

Abstract
Background and purpose:
A genome-wide clustered regularly interspaced short palindromic repeats- associated protein 9-based screen has revealed that the cell adhesion molecule matrix remodelling associated protein 8 (Mxra8) acts as an entry mediator for many alphaviruses including chikungunya virus. The first X-ray crystal structure reported for Mxra8 a few months ago has a low-resolution of 3.49Å.
Experimental approach:
Homology modelling of Mxra8 protein was done employing the SWISS-MODEL and PRIME module of Maestro. To design novel Mxra8 inhibitors pharmacophore guided fragment-based drug design and structure-based virtual screening of Food and Drug Administration approved drug libraries were undertaken. Molecular docking and molecular dynamics (MD) simulations study were carried out to validate the findings.
Findings / Results:
The molecule H1a (dock score: -6.137, binding energy: -48.95 kcal/mol, and PHASE screen score: 1.528816) was identified as the best hit among the fragment-based designed ligands. Structure- based virtual screening suggested histamine, epinephrine, and capreomycin as potential hits which could be repurposed as Mxra8 inhibitor. MD simulations study suggested that only small molecules like histamine could be a potential inhibitor of Mxra8. H-bond interaction with Arg58 and Glu200 amino acid residues seems to be crucial for effective binding.
Conclusion and implications:
To the best of our knowledge, this is the first report on the design of novel inhibitors against Mxra8 protein to tackle the menace of alphaviruses infections. This design strategy could be used for structure-based drug design against other apo-proteins. This study also advances the application of in silico tools in the field of drug repurposing.
Keywords: Alphavirus, Docking, Fragment-based drug design, Molecular dynamics simulations
INTRODUCTION
Chikungunya is a viral disease (genus Alphavirus) spread by mosquitoes like Aedes aegypti and Aedes albopictus. Chikungunya derives its name from a Swahili word, “kungunyala” which means to become contorted thus highlighting its arthritogenic potential. The first case of chikungunya virus (CHIKV) infection was reported in the year 1951-1952 in Tanzania, but even after six decades not much have been understood about the CHIKV. Researchers have solved the mystery of entry of many viruses like human immunodeficiency virus (HIV) and even developed drugs targeting their entry. But sadly, the same cannot be seen for CHIKV and other alphaviruses like Ross River, Mayaro, and O’nyong viruses. Recently in 2018, Zhang et al. have reported an interesting finding that matrix remodelling associated protein 8 (Mxra8) is a receptor for multiple arthritogenic alphaviruses (1). By using a genome-wide wide clustered regularly interspaced short palindromic repeats-associated protein 9-based screen, they identified the cell adhesion molecule Mxra8, which acts as an entry mediator for many alphaviruses.
They believe that Mxra8 could be a druggable target for the mitigation of infection and diseases caused by these alphaviruses (1). And more recently in May 2019, the first X-ray crystal structure of the complex structure of CHIKV envelope glycoprotein bound to human Mxra8 (PDB ID 6JO8) has been released by research collaborators for structural bioinformatics, protein data bank (2). These findings encouraged us to investigate Mxra8 protein and develop its small molecule inhibitors using state of the art computational techniques.
In the present study, we have employed a ligand-based drug design where structure-based virtual screening of the Food and Drug Administration (FDA) approved drugs library from the ZINC 15 database was undertaken (3). The basic hypothesis on which drug repurposing is believed to work is poly- pharmacology and the possibility of having off- target effects (4). Another big advantage with this approach is the reported and acceptable absorption, distribution, metabolism, excretion, and toxicity profile of approved drugs and this makes drug repurposing a viable and cost- effective approach. We have also used a pharmacophore-guided fragment-based drug design strategy (5). As we have used PDB ID 6JO8 (an apoprotein) and two other modelled 3D structures of Mxra8, a four-feature e- pharmacophore model based on predicted receptor cavity was developed and the model was employed for fragment-based drug design (6). To the best of our knowledge, this is the first report on the design of Mxra8 inhibitors employing molecular modelling techniques.
