analysis from 3 animals for each GCaMP6m. Each sample trace is normalized to its maximal intensity.
(H) Cumulative distribution function (CDF) of normalized fluorescence of ribo-GCaMP6m and GCaMP6m in the molecular layer, where DG dendrites are located.
(I) Comparison of regular GCaMP6m and ribo-GCaMP6m in CA3, where the axons from DG are located.
(J) Z projection of an individual striatal Ntrk1 neuron expressing mCherry and GCaMP6m (right) or ribo-GCaMP6m (left).
(K) Intensity of green fluorescence along the axon of Ntrk1 neurons expressing either GCaMP6m (black, n = 8) or ribo-GCaMP6m (green, n = 8). Tracking the axon was guided by mCherry fluorescence. **p < 0.01, ***p < 0.001, ns, p > 0.05, 2-way ANOVA, Holm-Sidak post hoc test.
All of the images are immunostained against GCaMP. Scale bar sizes are indicated in each panel.
We generated Cre-dependent adeno-associated viruses (AAVs) expressing GCaMP6f or ribo-GCaMP6f and then injected these viruses into the anterior hypothalamus of Sim1-Cre mice. Both viruses generated bright GCaMP expression that was enriched in the PVH, which is consistent with the expression pattern of endogenous SIM1 (Balthasar et al., 2005) (Figure 2C). However, the distribution of the fluorescence was very different, with GCaMP6f exhibiting a diffuse pattern distributed throughout the cell bodies and neuropil (Figure 2C, top), whereas ribo-GCaMP6f was tightly restricted to the neuronal soma (Figure 2C, bottom). This suggests that ribosome tethering can target GCaMP expression to neuronal cell bodies in vivo.
To confirm that this localization strategy works in different brain regions and cell types, we generated AAVs that constitutively express either GCaMP6m or ribo-GCaMP6m and then injected these into several sites in the mouse brain, including the dentate gyrus (DG), medial prefrontal cortex (mPFC), and superior colliculus (SC) (Figure 2D). We found that GCaMP6m exhibited a typical GFP expression pattern, with fluorescence filling the cell body, axons, and dendrites of the infected neurons (Figure 2E, top). By contrast, ribo-GCaMP6m fluorescence was tightly restricted to the neuronal somas at each injection site (Figure 2E, bottom). This was particularly evident in laminated structures such as the mPFC and DG, where ribo-GCaMP6m was confined to the layers containing cell bodies (layer 2/3 in the PFC and the granular layer in DG). Quantification of fluorescence within DG (Figure 2F) confirmed that ribo-GCaMP6m expression was strongly biased toward granular layers (Figure 2G) and virtually absent from the dendrites of the molecular layer (Figure 2H). Similarly, we observed no fluorescence in CA3 following the expression of ribo-GCaMP6m in DG (Figure 2I), indicating that ribo-tagging eliminates GCaMP expression from axons. Consistent with the data above, three-dimensional reconstructions of individual neurons showed that ribo-GCaMP6m is highly restricted to the soma, whereas GCaMP6m fills the entire cell (Figures 2J and 2K).
A Soma-Targeted GCaMP Faithfully Reports on Neural Activity Ex Vivo
We next investigated the ability of ribosome-tethered GCaMPs to report on neural activity. Acute slices were prepared from mice expressing GCaMP6m or ribo-GCaMP6m in DG, and these slices were then stimulated with field pulses (FPs) delivered through two parallel platinum electrodes in the presence of inhibitors of network activity (Figure 3A). During stimulation, slices were imaged at 20 Hz, and fluorescence signals from 30 to 80 polygon ROIs were extracted to enable the analysis of individual neurons. We found that GCaMP6m and ribo-GCaMP6m exhibited similar fluorescence responses to a single FP (ΔF/F = 6.6% versus 5.4%, p = 0.29), and the response of both reporters monotonically increased with the number of pulses (Figure 3B). Both reporters exhibited a similar rise time constant (Figure 3E), decay time constant (Figure 3D), and peak ΔF/F (Figure 3C) for GCaMP6m and ribo-GCaMP6m across a range of pulse numbers (1–30 FP). This indicates that the ability of GCaMP6m to faithfully report on neural activity is largely unaffected by tethering to the ribosome (linear regression of ΔF/F versus FP: R2: 0.5234 and 0.3825, p values: <0.0001 and <0.0001 for GCaMP6m and ribo-GCaMP6m).
Figure 3. Ribo-tagging Does Not Change the Kinetics of GCaMP in Response to Evoked Neural Activity.
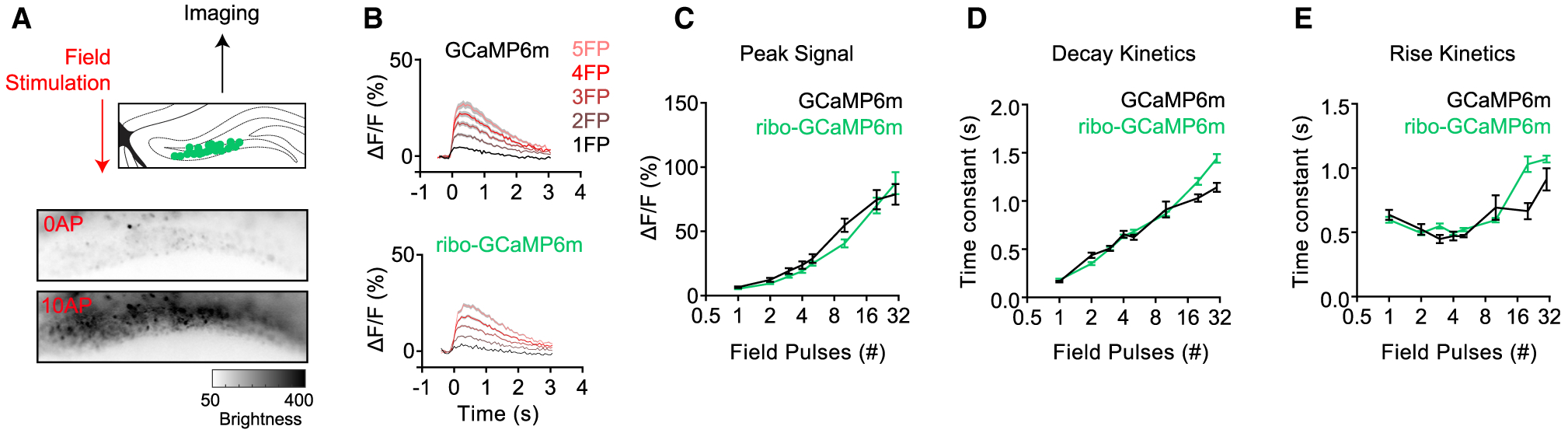
(A) Schematic describing the measurement of fluorescence in response to electrically evoked neural activity in DG. Grayscale heatmap shows the brightness of DG neurons expressing ribo-GCaMP6m before and during pulsed field stimulation.
(B) Response of ribo-GCaMP6m and regular GCaMP6m to different numbers of electrical field pulses (FPs). When calculating ΔF/F, the average fluorescence during the 300-ms time window before the start of the first FP was used as F0 (the denominator).
(C–E) Peak signal (C; p 0.71, 0.66, 0.65, 0.60, 0.67, < 0.01, 0.67, and 0.16), decay time constant (D; p 0.95, 0.27, 0.96, 0.95, 0.65, 0.87, < 0.01, and < 0.0001), and rise time constant (E; p 0.84, 0.84, 0.25, 0.84, 0.78, 0.43, < 0.0001, and 0.06) for ribo-GCaMP6m (green, n = 304 from 4 brain slices of 2 mice) and GCaMP6m (black, n = 137 from 4 brain slices of 2 mice) in response to the increasing numbers of FPs.
To corroborate this field stimulation data, we also measured the ability of both GCaMPs to report action potentials (APs) imaged with 2P microscopy during whole-cell recordings. We prepared acute coronal slices containing mPFC and made whole-cell current-clamp recordings from layer 5b thick tufted pyramidal cells. Ionotropic transmission was blocked with a cocktail of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), N-methyl-d-aspartate (NMDA), and γ-aminobutyric acid A (GABAA) receptor antagonists (see Method Details). We evoked trains of 100-Hz APs via somatic current injection and found that ribosome tagging of GCaMP6m did not significantly affect its peak fluorescence signal (Figure 4G, p > 0.05 comparing GCaMP6m to ribo-GCaMP6m for 1, 3, 5, and 10 APs), fluorescence rise kinetics (Figure 4J, p > 0.05 for 3, 5, and 10 APs), and decay time constant (Figure 4K, p > 0.05 for 3, 5, and 10 APs) at the soma. Consistently, the baseline noise level (Figure 4H, p = 0.67 comparing GCaMP6m to ribo-GCaMP6m) and peak signal:noise ratio (Figure 4I, p = 0.59) were also unaffected by ribo-tagging. Thus ribo-GCaMP6m exhibits fluorescence changes in response to neural activity that are broadly similar to GCaMP6m.
Figure 4. Subcellular Distribution and Kinetics of Ribo-GCaMP.
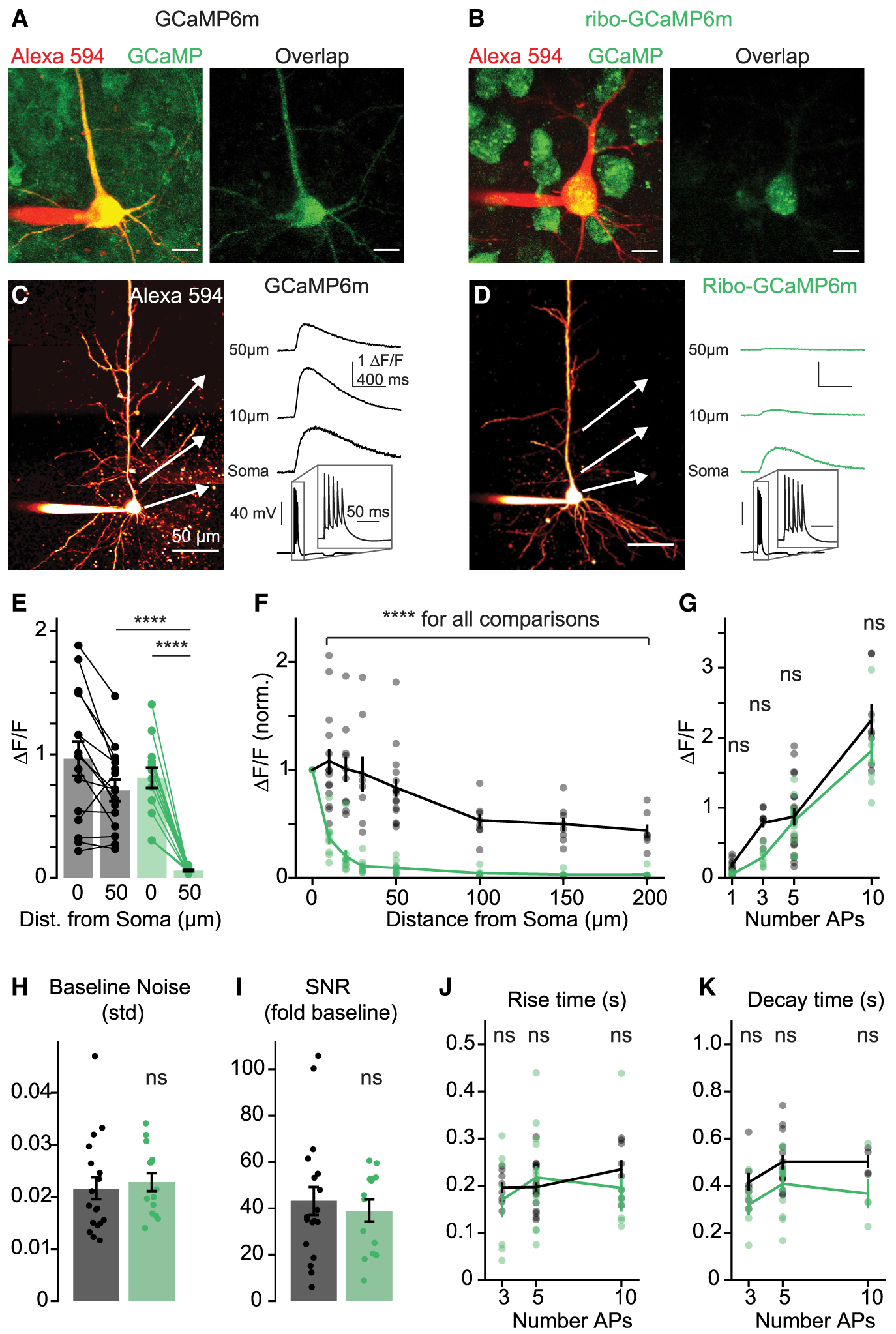
(A and B) 2P z stacks of layer 5 pyramidal neurons expressing GCaMP6m (A) or ribo-GCaMP6m (B). Left, overlay of Alexa 594 cell fill and and non-calcium-bound GCaMP fluorescence (810 nm imaging wavelength). Right, overlap of Alexa 594 and GCaMP fluorescence. Scale bars are 10 μm.
