

Abstract
Biological inflammation induced during penetrating cortical injury can disrupt functional neuronal and glial activity within the cortex, resulting in potential recording failure of chronically implanted neural interfaces. Oligodendrocytes provide critical support for neuronal health and function through direct contact with neuronal soma and axons within the cortex. Given their fundamental role to regulate neuronal activity via myelin, coupled with their heightened vulnerability to metabolic brain injury due to high energetic demands, oligodendrocytes are hypothesized as a possible source of biological failure in declining recording performances of intracortical microelectrode devices. To determine the extent of their contribution to neuronal activity and function, a cuprizone-inducible model of oligodendrocyte depletion and demyelination in mice was performed prior to microelectrode implantation. At 5 weeks of cuprizone exposure, mice demonstrated significantly reduced cortical oligodendrocyte density and myelin expression. Mice were then implanted with functional recording microelectrodes in primary visual cortex and neuronal activity was evaluated up to 7 weeks alongside continued cuprizone administration. Cuprizone-induced oligodendrocyte loss and demyelination was associated with significantly reduced recording performance at the onset of implantation, which remained relatively stable yet decreased over time compared to mice on normal diet. Further, electrophysiological analysis revealed deficits in multi-unit firing rates, frequency-dependent disruptions in neuronal oscillations, and altered laminar communication within the cortex of cuprizone-treated mice. Post-mortem immunohistochemistry revealed robust depletion of oligodendrocytes around implanted microelectrode arrays alongside comparable neuronal densities to control mice, suggesting that oligodendrocyte loss was a possible contributor to chronically impaired device performances. This study highlights potentially significant contributions from the oligodendrocyte lineage population concerning the biological integration and long-term functional performance of neural interfacing technology.
2.0. Introduction
Investigative and clinical neuroscience have the potential to benefit from the use of penetrating cortical neural interfaces, which can be applied as tools to reveal unknown neuroscientific phenomenon or alleviate neurological deficiencies following central nervous system (CNS) injury or disease [1–9]. Despite the wide versatility of implantable microelectrode arrays, overwhelming biological inflammation and large variability in recording performances debilitate the long-term applications of neural interfacing technology [10–20]. Investigations into proposed biological failure modes of microelectrode devices, such as chronic neurodegeneration or glial scar formation, have been unable to account for the characteristic degradation in device performances over time [21–24]. However, the brain parenchyma is not limited to neurons, microglia, and astrocytes, nor are these the only cells whose physiological activity can be compromised during CNS injury [10, 25–27]. Cells of the oligodendrocyte lineage, which have gone largely uninvestigated in regards to chronic device implantation, are intimately associated with neurons and may have a more direct contribution to the chronic degradation in recordable neuronal signals around intracortical microelectrode interfaces [26, 28].
Oligodendrocytes, a third type of glia alongside microglia and astrocytes, possess the ability to directly regulate neuronal health and function through the deposition of myelin membranes around axons [29–31]. Myelin sheaths support saltatory conduction of neuronal signals by providing electrical insulation of far-projecting axons within the white matter [32]. Both oligodendrocytes and myelin exist within gray matter structures, such as the cortex, although at relatively lower numbers compared to the white matter [33, 34]. Furthermore, a fraction of oligodendrocytes existing in the gray matter, termed satellite oligodendrocytes, maintain close contact with neuronal soma and may have a more critical role in providing metabolic and neurotrophic support [35–37]. However, the presence of monocarboxylate transporters in myelin suggest that myelinating oligodendrocytes are also a sustainable source of support for neurons [38, 39]. Oligodendrocyte densities are maintained by a self-renewing population of differentiating progenitor cells, called oligodendrocyte precursor cells, which replenish lost or damaged oligodendrocytes following demyelinating CNS injury [40–42]. This precursor population can become exhausted in their capacity to form new oligodendrocytes following persistent demyelinating injury or from experiencing differentiation block [43]. As a result, neurological symptoms can occur due to a loss of oligodendrocyte support or myelin encapsulation and are commonly observed during multiple sclerosis (MS) and other demyelinating diseases with the CNS [44, 45].
Due to their need for continuous production of lipid-dense myelin membranes, oligodendrocytes are considered one of the most energy demanding glial cells within the brain, and therefore have an increased susceptibility to metabolically damaging CNS injury [46–48]. Low levels of the antioxidant glutathione in oligodendrocytes and their precursors render them increasingly vulnerable to oxidative stresses that can occur during ischemia, traumatic brain injury (TBI), or autoimmune-mediated inflammatory diseases such as MS [49, 50]. Oligodendrocyte degeneration and demyelination are commonly observed during focal cerebral ischemia and white matter stroke [51]. Similarly, traumatic brain injury has demonstrated oligodendrocyte cell death and myelin loss in gray and white matter regions of the CNS [52–54]. Penetrating cortical injuries, such as microelectrode device insertion, demonstrate inflammation cascades similar to stroke and TBI and induce similar damage on the oligodendrocyte lineage population [26]. Winslow et al. demonstrated myelin loss as well as signs of blood-brain barrier leakage near chronically implanted single-shank electrodes [55, 56]. We have recently shown oligodendrocyte degeneration, myelin reorganization, and reactive oligodendrocyte precursor population preferentially around implanted electrode arrays [28, 57]. However, how oligodendrocyte loss or dysfunction affects the intrinsic electrophysiological properties of neuronal tissue and the functional performance of intracortical recording microelectrodes has yet to be investigated.
Cuprizone, a copper-chelating agent, can be administered as a highly reproducible model of oligodendrocyte depletion and demyelination within distinct brain regions, such as the corpus callosum and cortex [58]. It is often used as an alternative to experimental autoimmune encephalomyelitis (EAE), which induces activation of immunological and inflammatory processes to mimic biological symptoms of MS [59]. Cuprizone is most often administered via rodent chow, and acute exposure (<5–6 weeks) can effectively deplete a significant population of oligodendrocytes and myelin within the CNS [60, 61]. The effects of cuprizone administration can be reversed if mice are returned to normal diet following this acute period [62, 63]. A chronic state of oligodendrocyte depletion and demyelination can be achieved by administrating cuprizone continuously for up to 12 weeks, effectively exhausting the precursor population and incapacitating their ability to generate new oligodendrocytes and myelin [58]. Finally, cuprizone preferentially targets mature oligodendrocytes with no significant changes to neurons or axons within the brains of young mice, allowing for specific investigation of the effects of oligodendrocyte and myelin depletion on neuronal function [59, 64].