MATERIALS AND METHODS
Instrumentation
Homology modelling was performed using SWISS-MODEL, a fully automated protein structure homology-modelling server, accessed via the ExPASy web server (7). All the other molecular modelling studies were done employing Maestro, the small-molecule drug discovery suite of Schrödinger Inc (USA). These programmes were run using Linux Ubuntu 18.04.1 LTS platform. The hp desktop had 8 GB RAM, Intel Core i3-4160 processor and Intel Haswell graphics card.
Homology modelling
The only X-ray crystal structure of Mxra8 protein (PDB ID 6JO8) has been released in May 2019 (2). The reported structure is a complex of CHIKV envelope glycoprotein bound to human Mxra8 with a resolution of 3.49 Å. Generally, resolution of less than 2.5 Å is considered ideal for molecular modelling studies, but since we don’t have any other X- ray crystal structure of the Mxra8 protein, we decided to undertake homology modelling and use the modelled protein structure along with PDB ID 6JO8 for further studies. For homology modelling, two different types of modelling tools were used. In the first method, SWISS- MODEL an automated web server tool was used (8). In the second method, the Prime tool of the Maestro interface (Schrodinger) was employed (9).
SWISS-MODEL relies on ProMod3, which is an in-house comparative modelling engine based on Open Structure (10). ProMod3 employs the OpenMM library to perform the computations (11). It uses the CHARMM27 force field for parameterization (12). In the present work, homology modelling was done in an automated mode. The amino acid sequence of Mxra8 was obtained from the Uniprot database in FASTA format and used as the input sequence in SWISS-MODEL. Automated mode selects suitable templates based on BLAST (13), and HHblits (14).
The homology modelling workflow in the Prime suite of Maestro, incorporates template identification, alignment, and model building (15. In this method, the same FASTA format of the amino acid sequence of Mxra8 was used as the input sequence. Chain M (Mxra8) of PDB ID 6JO8 was used as a template to build the model. The energy-based model option was selected for the structure prediction of Mxra8 protein (16). The loops were then refined using Prime (17).
Protein preparation and active site prediction
In the present work total three structures of Mxra8 protein were used. The first structure (structure 1) was the chain M of PDB ID 6JO8, the second structure was the modelled structure generated using SWISS-MODEL (structure 2) and the third structure was the modelled structure generated using Prime tool of Maestro (structure 3). To design any potential inhibitor of Mxra8 protein, first, these three structures were refined using the protein preparation wizard tool of Maestro. This step included adding hydrogens, assignment of proper bond order, removal of water, and energy minimization of the input protein structures (18).
But all these structures were apoproteins, i.e. no ligand was bound to the protein and so, what could be the active site was the next question in front of us. We decided to use the SiteMap tool of Maestro for the prediction of possible active sites on all three structure (19). A set of potential sites was predicted by the SiteMap tool which was ranked based on site-score and D-score.
Structure-based high throughput virtual screening
Based on the amino acid residues present in the predicted active site of all the three Mxra8 structures, three different receptor grids were generated using the receptor grid generation tool of the Maestro interface. A PHASE library of FDA approved drugs (obtained from ZINC 15 database) was also prepared which essentially converted 2D structures of the drugs into 3D structures employing the LigPrep tool of Maestro (18,20). All the compounds were screened in high throughput virtual screening (HTVS) mode against all the three developed receptor grids (21). Screened compounds were ranked based on the docking score and three different hit lists were prepared for the three structures of Mxra8 protein. Compounds with a docking score above -5.0 were selected for molecular docking study in extra precision (XP) mode (22).