(C and D) Example 2P line scans of (C) GCaMP6m and (D) ribo-GCaMP6m fluorescence in response to trains of 5 APs (imaging wavelength 920 nm). Example transients are averages of 10 trials. Scale bars are 50 μm.
(E) Response to 5 APs measured at the soma and 50 μm along the apical dendrite for GCaMP6m (n = 15) and ribo-GCaMP6m (n = 14). Bars indicate means ± SEMs, ****p < 0.0001, 1-way ANOVA, Holm-Sidak correction.
(F) Normalized peak fluorescence imaged at varying distances along the apical dendrite in response to 5 APs (10, 20, 30, 50, 100, 150, and 200 μm; normalized to signal at 0 μm) for GCaMP6m (n = 7–15) and ribo-GCaMP6m (n = 6–14). Lines and bars indicate means ± SEMs. ****p < 0.0001 for all comparisons between GCaMP6m and ribo-GCaMP6m, 2-way ANOVA, Holm-Sidak correction.
(G) Response at the soma to 1, 3, 5, and 10 APs of GCaMPm (n = 8–18) and ribo-GCaMP6m (n = 7–14). p = 0.73, 0.06, 0.73, and 0.10 for 1, 3, 5, and 10 APs), 2-way ANOVA, Holm-Sidak correction.
(H) Standard deviation of the baseline fluorescence signal for GCaMP6m (n = 16) and ribo-GCaMP6m (n = 14). p = 0.67, 2-tailed unpaired t test.
(I) Fold increase of somatic fluorescence signal over the standard deviation of the baseline in response to 5 APs for GCaMP6m (n = 16) and ribo-GCaMP6m (n = 14). p = 0.59, 2-tailed unpaired t test.
(J) Time to peak after stimulus offset for GCaMP6m (n = 8–18) and ribo-GCaMP (n = 6–14). p = 0.56, 0.82, and 0.56 for 3, 5, and 10 APs, 2-way ANOVA, Holm-Sidak correction.
(K) Half-maximal decay time for GCaMP6m (n = 4–13) and ribo-GCaMP6m (n = 4–12). p = 0.14, 0.01, and 0.13 for 3, 5, and 10 APs, 2-way ANOVA, Holm-Sidak correction.
See also Figure S2.
We next characterized the subcellular dynamics of the AP-evoked fluorescence of ribo-GCaMP6m. To measure this at high spatial resolution, we loaded mPFC pyramidal neurons with the volume marker Alexa Fluor 594 (20 μM). We then evoked trains of 5 APs (100 Hz) via somatic current injection and imaged concomitant ribo-GCaMP6m or GCaMP6m fluorescent transients in the soma and along the apical dendrite. As expected, AP-evoked GCaMP6m transients were observed in both the soma and dendrites, with clear signals generated >200 μm along the apical dendrite trunk. By contrast, ribo-GCaMP6m-associated signals were largely restricted to the soma, with fluorescence declining as little as 10 μm from the cell body and becoming virtually undetectable at 50 μm (Figures 4C–4F, p < 0.0001 for all comparisons between ribo-GCaMP6m and GCaMP6m peak fluorescence at 10–200 μm from the cell body). This indicates that ribo-tagging is an effective strategy for the somatic localization of GCaMP expression.
Ribo-GCaMP6 Eliminates Artifactual Correlation between Neurons in 2P Imaging
We next evaluated the ability of ribo-GCaMPs to report on neural dynamics in vivo. To do this, we measured the response of neurons in the visual cortex to the orientation of drifting bars (Hubel and Wiesel, 1959). We injected AAVs expressing ribo-GCaMP6m or GCaMP6m into the visual cortex and installed a cranial window for imaging the L2/3 neurons of awake mice. After waiting 3 weeks for recovery, mice were head fixed and calcium dynamics were imaged using a 2P microscope. As observed in vitro, we found that the expression of ribo-GCaMP6m was tightly restricted to neuronal cell bodies, creating crisp boundaries between cells, whereas the expression of regular GCaMP6m resulted in saturation of the surrounding neuropil (Figure 5A). In contrast to slice experiments, we found that ribo-GCaMP6m was dimmer than GCaMP6m in vivo and therefore required higher laser power (67 versus 28 mW) for comparable imaging. We did not observe increased photo-bleaching of ribo-GCaMP6m compared to GCaMP6m in this setting (Figures S3D and S3E).
Figure 5. Ribo-GCaMP Reduces Neuropil Contamination during 2P Imaging.
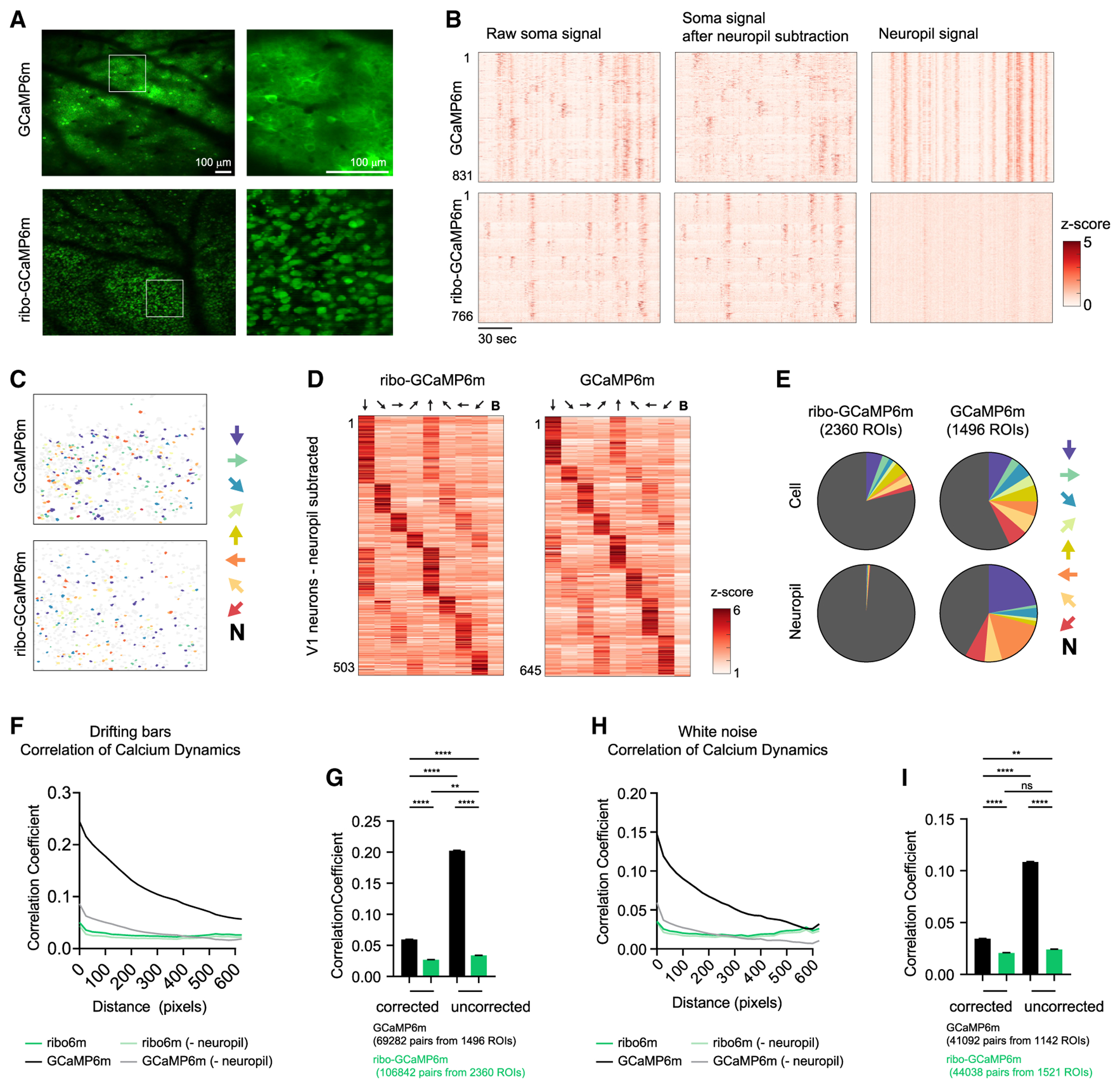
(A) Example fields of view of V1 neurons expressing GCaMP6m (top) or ribo-GCaMP6m (bottom).
(B) Calcium dynamics of neurons recorded from a single video. Calcium dynamics are either uncorrected (left) or with surrounding neuropil signal subtracted (right). Cells are sorted based on orientation tuning.
(C) Color coded ROIs based on orientation selectivity in V1.
(D) Peak response of angle-selective V1 neurons to drifting bars with different angles.
(E) Distribution of orientation selectivity. ROIs are either neurons or neuropils, which is defined as the area surrounding each identified neuron.
(F–I) Correlation coefficient analysis of neural signal for either GCaMP6m or ribo-GCaMP6m. The neuropil signal was either subtracted or not, as indicated. During recording, mice were exposed to either drifting bars (F and G) or white noise (H and I).
(F) Correlation coefficient of calcium dynamics between pairs of neurons plotted against the centroid distance. R2 = 0.92, 0.19, 0.84, and 0.10; p < 0.0001, 0.0208, < 0.0001, and 0.0970 for regular uncorrected, ribo-uncorrected, regular, and ribo, respectively, linear regression of averaged values.
(G) Correlation coefficient of calcium dynamics between pairs of neurons that are within 100 pixels. Regular corrected: 0.105 ± 0.010; ribo-corrected: 0.081 ± 0.010; regular uncorrected: 0.227 ± 0.010; ribo-uncorrected: 0.085 ± 0.008; values are means ± SEMs.
(H) Correlation coefficient of calcium dynamics between pairs of neurons plotted against centroid distance. R2 = 0.80, 0.003, 0.62, and 0.003; p < 0.0001, 0.79, < 0.0001, and 0.79 for regular uncorrected, ribo-uncorrected, regular, and ribo, respectively, linear regression of averaged values.
(I) Correlation coefficient of calcium dynamics between pairs of neurons that are within 100 pixels. Regular corrected: 0.034 ± 0.002; ribo-corrected: 0.021 ± 0.002; regular uncorrected: 0.109 ± 0.002; ribo-uncorrected: 0.024 ± 0.002; values are means ± SEMs.
In (C)–(E), arrows indicate the drifting bar orientation that the corresponding color (C and E) or column (D) represents. In (C) and (E), N denotes “nonselective,” which means that these neurons did not pass the criteria to be defined as orientation tuned (see Method Details). In (D), B denotes “blank,” in which no stimulus was given. In (G) and (I), **p < 0.01, ****p < 0.0001, ns, p > 0.05, 1-way ANOVA, Holm-Sidak post hoc test. Total numbers of cells were used to estimate degrees of freedom when calculating SEM; 1 pixel = ~1.9 μm.
We exposed mice to images of drifting bars in one of eight orientations and recorded calcium responses. Visual stimuli induced clear responses in many neurons for both ribo-GCaMP6m and GCaMP6m (Figures 5B, 5D, and 5E; Videos S1, S2, S3, and S4), and cells with different orientation tuning were anatomically intermingled (Figure 5C). We observed a greater percentage of cells that were orientation tuned in recordings from GCaMP6m versus ribo-GCaMP6 (43% versus 21%), which we believe reflects a combination of the enhanced detection of inactive cells and the reduced detection of neuropil-induced false positives with ribo-GCaMP6m (see Discussion).
In recordings from mice expressing GCaMP6m but not ribo-GCaMP6m, we observed strong neuropil fluorescence that was highly synchronized (Figure 5B) and showed orientation tuning (Figures 5B and 5E), suggesting that this neuropil fluorescence may induce artifactual correlation between neurons. To examine this, we analyzed the pairwise correlation between the activities of neurons separated by various distances, reasoning that neuropil fluorescence would be more likely to increase the correlation between neurons closer in space. We did find a strong pairwise correlation between neurons expressing GCaMP6m that declined with distance, whereas this correlation was dramatically reduced for ribo-GCaMP6m (Pearson correlation coefficient 0.20 ± 0.003 versus 0.03 ± 0.002 for GCaMP6m versus ribo-GCaMP6m; Figures 5F and 5G). Subtraction of this neuropil signal from GCaMP6m recordings reduced the correlation between neurons, but this correlation remained both distance dependent (Figure 5F) and elevated compared to ribo-GCaMP6m recordings without correction (0.06 ± 0.002 versus 0.03 ± 0.002 for GCaMP6m versus ribo-GCaMP6m; Pearson correlation coefficient of neuron pairs <100 pixels apart; Figure 5G). Similar results were obtained when we analyzed data from mice exposed to white noise (Figures 5H and 5I) and drifting gratings (Figure S3). Thus, ribo-GCaMP6m effectively reduces the neuropil-induced correlation between cells in 2P imaging experiments.