Since oligodendrocytes mediate neuronal health and activity via metabolic and trophic support as well as myelin ensheathment, we hypothesized that the presence of oligodendrocytes is critical for the neuronal signals acquired by intracortical microelectrode arrays. In this study, we used a cuprizone-induced oligodendrocyte depletion model to deplete oligodendrocytes and myelin within the cortex and evaluate the recording performance of chronically implanted intracortical devices. We demonstrate that cuprizone effectively depletes a majority of cortical oligodendrocytes resulting in myelin loss by 5 weeks of diet administration. We also show that the oligodendrocyte loss and demyelination due to cuprizone-treatment significantly reduces recording performance at the onset of device insertion and remains relatively steady over a 7-week implantation period compared to mice on control diets. Additional recording analyses revealed chronic alterations in multi-unit firing rate, neuronal oscillatory activity, and laminar coherence within the cortex of cuprizone-treated mice. Furthermore, histology at 7 weeks post-insertion and 12 weeks of cuprizone administration depicted robust loss of oligodendrocytes in cuprizone-treated mice coinciding with matched neuronal densities compared to controls. In summary, we establish that the presence of oligodendrocytes and myelin within the cortex is necessary to maintain normal electrophysiological properties of neurons and that their loss or dysfunction could underlie possible biological failure modes of chronically implanted neural interfaces.
3.0. Methods
Neural recording performances were compared between C57BL/6J mice fed with cuprizone diet (depleted oligodendrocyte) and C57BL/6J mice on a normal diet (normal oligodendrocyte). Post-mortem immunohistochemical analysis was performed for end-point analyses of different cellular markers following device implantation with and without cuprizone administration. All animal care and procedures were performed under approval of the University of Pittsburgh Institutional Animal Care and Use Committee and in accordance with regulations specified by the Division of Laboratory Animal Resources.
3.1. Animals and cuprizone administration
Cortical oligodendrocyte depletion and demyelination was induced by feeding 6–8 week old, male C57BL/6J mice (22–30g, Jackson Laboratory, Bar Harbor, ME) rodent chow infused with 0.2% cuprizone (bis-cyclohexanone oxaldihydrazone; Evigo, Cambridgeshire, United Kingdom). Mice were fed cuprizone diet for 5 weeks prior to electrode insertion, following which diet was maintained for another 7 weeks during recording experimentation for a total of 12 weeks of cuprizone administration, which is the maximum length of cuprizone exposure before mice begin to experience epileptic seizures in response to environmental stress [65]. A separate cohort of cuprizone-treated mice (n = 3) along with control mice (n = 3) were sacrificed after 5 weeks of cuprizone administration for immunohistological analysis in non-implanted brain tissue. Food and water were made available ad libitum, and since cuprizone is easily degraded due to environmental exposure [60], feeds were replaced every other day with fresh pellets.
3.2. Surgical electrode implantation
Single shank Michigan-style electrodes (A16–3 mm-50–703-CM15) were inserted into the left primary monocular (V1m) visual cortex of cuprizone-fed (n = 5) and control (n = 5) mice. Procedures for surgical device implantation were performed as described previously [21]. Briefly, an anesthetic cocktail of xylazine (7 mg/kg) and ketamine (75 mg/kg) was injected intraperitoneally (I.P.) for surgery sedation prior to fixing the animal onto a stereotaxic frame. Hair, skin, and connective tissue were removed from the top of the skull to reveal the site of surgical implantation. Vetbond adhesive was used to dry the surface of the skull prior to drilling and provide a supportive grip for a dental cement head cap. Three bone screw holes were drilled (two over both motor cortices and one over the contralateral visual cortex) prior to insertion of 4 mm long, 0.86 mm diameter stainless steel bone screws (Fine Science Tools, British Columbia, Canada) for wrapping of ground and reference wires. The ground wire was wrapped around the bone screw over the ipsilateral motor cortex while the reference wire was wrapped over the bone screws located above the contralateral motor and visual cortex. A drill-sized craniotomy positioned at 1 mm anterior to Lambda and 1.5 mm lateral to midline was made using a high speed dental drill and 0.7 mm drill bit. Saline was periodically applied to prevent thermal damage due to drilling. A microelectrode device was perpendicularly inserted at a speed of 15 mm/s using a DC motor-controller (C-863, Physik Instructmente, Karlsruhe, Germany). The electrode was inserted to a cortical depth of 800 μm, visually confirming that the last contact site disappeared beneath the pial surface. A silicone elastomer (Kwik-Sil) was used to fill the craniotomy around the electrode prior to sealing with a dental cement head cap. Body temperature was maintained and monitored using a rectangular heating pad (Deltaphase isothermal pad, Braintree Scientific Inc. Braintree, MA). Mice were given an I.P. injection of ketofen (5 mg/kg) on the day of surgery and up to two days after for post-operative recovery.
3.3. Electrophysiological recording
Electrophysiological recordings were conducted inside a grounded Faraday cage to prevent electrical interference from environmental noise as described previously [10, 11, 66–68]. Mice were situated on a rotating platform for awake, head-fixed recording. Trials to obtain spontaneous neural activity were recorded in a dark room. Visually-evoked neural activity was stimulated using the MATLAB-based Psychophysics toolbox on a 24” LCD (V243H, Acer. Xizhi, New Taipei City, Taiwan) monitor located 20 cm from the contralateral eye of the mouse covering a 60° wide by 60° high visual field. A drifting gradient of solid white and black bars were presented and synchronized with the neural recording system (RX7, Tucker-Davis Technologies, Alachua, FL) at a sample rate of 24,414 Hz. Each white and black grating was presented for 1 s (rotated in 135° increments), separated by 1 s of a dark screen, and repeated for a total of 64 trials per recording session.
3.4. Neural signal data processing
3.4.1. Current source density
Current source density (CSD) was used to identify layer IV along the length of the electrode following evoked activity within the visual cortex. CSD plots were generated by computing the average evoked (stimulus-locked) LFP for each electrode site, smoothing the signal across all electrode sites, and then calculating the second spatial derivative. CSDs were averaged across 64 stimulus trials and layer IV was identified as an inversion in LFP polarity within the first 100 ms following stimulus onset. Cortical drift and the magnitude change in drift of implanted arrays over time was reported as the average change in layer IV depth relative to depth at day 0 (day of surgery). All electrophysiological evaluation between different cortical depths occurred following alignment of all animals in each group for each day to their corresponding layer IV depth.