e-pharmacophore modelling and pharmacophore guided fragment-based drug design
Energetically minimized structure 1 of Mxra8 protein was taken up for the development of the pharmacophore model. Based on the amino acid residues present in the predicted receptor cavity, a four-feature e- pharmacophore model was generated employing the PHASE tool of the Maestro interface (23,24). The intention behind this exercise was to know the possible regions in the receptor cavity having different pharmacophoric features like H-bond donor, H-bond acceptor, aromatic feature, etc. Once these regions were identified, the amino acid residues in the vicinity of 5 Å were manually measured and identified for each region. This led to the generation of a smaller number of amino acid residues for each feature and these were used to generate four smaller receptor grids against which the golden fragment library from Enamine and our own in-house fragment library prepared from FDA drugs was screened in HTVS mode. All the screened fragments were ranked based on docking scores and fragments with a docking score of more than -3.0 were selected for the fragment-based design. The selected fragments were joined using the combined fragment tool of Maestro. The generated structures were put for pharmacophore-based virtual screening (PHASE) to identify the best hit which aligned with the developed e-pharmacophore model. The screened compounds were ranked based on the PHASE screen score which is computed using the following equation:
PHASE screen score
= (1.00)
× (1.0 – alignment score/1.2 Å)
+ (1.00) × vector score + (1.00)
× volume score + (1.00)
× included volume score
The highest-ranked compound was selected, and its more derivatives were designed using the R-group library enumeration tool of Maestro. It further led to the generation of many novel compounds, and these were again put for pharmacophore-based virtual screening. Top ten hits based on the PHASE screen score were taken up for molecular docking study using the receptor grid generated for the full receptor cavity of structure 1 of Mxra8 protein.
Molecular dynamics simulation study
Molecular dynamics (MD) simulation study was implemented using the Desmond module (25). It involved three steps with the system builder being the first one (26). A simple point charge was selected as the solvent model in an orthorhombic boundary box shape. The system was neutralized by adding Na+ and Cl- ions. The minimization of the generated system model was the second step followed by simulation for 20 ns.
RESULTS
Homology modelling
The SWISS-MODEL template library (SMTL version 2019-06-20, PDB release 2019-06-14) was searched with BLAST and HHBlits for evolutionary related structures matching the target sequence of Mxra8 protein retrieved from UniProt database. A total of 20 templates were found using BLAST search. Then an initial HHblits profile was built, followed by 1 iteration of HHblits against NR20. The obtained profile was then searched against all profiles of the SMTL. And this resulted in a total of 15627 templates. The first template i.e. 6JO8.1.C was used to build the model based on the target-template alignment employing ProMod3. Prime also utilizes BLAST for template search; again, in this case, 6jo8.1.C was used as a template, as this was the best template searched by BLAST. Energy-based homology modelling was performed, and a new structure was generated for Mxra8 protein.
Active site prediction
The SiteMap tool of Maestro was used to predict the possible active sites in all the three structures of Mxra8 protein. The predicted sites were ranked based on site-score and D-score and based on these parameters one site from each structure was selected for further studies (Fig. 1). All the structures were superimposed upon each other as shown in Fig. 1D. The Ramachandran plot was employed for the validation of all the three protein structures as shown in Fig. 2.
Fig. 1.
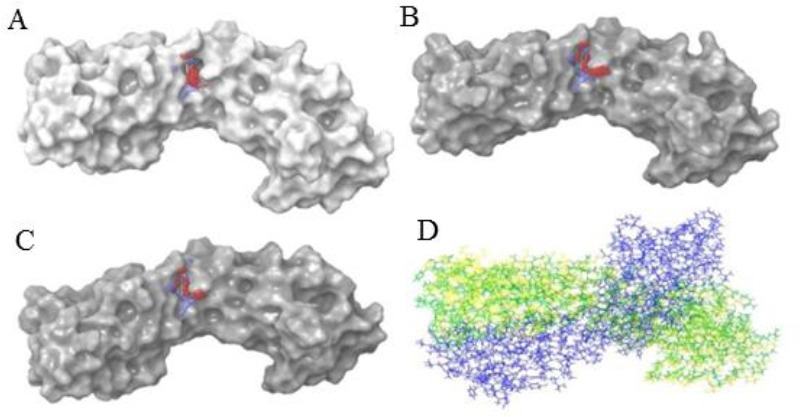
Predicted and selected active site of matrix remodelling associated protein 8 by SiteMap tool. (A) X-ray structure of PDB ID 6JO8; (B) model predicted by SWISS-MODEL; (C) model generated by PRIME, (D) all the three structures superimposed.
