

Abstract
Background and purpose:
Aberrant signaling by oncogenic RAS proteins occurs in almost all human tumors. One of the promising strategies to overcome such cancers is the inhibition of KRAS protein, a subtype of RAS family involved in cell growth, differentiation, and apoptosis, through preventing its effector, SOS1, from being attached to the protein.
Experimntal approach:
Herein, a virtual screening process was performed using pharmacophore search, molecular docking, and molecular dynamic simulations. A pharmacophore model was created to indicate essential features for a KRAS inhibitor and used for screening the National Cancer Institution (NCI) database to retrieve similar compounds to the pharmacophore model with more than 70% similarity. Chosen compounds were then docked into KRAS and four compounds were selected based on the highest binding scores. Next, a similarity search was done in the whole PubChem database to increase the number of potential inhibitors. The filtered compounds were docked again into KRAS and three of them were selected for molecular dynamic simulation.
Findings / Results:
Compounds 1a, 2d, and 3a can inhibit SOS-iKRASG12D interaction due to the higher number of interactions with the protein. Moreover, they achieved the equilibrium faster than the approved inhibitor.
Conclusion and implications:
Auriculasin, a polyphenol flavonoid, can be considered as a potential inhibitor of SOS1-KRAS interaction. This compound seems to be a stronger anticancer than 9LI, a known inhibitor of KRAS, due to its better docking scores. Moreover, this compound can be an appropriate candidate to be formulated as an oral drug.
Keywords: Auriculasin, Docking studies, Flavonoid, KRAS, Molecular dynamic simulations, Virtual Screening
INTRODUCTION
Cell genome mutations are believed to be the main cause of cancer. RAS is a family of genes (NRAS, HRAS, and KRAS) whose mutations are frequently involved in human tumors such as the pancreas (90%), colon (50%), lung (30%); thyroid (50%), and myeloid leukemia (30%) (1). These genes encode a group of GTPases, which involve a wide variety of signaling pathways such as cell growth and differentiation (2).
RAS proteins are seen in two structural states, active and inactive forms that are GTP- and GDP-bound, respectively.
The transformation between these states is regulated by two groups of molecules including guanine nucleotide exchange factors (GEFs), SOS1 and SOS2, which is necessary for the exchange of GDP to GTP; and GTPase- activating proteins (GAPs) which increase the intrinsic RAS GTPase activity and hydrolyze GTP to GDP (2,3).
Blocking RAS function includes several methods such as blocking the protein expression, post-translational processing, membrane anchorage, GTP affinity, effector interaction, and selective targeting of the cells with uncontrolled RAS signaling (4,5,6,7).
KRAS, a 21 kDa monomeric protein, is a subtype of RAS family which is believed to regulate cell growth, differentiation, and apoptosis through several signaling pathways including PI3K-PDK1-AKT, RAF-MEK-ERK, and TIAMI-RAC1 (8,9). Mutation at its codon 12 is one of the most frequent types of KRAS mutants (10) which disables the protein to hydrolyse GTP, resulting in a prolonged signal of cell proliferation (11,12). Pico molar binding affinity of KRAS for GTP and high concentration of cellular GTP are the reasons why designing a competitive inhibitor seems to be hardly practical (13). Therefore, it seems that interfering KRAS-SOS1 interaction might be an alternative approach to prevent RAS to be active by SOS1.
Flavonoids are a group of naturally occurring compounds with several biological activities including antitumor properties (14). They have been applied in RAS-targeted cancer treatment by controlling RAS gene expression and inhibiting RAS post-translational modifications (15,16).
Flavans are a subtype of flavonoids with common benzopyran backbone. These compounds are believed to be 93% of the total daily flavonoid intake in the USA (17). The beneficial effects of flavans are acknowledged in the treatment and prevention of cancer (18,19).
A pharmacophore model is a group of steric and electronic features, which are essential to reach the optimal interactions of a ligand with a protein active site. Today, pharmacophore screening is a well-known method that has two major advantages: first, it significantly increases the speed of the compounds filtering process and second, it allows retrieval of ligands with diverse structures (20).
The aim of this study is to investigate the potential anticancer flavonoids especially flavans that may disrupt KRAS-SOS1 interaction. To do this, the following methods were used: flavan similarity searches, pharmacophore filtering, molecular docking studies, and molecular dynamic (MD) simulations. Finally, a potential inhibitor was identified.
MATERIALS AND METHODS
Generation of pharmacophore model and structure selection
Both active and inactive forms of KRASG12D are unable to hydrolyze GTP, and a prolonged cell proliferation signal is observed after exchanging of GDP to GTP by SOS1 turning KRAS to its active form. Therefore, to interfere with KRAS-SOS1 interaction and hence prevent KRAS to be active by SOS1, inactive (GDP-bound) form of KRASG12D mutant (iKRASG12D) was used for discovering new potential inhibitors. Indeed, disturbing SOS1- iKRASG12D interaction necessarily depends on the ligand binding into the vacant SOS1 binding pocket, the state that is only observed in its inactive form.
In order to identify key interactions between the amino acid residues of SOS1 binding pocket and iKRASG12D potential inhibitors, benzimidazole (BZI) and 4,6- dichloro-2-methyl-3-aminoethyl-indole (9LI), a pharmacophore model was created by LigandScout (Ver. 3.1) software (Inte:Ligand GmbH, Austria). This program allows to a fully automated pharmacophore generation from protein-ligand complex, and also creates the best-matched pharmacophores to detect the number and type of primary existing ligand-residue interactions on the protein active site. These interactions contain H bond donor and acceptor, positive and negative ionizable area, hydrophobic areas, and aromatic ring. Then, prepared pharmacophoric map used for pharmacophore- based virtual screening.