3.4.2. Single-unit (SU) sorting and analysis
Processing of raw signal data occurred offline using a custom MATLAB script modified from previously published methods [11]. Data was passed through a Butterworth filter with a passband from 2 to 0.3 kHz to produce data containing information on local field potentials (LFP) and 0.3 to 5 kHz to isolate spiking information. Common average referencing was applied to the data as previously described [69]. A fixed threshold value of 3.5 standard deviations below the mean was used to identify potential neuronal single-unit (SU) and multi-unit (MU) activity. Only channels which exhibited a signal-to-noise ratio (SNR) >2 were considered for single-unit sorting. Signal-to-noise ratios (SNR) were calculated by dividing the peak-to-peak amplitude of each single unit by the noise and reported as average SNR and average SNR per active site (electrode channels reporting detection of SU) over time. Sortable single units were confirmed by observing the quality and shape of neuronal waveforms, auto-correlograms, and peri-stimulus time histograms (PSTH) with 50 ms bins. SU yield was calculated as the percentage of electrode sites (out of 16) with at least one identifiable single unit. The noise floor for each electrode site was taken as two times the standard deviation (2*STD) of the entire recorded data stream after removing all threshold crossing events. Finally, the SNR of any channels without a sortable SU waveform were reported as zero for the purposes of calculating averages.
3.4.3. Multi-unit (MU) analysis
Multi-unit activity was measured as any threshold crossing event that occurred within the 1 s period after each stimulus-locked trigger or pseudotrigger was recorded. Peri-stimulus time histograms (PSTHs) of 50 ms bin size were generated to gauge the dynamics of multi-unit activity. Multi-unit firing rate was measured as the average number of threshold crossing events within a 1-s period after each stimulus presentation or pseudotrigger. Evoked multi-unit activity was evaluated by calculating multi-unit yield and signal-to-noise firing rate ratio (SNFRR). Parameters for multi-unit spike counts involved varying the temporal bin size and latency after stimulus presentation from 0 to 1 seconds in length via 1 ms increments in order to evaluate multi-unit yield and SNFRR. Multi-unit yield was defined as the number of electrode sites that had a significantly different (p < 0.05) spike count for a given bin size and latency following stimulus presentation (stim ON) compared to spike counts within that same bin size immediately before stimulation (stim OFF). SNFRR measured the difference in the firing rate of multi-unit activity before and after stimulus relative to the average standard deviation between each stimulus condition:
where are the average firing rates (across 64 trials) during stimulus ON and OFF conditions and are the standard deviations of firing rates during ON and OFF conditions, respectively. Reported values were calculated as absolute SNFRR.
3.4.4. Local field potential analysis
LFP power spectra were calculated using a multitaper method of 1 s duration, 1 Hz bandwidth, and a taper number of 1. Relative power was calculated as the ratio of power within a specific frequency band over the entire frequency range of LFP power (broadband power). LFP power was normalized by subtracting the spontaneous power spectrum from the evoked power spectrum.
where R is relative power, S(f) is the LFP power spectrum, a and b are lower and upper values of the specific frequency range, N(f) is the normalized power spectrum, and SE(f) and SRS(f) are evoked and resting-state power spectra, respectively.
3.4.5. Laminar coherence analysis
Electrophysiological activity within and between different laminar depths was evaluated by calculating the magnitude-squared coherence, described previously [70]. Coherence is a quantitative description of the similarity between two signals based on their frequency-dependent responses. Coherence was reported as a value between 0 and 1, with 0 indicating no relationship and 1 indicating a perfect linear relationship between two corresponding signals. Coherence was calculated as follows:
where Cxy(f) is the coherence, Sxy(f) is the cross spectrum, and Sxx(f) and Syy(f) are the autospectra of LFP activity between electrode sites x and y, respectively. Normalized coherence was calculated by taking the difference between evoked coherence and resting-state coherence . Coherence calculations were performed within 1 s intervals following each stimulus presentation or pseudotrigger, at a half-bandwidth of 3 Hz and a taper number of 5, and then averaged across all trials. Coherence between different cortical regions was assessed by comparing the average coherence of all electrode sites located within each respective layer. Coherence values for a specific frequency band were reported as the average coherence within that frequency range across all animals within each group.
3.5. Electrochemical impedance spectroscopy
Electrochemical impedances were measured prior to each recording session. Mice were awake and head-fixed on a rotating platform while the implanted device was connected to an Autolab potentiostat along with a 16 channel multiplexer. Impedances were measured from each channel using a 10 mV RMS sine wave ranging from 10 Hz to 32 kHz. Impedances were reported as the average impedance at 1 kHz over all animals for each day unless otherwise noted.
3.6. Post-mortem immunohistochemistry
Mice were intracardially perfused with 4% paraformaldehyde (PFA), post-fixed in 4% PFA for 18 hours and equilibrated in 30% sucrose at 4°C. Brain s were frozen while embedding with optimum cutting temperature (OCT) media directly on the headpiece of the cryostat (Leica) and sectioned horizontally at 14 μm thickness starting from the convexity of the brain down to 800 μm below the cortex. Sections adhered to positively charged slides were permeabilized and blocked using 0.1% Triton-X with 10% normal goat serum in PBS at room temperature for 1 hour and incubated with primary antibodies to CC1 (1:100, Millipore, Burlington, Massachusetts USA, #OP80), Olig2 (1:200 Millipore, Burlington, Massachusetts USA, #Mabn50), NG2 (1:100, Millipore, Burlington, Massachusetts USA, #Ab5320), GFAP (1:1000 Abcam, Cambridge, Massachusetts USA, #Ab4674), Iba-1 (1:1000 Wako, Richmond, Virginia USA, #019–19741), MBP (1:500 Abcam, Cambridge, Massachusetts USA, #Ab40390), NeuN (1:200 Millipore, Burlington, Massachusetts USA, #MAB377), Neurofilament (NF-200) (1:200, Sigma-Aldrich, St. Louis, Missouri USA # N4142-.2ML), and phosphorylated NF-200 (SMI-31, Stenberger Monoclonals) overnight at 4°C. Alexa Fluor 488/594 conjugated secondary Ab (1:300 Jackson ImmunoResearch, West Grove, Pennsylvania USA # 115585–003, # 111–605-003, #111–545-003), IgG2b isotope specific (1:300 anti-mouse 488 Jackson ImmunoResearch, West Grove, Pennsylvania USA #115–545-207) and 633 anti-chicken (1:300 Thermo Fisher Scientific, Waltham, Massachusetts #a21103) were reacted for 1 hr at RT. Sections were washed and allowed to dry overnight in the dark before mounting with Fluoromount-G and DAPI (SouthernBiotech, Birmingham, Alabama USA, #0100-20).