Ribo-GCaMP6 Faithfully Reports on Neural Activity in Microendoscope Imaging
To investigate the utility of ribo-GCaMPs for 1P microscopy, we imaged mPFC neurons using a miniature head-mounted microscope (Ghosh et al., 2011). AAV-expressing ribo-GCaMP6m or GCaMP6m was injected into the mPFC, and a gradient refractive index (GRIN) lens was implanted above the mPFC to enable chronic imaging of neural dynamics (Figure 6A). In this configuration, the fluorescence of dozens of mPFC neurons could be readily resolved in each field of view (Figure 6B). We trained mice to consume a liquid diet (Ensure) from a lickometer and then correlated the response of individual neurons to behavioral events (Figure 6C). We found that the presentation of food to fasted mice triggered a rapid and sustained shift in the baseline fluorescence of a subset of mPFC neurons (Figure 6D). Overlaid on this tonic modulation, phasic responses of individual mPFC neurons that were time locked to the initiation and termination of individual licking bouts were observed (Figure 6E). These responses were heterogeneous, with some neurons activated by licking and others inhibited, and responses were similar between the two indicators (Figure 6E). This indicates that ribo-GCaMP6m can report on neural dynamics in the mPFC during behavior in a manner that is comparable to that of traditional GCaMPs.
Figure 6. Microendoscope Recording of mPFC Dynamics Using Ribo-GCaMP.
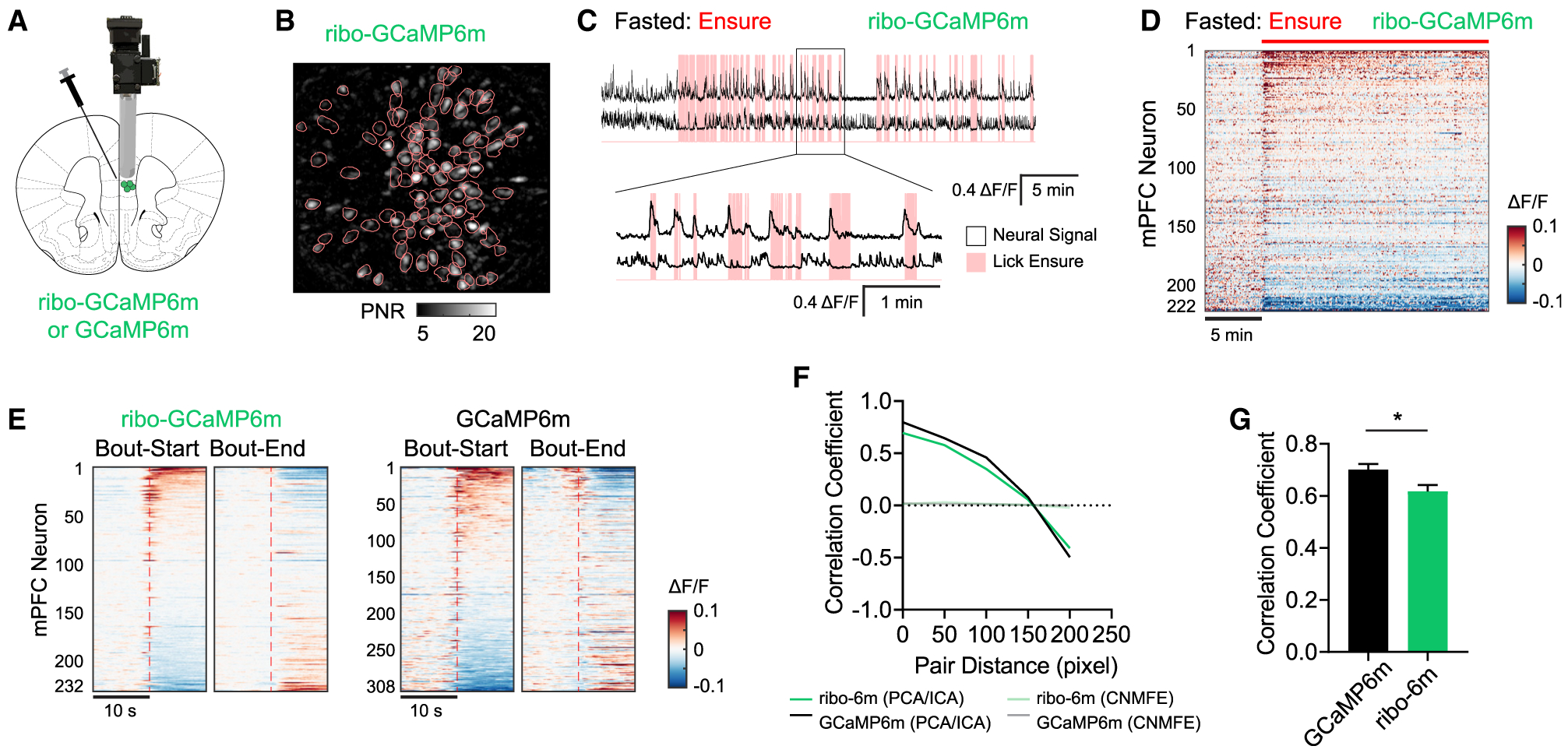
(A) Ribo-GCaMP6m expression was targeted to mPFC, and a GRIN lens was implanted for microendoscope recordings.
(B) Representative field of view of ribo-GCaMP6m imaged through a microendoscope, with ROIs (pink contours) indicated.
(C) Representative extracted calcium dynamics (black traces) time synchronized with licking events (pink shading) of liquid food in the fasted state.
(D) Response of mPFC neurons in fasted mice to the consumption of Ensure. Red bar indicates when food was made available.
(E) Peristimulus time histogram (PSTH) aligned to either the initiation or termination of licking bouts. Calcium dynamics were measured using either ribo-GCaMP6m (left) or GCaMP6m (right).
(F and G) Pearson correlation coefficient between the signal from different pairs of ROIs. mPFC neural dynamics were recorded in fed mice expressing either regular GCaMP6m (n = 154 from 5 mice) or ribo-GCaMP6m (n = 69 from 4 mice) during cage exploration. In (F), calcium dynamics were extracted using either PCA-ICA or CNMF_E methods, and the correlation coefficient is plotted against the distance between corresponding ROIs. In (G), calcium dynamics were extracted by PCA-ICA and the correlation coefficient between pairs within 100 pixels is compared for GCaMP6m and ribo-GCaMP6m. *p < 0.05, unpaired t test; cell number was used as degree of freedom when calculating SEMs.
We next compared the distance-dependent correlation between the activity of neurons expressing either indicator. To do this, we analyzed the data using principal-component analysis-independent-component analysis (PCA-ICA) segmentation, which extracts the fluorescence trace for each cell based on its spatial footprint. We found that cells expressing GCaMP6m had high pairwise correlation and that this correlation declined with increasing spatial distance between pairs (Figures 6F and 6G). The use of ribo-GCaMP6m reduced this correlation (Figure 6G), but the magnitude of this reduction was modest compared to the dramatic reduction observed in 2P imaging (Figures 5F–5I). This suggests that the “blurriness” of miniscope images is not primarily due to neuropil but instead arises from other sources (e.g., light scattering, out-of-focus fluorescence). Consistent with this, reanalysis of the data using CNMF_E (Zhou et al., 2018) completely eliminated the neural correlation for both indicators (Figure 6F).
Ribo-GCaMP6 Enables Long-Term Imaging Experiments In Vivo
Chronic or high-level expression of GCaMP has been associated with cellular toxicity (Tian et al., 2009). To characterize whether ribosome tagging of GCaMP6m interferes with neural function or viability, we performed three experiments. First, we measured in acute brain slices the electrophysiologic properties of layer 5 pyramidal cells expressing either GCaMP6m or ribo-GCaMP6m (Figure S4A). This revealed no significant difference in the resting membrane potential (−75 ± 0.7 for GCaMP6m versus −76 ± 1.0 mV for ribo-GCaMP6m, p = 0.65), input resistance(−77 ± 6.5 versus 70 ± 4.2 MU, p = 0.39), or spiking threshold (−54 ± 0.5 versus −55 ± 0.6 mV, p = 0.33) (Figures S4B–S4D). This suggests that ribo-GCaMP6m has no gross effect on cellular physiology relative to commonly used calcium reporters.
Second, we looked for evidence of local inflammation caused by the chronic overexpression of ribo-GCaMP6m. To do this, we injected a cohort of mice bilaterally with viruses expressing ribo-GCaMP6m and GCaMP6m, one in each hemisphere, and then harvested tissue from these animals after 4, 6, 8, and 12 weeks (Figure S4E). Staining of this tissue for GFP as well as the microglia marker IbaI revealed no signs of increased microglial infiltration on the side expressing ribo-GCaMP6m (Figures S4F and S4G). Thus, ribo-GCaMP6m expression does not trigger the inflammatory response that would be predicted if this protein were associated with significant toxicity.
Third, we performed longitudinal imaging of mPFC neurons expressing ribo-GCaMP6m to functionally evaluate the stability of the fluorescence signal over time. To do this, we targeted ribo-GCaMP6m to mPFC by AAV infection, implanted a GRIN lens for imaging, and then recorded the activity of neurons in the same mouse at 2, 4, and 6 months after infection (Figure S4H). We saw no evidence of deterioration of the fidelity or strength of calcium responses even after 6 months of imaging (Figure S4I). For example, the number of cells that responded to Ensure consumption was unchanged over time (34 ± 8 per field of view at 2 months versus 53 ± 14 at 6 months, p = 0.11) and the mean ΔF/F of the responses of these cells to licking increased between 2 and 6 months (1.22 ± 0.15 at 2 months versus 1.82 ± 0.16 at 6 months, p = 0.01). This robust stability may reflect natural quality control mechanisms that tightly regulate ribosomal protein expression (Sung et al., 2016), thereby preventing the accumulation of toxic concentrations of ribo-GCaMPs.
A Soma-Targeted GCaMP Enables Imaging of Neural Dynamics in Caenorhabditis elegans
To investigate the ability of ribosome-tethered GCaMP to report on neural activity in a different organism, we examined the nematode C. elegans. C. elegans has a transparent body that affords non-invasive optical access to its nervous system for imaging experiments, and many studies have recorded neuronal activity in the worm using genetically encoded calcium sensors. The worm homolog of mammalian ribosomal protein L10 is RPL-1 (Shaye and Greenwald, 2011), which shares 75% amino acid sequence identity with L10 (Figure S5A) and has previously been tagged with GFP for translational profiling (Gracida and Calarco, 2017). We therefore generated a worm-specific ribo-GCaMP by linking GCaMP6m to the N terminus of RPL-1 and then used this reagent to measure calcium transients in C. elegans neurons in response to sensory stimuli and spontaneous neural activity.
We generated matched transgenic lines expressing either GCaMP6m, ribo-GCaMP6m, or nls-GCaMP6m (nuclear-targeted GCaMP is commonly used in worm imaging) under the control of the sra-6 promoter, which directs expression to ASH nociceptive sensory neurons (Troemel et al., 1995) (Figure 7A). Polymodal ASH sensory neurons detect a variety of aversive stimuli (Bargmann et al., 1990; Hilliard et al., 2005; Kaplan and Horvitz, 1993; Troemel et al., 1997), and their activity can be reliably evoked by exposure to high concentrations of salt (Chatzigeorgiou et al., 2013). We imaged animals in a microfluidic device that allows repeatable salt stimulation via switching buffer streams across the nose combined with simultaneous optical recording of calcium transients in individual neurons (Chronis et al., 2007) (Figure 7B).
Figure 7. Stimulated In Vivo Calcium Imaging of C. elegans ASH and AFD Neuronal Activity with Ribo-GCaMP.