The crystal structure of the KRAS (PDB ID: 4DSU) with 1.7 Å resolution with its co-crystal inhibitor, BZI, was obtained from the Protein Data Bank (http://www.pdb.org). Although the available co-crystal ligand is principally applied for the pharmacophore elucidation, BZI is probably too small for appropriate pharmacophore generation. Therefore, the crystal structure of 4DST with 9LI (DCAI), another approved iKRASG12D inhibitor, was used to generate an appropriate pharmacophore (21). The overall process of our procedure is summarized in Fig. 1.
Fig. 1.
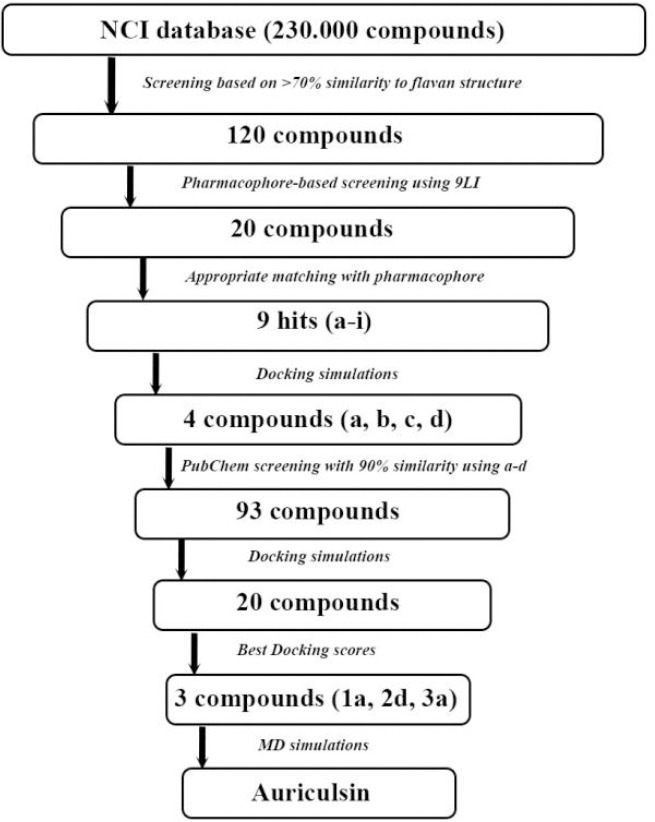
Flowchart illustrating the filtering process to select the final set of three compounds from approximately 120 hits from the National Cancer Institution and 93 hits from PubChem databases.
Docking simulation protocol
The ability of small molecules to interact with protein plays a major role in the dynamics of the protein, which may enhance/inhibit its biological function. The molecular docking method is used to study the spatial orientation of a ligand in an active site of the macromolecule. Besides, it is possible to calculate the binding affinity of a ligand on a defined protein active site.
The crystal structure of the KRAS (PDB ID: 4DSU) has been obtained from the RCSB protein data bank (http://www.pdb.org). The input files for docking of KRASG12D and all ligands were prepared by Auto Dock Tools (ADT) package (version 1.5.6, The Scripps Research Institute, Florida, USA). All Ligands and water molecules except GDP were removed from the protein PDB file and GDP was considered as a part of the receptor. Polar hydrogens were added and partial atomic charges computed by the Kollman-united charges method. The prepared protein structure was saved in PDBQT format as an input file for the docking process.
The two-dimensional (2D) structure of the BZI and DCAI ligands were drawn by MarvinSketch (Ver. 5.7, ChemAxon) and minimized by Chem3D Ultra (Ver. 8) and saved as a PDB format. To prepare the ligand input files, 3D structures of the ligands whether obtained from the National Cancer Institution (NCI) or PubChem databases with SDF format files were converted to the PDB format by the OpenBabel software. Non-polar hydrogens of ligands were merged and the Gasteiger charges were computed by AutoDock Tools. Finally, prepared files were saved as a PDBQT format.
In this study, docking simulation by AutoDock Vina (version 1.1.2, The Scripps Research Institute, Florida, USA) was used to calculate the free energies of binding of inhibitors and to determine the positions of the ligands on the defined active sites. A grid box of 25 × 25 × 25 Å with a grid point with a spacing of 0.375 Å was created around the SOS1 binding pocket, in a 100-run job. The center of the grid box was set to the co-crystallized ligand. Other docking parameters were set to the default values. The best binding mode of each ligand was selected based on its binding energy with reasonable spatial orientation inside of the binding pocket.
Validation
In validation step, the BZI co-crystal ligand of KRAS is redocked into the protein active site. The docking simulations are convincing if root mean square deviation (RMSD) value between the simulated ligand pose and experimentally known orientation is lower than 2 Å. For this purpose, BZI was docked, RMSD value calculated, and then superimposed over the co-crystallized ligand inside the iKRASG12D binding pocket (Fig. 2A).
Fig. 2.
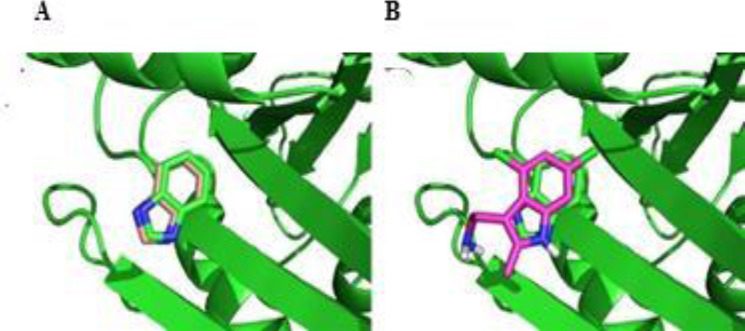
Superimposed perspective of (A) benzimidazole and (B) 4,6-dichloro-2-methyl-3-aminoethyl-indole docking poses over the co-crystallized ligand (benzimidazole) of the crystal structure of the KRAS. As it can be seen, docking poses of both ligands accurately mimic the spatial orientation of co-crystallized ligand.