Fig. 2.

Ramachandran plot of all three structures of matrix remodelling associated protein 8. (A) Structure 1 (PDB ID 6JO8), (B) structure 2 (modelled by SWISS-MODEL), (C) structure 3 (modelled by PRIME).
Structure-based high-throughput virtual screening
Based on the amino acid residues present in the predicted active site of the three structures used for the study, three different receptor grids were generated. Using the GLIDE module of Maestro, molecular docking in HTVS mode was carried out against the generated PHASE library of FDA approved drugs. The compounds were ranked based on dock score and the compounds with a docking score above -5.0 were selected for molecular docking in XP mode. This exercise led to shortlisting of 44, 83, and 65 compounds as potential hits for structures 1-3 respectively. The free binding energy of association of hits and protein (MMGBSA dG bind) was also calculated for all the predicted hits employing the PRIME module of Maestro. The results of the top five hits for all three structures are compiled in Table 1.
Table 1.
Molecular docking and binding energy calculation of top five hits for all the three structures of matrix remodelling associated protein 8.
| Sr. no. | Structure 1 | Structure 2 | Structure 3 | |||
|---|---|---|---|---|---|---|
| Compound code | Docking score MMGBSA dG bind (kcal/mol) | Compound code | Docking score MMGBSA dG bind (kcal/mol) | Compound code | Docking score MMGBSA dG bind (kcal/mol) | |
| 1 | ZINC000150338698 | -9.006 -41.09 | ZINC000000057624 | -7.423 -29.89 | ZINC000000388081 | -7.379 -42.63 |
| 2 | ZINC000000057320 | -7.306 -45.73 | ZINC000150338698 | -7.179 -33.35 | ZINC000150338698 | -7.369 -36.76 |
| 3 | ZINC000001530775 | -7.018 -52.88 | ZINC000000388081 | -6.366 -42.63 | ZINC000013585233 | -5.974 -34.15 |
| 4 | ZINC000000057319 | -6.224 -45.82 | ZINC000002539827 | -6.278 -36.03 | ZINC000003806262 | -5.227 -40.01 |
| 5 | ZINC000000388081 | -6.143 -42.63 | ZINC000001530775 | -5.404 -45.36 | ZINC000000001644 | -4.914 -26.71 |
e-pharmacophore modelling and pharmaco- phore guided fragment-based drug design
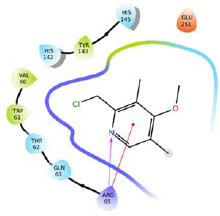
A four-feature e-pharmacophore model (RRDN) was developed based on the amino acid residues present in the predicted active site of MXRA8 protein (structure 1). For donor feature i.e. D6, three amino acid residues (Val60, Trp61, and His145) were identified. For negative ionic feature (N10), three amino acid residues (Trp61, Thr62, and Arg65) were identified. For aromatic feature R14, three amino acid residues (Gln63, Thr62, and Arg65) were identified. For aromatic feature R15, five amino acid residues (Trp61, Arg65, His142, Tyr143, and Glu200) were identified. Based on these amino acid residues four different smaller receptor grids were generated. A combined fragment library of Enamine golden fragments and our in-house fragment library were docked in HTVS mode against these receptor grids. The docking score and 2D interaction diagram of top two fragments for each pharmacophoric feature have been reported in Table 2. The fragments were ranked based on dock score and fragments with docking score of more than -3.0 were selected as potential hits. All the hits were combined using Combine fragments tool to give a series of novel compounds. All the generated structures were put for pharmacophore-based screening and molecule H1 was identified as the best hit based on phase screen score. H1 was selected for library enumeration and this exercise led to the generation of forty-three derivatives. These derivatives were again put for pharmacophore-based screening and the top ten ranked compounds were then put for molecular docking study (XP mode), using the receptor grid generated previously for the complete predicted the active site of structure 1.