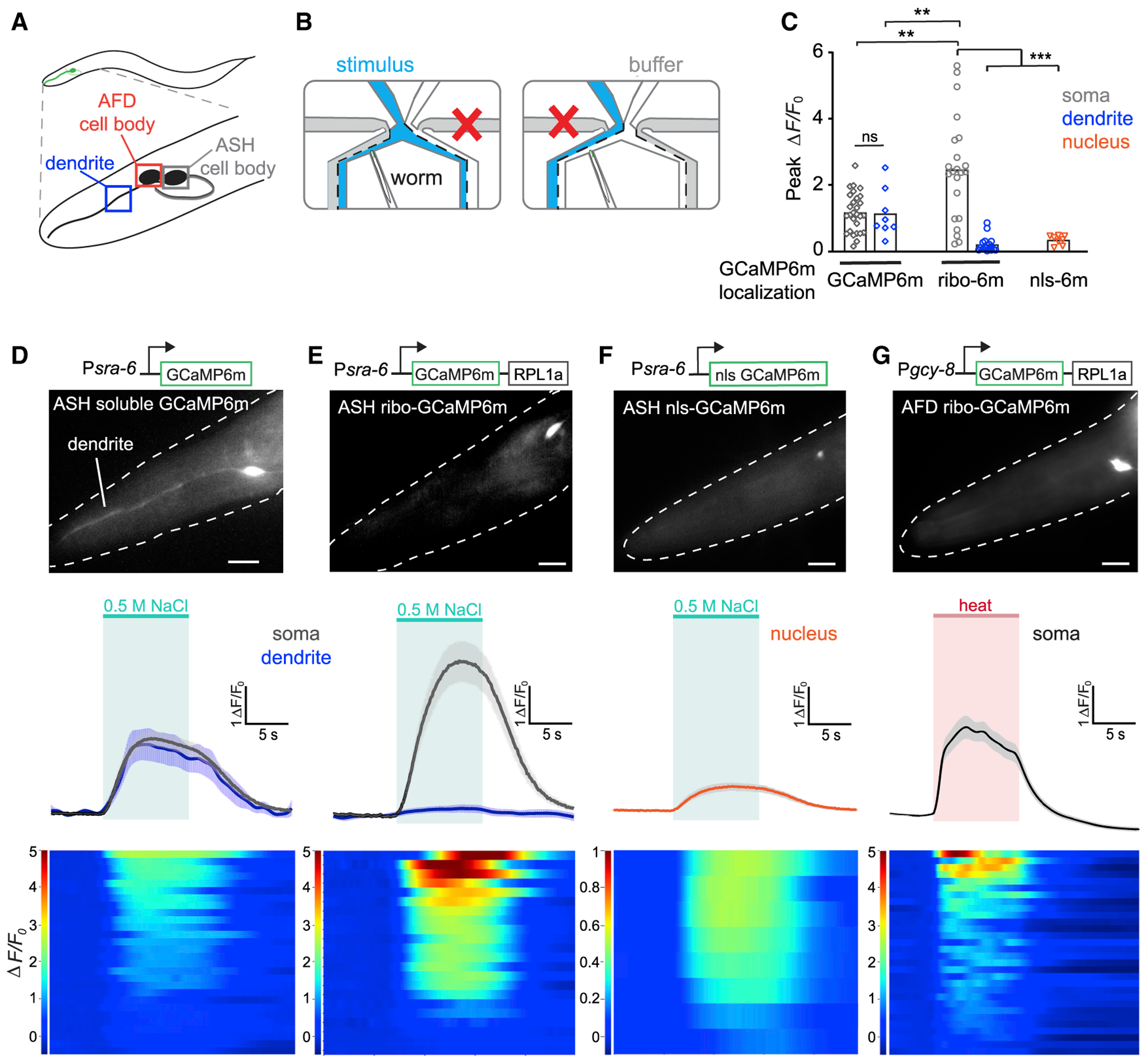
(A) Schematic representation of ASH and AFD neurons in the worm head. Boxes indicate ASH cell body, AFD cell body, and ASH dendrite, respectively.
(B) Animals were imaged in a microfluidic device that allows switchable fluid flow past the nose.
(C) Response amplitude ΔF/F0 for imaging traces shown in (D)–(F); GCaMP6m localization for soma in gray, dendrite in blue, and nucleus in orange.
(D–G, top) Diagram of constructs used to make transgenic worm strains. sra-6 promoter drives expression in ASH; gcy-8 promoter drives expression in AFD. Representative images showing fluorescent responses in ASH and AFD; dotted white lines denote worm head; scale bars, 10 μm.
(D–G, center) Mean fluorescence transients in ASH neurons expressing GCaMP6m (D), ribo-GCaMP6m (E), or nuclear-localized (nls)-GCaMP6m (F), in response to a 10-s 0.5 M NaCl pulse (green area denotes stimulus; means ± SEMs). Black and gray traces denote responses in cell soma; blue traces denote responses in dendrites. (G) Mean fluorescence transients in AFD neurons expressing ribo-GCaMP6m in response to a 10-s pulse of warm (~30°C) buffer (red area denotes stimulus).
(D–G, bottom) Heat plots of individual soma responses, 1 neuron per row. Change in fluorescence from baseline over time (ΔF/F0) is represented in the color bars at left. n = 11 worms responding to 2 consecutive salt pulses for ASH ribo-GCaMP6m; 10 for ASH soluble GCaMP6m; 5 for ASH nls-GCaMP6m; 11 worms responding for 3 consecutive heat pulses for AFD ribo-GCaMP6m. **p < 0.01, ***p < 0.001, ns, not significant using unpaired t test with Welch’s correction. See also Figure S5.
In animals expressing GCaMP6m, we observed calcium transients in ASH in response to a 10-s pulse of 0.5 M NaCl, as has been previously reported for other GCaMPs (Chatzigeorgiou et al., 2013; Davis et al., 2018), and the magnitude of these responses was similar between the soma and dendrite (Figures 7C and 7D). In a matched strain expressing ribo-GCaMP6m in the same cells, we observed large calcium transients in the soma, but not the processes, that were reproducible across animals and trials (Figure 7C). Of note, these somatic signals were larger for ribo-GCaMP6m than GCaMP6m (ΔF/F0 = 242% ± 35% versus 117% ± 11%; Figures 7D and 7E, gray lines). As expected, nls-GCaMP6m showed nuclear localized fluorescence, but the magnitude of this fluorescence change was considerably less than traditional GCaMP6m (ΔF/F0 = 117% ± 11% versus 33% ± 5%; Figures 7C and 7F). This indicates that ribo-GCaMPm can robustly report on neural activity in response to a stimulus in C. elegans and that its expression is tightly restricted to the soma in the worm as observed in the mouse.
To confirm that soma-targeted GCaMP is compatible with different cell types and stimuli, we expressed ribo-GCaMP6m in AFD sensory neurons using the ntc-1 promoter (Beets et al., 2012; Garrison et al., 2012) (Figure 7G). AFD responds to changes in temperature that enable worms to detect and navigate environmental temperature gradients (Biron et al., 2006; Hawk et al., 2018; Kimura et al., 2004; Luo et al., 2014). We found that ribo-GCaMP6m was soma localized in AFD sensory neurons and faithfully reported on oscillatory calcium transients in AFD following a 10-s pulse of warm buffer across the animal’s nose (Figure 7G). While previous imaging studies in AFD have relied on warming the entire worm body, our data suggest that temperature changes only at the tip of the nose, where sensory cilia are located (Perkins et al., 1986; Ward et al., 1975), are sufficient to induce AFD activation.
Ribo-GCaMP6m Enables Whole-Brain Imaging in the Worm with High Temporal Precision
The compact nervous system of C. elegans makes it possible to image the calcium dynamics of most or all neurons simultaneously in a single animal. Previous whole-brain imaging studies in the worm have required the use of nls-GCaMPs to enable segmentation of cells in densely packed ganglia (Schrödel et al., 2013; Prevedel et al., 2014; Kato et al., 2015; Nguyen et al., 2016). We therefore investigated whether ribo-GCaMPs could also be used for whole-brain imaging and, if so, whether this would have any advantages over nls sensors.
We generated transgenic animals expressing ribo-GCaMP6m or nls-GCaMP6m in all neurons (Figures 8A and 8B; Videos S5 and S6) and then imaged the head ganglia of anesthetized
Figure 8. Whole-Brain Calcium Imaging of C. elegans Basal Neuronal Activity with Ribo-GCaMP.
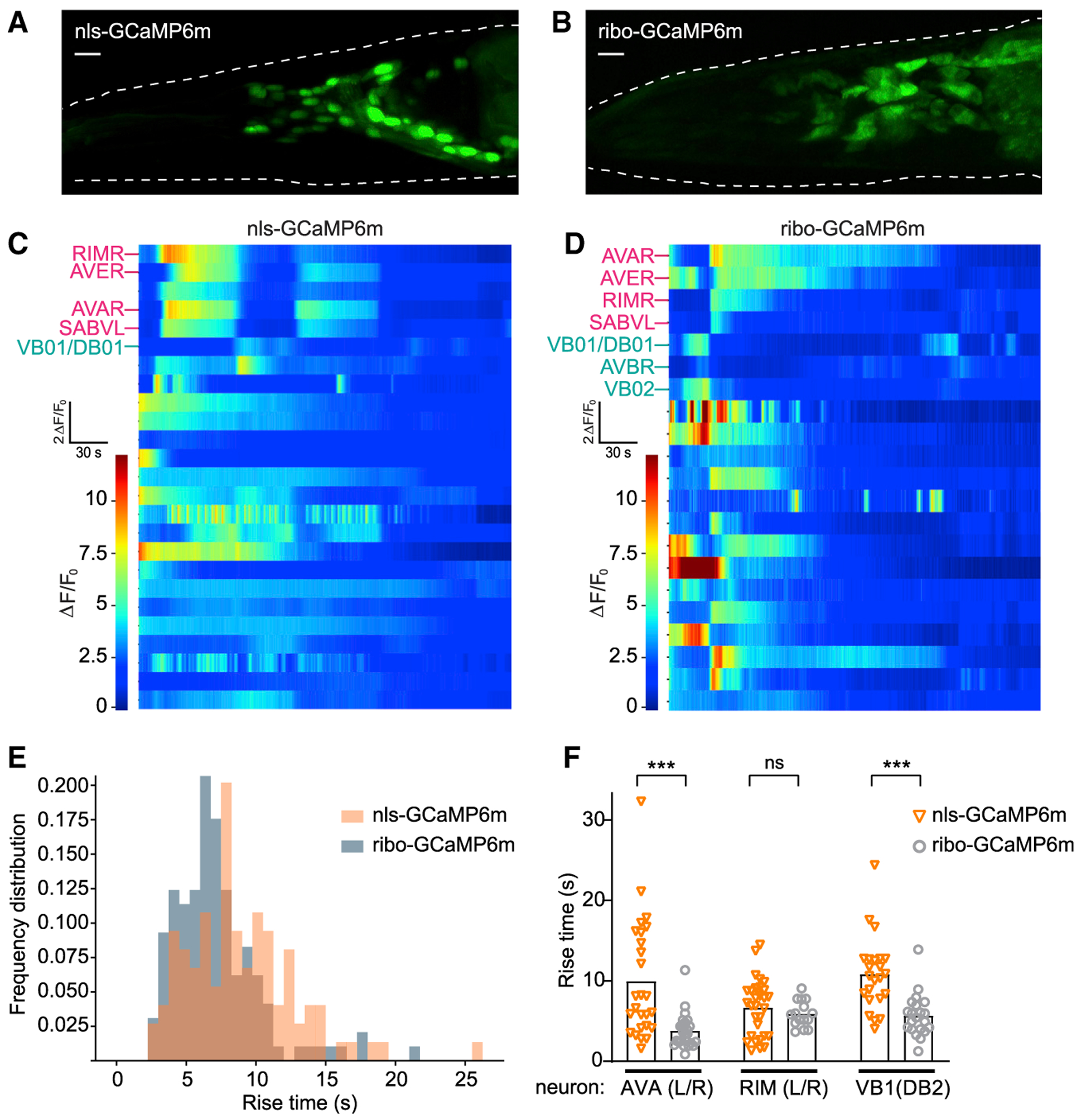
(A and B) Maximum intensity projection of representative worms recorded under constant conditions expressing pan-neuronal nls-GCaMP6m (A) or ribo-GCaMP6m (B). Dotted white lines denote worm head. Scale bars, 10 μm.
(C and D) Representative heat plots of fluorescence (ΔF/F0) 5-min time series for pan-neuronal nls-GCaMP6m (C) or ribo-GCaMP6m (D), 1 neuron per row. Labeled neurons indicate putative cell IDs, PC1+ neurons in pink and PC1− neurons in cyan.
(E) Histogram of frequency distributions for all rise times of peaks in calcium transients measured for 47 ribo-GCaMP6m traces (from 5 time series recordings) and 60 nls-GCaMP6m traces (from 8 time series recordings).
(F) Rise times of peaks in calcium transients measured from the same neuron across recordings expressing either nls-GCaMP6m or ribo-GCaMP6m. ***p < 0.001, ns, not significant using Kolmogorov-Smirnov test.
C. elegans adults at single-cell resolution. We recorded spontaneous calcium dynamics under environmentally constant conditions in unstimulated and immobilized worms (8 nls-GCaMP6m, 5 ribo-GCaMP6m) for 5–18 min (Figures 8C and 8D) and observed Ca2+ dynamics in most active neurons that matched one of the first 3 temporal principal components (PC1–PC3) described in previous studies (Kato et al., 2015). The location, morphology, and neuronal activity patterns of some of these neurons allowed us to reliably identify them across recordings using either sensor (Figure 8F). This indicates that the soma restriction of ribo-GCaMP6m makes it possible to distinguish individual cells during whole-brain imaging in the worm.
Calcium reporters can show slower responses in the nucleus compared to the cytosol (Kim et al., 2014). We therefore directly compared the temporal resolution of ribo-GCaMPm and nls-GCaMP6m during whole-brain imaging. To do this, we manually selected individual ROIs from each soma or nucleus and then automated the quantification of large-magnitude rise transients in all whole-brain recordings. This revealed that the distribution of rise times for nls-GCaMP6m was shifted to slower responses compared to ribo-GCaMP6m (Figure 8E). To quantify this slowing, we modeled an effective transformation between nls-GCaMP6m and ribo-GCaMP6m traces as a single-exponential linear filter, then estimated the time constant of the best-fit filter across both distributions of rise transients. This suggested a delay of ~3 s for nls-GCaMP6m compared to ribo-GCaMP6m (see Method Details and Figure S5B). To validate this population analysis at the level of identified neurons, we measured rise times across recordings in three specific neuron pairs: AVA, RIM, and VB1. This confirmed that ribo-GCaMP6m displayed faster rise time kinetics in AVA (3.8 ± 0.5 s ribo versus 9.9 ± 1.4 s nls) and VB1 (5.7 ± 0.6 s ribo versus 10.8 ± 0.9 s nls) neurons (Figure 8F). These findings reveal that ribosome-tethered GCaMPs support whole-brain imaging in the worm with enhanced signal and temporal fidelity.