In the same manner, the best docking pose of 9LI was superimposed over co-crystallized ligand to make validation stronger (Fig. 2B).
Filtering strategy
In the first step, we started with enhanced NCI database browser 2.2 (https:// cactus.nci.nih.gov/ncidb2.2/) which is an open compilation of the experimentally approved anticancer compounds (> 250,000 structures) of which, the flavan-like structures were retrieved to minimize the cytotoxicity and adverse effects of inhibitors. The general structure of flavan (Fig. 3) was used as a query in NCI database and a 70% similarity search was chosen. The result structures were downloaded in a SDF format. Afterward, pharmacophore filtering was conducted by LigandScout to find natural compounds with similar binding features of DCAI inhibitor against iKRASG12D. Compounds retrieved from pharmacophore filtering were then docked inside the iKRASG12D-SOS1 binding pocket and the ligands with the best spatial orientations and lowest docking energies were chosen, and their 2D structures were then used for > 90% similarity search by PubChem database to increase the number of capable anti iKRASG12D compound. The final compounds were further analyzed regarding Lipenski’s Rule of Five to determine the structures, whose chemical and physical properties make them a potential orally active drug. Finally, the most potent compounds retrieved from PubChem were chosen to be considered for MD simulations after being docked and screened based on the lowest binding energy and mode of interaction.
Fig. 3.
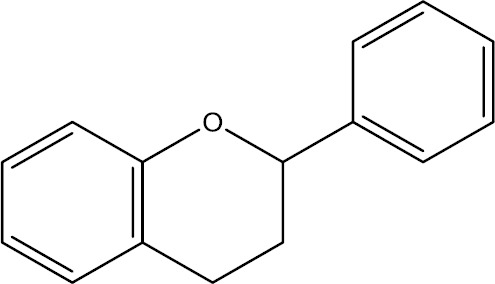
Chemical structure of flavan as a general structure has been found in many flavonoids.
Classical molecular dynamic simulations
MD simulations were applied to provide a better insight into the probable behavior of final ligands inside the SOS1 binding pocket. GROMACS package 5.0.1 was used to study ligands-protein interactions in a dynamic environment. The best docking posing the final compounds were chosen and, were then fixed inside the iKRASG12D-SOS1 binding pocket and used as the starting point of MD simulations.
The topology file of iKRASG12D was generated by PRODRG server (http://prodrg1.dyndns.org/). The GROMACS parameters and values were set similar to our previous study (22). Since it is essential to define the correct protonation of the amino acids especially inside the active site for MD simulations, pKa values of ionizable groups for iKRASG12D residues were predicted by PROPKA (http://propka.org/) server using AMBER force field in pH 7.4.
Afterward, protein complexes were separately immersed in a dodecahedron-shaped box and the minimum distances between the protein surface and box walls were set to 1 nm. This box was filled with 6368 water model molecules through GROMACS’ solvation method. The total charges of systems were negative. Therefore, seven Na+ atoms were added to neutralize the complexes. In the next step, complexes were energy minimized by the steepest descent algorithm with the tolerance of 1000 kJ/mol/nm. The system went through an NVT and NPT condition for 20 ps and 2 fs, respectively. The pressure and temperature constant at 1 bar and 300 K. Then, two equilibration states were performed to optimize the solvent and ions around the protein for a period of 20 ps. Finally, the MDs were continued for a period of 100 ns.
Data analysis and presentation software
The ligand-protein interactions were analyzed and visualized employing LigandScout and LigPlus softwares. Docking poses were depicted by PyMol (Schrödinger, Inc), and visual MD software (VMD; University of Illinois at Urbana-Champaign, USA) was used for visualizing MD results.
RESULTS
Pharmacophore map
The validation step showed that 9LI binds to iKRASG12D exactly in SOS1-binding pocket as BZI does but with lower binding energy (Fig. 4) suggesting its higher potency. Both 9LI and BZI interact with similar amino acid residues. In addition, Asp54 and Ser39 are responsible residues for making hydrogen bonds with BZI and 9LI, respectively (Fig. 4A and B). These residues are located at the outer side of the binding pocket suggesting that outer residues have a significant role in ligand binding, and could be targeted for drug design.
Fig. 4.
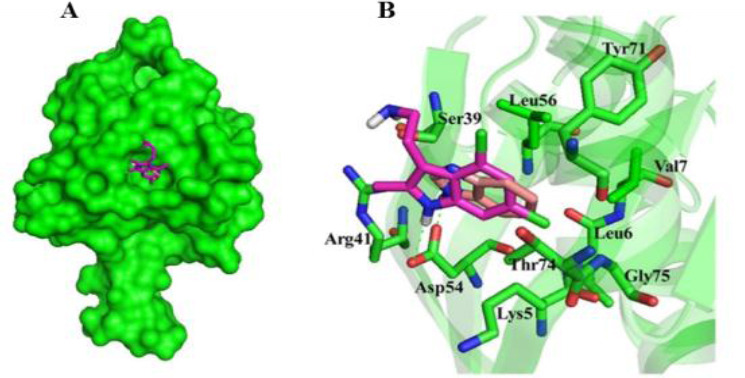
Molecular basis for the interaction of compounds 9LI and benzimidazole into SOS1-binding pocket of KRASG12D mutant. (A) The superimposed structure of compounds 9LI and benzimidazole are shown in stick and KRAS mutant in surface model. (B) Spatial arrangements of the residues involved in binding of compounds 9LI and benzimidazole with binding pocket. The models were generated using PyMOL (DeLano Scientific, LLC). 9LI, 4,6-dichloro-2-methyl-3-aminoethyl-indole.
Moreover, the calculated RMSD value was 0.337 Å for BZI and co-crystal bound BZI and this value was 1.85 Å for aromatic rings of 9LI and BZI. This suggests that docking simulation was successfully performed the calculations.