3.7. Imaging and data analysis
For cellular markers, 20x TIFF images were captured over the probe site and at an equivalent area in the contralateral hemisphere or baseline tissue with an Olympus (Center Valley, Pennsylvania USA) BX-51 epifluorescent microscope with MicroSuite software. Grey scale, individual layer images were cropped to square and loaded into using a previously published I.N.T.E.N.S.I.T.Y. MATLAB script where the binning was applied around the probe site [71, 72]. Three bins were applied 0–50 μm, 50100 μm and 100–150 μm extending from the center of the probe site or image of non-implanted tissue. Labeled cells were counted per bin at 400–800 μm through the cortical depth per brain. For intensity analysis, images were acquired by collecting 20× z-stack fields with 9 steps (1 μm step size) using Nikon A1R Confocal microscope (Melville, NY USA) with NIS Elements software. The sum intensity projections (SUMIP) were processed through MATLAB by applying 10 μm bins up to 15 bins concentrically around the site of probe insertion, contralateral tissue, or baseline tissue in non-implanted mice. Data were expressed as the fold intensity change at the probe site over the intensity in the corresponding area in the contralateral hemisphere [71].
3.8. Statistics
Changes in recording metrics with respect to time were modeled using a linear mixed effects model, described previously [73]. To fit the model to nonlinear relationships, a restricted cubic spline was implemented by placing 4 knots at the 5th, 35th, 65th, and 95th percentiles of the data. Group (cuprizone versus control) and group-by-time interactions were included as fixed effects. A likelihood ratio test was performed on the estimated model. Confidence intervals were determined using a bootstrapping method with 1000 iterations. A 95% confidence interval was taken as 1.96 times the standard error of the model output. For histological analysis, a two-way ANOVA (p < 0.05) was used to determine significant differences in tissue stains between cuprizone-treated and control mice. Pairwise significances were determined using a post hoc t-test followed by a Bonferroni correction, to reduce the probability of type I error when performing multiple comparisons.
4.0. Results
4.1. Cuprizone administration induces oligodendrocyte loss and demyelination within the mouse cortex
For safety reasons, cuprizone could only be administered consecutively for 12 weeks [65]. Therefore, C57BL/6J mice (n = 8) were pre-treated with cuprizone for 5 weeks to deplete oligodendrocytes and myelin within the visual cortex prior to electrode insertion while longitudinal electrophysiological recordings were carried out during the last 7 weeks of cuprizone administration- (Fig. 1a). To determine the extent of oligodendrocyte loss and demyelination at the time of implantation, cortical tissue was examined in experimental mice following 5 weeks of cuprizone treatment (n = 3) as well as age-matched mice on control diet (n = 3) without electrode implantation to confirm success of the cuprizone depletion model. The CC1 marker was used to measure the survival of mature oligodendrocytes after cuprizone administration. At five weeks following cuprizone exposure, mice demonstrated significant reductions in CC1+ oligodendrocyte density (~85–90%) within the visual cortex (Fig. 1b). Similarly, MBP, a marker used to stain myelin fibers, showed significant decreases in MBP+ fluorescence intensity (~65–70%) compared to control mice (Fig. 1c). Staining for the NG2+ oligodendrocyte precursor population demonstrated a slight, yet not significantly different, elevation in NG2 glial densities (Fig. 1d). This reported increase in NG2+ glial density following cuprizone administration is expected after five weeks of treatment alongside a reduced oligodendrocyte population, since NG2 glia proliferate in an attempt to repopulate lost oligodendrocyte densities. Since glial cell activation is associated with cuprizone-induced oligodendrocyte and myelin loss [60, 74], mice were also examined for microglia and astrocyte density within the cortex following 5 weeks of cuprizone administration. Cuprizone-treated mice demonstrated comparable Iba-1+ fluorescence intensities relative to control animals, indicating no activation of microglia cells following cuprizone treatment (Fig. 1e). However, cuprizone-treated mice did exhibit a slight increase in GFAP+ staining intensity, indicating partial activation of astrocyte cells following cuprizone toxicity, although there were no significantly reported differences in the GFAP+ fluorescence intensities between cuprizone-treated and control groups (Fig. 1f). Overall, cuprizone demonstrated effective depletion of oligodendrocytes and myelin with little influence on glial activation within the cortex by 5 weeks of diet administration.
Figure 1. Depletion of oligodendrocytes and demyelination in the visual cortex at 5 weeks following cuprizone administration without electrode implantation.

(a) Experimental timeline of cuprizone administration, device implantation, electrophysiological and impedance recording, and pre- and post-implant histology. (b) CC1+ staining reveals marked reduction in oligodendrocyte density in the visual cortex following 5 weeks of cuprizone administration. (c) MBP+ stain demonstrates reduced myelin fluorescence intensity in the visual cortex following 5 weeks of cuprizone treatment. (d) Staining for NG2+ cells shows elevated oligodendrocyte precursor cell density following 5 weeks of cuprizone administration. (e) Iba-1+ staining demonstrates relatively comparable microglia fluorescence intensities between cuprizone-treated and control mice. (f) GFAP+ astrocyte staining similarly shows comparable fluorescence intensities between cuprizone-treated and control mice. Scale bars = 50 μm. *** indicates p < 0.001. **** indicates p < 0.0001.
4.2. Electrode implantation and neural recording setup
Five weeks of continuous cuprizone administration can induce near-complete depletion of oligodendrocytes and myelin within the visual cortex. To test whether oligodendrocyte and myelin depletion impairs neuronal electrophysiology, cuprizone-treated mice (n =5) and mice on normal rodent diet (n = 5) were implanted with a 16 channel single-shank Michigan-style microelectrode array (50 μm site spacing) to a depth of 800 μm below the visual cortex. For electrophysiological data acquisition, mice were head-fixed on a rotating platform inside a grounded, electrically-isolated Faraday cage for awake recording (Fig 2a–b). The cage was enclosed in a blackout curtain to record spontaneous visual activity in a dark environment. To record visually-evoked activity, mice were positioned in front of an LED screen displaying a drifting bar gradient. Electrophysiological signals were passed through a biological amplifier before being collected by an external recording system for further signal processing. Raw data streams were passed through a high-frequency filter to isolate neuronal spiking events, which were aligned to the onset of visual stimulation (Fig. 2c). Spike sorting was performed using principal component analysis to isolate distinct recorded signals and assign them to individual clusters (Fig. 2d). For each recording session, waveforms on each channel were aligned to the onset of visual stimulation or to pseudotriggers assigned to spontaneously recorded data for single-unit and multi-unit classification (Fig. 2e–f). Authentic neuronal units were confirmed based on average waveform shape and stimulus-evoked firing properties (Fig. 2g–h).
Figure 2. Device implantation and electrophysiological recording setup.