DISCUSSION
We have described here two strategies that use the ribosome to target fluorescent proteins to the cell body of neurons. We have shown that these reagents eliminate fluorescence from neuropil while enhancing the signal from the soma, thereby enabling crisp visualization of individual neurons and their calcium dynamics in mice and worms. These soma-targeted reagents fill an important gap in the toolbox of imaging reagents available to neuroscience.
A Nanobody Enhancer of GFP Fluorescence
These studies were initially motivated by the observation that the expression of Nano-L10 in neurons dramatically enhanced the somatic fluorescence of co-expressed GFP. Nano-L10 produced a soma-localized GFP signal that could be easily visualized by native fluorescence in every GFP transgenic line we tested. In contrast, the same GFP reporter mice without Nano-L10 often displayed fluorescence that was undetectable, even after immunostaining, or alternatively so diffuse that it was difficult to localize to individual cells (Figure 1). This suggests that the Nano-L10 mouse could be broadly useful for enhancing the performance of GFP reporter mice, particularly for applications that require visualization of the labeled cells by native fluorescence (e.g., slice electrophysiology).
The increase in somatic fluorescence caused by Nano-L10 likely involves multiple mechanisms. Some of the enhancement is presumably mediated by trapping and concentrating GFP in the cell body, which would increase the fluorescence from the soma while reducing the background from neuropil. The concentration factor achieved in this way could be dramatic, given that the axonal volume is >100 times larger than the somatic volume in some neurons (Pannese, 2014). In addition to this localization effect, nanobody binding has been shown to increase the intrinsic fluorescence of GFP by ~50% in vitro (Kirchhofer et al., 2010; Kubala et al., 2010). Finally, nanobody binding may protect GFP from degradation and thereby increase its half-life in vivo. This is possible because the nanobody and GFP form a high-affinity, stable complex (Kd = 0.59 nM; Kirchhofer et al., 2010), and the ribosome is recycled slowly in the brain (e.g., half-life of 9 days in the rat brain; Retz and Steele, 1980).
A Ribo-GCaMP for Soma-Targeted Calcium Imaging
Inspired by the effect of ribosome tethering on GFP fluorescence, we investigated whether a similar strategy could be used for soma targeting of calcium reporters. For this purpose, we linked GCaMP6m directly to ribosomal protein L10, analogous to the tagging strategy that has been used for ribosome profiling (Heiman et al., 2008). We then compared the performance of this ribo-GCaMP6m with traditional GCaMP6m in mouse brain slices, by 1P and 2P imaging in behaving mice and by widefield single neuron and whole brain imaging in worms.
The expression of ribo-GCaMP6m was tightly restricted to the cell body in every context we tested. For example, we found that ribo-GCaMP6m fluorescence in striatal interneurons (Figure 2K) and cortical pyramidal cells (Figure 4F) began to decline as little as 10 μm from the cell body and became undetectable within 50 μm, effectively eliminating neuropil fluorescence. This soma restriction did not alter the electrophysiologic properties of the target cells (Figures 3 and 4) or induce detectable toxicity in imaging experiments lasting up to 6 months in vivo (Figure S4). In functional experiments, we found that ribo-GCaMP6m faithfully reported on the neural responses to drifting bars in the visual cortex (Figure 5), food ingestion in the PFC (Figure 6), and osmotic and heat stimulation in C. elegans sensory neurons (Figure 7). It also enabled large-scale imaging of spontaneous network activity in the worm head (Figure 8). Thus, ribo-GCaMP appears to be a broadly useful reagent for soma-targeted calcium imaging.
Soma Targeting Enables Facile Segmentation at High Expression Densities
Eliminating neuropil fluorescence should clarify the boundaries between neurons, and we found that ribo-GCaMP6m enabled straightforward segmentation of densely intermingled neurons in the mouse brain (Figure 5) and the worm head (Figure 8). This was particularly useful in the worm because it enabled the use of ribo-GCaMP6m, rather than nuclear GCaMPs, for whole-brain imaging. We generated transgenic animals expressing ribo-GCaMP6m in every neuron and showed that we could monitor the spontaneous activity of individual neurons in these animals. While soma-targeted GCaMPs are inherently less spatially segregated than nuclear GCaMPs (which have dark space surrounding each nucleus), ribo-GCaMP6m supported accurate cell identification along with faster kinetics and stronger peak fluorescence. Thus ribo-GCaMP6m enables whole-brain monitoring of somatic calcium dynamics in the worm.
In the mouse brain, the use of ribo-GCaMP6m made it possible to perform accurate segmentation of ~2,000 neurons from a static image of a 1-mm2 2P field of view (Figures S3G and S3H). This was not possible with GCaMP6m, because many cells were indistinguishable from neuropil in the absence of activity (Figures S3G and S3H). While this problem can be addressed using algorithms that incorporate information about dynamics (e.g., CNMF, Suite2P, PCA-ICA), it is important to note that these algorithms are inherently biased toward the identification of cells that are active during the experiment (Pachitariu et al., 2017). In this regard, we believe that the increased number of silent cells identified with ribo-GCaMP6m is responsible in part for the lower percentage of orientation tuned cells observed in V1 (Figure 5E). This ability of ribo-GCaMP6m expression to clearly delineate every cell at baseline may be useful in contexts in which it is critical to obtain an unbiased census of activity within a brain region.
Soma Targeting Eliminates Correlated Activity Induced by Neuropil Contamination
Fluctuations in neuropil fluorescence can lead to the spurious appearance of correlated activity between neurons (Harris et al., 2016). We found during 2P imaging of the visual cortex that cells expressing GCaMP6m showed a clear distance-dependent correlation in their activity. The use of ribo-GCaMP6m eliminated neuropil fluorescence and abolished this correlation (Figures 5F–5I). As a comparison, we also performed post hoc subtraction of the neuropil signal from traditional GCaMP6m recordings (see Method Details). This reduced the correlated activity between neurons, but residual correlation persisted and was both distance dependent and elevated compared to ribo-GCaMP6m recordings analyzed without any correction. Thus, soma targeting is highly effective at removing neuropil-induced correlations between neurons in 2P imaging experiments.
Signal contamination is also problematic during recordings with microendoscopes and other forms of widefield imaging. To investigate this, we directly compared GCaMP6m and ribo-GCaMP6m during GRIN lens imaging of mPFC with head-mounted miniscopes. We found that soma targeting did reduce the distance-dependent correlation in activity between pairs of neurons (Figure 6G), but the magnitude of this reduction was modest compared to what we observed in 2P imaging (12% for 1P versus 83% for 2P). This suggests that most of the signal contamination in microendoscope recordings results from sources other than neuropil fluorescence, such as out-of-focus light and light scattering.
Limitations and Future Directions
We have shown that ribo-GCaMP6m has several advantages relative to traditional GCaMPs, but there are trade-offs that should be considered. In general, we found that ribo-GCaMP6m was not as bright as GCaMP6m in side-by-side recordings performed in the mouse brain by 1P and 2P imaging. We could compensate for this difference by increasing the illumination power (discussed further in Method Details), but in some contexts this will be a disadvantage. Of note, this difference in brightness was not observed in vitro (Figures 3 and 4), and in the worm, we found that ribo-GCaMP6m displayed stronger peak fluorescence than traditional GCaMP6m (Figure 7). The source of this variability is unknown, but it may reflect the variation in expression levels between samples that cannot be fully controlled (despite using matched constructs and viral titers).
Previous studies have described efforts to localize opsins to the cell body using targeting motifs derived from the potassium channel Kv2.1 (Baker et al., 2016; Mahn et al., 2018; Messier et al., 2018), the kainate receptor subunit 2 (Shemesh et al., 2017), and ankyrin-binding proteins (Greenberg et al., 2011; Grubb and Burrone, 2010; Zhang et al., 2015). While these soma-targeting motifs have been useful, the ribosome has several properties that make it particularly well suited to serve as a localization tag for fluorescent proteins. These include the fact that the ribosome is expressed at high levels throughout the volume of the cell body of every neuron, yet it is tightly restricted from axons and dendrites. This enables a high level of transgene expression with exquisite soma localization. In this study, we have described two methods for tethering fluorescent proteins to the ribosome and shown that they have distinct advantages for biological imaging. However, there remains a significant opportunity to optimize these constructs to improve their properties for certain applications. Moreover, it should be possible to combine this localization strategy with ongoing improvements in hardware and optical reporters to enable new levels of sensitivity and resolution.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information about resources, reagents, and code should be directed to and will be fulfilled by the Lead Contact, Zachary Knight (zachary.knight@ucsf.edu).
Materials Availability
Plasmids generated in this study will be deposited at Addgene. Transgenic worm strains generated in this study will be deposited at the Caenorhabditis Genetics Center (CGC) and will be available within six months of publication.
Data and Code Availability
The datasets and code generated during this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
We obtained SIM1-Cre transgenic mice (Tg(Sim1-cre)1Lowl/J, #006395) and wild-type mice (C57BL/6J, #000664) from Jackson Laboratory. We obtained Agtr1a-GFP transgenic mice (Tg(Agtr1a-EGFP)NZ44Gsat, MGI:4846843) from the GENSAT project. GCG-GFP, GAD67-GFP, TrkB-tauGFP, TRPV1-GFP-DTR, TRPM8-GFP-DTR, and Nano-L10 mice have been described previously (Ekstrand et al., 2014; Hayashi et al., 2009; Li et al., 2011; Pogorzala et al., 2013; Tamamaki et al., 2003). We obtained Ntrk1-Cre knockin mice (B6;129S4-Ntrk1tm1(cre)Lfr/Mmucd, RRID: MMRRC_015500-UCD) from MMRRC. Adult mice (> 6 weeks old) of both sexes were used for experiments. All animals were maintained on a 12-h light/dark cycle and given ad libitum access to chow (PicoLab Rodent Diet 5053) and water. All procedures were conducted during the light cycle unless otherwise noted. All experimental protocols were approved by the University of California, San Francisco IACUC following the National Institutes of Health guidelines for the Care and Use of Laboratory Animals.
Worms
All C. elegans strains were cultivated using standard protocols in a 20°C incubator on nematode growth media (NGM) plates seeded with Escherichia coli OP50 bacteria as a food source. Young adult (Day 1 or 2) hermaphrodites were used for all experiments. Transgenic lines were constructed by injecting plasmids using standard techniques.
METHOD DETAILS
Protein engineering
To tether GCaMP6 and the GFP nanobody to ribosomes, GCaMP6m (Chen et al., 2013) or the GFP nanobody (Ekstrand et al., 2014; Rothbauer et al., 2006) was linked to ribosomal subunit protein RPL10 through a short linker of amino acid sequence SGRTQISSSS-FEF (Heiman et al., 2008). The resultant construct is GCaMP6-RPL10 and is referred to as ribo-GCaMP for simplicity in the paper. All constructs were designed using a combination of restriction cloning, Gibson Assembly and gBlock gene fragments (Integrated DNA Technologies). For C. elegans constructs, the sequence of rpl-1 was fused to the C-terminus of GCaMP6m using an overlap PCR strategy. All regions that underwent PCR amplification were checked through sanger sequencing (GeneWitz; Elim Biopharm) following RCA-based amplification (GE Templiphi). Constructs were made into custom AAV through Stanford Vector Core:
AAV8-hSyn-GCaMP6m/f/s (Restriction Cloning: AscI & NheI)
AAV5/8-hSyn-riboGCaMP6m/f/s (Gibson Assembly + gBlock)
AAV5/8-hSyn-DIO-riboGCaMP6m/f/s (Restriction Cloning: AscI & NheI)
Linker-RL10 amino acid sequence: SGRTQISSSSFEFSSKVSRDTLYEAVREVLHGNQRKRRKFLETVELQISLKNYDPQKDKRFSGTVRLKSTPRPKFSVCVLGDQQHCD EAKAVDIPHMDIEALKKLNKNKKLVKKLAKKYDAFLASESLIKQIPRILGPGLNKAGKFPSLLTHNENMVAKVDEVKSTIKFQMKKVLC LAVAVGHVKMTDDELVYNIHLAVNFLVSLLKKNWQNVRALYIKSTMGKPQRLY
General surgical procedures
All surgical procedures were performed in accordance with institutional guidelines for anesthesia and analgesia. In brief, mice were anesthetized with isoflurane and placed in a stereotaxic device with eyes covered with ophthalmic ointment. Buprenorphine SR (1.5 mg/kg), meloxicam (5 mg/kg), and dexamethasone (0.6 mg/kg) were administered systemically to prevent pain and brain edema. Bupavicaine was applied at the surgical site. Specific surgical procedures are described in the sections that follow.
Immunohistochemistry
General procedure: Mice were transcardially perfused with PBS followed by 10% formalin, brains were dissected, post-fixed in 10% formalin overnight at 4°C, and then washed 3×20 minutes with PBS at RT. Tissue was then cryoprotected with 30% sucrose in PBS overnight at 4°C, embedded in OCT and frozen at −20°C. For brain tissue, sections (40 um) were prepared with a cryostat. Sections were then washed and mounted on slides with DAPI fluoromount-G (Southern Biotech) or stained. For staining, free-floating sections were blocked (5% NGS in 0.1% PBST (0.1% Triton X-100 in PBS)) for 30 min at RT and incubated with primary antibodies overnight at 4°C. The next day, sections were washed 3×10 min with 0.1% PBST, incubated with secondary antibodies for 2 hours at RT, washed again 3 × 10 minutes with 0.1% PBST, and then mounted as above.