The structure-based pharmacophore model of the 9LI-iKRASG12D binding pocket was generated using LigandScout software. The pharmacophore map included three hydro- phobic regions surrounded by Val7, Leu56, and Thr74; and formed by two chlorine atoms (5.4 Å away from each other) and one with a methyl substituent (with the distances of 5.9 Å and 7.6 Å from Cl atoms) (Fig. 5). This information provided basic features for designing new inhibitor which is able to disturb the desired pocket.
Fig. 5.

The generated pharmacophore model includes three hydrophobic regions which are generated by its two chlorine atoms and a methyl substituent. (A and B) It is surrounded by Val 7, Leu56, and Thr74. The distance between the three regions are shown in part C. Yellow spheres indicate the hydrophobic regions.
Identifying potential inhibitors
The compounds’ library of NCI was screened based on 70% similarity to the flavan structure. A total of 120 compounds were retrieved and screened based on the DCAI- iKRASG12D pharmacophore. As a result, twenty ligands proved to fit with the pharmacophore.
Among them, nine hits (a-i) were chosen since they had appropriate matchings with the pharmacophore (Table 1). After docking to the iKRASG12D SOS1-binding pocket, four ligands a, b, c, and d were selected due to their higher affinities (lower free binding energies) and their positions in SOS-iKRASG12D binding pocket. Therefore, compounds a-d proved to have the best pharmacophores and affinities.
Table 1.
Two dimensional representations of compounds retrieved from the National Cancer Institution database and further docking simulations. Nine hits with the best matching with KRASG12D-9LI pharmacophore. Compounds a, b, c, and d were selected due to their higher affinities (lowest free energies of binding).
| Compounds | PubChem ID | Structures | Binding energies (kcal/mol) | ||
|---|---|---|---|---|---|
| a | 317611 | 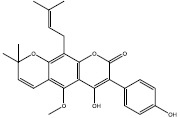 |
-8.6 | ||
| b | 54683839 | 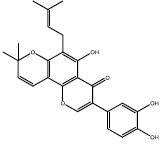 |
-8.6 | ||
| c | 4871 | 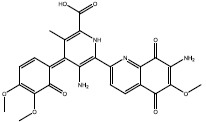 |
-10.2 | ||
| d | 2298 | 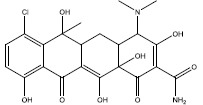 |
-8.5 | ||
| e | 54677440 | 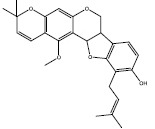 |
-6.4 | ||
| f | 1205 | 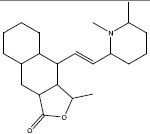 |
-6.7 | ||
| g | 247673 | 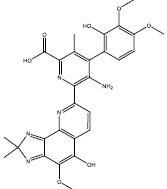 |
-7.9 | ||
| h | 3213 | 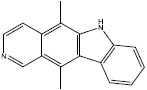 |
-8.2 | ||
| i | 54683835 |  |
-5.5 | ||
To increase the number of lead compounds, 2D chemical structures of a, b, c, and d compounds were used as queries for similarity search provided at PubChem compounds database. Ninety-three compounds with more than 90% structural similarity to compounds a-d were identified and docked for further evaluations. Nineteen compounds with lowest binding free energies and the best modes of interactions with the SOS1-iKRASG12D binding pocket are presented in Table 1.
Among them, three compounds (1a, 2d, and 3a) were chosen as candidates for further MD studies based on the following criteria:
-
(1)
A chemically match between the atoms in the ligands and the receptor. For example, it is preferred that the ligand’s carbon atoms are close to the receptor’s hydrophobic atoms, and nitrogens and oxygens in the ligand are close to chemically similar atoms in the binding pocket.
-
(2)
Charge complementarity between the ligands and binding sites of the proteins at physiological pH. In fact, charge complementary defines a reasonable orientation of a protonated ligand docking poses inside of protonated iKRASG12D active site in physiological pH. For example, in physiological pH it is possible that an OH group turns to O- and therefore it should be located near a positive group to have a proper electrostatic interaction. In docking methods sometimes, the orientation of a ligand is improper, and therefore it is essential to visualize interactions between ligand and receptor.
-
(3)
The estimated binding energy should be less than or equal to -6 kcal/mol since the binding energy of 9LI was -5.2 kcal/mol and more potent inhibitors are considered in this study.
Interactions of 1a, 2d, and 3a compounds with iKRASG12D protein are shown in Figs. 6 and 7. Besides, the calculated free energies of binding and modes of their interactions are listed in Tables 2 and 3. It can be observed that compounds 1a, 2d, and 3a have better free binding energies in comparison with 9LI (-9.5, -9, and -9.8 Kcal/mol for 1a, 2d, and 3a, respectively; and -5.2 for 9LI). This issue suggests that these compounds may interfere SOS1 and iKRASG12D interaction with higher potencies than 9LI. As seen in Fig. 7C and E, Met67 and Asp54 can establish a hydrogen bond with the OH substituents of 1a and 3a, respectively, while no hydrogen bond is seen in 2d-iKRASG12D (Fig. 7D).
Fig. 6.