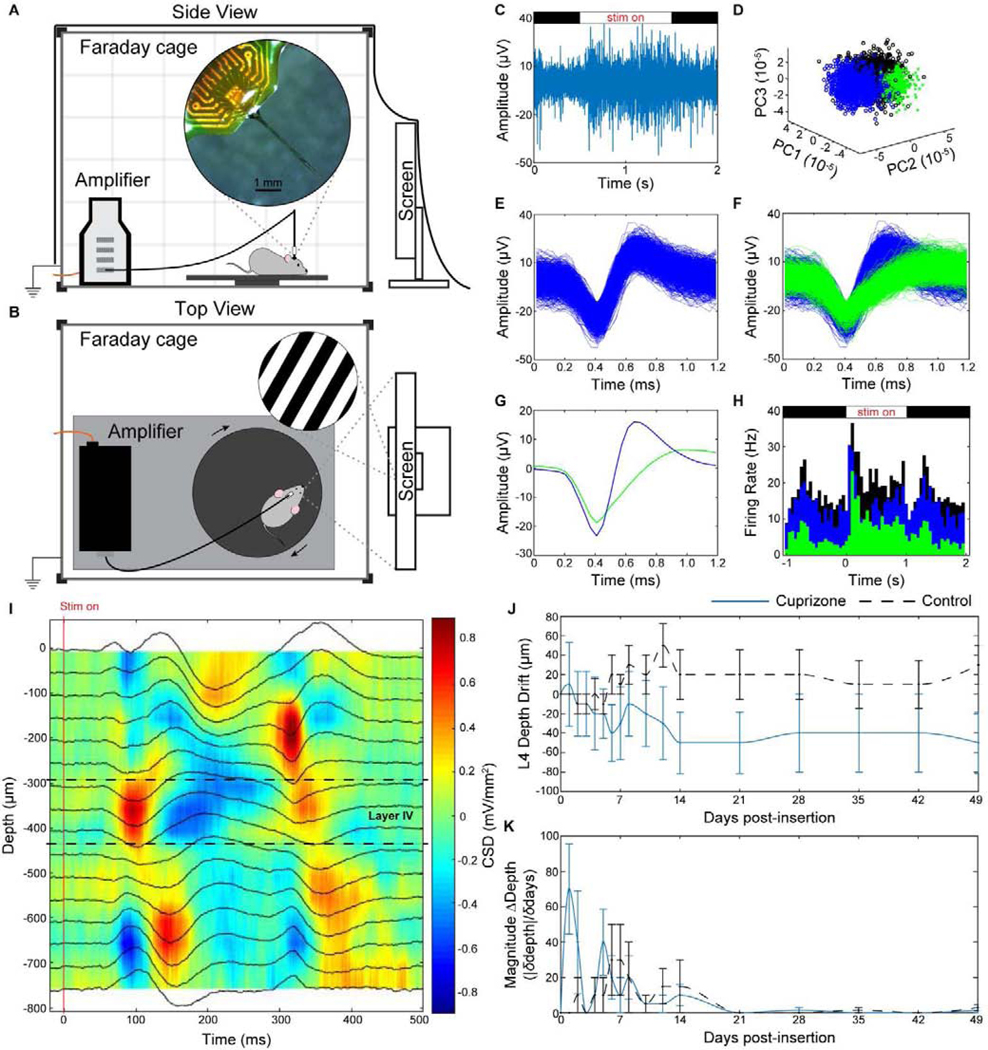
(a) Side view and (b) top view of electrophysiological recording setup in an electrically-isolated Faraday cage. Implanted mice were head-fixed using a stand (not shown) magnetically attached to a base plate on a movable platform for awake recording. An LED monitor displaying drifting bar gradients was used for visual stimulation. Recorded signals were filtered through a biological amplifier before transmission to an external recording system via a non-conductive fiber optic cable (orange wire). (c-h) Representative examples of one electrode channel in control mice demonstrating (c) stimulus-evoked raw spiking data, (d) unit sorting using principal component analysis, pile plot of (e) single-unit and (f) multi-unit activity, (g) average waveforms, and (h) evoked firing rate activity. (i) Representative current source density plot demonstrating 500 ms of electrical current activity versus electrode implant depth following visual stimulus presentation (‘stim on’) in the visual cortex used to identify layer IV depth (sink = red, source = blue). (j) Average drift of layer IV depth along implanted microelectrode shank compared to day 0 of device insertion between cuprizone (blue solid) and control (black dashed) mice. (k) Magnitude change in depth over change in time demonstrating most fluctuations in layer IV depth occur with the first two weeks before stabilizing.
Current source density (CSD) plots were generated to identify the position of cortical layer IV along the depth of the implanted microelectrode shank following visual stimulus presentation (Fig. 2i). Electrical activity was evoked at the onset of visual stimulation (‘stim on’), causing neurons to depolarize (current sink) within the input layer (layer IV) of the visual cortex. This appears as a negative deflection in LFP polarity within CSD plots. Layer IV depth can also be confirmed by the temporally delayed activity in cortical layer II/III above and layer V below. For drift analysis, the depth of layer IV on the day of surgery was defined as the starting point (0 μm) to compare subsequent changes in electrode drift between groups. Cuprizone-treated mice experienced a gradual decrease in layer IV depth along the microelectrode array for the first two weeks post-insertion before remaining stable throughout the remainder of device implantation (Fig. 2j). In contrast, mice on control diet demonstrated an upward shift in layer IV position along the microelectrode array for the first two weeks before stabilizing until experimental end-point. Analysis of the magnitude change in depth demonstrated that most of the drift in layer IV position occurred within the first two weeks post-insertion (Fig. 2k). These patterns in layer IV drift are similarly supported by previously published results [21]. An electrode site spacing of 50 μm and an average change in layer IV depth of only ~30–50 μm beyond the first two weeks of implantation indicates the implanted microelectrode experienced minimal drift within the range of 1 electrode site throughout the duration of implantation. As a result, we employed a robust and reproducible model of oligodendrocyte and myelin depletion around chronically implanted microelectrode arrays to study the electrophysiological properties of neurons within the demyelinated cortex.