For peripheral sensory ganglia, ventral aspect of skulls and vertebral columns were dissected, post-fixed in 10% formalin overnight at 4°C, and washed 3 × 20 minutes with PBS at RT. Nodose ganglion and dorsal root ganglion were then dissected out and cryoprotected as described above. Ganglion sections (20 μm) were prepared with a cryostat, collected on slides, and dried overnight at RT before staining. Slides were washed 3 × 10min with 0.1% PBST and then stained as described above.
The following antibodies were used: chicken anti-GFP (Abcam, ab13970), 1:1000 in blocking solution; rabbit anti-Iba1 (Wako, 019–19741), 1:1000 in blocking solution; goat anti-chicken Alexa 488 (Invitrogen, A-11039), 1:500 in blocking solution; goat anti-rabbit Alexa 568 secondary antibody (Invitrogen, A-11036), 1:1000 in blocking solution.
For histologic characterization of Cre-dependent ribo-GCaMP in PVH, AAV5 encoding DIO-riboGCaMP6f or DIO-GCaMP6f (200 nL) was bilaterally injected into the PVH (−0.75 mm AP, ± 0.30 mm ML, −4.85 mm DV) of SIM1-Cre mice. For histologic characterization ribo-GCaMP in medial prefrontal cortex (mPFC), hippocampus (HPC) and superior colliculus (SC), AAV encoding ribo-GCaMP6m or GCaMP6m (200–300 nL) was unilaterally injected into the mPFC (+1.5 mm AP, 0.35 mm ML, 2.6 mm DV), HPC(−1.8 mm AP, −1.0 mm ML, −2.2 mm DV), SC (0 mm AP (lambda), −~0.6mm ML, −1.5 mm DV) and PVH (−0.75 mm AP, −0.25 mm ML, −4.8 mm DV). Mice were transcardially perfused 4 weeks after surgery.
For histologic characterization of the subcellular distribution of ribo-GCaMP in striatal Ntrk1 neurons, AAV9 encoding hSyn-DIO-riboGCaMP6m or hSyn-DIO-GCaMP6m (100 nL) was co-injected with AAV5 encoding EF1a-DIO-ChR2(H134R)-mCherry (100 nL) into the striatum (0.6 mm AP, ± 2.5 mm ML, 2.9 mm DV) of Ntrk1-Cre mice. Mice were transcardially perfused 2–4 weeks after viral injection. To reconstruct single Ntrk1-Cre neurons, a tiled Z stack image was taken for both the red (ChR2(H134R)-mCherry) and green (GCaMP6m or ribo-GCaMP6m) channels. The red channel was used to create a 3D mask based on pixel threshold and connectivity using FIJI (Schindelin et al., 2012). Then both the green and red fluorescence inside but not outside the mask were retained to show the relationship between the green channel and the shape of single cells. To trace the intensity of GCaMP6m or ribo-GCaMP6m along individual axons, we manually traced the axon of selected neurons in the red channel starting from the beginning of the axon. We then extracted the pixel intensity along the traced line to generate plot of green intensity versus distance from soma.
For histologic characterization of ribo-GCaMP after long-term expression in mPFC, AAV8 encoding ribo-GCaMP6m and GCaMP6m (200 nL each) were each injected into opposing hemispheres of mPFC (+1.5 mm AP, ± 0.35 mm ML, −2.6 mm DV). Mice were transcardially perfused 4, 6, 8 or 12 weeks after surgery.
Slice field stimulation
AAV encoding ribo-GCaMP6m or GCaMP6m (200 nL) was bilaterally injected into the HPC (−1.8 mm AP, ± 1.0 mm ML, 2.2 mm DV). Brains were sliced and in recovered NMDG solution (Ting et al., 2014). Recordings were made with standard ACSF (2−mM calcium) containing a cocktail of inhibitors to repress neurotransmission and thus network activity: 10 μM CNQX (Tocris, 0190), 10 μM (R)-CPP (Tocris, 0247), 10 μM Gabazine (Tocris, 1262) and 1000 μM (S)-MCPG (Tocris, 0337) (Wardill et al., 2013). Fluorescence signals were recorded using a digital CCD camera (Hamamatsu, ORCA-ER) mounted on an Olympus upright microscope (BX51WI). Micro-manager software (version 1.4) was used as microscope control interface (Edelstein et al., 2014). Slices were imaged (5 ms exposure time; 20 Hz) with 470 nm excitation through a filter set (U-N41 017, E.X. 470 nm, B.S. 495 nm, E.M. 5, Olympus). Field stimulation (30–40V) was delivered through parallel platinum wires (Warner Instruments, RC-49MFS) from a stimulation isolator (A.M.P.I, ISO-Flex). The stimulation pattern (80 Hz, 5 ms pulse width) was generated by Digidata 1550 (Molecular Devices) and pClamp 10.5 software (Molecular Devices). The same stimulation and recording parameters were used to evaluate the response of ribo-GCaMP6m and GCaMP6m. Rise time constant and decay time constant were defined, respectively, as the time that it takes fluorescence to reach 50% of peak fluorescence from the onset of stimulation, and the time to decay to 50% of peak fluorescence from the peak of fluorescence.
Slice two-photon recording
All cells recorded were Type 2 L5 pyramidal neurons as determined by Ih mediated sag and rebound (Gee et al., 2012). Recordings were made with standard ACSF (2 mM calcium) and k-gluconate internal with EGTA omitted. Internal contained 20 μM for cell visualization. Cells were held a −68 mV and stimulated with variable number of spikes at 100 Hz while simultaneously imaging with 2-Photon linescans using 920 nm stimulation. Imaging was first conducted at the soma, then the apical dendrite at distances of 20, 50, 100, 150, and 200 μm, and then the basal dendrites at 20, 50, and 100 μm. Z stacks of the entire cell were then taken to recover the full dendritic morphology. Baseline noise was calculated from the standard deviation of the fluorescent signal 100 ms before stimulus onset. Signal to noise was calculated as the peak dF/F divided by the baseline noise.
Cranial window surgery and headfixed two-photon imaging
Experiments were conducted on adult C57BL6/J mice (males, age > 8 weeks). During surgery a custom titanium head plate (eMachineshop) was attached to the skull using dental cement (Metabond, Parkell), and a 3 mm diameter craniotomy was made over visual cortex (0.8 mm anterior from lambda and 2.5 mm lateral from bregma). In the middle of the craniotomy, virus (AAV9-ri-boGCaMP6m or AAV9-GCaMP6m) was injected at two sites (−0.6 mm ventral from skull surface, 0.8 and 1.0 mm anterior from lambda, volume: 150 nL at each site, rate: 15 nL/min). A window plug made from two 3 mm diameter coverglasses glued to a 5 mm diameter coverglass was placed over the craniotomy and fixed in place using dental cement. Imaging experiments began 3 weeks after window implantation.
Mice were headfixed in a body tube under a Nikon 16X objective and imaged using a resonant scanning two-photon microscope (Neurolabware). Images were acquired at a rate of 15.49 Hz with a field of view of 1.2 mm by 0.9 mm mm. We found that ribo-GCaMP6m was dimmer than GCaMP6m in vivo under the same laser setting. To match the fluorescence intensity and thus enable fair cross-correlation comparison, laser power was set to be higher when imaging ribo-GCaMP6m than GCaMP6m preps (67 mW versus 28 mW). Pixel size was calibrated to be ~1.9 μm ×7 1.9 μm. Imaging in layer 2/3 was performed 200 μm below the pial surface. Moving bar stimuli were generated using Psychtoolbox3 (MATLAB) and presented on an LCD screen positioned 20 cm away from the mouse (Kleiner et al., 2007). Bars were 5–7° wide and drifted at 18–22° s-1. Bars were white against a gray background. In a separate series of experiments, full-screen moving grating stimuli were presented with a cycle length of 10–15° and a temporal frequency of 2 Hz. All stimuli were presented in a pseudorandom sequence (sampling without replacement) with interspersed breaks (8.4 s).
For data analysis, lateral brain motion was corrected using NoRMCorre (MATLAB) (Pnevmatikakis and Giovannucci, 2017). Neuron identification, segmentation and fluorescent signal extraction were performed using Suite2P (Python) (Pachitariu et al., 2017). Identified ROIs were further manually screened. Suite2P computed the neuropil signal for each ROI, which is defined as the weighted average signal of all pixels surrounding each ROI; the minimal width of the donut-shaped surrounding neuropil area is defined as 100 pixels (~190 μm).
To subtract the neuropil signals from the soma signal, we used the equation suggested by Suite2P, that is Fsubtracted = Fsoma − 0.6 × Fneuropil. Data shown in the following figures underwent neuropil subtraction for both ribo-GCaMP6m and GCaMP6m: Figures 5C–5E and S3B. Some data shown in those figures also underwent neuropil subtraction, and the details are indicated in the following figure legends: Figures 5B, 5F–5I, S3C, and S6D. Note that the neuropil subtraction has little effect on the ribo-GCaMP6m results and is applied primarily so that ribo-GCaMP6m can be compared to GCaMP6m using a standard two-photon data analysis procedure.
To normalize the fluorescent signal of each ROI, the signal of each frame was z-scored as Fz = (Feachframe-mean(Flocal))/std(Flocal). Flocal is the fluorescence signal of a 1-minute time window centering each frame. When calculating z-score for the first or last N frames (N < 60), a moving window with size 60-N was used.
To compute the response of a neuron to each visual stimulus, a time window of two seconds before the onset of each stimulus was defined as the pre-stim period, and a 5 s (for drifting grating stimuli) or 7 s (for drifting bar stimuli) time window after each stimulus was defined as the post-stim period. The response of the neuron to the stimulus was calculated as the signal maximum minus the median signal during the pre-stim period. To identify orientation-tuned neurons, we used 1-way ANOVA to test whether a neuron responds differentially to visual stimuli with different orientations. Neurons with P value < 0.01 were defined as orientation-selective (Dana et al., 2019). The stimuli that triggered the maximal neural response are defined as having the preferred angle.
To perform segmentation based on static images, we first averaged z-projection of each video of drifting bar experiment and enhanced the local contrast by applying adapthisteq function (MATLAB). We then used NeuroSeg (MATLAB), a segmentation algorithm that solely based on static image, to generate ROIs (Min sigma = 1; Max sigmal = 3; Min area = 12) (Guan et al., 2018). All automatically generated ROIs are used in the comparison without manual selection.
Microendoscope imaging and ingestive behavior
For microendoscope experiments, mice were prepared based on published protocols (Flusberg et al., 2008; Ghosh et al., 2011; Resendez et al., 2016). In brief, AAV encoding ribo-GCaMP6m (200 nL) was unilaterally injected into the mPFC (−1.5 mm AP, −0.35 mm ML, −2.6 mm DV). A Ø500 μm gradient index (GRIN) lens (6.1 mm length; Inscopix) was then placed 0.10 mm above the injection site in the same surgery. Baseplates (Inscopix) were then mounted 4–8 weeks after initial surgery. Mice were allowed to recover for 2 weeks after baseplating and were habituated to handling and behavioral apparatus for another week. During the experiment, mPFC neurons of each mouse were recorded through a miniature microscope (Inscopix) using nVista software (www.inscopix.com/nvista) with identical settings (20 Hz, 20% LED power, 3.0 gain). After a 10-minute baseline measurement, the mice were allowed to consume liquid food (Ensure). The mice were either fed or food deprived for 24 hours before the start of each experiment.
To analyze microendoscope data, videos were first pre-processed (spatial downsample by a factor of 2; temporally downsample by a factor of 5) and motion-corrected using Mosaic software MATLAB API suite (https://support.inscopix.com/mosaic-workflow). Activity traces for individual neurons were then extracted from these videos using the constrained nonnegative matrix factorization - endoscope (CNMF_E) pipeline (MATLAB) (Zhou et al., 2018). Consummatory events were recorded by a capacitance-based (https://www.arduinolibraries.info/libraries/capacitive-sensor) contact lickometer custom build with microcontrollers (Arduino Uno) connected to the data acquisition box of mini-endoscope.