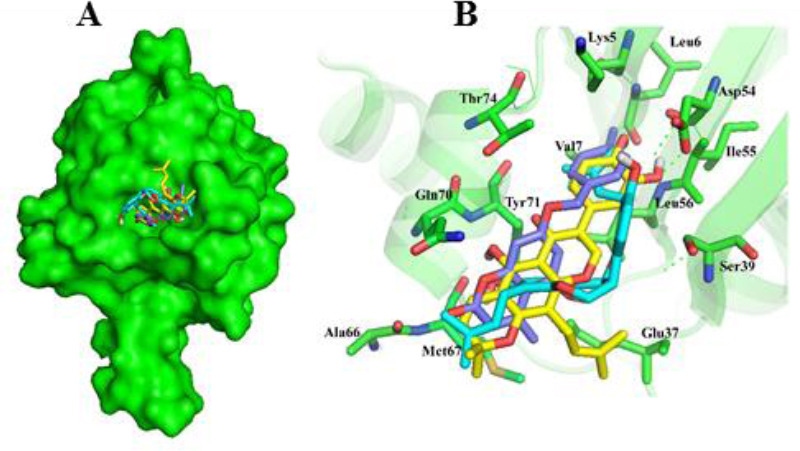
Molecular basis for the interaction of compounds 1a, 2d, and 3a into SOS1-binding pocket of iKRASG12D. (A) The superimposed structure of compounds 1a, 2d, and 3a are shown in stick and iKRAS in surface model. (B) Spatial arrangements of the residues involved in binding of compounds 1a, 2d, and 3a with binding pocket. The models were generated using PyMOL (DeLano Scientific, LLC).
Fig. 7.

Tow and 3 dimensional descriptions of compounds (A) 9LI, (B) benzimidazole, (C) 1a, (D) 2d, and (E) 3a (auriculasin) binding modes. 9LI and benzimidazole are depicted in magenta and salmon sticks while cyan, deep blue and yellow sticks imply compounds 1a, 2d, and 3a.
Table 2.
Two dimensional representations of 20 compounds retrieved from PubChem database and following docking simulations. Among them, 1a, 2d, and 3a were chosen for further evaluations.
| Compounds | PubChem IDs | Structures | Binding energies |
|---|---|---|---|
| 1a | 362562 |  |
-9.5 |
| 1b | 1680 |  |
-8.1 |
| 1c | 3322970 | 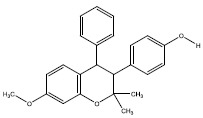 |
-7.4 |
| 1d | 454890 | 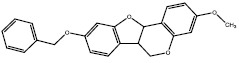 |
-8.8 |
| 1e | 500224 | 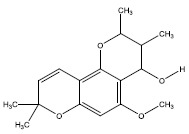 |
-7.8 |
| 1f | 390101 | 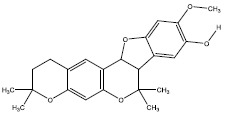 |
-7.8 |
| 2a | 5357627 | 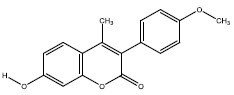 |
-8.3 |
| 2b | 375234 | 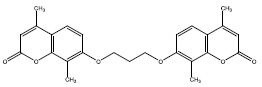 |
-9.3 |
| 2c | 228560 | 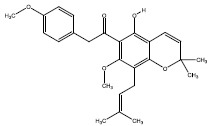 |
-8.3 |
| 2d | 5381483 | 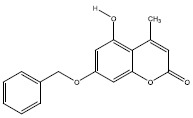 |
-9 |
| 2e | 5382848 | 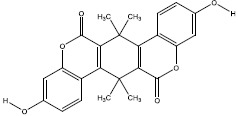 |
-10 |
| 2f | 54676828 | 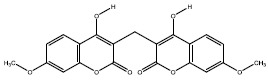 |
-7.5 |
| 2g | 54678441 | 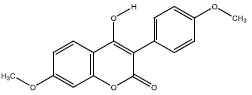 |
-7.5 |
| 3a | 5358846 | 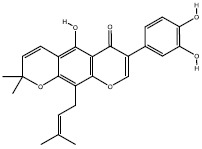 |
-9.8 |
| 3b | 403815 | 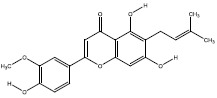 |
-8.8 |
| 3c | 403814 | 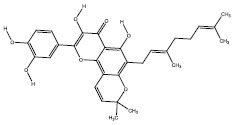 |
-9.3 |
| 4a | 18834 | 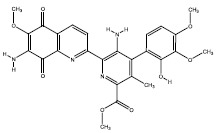 |
-7.3 |
| 4b | 135457174 | 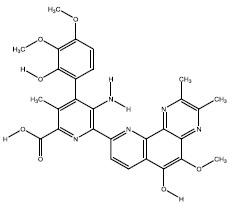 |
-8.4 |
| 4c | 312768 | 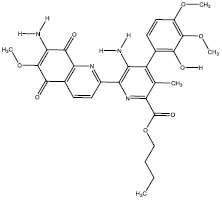 |
-7.2 |
| 4d | 5298 | 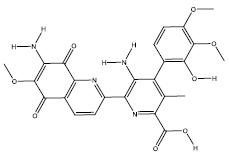 |
-8 |
Table 3.
Binding energy values and residues of interaction for compounds 1a, 2d, and 3a in comparison with previously examined compounds (9LI and benzimidazole).
| Compounds | Chemical structures | Binding energies (kcal/mol) | Residues involved in interaction | |
|---|---|---|---|---|
| 9LI |  |
-5.2 | Lys5, Val7, Ser39, Arg41, Asp54, Leu56, Tyr71, Gly75 | |
| Benzimidazole |  |
-5 | Lys5, Leu6, Val7, Ser39, Asp54, Leu56, Tyr71, Thr74 | |
| 1a |  |
-9.5 | Val7, Lys5, Leu56, Tyr71, Asp54, Ser39, Thr74, Leu6, Ala66, Gln70, Met67 | |
| 2d |  |
-9 | Val7, Lys5, Leu56, Tyr71, Asp54, Thr74, Leu6, Gln70, Met67, Glu37 | |
| 3a (Auriculasin) | 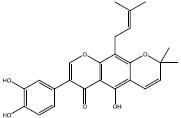 |
-9.8 | Lys5, Leu56, Tyr71, Asp54, Ser39, Thr74, Leu6, Ala66, Gln70, Met67, Glu37, Ileu55 | |
9LI, 4,6-dichloro-2-methyl-3-aminoethyl-indole.