4.3. Cuprizone-induced oligodendrocyte loss and demyelination impairs neuronal single-unit activity at the onset of device implantation
The use of microscale recording arrays with high spatial and temporal resolution allowed observation of how oligodendrocyte loss and demyelination influences the electrophysiological characteristics of individual neurons. Due to the dense distribution of oligodendrocytes and myelin fibers, particularly within cortical input (layer IV) and output layers (layer V) [75], cuprizone-induced oligodendrocyte depletion and demyelination was expected to significantly alter the health and strength of detectable neural signals within the visual cortex. To demonstrate the impact of oligodendrocyte loss and demyelination on electrophysiological recording quality, comparison of recording metrics was first examined independent of cortical depth by averaging across all 16 microelectrode channels. Due to the residual effects of anesthesia, specifically the influence of ketamine/xylazine on evoked activity within the visual cortex on the day of surgery [70], acute recording metrics were evaluated with respect to data collected on day 1 post-implantation. One day after electrode insertion following 5 weeks of cuprizone administration, oligodendrocyte-depleted mice demonstrated a significant reduction of nearly 36% in electrode yield compared to mice on normal diets (cuprizone: 40 ± 5%, control: 62.5 ± 8.6%, Fig. 3a). By 7 weeks post-insertion, SU yield of cuprizone mice was reduced to 30 ± 9.1%. Control mice, however, experienced a greater and more pronounced decline in yield over time, reducing to 42.5 ± 4.1% and demonstrating no significant differences in yield compared to cuprizone-treated animals by 7 weeks post-insertion. Similar patterns of significant difference between cuprizone-treated and control mice were reported for SNR (Fig. 3b). Average SNR on day 1 following implantation was significantly reduced by nearly 52% in cuprizone-treated animals compared to control mice (cuprizone: 1.3 ± 0.62, control: 2.7 ± 0.44). Over time, SNR in control mice gradually declined to the level of cuprizone-treated animals (cuprizone: 1.75 ± 0.6, control: 1.75 ± 0.29). Evaluation of only active electrode sites (channels which detected SU activity) revealed no significant differences in SNR between cuprizone-treated and control mice over time, although cuprizone-treated animals demonstrated overall reduced values of SNR (Fig. 3c). The average amplitude of cuprizone-treated mice on day 1 post-insertion was 7.5 ± 1.1 μV compared to 14.3 ± 1.4 μV in control mice (Fig. 3d). Over the course of the subsequent 4 weeks, signal amplitude in control mice remained elevated before declining to the level of cuprizone-treated mice by 7 weeks post-insertion (cuprizone: 8.8 ± 2.8 μV, control: 10.6 ± 2.0 μV). Similarly, cuprizone-treated mice demonstrated reduced amplitudes of the noise floor compared to control mice on day 1 post-insertion (Fig. 3e, cuprizone: 1.69 ± 2.1 μV, control: 3.4 ± 4.6 μV). Control mice maintained elevated noise amplitudes until declining to the level of cuprizone-treated animals by 7 weeks post-insertion (cuprizone: 1.93 ± 5.8 μV, control: 2.5 ± 5.3 μV). Device impedances in cuprizone-treated animals were reduced significantly compared to control mice for the first week post-insertion, slightly fluctuating until 14 days post-insertion, and then stabilized after 3 weeks (Fig. 3f).
Figure 3. Cuprizone-induced oligodendrocyte loss and demyelination reduces electrode recording performance over time.

Chronic electrophysiological metrics over time between cuprizone-treated (blue solid) and control mice (black dashed). a) Single-unit yield over time. b) Single-unit SNR (mean peak-peak amplitude over 2*STD of noise floor). c) SNR per electrode channel detecting a single unit (active site). d) Single-unit amplitude over time. e) Amplitude of noise floor over time. f) Average impedance reported at 1 kHz over time. * indicates non-overlapping 95% confidence intervals at each time point as determined by likelihood ratio test applied to a linear mixed effects model for cuprizone-treated and control mice.
Reduction of single-unit recording metrics in cuprizone-treated mice compared to mice on control diets was also observed in a depth-dependent manner (Fig. 4a–d). Animals at each time point were aligned to their average layer IV depth, determined from their CSD analysis. Recording performance within different cortical depths along the electrode shank as well as over time post-insertion were demonstrated in the form of averaged heat maps. Smoothing was applied to easily visualize gradual changes in recorded metrics over time and depth. Cuprizone-treated animals demonstrated visually apparent reductions in electrode recording yield compared to control mice specifically in cortical layers II/III, IV, and V (Fig. 4a). Control mice demonstrated a reduction in yield over time, most notably decreasing in cortical layer II/III by 6 and 7 weeks post-insertion. Average SNR was reduced in cuprizone-treated mice vs. control mice within layer II/III, IV, and V, demonstrating the most loss in SNR values within layer II/III and IV (Fig. 4b). Furthermore, amplitudes were most visibly reduced in layer II/III and IV following cuprizone-administration compared to control mice (Fig. 4c). Single-unit amplitudes in control mice were particularly reduced in cortical layer IV after 4 weeks post-insertion. Finally, cuprizone-treated mice demonstrated reduced noise amplitudes within each cortical layer (II/III, IV, and V) compared to control mice (Fig. 4d). Control mice demonstrated a decline in noise amplitude in all cortical layers after 5–6 weeks post-insertion. Collectively, the results indicate that a deficiency in oligodendrocyte function and myelination within the cortex can alter the detectability and recording strength of individual neurons around implanted microelectrode arrays.
Figure 4. Cuprizone administration reduces recording performance metrics in a depth-dependent manner.