Table 2.
Two dimensional interaction diagram, dock score, and non-bonding interaction of top two ranked fragments for each pharmacophore feature.
| Pharmacophore feature | 2 Dimensional interaction diagram | Docking score | Possible non-bonding interaction | |
|---|---|---|---|---|
| D6 | 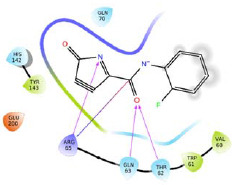 |
-4.762 | H-bond: Thr62, Gln63, Arg65. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, Gln70, His142.. | |
 |
-4.730 | H-bond: Arg65. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, His142, His145 | ||
| N10 |  |
-5.045 | H-bond: Thr62, Gln63, Arg65. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, Gln70, His142.. | |
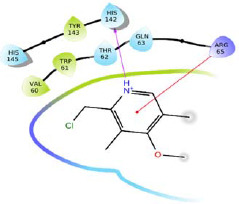 |
-4.618 | H-bond: His142. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, His142, His145 | ||
| R14 | 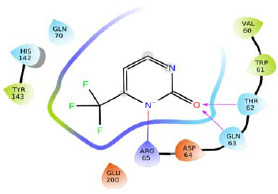 |
-5.184 | H-bond: Thr62, Gln63, Arg65. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, Gln70, His142. | |
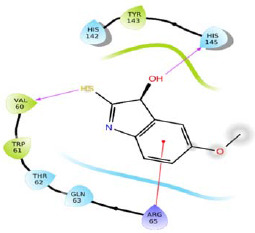 |
-5.184 | H-bond: Val60, His145. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, His142, His145. | ||
| R15 | 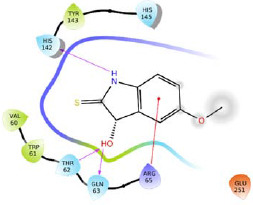 |
-5.220 | H-bond: Thr62, Gln63, His142. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, His142. | |
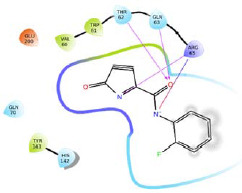 |
-4.932 | H-bond: Thr62, Gln63, Arg65. Hydrophobic: Val60, Trp61, Tyr143. Polar: Thr62, Gln63, Gln70, His142. | ||
Molecular dynamics simulation study
In order to understand the stability of identified hits with MXRA8 (structure 1) protein in the dynamic state (as would be the case inside the human body), MD simulations study was performed. A-frame was captured every 25 ps and saved in a trajectory. As a result, 800 frames were generated over a period of 20 ns simulation time. The root mean square deviation (RMSD) for the C-alpha atoms of the E6 protein and ‘Lig fit Prot’ for the ligand were computed. The first frame was selected as the reference frame and by aligning the rest of the frames above it RMSD for the C-alpha atoms of the Mxra8 protein and ‘Lig fit Prot’ for the ligand were calculated.