Compound 1a is located into a hydrophobic cage surrounded by Val7, Lys5, Leu56, Tyr71, Asp54, Ser39, Thr74, Leu6, Ala66, and Gln70; and the hydroxyl groups belong to one of the phenols made a hydrogen bond with Met67. The benzyl ring of compound 2d was located between Val7, Leu6, Leu56, and Asp54, and its methyl group made hydrophobic interaction with Glu37 and Met67 (Fig. 7D). All residues involving in 3a-protein interactions formed hydrophobic interactions except that Asp54 made a hydrogen bond with 3a’s hydroxyl group of prenyl substituent (Fig. 7C).
Lipenski’s Rule of Five
Table 4 shows the physicochemical features of compounds 1a, 2d, and 3a, all of which have the potential to be used as oral drugs based on Lipinski’s criterion.
Table 4.
Physicochemical features of compounds 1a, 2d and 3a based on Lipenski’s Rule of Five.
| Compounds | Molecular weight (Da) | No. Hbond donor | No. Hbond acceptor | LogP | Molecular refractivity |
|---|---|---|---|---|---|
| Lipenski’s rule | ≤ 500 | ≤ 5 | ≤ 10 | ≤ 5 | 40-130 |
| 1a | 346 | 0 | 4 | 0.94 | 81 |
| 2d | 282 | 1 | 4 | 2.34 | 70.43 |
| 3a | 420 | 3 | 6 | 3.6 | 111.79 |
Molecular dynamic studies
To provide a closer view of compounds stability inside of KRAS active pocket, MD simulations were performed for compounds 1a, 2d, and 3a as well as 9Li for a period of 100 ns (Fig. 8). The RMSD plot of Ca of the ligands shows that all compounds reached an equilibrium and made stable complexes with the protein during the span of 100 ns. However, comparing RMSD graphs of 9LI with 1a, 2d, and 3a suggests that 9LI needs a longer time to form a stable complex with KRAS (about 25 ns) while others achieved the equilibrium in a shorter period after start point.
Fig. 8.
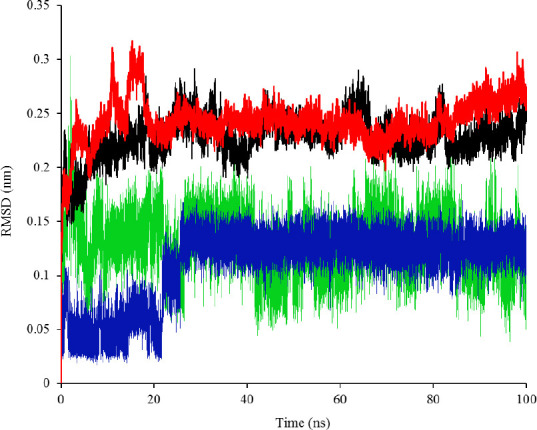
RMSD plot of KRASG12D in complex with a, 1a (black), 2d (red), 3a (auriculasin, green), and 9LI (blue).
DISCUSSION
This study focuses on finding the potential inhibitors of iKRASG12D, a subtype of RAS GTPase proteins, whose mutations have been previously demonstrated in several types of cancers (1). KRAS serves as an important modulator of cell growth signaling pathway (2). Therefore, its inhibition may be an appropriate target for the treatment of KRAS dependent- tumors. However, the picomolar affinity of GTP to KRAS and micromolar concentration of cellular GTP are the reasons why designing a competitive inhibitor seems to be hardly practical (13). Therefore, this study has been planned in order to assess the interfere SOS1 interaction, the enzyme which catalyzes the exchange of GDP to GTP allowing KRAS to be active which further sends the cell growth signal to downstream proteins (2).
We focused on the mutation on G12 residue (KRASG12D) which suppresses GAP-induced GTP hydrolysis (23) and makes the protein constitutively inactive form. The persistence of active GTP-bound leads to the constant activation of its downstream pathways and hence, abnormal cell growth happens. Moreover, iKRASG12D was preferred for discovering new potential inhibitors since this form is unable to hydrolyze GTP and hence returning to its inactive form.
Flavonoids are appropriate candidates to find potential lead compounds for iKRASG12D inhibition since they have been demonstrated to inhibit the activity of enzymes involving in cancer, RAS gene expression, and RAS post- translational modification (15,16,24,25). Herein, the in silico results indicated that several flavonoids are capable of iKRASG12D inhibition as their binding modes and pharmacophoric features are coincident with the KRASG12D-9LI complex (Table 1). This could be another promising mechanism for their anticancer properties, however, further evaluations are required.
Based on molecular docking and MD results, compounds 1a, 2d, and 3a might be potential candidates to interfere with iKRASG12D-SOS1 interaction. The lower binding energy of the protein in complex with 1a, 2d, and 3a suggests that these compounds are hypothetically more potent than 9LI against iKRASG12D (Table 3). Forming more interaction with the binding pocket may be an explanation for their higher affinity to iKRASG12D (Table 3 and Fig. 7).
Furthermore, compound 3a seems more promising than 1a and 2d due to its lower binding energy (Tables 2 and 3). However, a comparison of MD results for 1a, 2d, and 3a showed that these compounds inhibit KRAS in a similar manner according to the time they need to achieve the equilibrium (Fig. 8).
Compound 3a, known as auriculasin, has been associated with anticancer effects in humans and mice. This feature is partly attributed to its ortho-located dihydroxyphenyl (catechol) group and its prenyl side chain (26,27). Since it has been demonstrated that auriculasin has the potential to inhibit other types of hydrolases such as phosphodiesterase enzyme (28), it is plausible to suggest that compound 3a may exert its impact by acting as a direct inhibitor of KRAS during its activation by SOS, another mechanism for its anticancer activity.