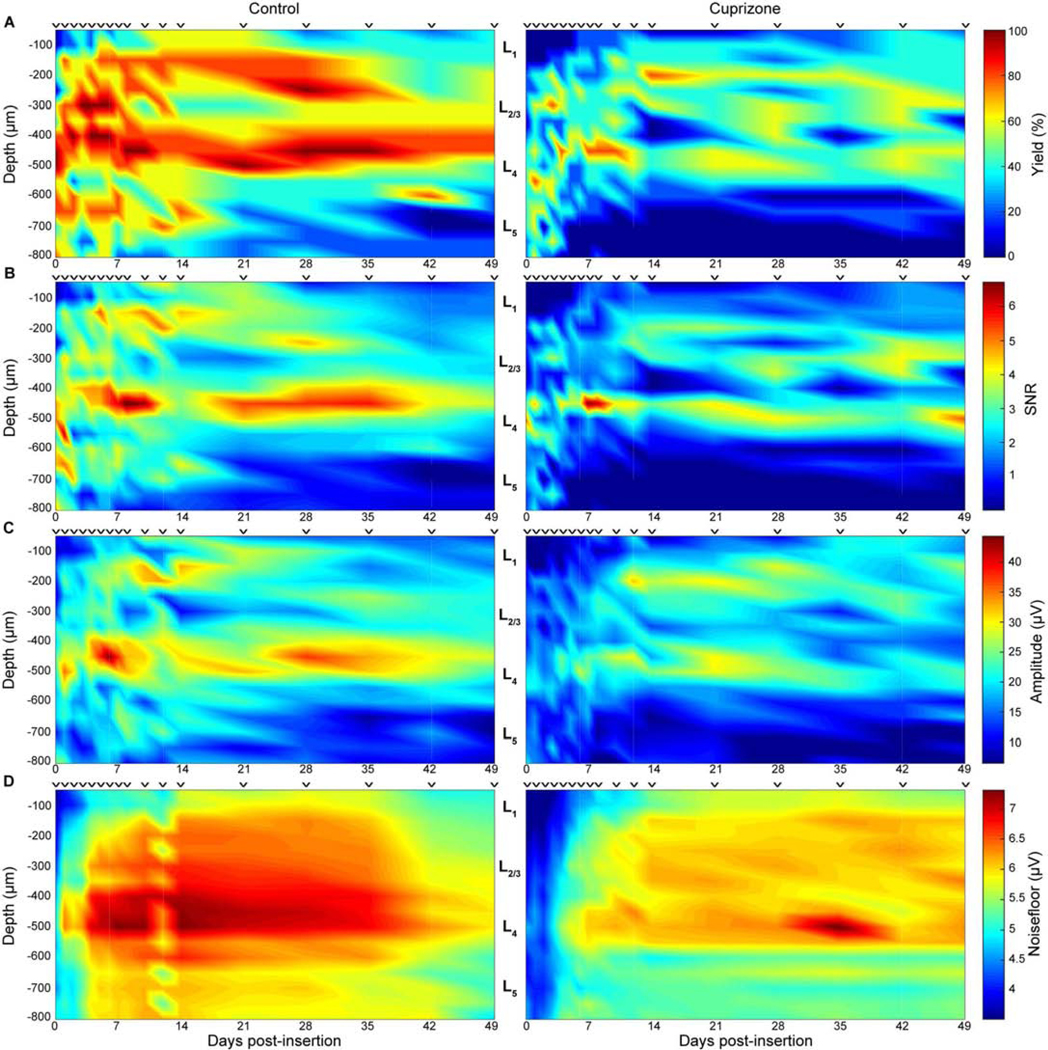
Recording performance metrics between cuprizone-treated and control mice demonstrated as a function of depth and time. ⱽ indicate discrete sampling time points. a) Recording yield. b) SNR. c) Peak-to-peak amplitude. d) Noise amplitude.
4.4. Cuprizone administration reduces spontaneous and evoked neuronal firing rate
After demonstrating that oligodendrocyte and myelin depletion impairs single-unit recordings, we asked whether cuprizone administration would affect the functional multiunit firing properties of local neuronal populations within the visual cortex. Given the dual purpose of myelin as electrical insulation for fast propagation of action potentials as well as being a conduit for metabolic and neurotrophic support to satisfy energetic demands of neurons, cuprizone-induced demyelination was expected to reduce overall neuronal firing rates within the vicinity of microelectrode arrays. Multi-unit activity was compared between cuprizone-treated and control mice by evaluating all threshold-crossing events (non-sorted spiking activity) that occurred during resting-state and evoked recording sessions. Average firing rate was defined as the number of threshold-crossing events that occurred within a 1 second period after each visual stimulus trial (or 1 second after each pseudotrigger assigned to spontaneous activity). As expected, neuronal firing rate was increased following visually evoked stimulation compared to spontaneous (resting-state) activity in both cuprizone-treated and control mice (Fig. 5a–b). However, cuprizone administration effectively reduced firing rate compared to control mice under both spontaneous and visually-evoked recording conditions, albeit not significantly. Neuronal firing rate during resting-state activity was highest on day 1 post-insertion for both cuprizone (12.1 ± 0.6 spikes/s) and control mice (13.9 ± 0.7). By 7 weeks post-insertion, spontaneous firing rate decreased in both groups (cuprizone: 9.5 ± 0.9 spikes/s, control: 9.81 0.8 spikes/s). Similarly, firing rate was highest during visually-evoked stimulation on day 1 post-insertion for cuprizone-treated mice (17.4 ± 1.6 spikes/s) and control mice (18.5 ± 1.0 spikes/s) before firing rate in both groups gradually declined by 7 weeks post-insertion (cuprizone: 12.9 ± 1.3 spikes/s, control: 13.9 ± 1.4 spikes/s). Changes in neuronal firing rate for cuprizone-treated and control mice were also observed in a depth-dependent manner (Fig. 5c–d). Reductions in firing rate over time during resting-state and visually-evoked activity for both cohorts occurred predominantly in cortical layer II/III, matching depth-dependent patterns of single-unit recording. Interestingly, evoked firing rate for both groups increased within layer II/III following 2 weeks post-insertion. Together, these findings establish that oligodendrocytes and myelin modulate the firing properties of neuronal networks during resting-state and evoked cortical activity.
Figure 5. Oligodendrocyte loss reduces neuronal firing rate over time and depth around chronically implanted microelectrode arrays.

Average neuronal firing rate during resting-state (a) and visually-evoked (b) activity between cuprizone-treated and control mice over time. Firing rate between cuprizone-treated and control mice demonstrated as a function of depth and time during resting-state (c) and visually evoked (d) activity. ⱽ indicate discrete sampling time points.
4.5. Cuprizone-induced oligodendrocyte loss and demyelination reduces multi-unit activity in a latency-dependent manner
Multi-unit activity was evaluated further to determine how oligodendrocytes and myelin, whose function is to regulate conduction velocities of neuronal signals, affect electrophysiological latency following evoked visual stimulation within the cortex. Contribution of oligodendrocytes and myelin on evoked neuronal population responses were further evaluated by observing dynamic changes in multi-unit activity (MUA) following implantation. Multi-unit yield and SNFRR were metrics generated by comparing spike counts before and after visual stimulation, whose responses can differ based on parameters such as bin duration and temporal latency from stimulation onset [70]. Therefore, a range of bin sizes and latencies post-stimulation were evaluated to observe patterns in MUA responses between cuprizone-treated and control mice over time (Fig. 6a). Bin sizes and latencies varied from 1 ms to 1000 ms with a temporal bin resolution of 1 ms, generating heat maps that highlight hotspots of MU yield and SNFRR for certain durations of bin size and latency (Fig. 6b). Naturally, bin sizes and latencies less than 50 ms produced low or virtually no evoked responses in MUA due to the time required for evoked responses to be recognized by the recording electrode within the visual cortex (Fig. 2i). Most dynamic changes occurred within a bin size duration up to 200 ms and latencies up to 800 ms, which were further observed as the difference in MU yield and SNFRR between cuprizone-treated and control mice over time (Fig. 6c–d). On the day of surgery, control mice demonstrated greater MU yield and SNFRR compared to cuprizone-treated mice for all bin durations and latencies. Interestingly, cuprizone-treated mice exhibited higher MU yield and SNFRR compared to control mice on day 1 post-implantation. Beyond this first day after device insertion, MU yield and SNFRR were elevated in mice on control diets for latencies greater than 200 ms. Note that most significant differences in MU yield and SNFRR were observed only when values were higher in control mice and when latencies were greater than ~400 ms. Significant differences that did occur in cuprizone-treated animals generally occurred less than ~400 ms after stimulus presentation. In line with aforementioned trends in single-unit activity and firing rate, control mice demonstrated virtually no significant differences in MU yield or SNFRR compared to cuprizone-treated mice at 6 and 7 weeks post-insertion, indicating a reduction in evoked MUA of control mice over time. To summarize, loss of cortical oligodendrocytes and myelin following cuprizone administration impaired evoked multi-unit activity with increasing latency post-stimulation, a difference which deteriorated in mice fed on control diets over time.