DISCUSSION
Homology modelled structures employing both SWISS-MODEL and Prime were of similar quality as was evident from Ramachandran outliers. There are some advantages using Prime like loop refinement and adding missing loops, but the final structure was of similar quality to the freely available web server-based tool SWISS-MODEL. Molecular docking results showed no possible solvent exposure for histamine but capreomycin showed solvent exposure even though it had a good dock score. It was interesting to find capreomycin, as the best hit for structure 1, while second-best hit for structure 2. It showed a maximum dock score of -9.006 with structure 1. The probable non- bonding interactions shown by capreomycin were six H-bond interactions with Gln55, Thr62, Arg65, His145, Asp248, and Glu251; hydrophobic interactions with Pro57 and Val60; polar interactions with Gln55, Thr62, His142, and His145, charged (negative) interactions with Asp248 and Glu251; and charged (positive) interactions with Arg65 and Arg252 (Fig. 3A). Maximum binding energy was found to be -41.09 kcal/mol for structure 1 as calculated by the PRIME module. It was an interesting finding as the active pockets predicted by the SiteMap tool were smaller in size and we were not expecting any big molecule like capreomycin to fit into the pocket. The best hit for structure 2 was norepinephrine and it showed a docking score of -7.423 but binding energy was comparatively lower than capreomycin (Fig. 3B). It showed probable H-bond interactions with Thr62, Gln63, and Tyr143. π-π interaction was also observed with Arg65. As expected, this compound was a smaller molecule in comparison with capreomycin. The best hit for structure 3 and third-best hit for structure 2 was histamine. It showed a docking score of - 7.379 and binding energy of -42.63 kcal/mol which was better than norepinephrine. It showed four possible H-bond interactions with His142, Tyr143 and Glu200 (Fig. 3C).
Fig. 3.

Two dimensional ligand interaction diagram (A) ZINC000150338698 with structure 1; (B) ZINC000000057624 with structure 2; and (C) ZINC000000388081 with structure 3.
The library of novel compounds enumerated from the molecule H1 showed a maximum dock score of -5.182. The methoxy group of pyridine ring system of H1 showed H-bond interaction with Gln63, pyridine ring system itself showed possible pi-cationic interaction with Arg65, nitrogen of pyridine ring showed possible H- bond interaction with His145, therefore, pyridine ring system was considered as core moiety and no modification was done to it. The methoxy group on the fused ring system was selected for modifications and library enumeration. The results of best-designed molecule H1, by combining fragments and the top five derivatives of H1 enumerated have been reported in Table 3. The top-ranked compound H1a showed possible H-bond interactions with Thr62, Gln63, and Glu252. It also showed a possible salt bridge with Asp248 and Glu251 residues. π-π interaction was observed with Trp61 and pi-cationic interaction with Arg65.
Table 3.
Dock score, phase screen score, and free binding energy of the designed compounds based on fragment design.
| Codes | Structures | Docking scores | MMGBSA dG bind(kcal/mol) | Phase screenscores |
|---|---|---|---|---|
| H1 | 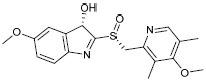 |
5.182 | -48.10 | 1.528816 |
| H1a | 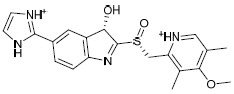 |
-6.137 | -48.95 | 1.528816 |
| H1b | 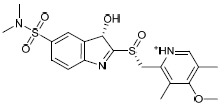 |
-5.659 | -25.84 | 1.528816 |
| H1c | 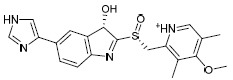 |
-5.458 | -38.87 | 1.528816 |
| H1d | 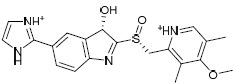 |
-4.951 | -44.12 | 1.528816 |
| H1e | 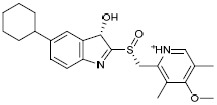 |
-4.678 | -45.09 | 1.528816 |
MMGBSA, The free binding energy of association of hits and protein.