Other inhibitors aiming to disturb KRAS- SOS1 interaction have been identified (29). However, their emphasis was put on SOS1 protein rather than KRAS itself. For instance, Hiling et al. found an inhibitor by a series of biophysical techniques. However, the mechanism by which their inhibitor prevents KRAS-SOS1 complex was different, so that it bound to SOS1 protein not KRAS, another mechanism with the same final result (30). Moreover, a peptide mimicking the α-helix of SOS1 was synthesized by Patgiti et al. which was able to attach KRAS and inhibit its activity (31). After testing more than 160,000 molecules, Burns et al. found small molecules whose anti- KRAS inhibition was due to their attachment to SOS1 binding domain (32).
Finding new compounds with the capability of interfering with protein-protein interactions is complicated since a large surface is needed for effector recognition and contact. Therefore, a bulky compound is needed to overlap with all parts of the interfacial surface area, and this doesn’t match with drug-likeness properties (33). However, auriculasin has a low molecular weight (420.406 g/mol) and therefore, it is appropriate for being considered as a drug.
Although values obtained by docking analysis should be considered as a theoretical approximation, this information could be useful to explore possible mechanisms by which these natural-derived ligands behave as anticancer compounds, in particular, if they directly bind proteins such as RAS. In addition, the proposed compounds could be considered as potential leading compounds to design new and safe drugs for the treatment of cancers related to KRASG12D.
CONCLUSION
Although three final compounds (1a, 2d, and 3a) had similar RMSD plots, which implies their similar behaviors reaching the equilibrium, docking simulations suggested that auriculasin (3a) formed the most favorable complex with the protein due to its lowest binding energy compared with 1a and 2d. Moreover, this compound can be an appropriate candidate to be formulated as an oral drug.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest for this study.
AUTHORS’ CONTRIBUTION
S. Hashemi, A. Sharifi, and S. Zareei performed the experiments and acquisition of data and analysis of the results. S. Zareei and Gh. Mohamedi wrote the manuscript draft and M. Biglar and M. Amanlou proposed the experiments and research design and approval of the final version for published.
ACKNOWLEDGMENTS
This study was financially (Grant No. 87-0233-7118) supported by the Research Council of Tehran University of Medical Sciences, Tehran, I.R. Iran.
REFERENCES
- 1.Cox AD, Der CJ. Ras history: the saga continues. Small GTPases. 2010;1(1):2–27. doi: 10.4161/sgtp.1.1.12178. DOI: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schubbert S, Shannon K, Bollag G. Hyperactive ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi: 10.1038/nrc2109. DOI: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 3.Yu Y, Nie Y, Feng Q, Qu J, Wang R, Bian L, et al. Targeted covalent inhibition of Grb2-Sos1 interaction through proximity-induced conjugation in breast cancer cells. Mol Pharm. 2017;14(5):1548–1557. doi: 10.1021/acs.molpharmaceut.6b00952. DOI: 10.1021/acs.molpharmaceut.6b00952. [DOI] [PubMed] [Google Scholar]
- 4.Ahmadian MR, Zor T, Vogt D, Kabsch W, Selinger Z, Wittinghofer A, et al. Guanosine triphosphatase stimulation of oncogenic ras mutants. Proc Natl Acad Sci U S A. 1999;96(12):7065–7070. doi: 10.1073/pnas.96.12.7065. DOI: 10.1073/pnas.96.12.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoki K, Yoshida T, Sugimura T, Terada M. Liposome-mediated in vivo gene transfer of antisense K-ras construct inhibits pancreatic tumor dissemination in the murine peritoneal cavity. Cancer Res. 1995;55(17):3810–3816. [PubMed] [Google Scholar]
- 6.Kloog Y, Gana-Weisz M, Niv H, Elad G, Marciano D, Haklai R. Dislodgment and accelerated degradation of ras. Neurosci Lett. 1997;237:S28–S28. doi: 10.1021/bi972032d. DOI: 10.1016/S0304-3940(97)90115-4. [DOI] [PubMed] [Google Scholar]
- 7.Marom M, Haklai R, Ben-Baruch G, Marciano D, Egozi Y, Kloog Y. Selective inhibition of ras- dependent cell growth by farnesylthiosalisylic acid. J Biol Chem. 1995;270(38):22263–22270. doi: 10.1074/jbc.270.38.22263. DOI: 10.1074/jbc.270.38.22263. [DOI] [PubMed] [Google Scholar]
- 8.McCormick F. Ras GTPase activating protein: signal transmitter and signal terminator. Cell. 1989;56(1):5–8. doi: 10.1016/0092-8674(89)90976-8. [DOI] [PubMed] [Google Scholar]
- 9.Cully M, Downward J. SnapShot: ras signaling. Cell. 2008;133(7):1292–1292. doi: 10.1016/j.cell.2008.06.020. e1. DOI: 10.1016/j.cell.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 10.Mariyama M, Kishi K, Nakamura K, Obata H, Nishimura S. Frequency and types of point mutation at the 12th codon of the c-Ki-ras gene found in pancreatic cancers from Japanese patients. Jpn J Cancer Res. 1989;80(7):622–626. doi: 10.1111/j.1349-7006.1989.tb01687.x. DOI: 10.1111/j.1349-7006.1989.tb01687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbacid M. Ras genes. Annu Rev Biochem. 1987;56(1):779–827. doi: 10.1146/annurev.bi.56.070187.004023. DOI: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 12.Ellis CA, Clark G. The importance of being K-ras. Cell Signal. 2000;12(7):425–434. doi: 10.1016/s0898-6568(00)00084-x. DOI: 10.1016/s0898-6568(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 13.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2(3):359–372. doi: 10.1177/1947601911412376. DOI: 10.1 177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhan G, Pan L, Tu K, Jiao S. Antitumor, antioxidant, and nitrite scavenging effects of Chinese water chestnut (Eleocharis dulcis) peel flavonoids. J Food Sci. 2016;81(10):H2578–H2586. doi: 10.1111/1750-3841.13434. DOI: 10.1111/1750-3841.13434. [DOI] [PubMed] [Google Scholar]