Calcium imaging in Caenorhabditis elegans
For ASH and AFD imaging, well-fed young adult worms were immobilized in custom-built microfluidic devices (Chronis et al., 2007) in S buffer (100 mM NaCl, 50 mM KH2PO4, pH 6.0) with addition of 0.02% (m/v) tetramisole hydrochloride (Sigma-Aldrich, St. Louis, MO). Ribo-GCaMP6m (GCaMP6m C-terminal fusion with RPL-1) or soluble GCaMP6m was expressed in the C. elegans ASH sensory neuron under control of the sra-6 promoter. Worms were stimulated with 0.5 M sodium chloride delivered to the nose of the animal for 10 s (one pulse). ASH imaging was preceded by a 30 s exposure to blue light to reduce intrinsic light response (Hilliard et al., 2005; Ward et al., 2008). Each worm was subjected to two stimulatory pulses of 0.5 M sodium chloride with a 1 min interval between the pulses. For AFD imaging, GCaMP6m-RPL-1 fusion was expressed in the AFD sensory neuron under control of the ntc-1 promoter. Heat stimulation was performed by delivering pre-warmed buffer (~30°C) to the nose of the animal for 10 s (one pulse). An in-line solution heater (Warner Instruments, SF-28) was incorporated into the setup to provide pre-warmed buffer flow and buffer temperature at the device was measured using a handheld thermocouple thermometer. Each worm was subjected to three stimulatory warm buffer pulses with a 1 min interval between the pulses. GCaMP fluorescence was visualized by illumination with a LED lamp (X Citeª 120LED, Excelitas Technologies, 480/30 nm excitation filter, 535/40 nm emission filter, Chroma). Images were collected on Axio Observer A1 microscope (Zeiss) at 10 frames per second using a 63x, 1.4 NA objective and a CMOS camera (ORCA-Flash4.0, Hamamatsu). Fluorescent signal modulation over the period of acquisition was analyzed using Fiji software by defining the soma or dendrite and subtracting the background fluorescence (Reilly et al., 2017). Fluorescence intensity was normalized to the baseline signal (F0) during the last 1 s in control buffer prior to stimulus delivery and further analyzed using a custom written Python script. Data are plotted as mean ± SEM (shaded region).
For whole-brain imaging of head ganglia neurons, ribo-GCaMP6m or nls-GCaMP6m were expressed using the pan-neuronal tag-168 and rab-3 promoters, respectively, in the lite-1 genetic background with pan-neuronally expressed nuclear RFP markers.
Calcium imaging recordings were made using a Nikon Ti Microscope with Andor Zyla 5.5 sCMOS camera. The worm was immobilized in a microfluidic chip with addition of 1% tetramisole as described (Kato et al., 2015). Recordings were started within 5 min after removal from food. The head of the worm was imaged for 5–18 minutes using a 40x objective and collecting 10–24 z stacks 2 μm step size to cover the thickness of the worm, with 33.3 or 50 frames per second. The center of the individual regions of interest (ROIs) were manually selected from each soma (ribo-GCaMP6m) or nucleus (nls-GCaMP6m) using Fiji software, and subsequent analysis was performed by a custom-written Python script. The fluorescence intensity from a square of 3×3 pixel areas surrounding the center of the ROI was measured and delta F/F0 was calculated by defining baseline signal (F0) as the mean intensity over the whole trace.
Rise time measurement and comparison
Rising transients in extracted neural timeseries were computationally detected using a peak finding algorithm followed by detection of onset and termination frames of rise periods using first and second derivative threshold criteria on smoothed timeseries, as implemented in the Python notebook included in Key Resources Table. To estimate the temporal differences between the ribo-GCaMP6m and nls-GCaMP6m indicators, epochs from ribo-GCaMP6m recordings containing rise transients were extracted and convolved with a first-order linear filter with a time constant of tau. Rise times were then measured in the resulting simulated trace excerpts. An optimal tau was determined by minimizing the error between a histogram of the simulated trace excerpts and a histogram of the nls-GCaMP6m rise times. This optimal tau gives an estimate of the temporal blurring effects of the nls-GCaMP6m versus the ribo-GCaMP6m indicator.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses and linear regressions were performed using Prism 7 (https://www.graphpad.com/scientific-software/prism Values are reported as mean ± s.e.m. (error bars or shaded area), represented as black brackets in bar graphs and shaded areas in PSTH plots. In figures with linear regressions, the dotted lines represent the 95% confidence interval for the line-of-best-fit. P values for pairwise comparisons were performed using a two-tailed Student’s t test. P values for comparisons across multiple groups were performed using ANOVA and corrected for multiple comparisons using the Holm-Šídák method. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. N values and definitions can be found in the figure legends. No statistical method was used to predetermine sample size. Randomization and blinding were not used. Here is a full list of published open-source packages/software used in this study:
FIJI (Figures 1, 2, 4, 7, S1, S3, and S4): https://imagej.net/Fiji/Downloads (Schindelin et al., 2012)
Psychtoolbox3 (Figures 5 and S3): https://github.com/Psychtoolbox-3/Psychtoolbox-3 (Kleiner et al., 2007) NormCorre (Figures 5 and S3): https://github.com/flatironinstitute/NoRMCorre (Pnevmatikakis and Giovannucci, 2017) Suite2P (Figures 5 and S3): https://github.com/cortex-lab/Suite2P (Pachitariu et al., 2017)
Capacitive Sensing Library (Figure 6): https://playground.arduino.cc/Main/CapacitiveSensor/
CNMF_E (Figure 6): https://github.com/zhoupc/CNMF_E (Zhou et al., 2018)
PCA/ICA (Figure 6): https://github.com/mukamel-lab/CellSort (Mukamel et al., 2009)
NeuroSeg (Figure S3): https://github.com/baidatong/NeuroSeg (Guan et al., 2018)
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-GFP | Abcam | Abcam Cat# ab13970, RRID:AB_300798 |
| Rabbit anti-Iba1 | Wako | Wako Cat# 019–19741, RRID:AB_839504 |
| Goat anti-chicken Alexa 488 | Invitrogen | Thermo Fisher Scientific Cat# A-11039, RRID:AB_2534096 |
| Goat anti-rabbit Alexa 568 | Invitrogen | Thermo Fisher Scientific Cat# A-11036, RRID:AB_10563566 |
| Bacterial and Virus Strains | ||
| AAV5- EF1a-DIO-hChR2(H134R)-mCherry-WPRE-HGHpA | Gift from Karl Deisseroth; unpublished | Karl Deisseroth Addgene plasmid #20297 |
| Plasmid: pAAV-hSyn-GCaMP6m-RPL10a-WPRE-HGH | This paper; AAV8 produced by Stanford Vector Core | N/A |
| Plasmid: pAAV-hSyn-GCaMP6f-RPL10a-WPRE-HGH | This aper; AAV8 produced by Stanford Vector Core | N/A |
| Plasmid: pAAV-hSyn-GCaMP6s-RPL10a-WPRE-HGH | This paper; AAV8 produced by Stanford Vector Core | N/A |
| Plasmid: pAAV-hSyn-GCaMP6m-WPRE-HGH (control vector) | This paper; AAV8 produced by Stanford Vector Core | N/A |
| Plasmid: pAAV-hSyn-DIO-GCaMP6m-RPL10a-WPRE-HGH | This paper; AAV8 produced by Stanford Vector Core | N/A |
| Plasmid: pAAV-hSyn-DIO-GCaMP6f-RPL10a-WPRE-HGH | This paper; AAV5 produced by Stanford Vector Core | N/A |
| Plasmid: pAAV-hSyn-DIO-GCaMP6s-RPL10a-WPRE-HGH | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CNQX | Tocris | Cat# 0190 |
| (R)-CPP | Tocris | Cat# 00247 |
| Gabazine | Tocris | Cat# 01262 |
| (S)-MCPG | Tocris | Cat# 00337 |
| Tetramisole Hydrochloride | Sigma-Aldrich | CAS# 5086-74-8 |
| DAPI fluoromount-G | Southern Biotech | Cat# 0100–20 |
| Experimental Models: Organisms/Strains | ||
| M. musculus: C57BL/6J (WT) | JAX | JAX: 000664, RRID: IMSR_JAX:000664 |
| M. musculus: Tg(Sim1-cre)1Lowl/J (SIM1-Cre) | JAX | JAX: 006395, RRID: IMSR_JAX:006395 |
| M. musculus: Tg(Agtr1a-EGFP)NZ44Gsat (Agtr1a-EGFP) | GENSAT | MGI:4846843 |
| GCG-GFP | Gift from Hayashi Yoshitaka; Hayashi et al., 2009 | N/A |
| GAD67-GFP | Gift from Allan Basbaum, Tamamaki et al., 2003 | N/A |
| TrkB-tauGFP | Gift from David Ginty; Lietal., 2011 | N/A |
| TRPV1-GFP-DTR | Gift from Mark Hoon; Pogorzala et al., 2013 | N/A |
| TRPM8-GFP-DTR | Gift from Mark Hoon; Pogorzala et al., 2013 | N/A |
| Nano-L10 | Ekstrand et al., 2014 | N/A |
| M. musculus: B6;129S4-Ntrk1tm1(cre)Lfr/Mmucd (Ntrk1-Cre) | MMRRC | RRID: MMRRC_015500-UCD |
| C. elegans: Strain N2 | Caenorhabditis Genetics Center | WB Strain: 00000001 |
| C. elegans: Strain KG1180: lite-1(ce314) X. | Caenorhabditis Genetics Center | WB Strain: 00023485 |
| C. elegans: Strain JAZ279: jlgEx289[Psra6::GCaMP6m:: rpl-1a;Punc-122::dsRed2] | This study | N/A |
| C. elegans: Strain JAZ312: jlgEx301[Psra6::GCaMP6m; Punc-122::GFP] | This study | N/A |
| C. elegans: Strain JAZ275: jlgEx285[Pntc-1::GCaMP6m:: rpl-1a;Punc-122::dsRed2] | This study | N/A |
| C. elegans: Strain JAZ276: lite-1(ce314) X; jlgEx286[Ptag-168::GCaMP6m::rpl1a;Punc-122::dsRed2];otIs355 [Prab3::2xNLS::tagRFP] | This study | N/A |
| C. elegans: Strain JAZ313: lite-1(ce314) X; jlgEx302[Prab-3::GCaMP6m::rpl1a;Pelt-2::2xNLS::GFP];otIs355 [Prab3::2xNLS::tagRFP] | This study | N/A |
| Software and Algorithms | ||
| FIJI | Schindelin et al., 2012 | https://imagej.net/Fiji/Downloads |
| Psychtoolbox3 | Kleiner et al., 2007 | https://github.com/Psychtoolbox-3/Psychtoolbox-3 |
| NormCorre | Pnevmatikakis and Giovannucci, 2017 | https://github.com/flatironinstitute/NoRMCorre |
| Suite2P | Pachitariu et al., 2017 | https://github.com/cortex-lab/Suite2P |
| Capacitive Sensing Library | Arduino Playground | https://playground.arduino.cc/Main/CapacitiveSensor/ |
| CNMF_E | Zhou et al., 2018 | https://github.com/zhoupc/CNMF_E |
| PCA/ICA | Mukamel et al., 2009 | https://github.com/mukamel-lab/CellSort |
| NeuroSeg | Guan et al., 2018 | https://github.com/baidatong/NeuroSeg |
| PRISM 7 | GraphPad |
https://www.graphpad.com RRID:SCR_005375 |
| Mosaic | Inscopix |
https://support.inscopix.com/mosaic-workflow RRID:SCR_017408 |
| MATLAB R2019a | MathWorks | https://www.mathworks.com |
| Python (Custom Code) | This study | https://github.com/focolab/ribo-gcamp/ |
| Other | ||
| Ø500 μm gradient index (GRIN) lens (6.1 mm length) | Inscopix | N/A |
In Brief.
Chen et al. show that ribosome tethering can restrict fluorescent proteins to the soma, thereby eliminating neuropil fluorescence. This enables ultrasensitive detection of GFP in brain slices and eliminates the cross-contamination of somatic GCaMP signals during calcium imaging in mice and worms.
Highlights.