The molecular dynamics simulation study was undertaken for three compounds namely histamine, capreomycin and the designed compound H1a. Interestingly, MD simulation studies suggest that only histamine might remain in the active site of the protein (Fig. 4A). The RMSD of MXRA8-histamine complex stabilizes after 2.5 ns and then there is minimal drift. But for the other two compounds (Fig. 4D and G) there is significant drift in RMSD values, and it suggests that the ligand might have diffused away from its initial binding site. Protein-ligand contact (Fig. 4B, C, E, F, H, I) was also monitored for all the three ligands for 20 ns and it was evident that the binding pocket is small and only a small ligand like histamine, could fit and form a stable complex with the MXRA8 protein. Histamine showed H-bond interaction predominantly with Arg58 and Glu200 amino acid residues. Hydrophobic interaction fraction with Trp61 residue was more than 1.6 which indicates multiple interactions. Many amino acid residues like Pro57, Arg58, Val60, Thr62, Gln63, His142, Tyr143, His145, Gln203, and His276 were found to make interaction with the ligand through water bridges. For capreomycin and H1a most of the H-bond interactions as found in XP docking was lost during MD simulations study.
Fig. 4.

Results of molecular dynamic studies. (A) RMSD plot of Mxra8-histamine; (B) histogram plot of protein-histamine; (C) Ligand atom interactions (histamine) with the protein residues (Mxra8); (D) RMSD plot of Mxra8-H1a; (E) histogram plot of protein-H1a; (F) ligand atom interactions (H1a) with the protein residues; (G) RMSD plot of Mxra8-capreomycin; (H) histogram plot of protein-capreomycin; (I) ligand atom interactions (capreomycin) with the protein residues. In parts B, E, and H the colors green, grey, pink, and blue represent H-binding, hydrophobic, ionic interaction, and water bridges. Mxra8, matrix remodelling associated protein 8; RMSD, root mean square deviation
CONCLUSION
To the best of our knowledge, this is the first study reported for the identification of potential small-molecule inhibitors against the Mxra8 protein of CHIKV. Drug repurposing approach was used with the help of in silico tools and small molecules like histamine and norepinephrine seem to be potential inhibitors. It might be the first report where histamine and norepinephrine have been predicted to have antiviral properties especially against CHIKV. In vitro evaluation will be interesting but even if these molecules don’t show activity, the scaffold from these compounds can be used for further design of Mxra8 inhibitors. As per the predictions of the SiteMap tool, the possible active site may be a small pocket with a maximum volume of 120.736 Å2, hence bigger molecules like capreomycin and the designed ligand H1a might not be potential Mxra8 inhibitors. Further in vitro testing is required to validate these findings. This approach of e- pharmacophore development for apo-protein structures can be applied for other targets and used not only for pharmacophore guided fragment-based design but also for high throughput virtual screening programmes.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest for this study.
AUTHORS’ CONTRIBUTION
A. Kumar conceptualized, conducted and drafted the manuscript. E. Rathi and S.G. Kini critically evaluated the results. All the authors revised the manuscript.
ACKNOWLEDGEMENTS
The authors are thankful to Manipal - Schrödinger Centre for Molecular Simulations, Manipal Academy of Higher Education, and Manipal College of Pharmaceutical Sciences for providing necessary supports and facilities to carry out the present research work.