- 15.Ranelletti FO, Maggiano N, Serra FG, Ricci R, Larocca LM, Lanza P, et al. Quercetin inhibits p21- ras expression in human colon cancer cell lines and in primary colorectal tumors. Int J Cancer. 2000;85(3):438–445. DOI: 10.1002/(SICI)1097-0215(20000201)85:3<438:: AID-IJC22>3.0.CO;2-F. [PubMed] [Google Scholar]
- 16.Kang HM, Kim JH, Lee MY, Son KH, Yang DC, Baek NI, et al. Relationship between flavonoid structure and inhibition of farnesyl protein transferase. Nat Prod Res. 2004;18(4):349–356. doi: 10.1080/14786410310001622022. DOI: 10.1080/14786410310001622022. [DOI] [PubMed] [Google Scholar]
- 17.Sebastian RS, Goldman JD, Enns CW, Moshfegh AJ. Usual intakes of flavonoids: estimates from what we eat in America, NHANES 2007-2010. FASEB J. 2017;31(1 Supplement):647. 3. [Google Scholar]
- 18.Azuine MA, Bhide SV. Adjuvant chemoprevention of experimental cancer: catechin and dietary turmeric in forestomach and oral cancer models. J Ethnopharmacol. 1994;44(3):211–217. doi: 10.1016/0378-8741(94)01188-5. DOI: 10.1016/0378-8741(94)01188-5. [DOI] [PubMed] [Google Scholar]
- 19.Kuzuhara T, Suganuma M, Fujiki H. Green tea catechin as a chemical chaperone in cancer prevention. Cancer lett. 2008;261(1):12–20. doi: 10.1016/j.canlet.2007.10.037. DOI: 10.1016/j. canlet.2007. 10.037. [DOI] [PubMed] [Google Scholar]
- 20.Markt P, Schuster D, Langer T. Pharmacophore models for virtual screening. In: Sotriffer C, Mannhold R, Kubinyi H, Folkers G, editors. Virtual screening: principles, challenges, and practical guidelines: Wiley-VCH. 2011. pp. 115–152. DOI: 10.1002/9783527633326.ch5. [Google Scholar]
- 21.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, et al. Small-molecule ligands bind to a distinct pocket in ras and inhibit Sos-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109(14):5299–5304. doi: 10.1073/pnas.1116510109. DOI: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagherzadeh K, Shirgahi Talari F, Sharifi A, Ganjali MR, Saboury AA, Amanlou M. A new insight into mushroom tyrosinase inhibitors: docking, pharmacophore-based virtual screening, and molecular modeling studies. J Biomol Struct Dyn. 2015;33(3):487–501. doi: 10.1080/07391102.2014.893203. DOI: 10.1080/07391102.2014.893203. [DOI] [PubMed] [Google Scholar]
- 23.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. Ras oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–774. doi: 10.1038/nrc3106. DOI: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang NJ, Shin SH, Lee HJ, Lee KW. Polyphenols as small molecular inhibitors of signaling cascades in carcinogenesis. Pharmacol Ther. 2011;130(3):310–324. doi: 10.1016/j.pharmthera.2011.02.004. DOI: 10.1016/j.pharmthera.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Lee KW, Kang NJ, Heo YS, Rogozin EA, Pugliese A, Hwang MK, et al. Raf and MEK protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer Res. 2008;68(3):946–955. doi: 10.1158/0008-5472.CAN-07-3140. DOI: 10.1158/0008-5472.CAN-07-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito C, Itoigawa M, Tan HT, Tokuda H, Mou XY, Mukainaka T, et al. Anti-tumor-promoting effects of isoflavonoids on Epstein-Barr virus activation and two-stage mouse skin carcinogenesis. Cancer Lett. 2000;152(2):187–192. doi: 10.1016/s0304-3835(00)00331-1. DOI: 10.1016/s0304-3835(00)00331-1. [DOI] [PubMed] [Google Scholar]
- 27.Ito C, Itoigawa M, Kojima N, Tokuda H, Hirata T, Nishino H, et al. Chemical constituents of Millettia taiwaniana: structure elucidation of five new isoflavonoids and their cancer chemopreventive activity 1. J Nat Prod. 2004;67(7):1125–1130. doi: 10.1021/np030554q. DOI: 10.1021/np030554q. [DOI] [PubMed] [Google Scholar]
- 28.Ribaudo G, Vendrame T, Bova S. Isoflavones from Maclura pomifera: structural elucidation and in silico evaluation of their interaction with PDE5. Nat Prod Res. 2017;31(17):1988–1994. doi: 10.1080/14786419.2016.1269101. DOI: 10.1080/14786419.2016.1269101. [DOI] [PubMed] [Google Scholar]
- 29.Cruz-Migoni A, Canning P, Quevedo CE, Bataille CJR, Bery N, Miller A, et al. Structure-based development of new ras-effector inhibitors from a combination of active and inactive ras-binding compounds. PNAS. 2019;116(7):2545–2550. doi: 10.1073/pnas.1811360116. DOI: 10.1073/pnas.1811360116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent Sos1 inhibitors that block ras activation via disruption of the Ras-Sos1 interaction. PNAS. 2019;116(7):2551–2560. doi: 10.1073/pnas.1812963116. DOI: 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7(9):585–587. doi: 10.1038/nchembio.612. DOI: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burns MC, Howes JE, Sun Q, Little AJ, Camper DV, Abbott JR, et al. High-throughput screening identifies small molecules that bind to the Ras:Sos:Ras complex and perturb ras signaling. Anal Biochem. 2018;548:44–52. doi: 10.1016/j.ab.2018.01.025. DOI: 10.1016/j.ab.2018.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fletcher S, Hamilton AD. Targeting protein-protein interactions by rational design: mimicry of protein surfaces. J R Soc Interface. 2006;3(7):215–233. doi: 10.1098/rsif.2006.0115. DOI: 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]