Ribosome tethering restricts fluorescent proteins to the cell soma
A ribo-nanobody mouse boosts the fluorescence of existing GFP reporters
Ribo-GCaMP reduces fluorescence cross-contamination during 2P calcium imaging
Ribo-GCaMP enables whole-brain imaging of somatic calcium dynamics in the worm
ACKNOWLEDGMENTS
We thank Yuhan Yang and Na Ji for advice on 2P imaging and analysis, Andrew Gordus for microfluidic devices, David Ginty for TrkB-tauGFP mice, Hayashi Yoshitaka for GCG-GFP mice, Mark Hoon for TRPV1-GFP-DTR and TRMP8-GFP-DTR mice, the Nikon Imaging Center at the University of California, San Francisco (UCSF) (S10OD017993), and the Caenorhabditis Genetics Center (CGC) for some worm strains (P40OD010440). Y.C. is supported by a Howard Hughes Medical Institute International Student Fellowship. H.J. is supported by an NIH Institutional National Research Service Award (T32 AG000266) training grant at the Buck Institute. This work was supported by the New York Stem Cell Foundation, the American Diabetes Association Pathway Program, and NIH grants DP2DK109533, R01DK106399, and R01NS094781 (to Z.A.K.); the American Federation for Aging Research, the Glenn Foundation for Medical Research, and NIH grants R35GM119828 and S10OD017993 (to J.L.G.); the E. Matilda Ziegler Foundation for the Blind, the Sandler Foundation, the Esther A. and Joseph Klingenstein Fund, the Brain and Behavior Research Foundation, the Whitehall Foundation, the Simons Foundation, and NIH grants DP2MH119426 and R01 NS109060 (to E.H.F.) and R01DA035913, R01MH112729, and R21MH112117 (to K.J.B.). Z.A.K. is a Howard Hughes Medical Institute Investigator.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2020.05.005.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Baker CA, Elyada YM, Parra A, and Bolton MM (2016). Cellular resolution circuit mapping with temporal-focused excitation of soma-targeted channelrhodopsin. eLife 5, e14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, et al. (2005). Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123, 493–505. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Thomas JH, and Horvitz HR (1990). Chemosensory cell function in the behavior and development of Caenorhabditis elegans. Cold Spring Harb. Symp. Quant. Biol 55, 529–538. [DOI] [PubMed] [Google Scholar]
- Beets I, Janssen T, Meelkop E, Temmerman L, Suetens N, Rademakers S, Jansen G, and Schoofs L (2012). Vasopressin/oxytocin-related signaling regulates gustatory associative learning in C. elegans. Science 338, 543–545. [DOI] [PubMed] [Google Scholar]
- Biron D, Shibuya M, Gabel C, Wasserman SM, Clark DA, Brown A, Sengupta P, and Samuel AD (2006). A diacylglycerol kinase modulates long-term thermotactic behavioral plasticity in C. elegans. Nat. Neurosci 9, 1499–1505. [DOI] [PubMed] [Google Scholar]
- Chatzigeorgiou M, Bang S, Hwang SW, and Schafer WR (2013). tmc-1 encodes a sodium-sensitive channel required for salt chemosensation in C. elegans. Nature 494, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis N, Zimmer M, and Bargmann CI (2007). Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat. Methods 4, 727–731. [DOI] [PubMed] [Google Scholar]
- Dana H, Sun Y, Mohar B, Hulse BK, Kerlin AM, Hasseman JP, Tsegaye G, Tsang A, Wong A, Patel R, et al. (2019). High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657. [DOI] [PubMed] [Google Scholar]
- Davis KC, Choi YI, Kim J, and You YJ (2018). Satiety behavior is regulated by ASI/ASH reciprocal antagonism. Sci. Rep 8, 6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using mManager software. J. Biol. Methods 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Nectow AR, Knight ZA, Latcha KN, Pomeranz LE, and Friedman JM (2014). Molecular profiling of neurons based on connectivity. Cell 157, 1230–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, Nimmerjahn A, Cocker ED, Mukamel EA, Barretto RPJ, Ko TH, Burns LD, Jung JC, and Schnitzer MJ (2008). High-speed, miniaturized fluorescence microscopy in freely moving mice. Nat. Methods 5, 935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison JL, Macosko EZ, Bernstein S, Pokala N, Albrecht DR, and Bargmann CI (2012). Oxytocin/vasopressin-related peptides have an ancient role in reproductive behavior. Science 338, 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, and Sohal VS (2012). Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J. Neurosci 32, 4959–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh KK, Burns LD, Cocker ED, Nimmerjahn A, Ziv Y, Gamal AE, and Schnitzer MJ (2011). Miniaturized integration of a fluorescence microscope. Nat. Methods 8, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez AD, Wang G, Waters EM, Gonzales KL, Speth RC, Van Kempen TA, Marques-Lopes J, Young CN, Butler SD, Davisson RL, et al. (2012). Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice. Neuroscience 226, 489–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracida X, and Calarco JA (2017). Cell type-specific transcriptome profiling in C. elegans using the Translating Ribosome Affinity Purification technique. Methods 126, 130–137. [DOI] [PubMed] [Google Scholar]
- Greenberg KP, Pham A, and Werblin FS (2011). Differential targeting of optical neuromodulators to ganglion cell soma and dendrites allows dynamic control of center-surround antagonism. Neuron 69, 713–720. [DOI] [PubMed] [Google Scholar]
- Grubb MS, and Burrone J (2010). Channelrhodopsin-2 localised to the axon initial segment. PLoS One 5, e13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Li J, Liang S, Li R, Li X, Shi X, Huang C, Zhang J, Pan J, Jia H, et al. (2018). NeuroSeg: automated cell detection and segmentation for in vivo two-photon Ca2+ imaging data. Brain Struct. Funct 223, 519–533. [DOI] [PubMed] [Google Scholar]
- Harris KD, Quiroga RQ, Freeman J, and Smith SL (2016). Improving data quality in neuronal population recordings. Nat. Neurosci 19, 1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk JD, Calvo AC, Liu P, Almoril-Porras A, Aljobeh A, Torruella-Suarez ML, Ren I, Cook N, Greenwood J, Luo L, et al. (2018). Integration of Plasticity Mechanisms within a Single Sensory Neuron of C. elegans Actuates a Memory. Neuron 97, 356–367.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Yamamoto M, Mizoguchi H, Watanabe C, Ito R, Yamamoto S, Sun XY, and Murata Y (2009). Mice deficient for glucagon gene-derived peptides display normoglycemia and hyperplasia of islet alpha-cells but not of intestinal L-cells. Mol. Endocrinol 23, 1990–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard MA, Apicella AJ, Kerr R, Suzuki H, Bazzicalupo P, and Schafer WR (2005). In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. EMBO J. 24, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, and Wiesel TN (1959). Receptive fields of single neurones in the cat’s striate cortex. J. Physiol 148, 574–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JM, and Horvitz HR (1993). A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 90, 2227–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Kaplan HS, Schrödel T, Skora S, Lindsay TH, Yemini E, Lockery S, and Zimmer M (2015). Global brain dynamics embed the motor command sequence of Caenorhabditis elegans. Cell 163, 656–669. [DOI] [PubMed] [Google Scholar]
- Keemink SW, Lowe SC, Pakan JMP, Dylda E, van Rossum MCW, and Rochefort NL (2018). FISSA: a neuropil decontamination toolbox for calcium imaging signals. Sci. Rep 8, 3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CK, Miri A, Leung LC, Berndt A, Mourrain P, Tank DW, and Burdine RD (2014). Prolonged, brain-wide expression of nuclear-localized GCaMP3 for functional circuit mapping. Front. Neural Circuits 8, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Miyawaki A, Matsumoto K, and Mori I (2004). The C. elegans thermosensory neuron AFD responds to warming. Curr. Biol 14, 1291–1295. [DOI] [PubMed] [Google Scholar]
- Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, et al. (2010). Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol 17, 133–138. [DOI] [PubMed] [Google Scholar]
- Kleiner M, Brainard D, and Pelli D (2007). What’s new in Psychtoolbox-3? Perception 36, 1–16. [Google Scholar]
- Kubala MH, Kovtun O, Alexandrov K, and Collins BM (2010). Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 19, 2389–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Rutlin M, Abraira VE, Cassidy C, Kus L, Gong S, Jankowski MP, Luo W, Heintz N, Koerber HR, et al. (2011). The functional organization of cutaneous low-threshold mechanosensory neurons. Cell 147, 1615–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Cook N, Venkatachalam V, Martinez-Velazquez LA, Zhang X, Calvo AC, Hawk J, MacInnis BL, Frank M, Ng JH, et al. (2014). Bidirectional thermotaxis in Caenorhabditis elegans is mediated by distinct sensorimotor strategies driven by the AFD thermosensory neurons. Proc. Natl. Acad. Sci. USA 111, 2776–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn M, Gibor L, Patil P, Cohen-Kashi Malina K, Oring S, Printz Y, Levy R, Lampl I, and Yizhar O (2018). High-efficiency optogenetic silencing with soma-targeted anion-conducting channelrhodopsins. Nat. Commun 9, 4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messier JE, Chen H, Cai ZL, and Xue M (2018). Targeting light-gated chloride channels to neuronal somatodendritic domain reduces their excitatory effect in the axon. eLife 7, e38506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel EA, Nimmerjahn A, and Schnitzer MJ (2009). Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, and Imoto K (2001). A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol 19, 137–141. [DOI] [PubMed] [Google Scholar]
- Nguyen JP, Shipley FB, Linder AN, Plummer GS, Liu M, Setru SU, Shaevitz JW, and Leifer AM (2016). Whole-brain Calcium Imaging With Cellular Resolution in Freely Behaving Caenorhabditis Elegans. Proc. Natl. Acad. Sci. U S A 113, E1074–E1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachitariu M, Stringer C, Dipoppa M, Schröder S, Rossi LF, Dalgleish H, Carandini M, and Harris KD (2017). Suite2p: beyond 10,000 neurons with standard two-photon microscopy. BioRxiv. 10.1101/061507. [DOI] [Google Scholar]
- Pannese E (2014). Neurocytology: Fine Structure of Neurons, Nerve Processes, and Neuroglial Cells (Springer). [Google Scholar]
- Perkins LA, Hedgecock EM, Thomson JN, and Culotti JG (1986). Mutant sensory cilia in the nematode Caenorhabditis elegans. Dev. Biol 117, 456–487. [DOI] [PubMed] [Google Scholar]
- Pnevmatikakis EA, and Giovannucci A (2017). NoRMCorre: an online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 291, 83–94. [DOI] [PubMed] [Google Scholar]
- Pnevmatikakis EA, Soudry D, Gao Y, Machado TA, Merel J, Pfau D, Reardon T, Mu Y, Lacefield C, Yang W, et al. (2016). Simultaneous Denoising, Deconvolution, and Demixing of Calcium Imaging Data. Neuron 89, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogorzala LA, Mishra SK, and Hoon MA (2013). The cellular code for mammalian thermosensation. J. Neurosci 33, 5533–5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevedel R, Yoon Y, Hoffmann M, Pak N, Wetzstein G, Kato S, Schrödel T, Raskar R, Zimmer M, Boyden ES, et al. (2014). Simultaneous whole-animal 3D imaging of neuronal activity using light-field microscopy. Nat. Methods 11, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly DK, Lawler DE, Albrecht DR, and Srinivasan J (2017). Using an Adapted Microfluidic Olfactory Chip for the Imaging of Neuronal Activity in Response to Pheromones in Male C. Elegans Head Neurons. J. Vis. Exp (127) 10.3791/56026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resendez SL, Jennings JH, Ung RL, Namboodiri VMK, Zhou ZC, Otis JM, Nomura H, McHenry JA, Kosyk O, and Stuber GD (2016). Visualization of cortical, subcortical and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat. Protoc 11, 566–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retz KC, and Steele WJ (1980). Ribosome turnover in rat brain and liver. Life Sci. 27, 2601–2604. [DOI] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, Backmann N, Conrath K, Muyldermans S, Cardoso MC, and Leonhardt H (2006). Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 3, 887–889. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödel T, Prevedel R, Aumayr K, Zimmer M, and Vaziri A (2013). Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light. Nat. Methods 10, 1013–1020. [DOI] [PubMed] [Google Scholar]
- Shaye DD, and Greenwald I (2011). OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One 6, e20085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh OA, Linghu C, Piatkevich KD, Goodwin D, Gritton HJ, Romano MF, Siciliano C, Gao R, Yu C-C, Tseng H-A, et al. (2020). Precision calcium imaging of dense neural populations via a cell body-targeted calcium indicator. Neuron 107 Published online June 22, 2020 10.1016/j.neuron.2020.05.029. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh OA, Tanese D, Zampini V, Linghu C, Piatkevich K, Ronzitti E, Papagiakoumou E, Boyden ES, and Emiliani V (2017). Temporally precise single-cell-resolution optogenetics. Nat. Neurosci 20, 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MK, Reitsma JM, Sweredoski MJ, Hess S, and Deshaies RJ (2016). Ribosomal proteins produced in excess are degraded by the ubiquitin-proteasome system. Mol. Biol. Cell 27, 2642–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, and Kaneko T (2003). Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knockin mouse. J. Comp. Neurol 467, 60–79. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, et al. (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, and Looger LL (2012). Imaging neuronal activity with genetically encoded calcium indicators. Cold Spring Harb. Protoc 2012, 647–656. [DOI] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Chou JH, Dwyer ND, Colbert HA, and Bargmann CI (1995). Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell 83, 207–218. [DOI] [PubMed] [Google Scholar]
- Troemel ER, Kimmel BE, and Bargmann CI (1997). Reprogramming chemotaxis responses: sensory neurons define olfactory preferences in C. elegans. Cell 91, 161–169. [DOI] [PubMed] [Google Scholar]
- Tsien RY (1998). The green fluorescent protein. Annu. Rev. Biochem 67, 509–544. [DOI] [PubMed] [Google Scholar]
- Ward S, Thomson N, White JG, and Brenner S (1975). Electron micro-scopical reconstruction of the anterior sensory anatomy of the nematode Caenorhabditis elegans. J. Comp. Neurol 160, 313–337. [DOI] [PubMed] [Google Scholar]
- Ward A, Liu J, Feng Z, and Xu XZ (2008). Light-sensitive neurons and channels mediate phototaxis in C. elegans. Nat. Neurosci 11, 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardill TJ, Chen TW, Schreiter ER, Hasseman JP, Tsegaye G, Fosque BF, Behnam R, Shields BC, Ramirez M, Kimmel BE, et al. (2013). A neuron-based screening platform for optimizing genetically-encoded calcium indicators. PLoS One 8, e77728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Feng J, Wu C, Lu Q, and Pan ZH (2015). Targeted Expression of Channelrhodopsin-2 to the Axon Initial Segment Alters the Temporal Firing Properties of Retinal Ganglion Cells. PLoS One 10, e0142052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Resendez SL, Rodriguez-Romaguera J, Jimenez JC, Neufeld SQ, Giovannucci A, Friedrich J, Pnevmatikakis EA, Stuber GD, Hen R, et al. (2018). Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. eLife 7, e28728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets and code generated during this study are available from the Lead Contact without restriction.