REFERENCES
- 1.Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature. 2018;557(7706):570–574. doi: 10.1038/s41586-018-0121-3. DOI: 10.1038/s41586-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song H, Zhao Z, Chai Y, Jin X, Li C, Yuan F, et al. Molecular basis of arthritogenic alphavirus receptor Mxra8 binding to chikungunya virus envelope protein. Cell. 2019;177(7):1714–1724. doi: 10.1016/j.cell.2019.04.008. e12. DOI: 10.1016/j.cell.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Sterling T, Irwin JJ. Zinc 15-ligand discovery for everyone. J Chem Inf Model. 2015;55(11):2324–2337. doi: 10.1021/acs.jcim.5b00559. DOI: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar A, Rathi E, Kini SG. Drug repurposing approach for the identification and designing of potential E6 inhibitors against cervical cancer: an in silico investigation. Struct Chem. 2020;31:141–153. DOI: 10.1007/s11224-019-01378-x. [Google Scholar]
- 5.Kirsch P, Hartman AM, Hirsch AKH, Empting M. Concepts and core principles of fragment-based drug design. Molecules. 2019;24(23):e4309,1–22. doi: 10.3390/molecules24234309. DOI: 10.3390/molecules24234309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar A, Rathi E, Kini SG. E-pharmacophore modelling, virtual screening, molecular dynamics simulations and in-silico ADME analysis for identification of potential E6 inhibitors against cervical cancer. J Mol Struct. 2019;1189:299–306. DOI: 10.1016/j.molstruc.2019.04.023. [Google Scholar]
- 7.Guex N, Peitsch MC, Schwede T. Automated comparative protein structure modeling with Swiss- model and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(S1):S162–S173. doi: 10.1002/elps.200900140. DOI: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 8.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. Swiss-model: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. DOI: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, et al. A hierarchical approach to all- atom protein loop prediction. Proteins. 2004;55(2):351–367. doi: 10.1002/prot.10613. DOI: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 10.Biasini M, Schmidt T, Bienert S, Mariani V, Studer G, Haas J, et al. OpenStructure: an integrated software framework for computational structural biology. Acta Crystallogr D Biol Crystallogr. 2013;69(5):701–709. doi: 10.1107/S0907444913007051. DOI: 10.1107/S0907444913007051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, et al. OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol. 2017;13(7):e1005659,1–17. doi: 10.1371/journal.pcbi.1005659. DOI: 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. DOI: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 13.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421–429. doi: 10.1186/1471-2105-10-421. DOI: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2011;9(2):173–175. doi: 10.1038/nmeth.1818. DOI: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 15.Cappel D, Hall ML, Lenselink EB, Beuming T, Qi J, Bradner J, et al. Relative binding free energy calculations applied to protein homology models. J Chem Inf Model. 2016;56(12):2388–2400. doi: 10.1021/acs.jcim.6b00362. DOI: 10.1021/acs.jcim.6b00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320(3):597–608. doi: 10.1016/s0022-2836(02)00470-9. DOI: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 17.Zhu K, Pincus DL, Zhao S, Friesner RA. Long loop prediction using the protein local optimization program. Proteins. 2006;65(2):438–452. doi: 10.1002/prot.21040. DOI: 10.1002/prot.21040. [DOI] [PubMed] [Google Scholar]
- 18.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. doi: 10.1007/s10822-013-9644-8. DOI: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Rathi E, Kini SG. Identification of E6 inhibitors employing energetically optimized structure-based pharmacophore modelling, ligand docking and molecular dynamics simulations studies. ChemistrySelect. 2019;4(36):10701–10708. DOI: 10.1002/slct.201902105. [Google Scholar]
- 20.Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1 methodology and preliminary results. J Comput Aided Mol Des. 2006;20(10-11):647–671. doi: 10.1007/s10822-006-9087-6. DOI: 101007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- 21.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring 1 method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. DOI: 101021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 22.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. DOI: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 23.Dixon SL, Smondyrev AM, Rao SN. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem Biol Drug Des. 2006;67:370–372. doi: 10.1111/j.1747-0285.2006.00384.x. DOI: 101111/j1747-0285200600384x. [DOI] [PubMed] [Google Scholar]
- 24.Salam NK, Nuti R, Sherman W. Novel method for generating structure-based pharmacophores using energetic analysis. J Chem Inf Model. 2009;49(10):2356–2368. doi: 10.1021/ci900212v. DOI: 10.1021/ci900212v. [DOI] [PubMed] [Google Scholar]
- 25.Association for Computing Machinery, Sigarch, IEEE Computer Society. Proceedings: SC 06: powerful beyond imagination: proceedings: November 11-17, 2006. Tampa Florida: Tampa Convention Center; [Google Scholar]
- 26.Shaw DE. Schrödinger: 2019. Desmond molecular dynamics systems. Version 2019-4 [Desmond] Available from https://schrodinger.com/-desmond/ [Google Scholar]