Figure 6. Oligodendrocyte depletion and demyelination following cuprizone administration impairs multi-unit recording activity.
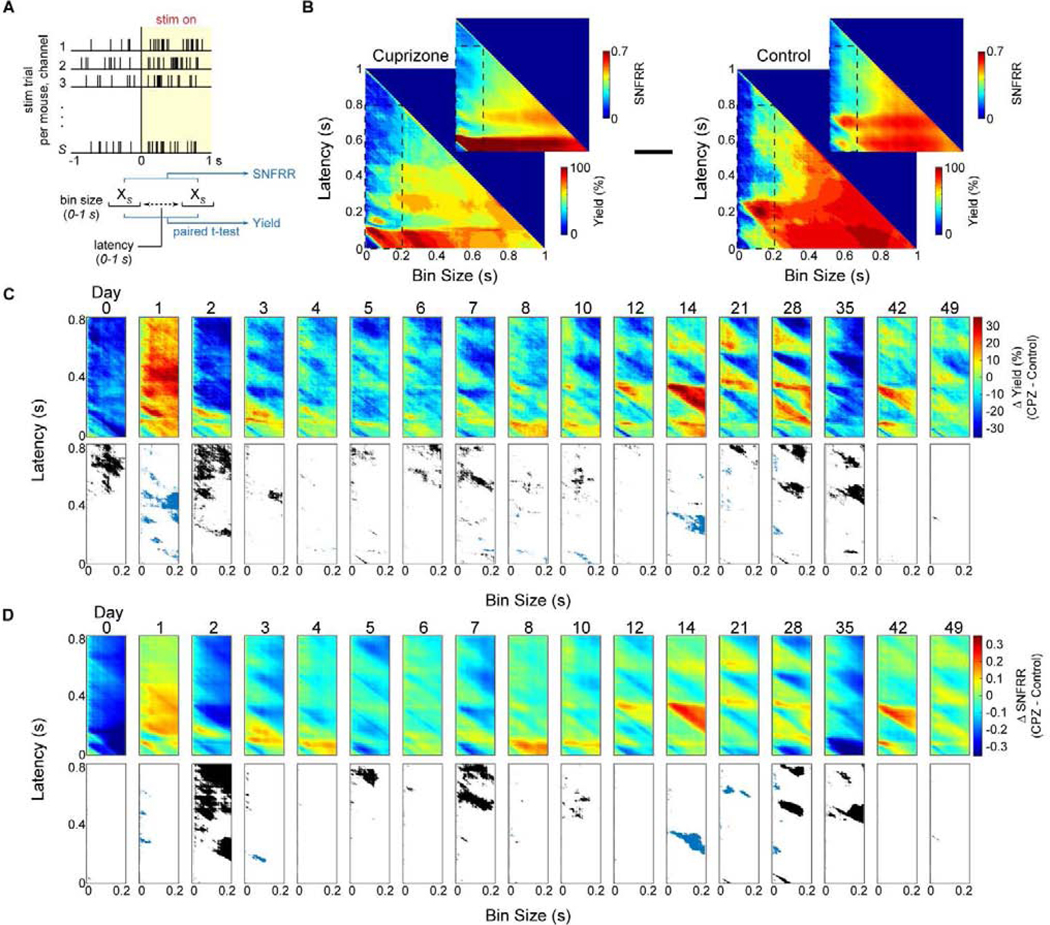
(a) Diagram demonstrating how dynamic changes in multi-unit yield and SNFRR during evoked electrophysiological recordings were measured. Spike counts, XS, were collected within a temporal bin size and latency ranging from 0 to 1 s following stimulus presentation and compared to counts within the same bin size before stimulus presentation using a paired t-test. MU yield was calculated as the percentage of electrode channels (out of 16 sites) that had a significantly different spike count for a given bin size and latency after stimulus presentation compared with the same bin size before stimulus presentation. MU SNFRR was calculated as the difference in average spike counts relative to the standard deviation in spike counts before and after stimulus presentation. (b) Representative plots of MU yield and SNFRR taken at varying bin sizes and latency between 0 and 1 s at 1 ms increments in cuprizone-treated and control mice. MU yield and SNFRR plots within a window of 200 ms bin size and 800 ms latency (inset) were averaged within each group and control data was subtracted from cuprizone mice to produce (c) and (d). (c-d) Difference in MU yield or SNFRR between cuprizone-treated and control mice over time (top panel). A paired t-test (p < 0.05) was performed at each bin size and latency between cuprizone-treated and control mice. Significant differences where yield or SNFRR was greater in control mice are reported in black, where yield or SNFRR was greater in cuprizone mice are reported in blue, and no significant difference between the two groups are reported in white (bottom panel).
4.6. Cuprizone-induced oligodendrocyte loss and demyelination alters evoked oscillatory activity within the visual cortex
The previous findings demonstrated that oligodendrocyte and myelin depletion alters electrophysiological properties within local neuronal populations. Knowing that oligodendrocytes and myelin regulate neuronal activity in a time-dependent manner, we then asked how the frequency-dependent oscillations in neuronal signaling are influenced by cuprizone administration. The contribution of oligodendrocytes and myelin on evoked oscillatory neuronal activity within the cortex was evaluated by performing power spectral analysis. Power was calculated by taking the base 10 logarithm of recorded local field potentials. Evoked power was normalized by subtracting spontaneous power across a frequency range of 0.5 to 125 Hz (Fig. 7a–b). While both cuprizone-treated and control mice experienced similar evoked power across frequencies, cuprizone-treated mice demonstrated greater fluctuations in power within the first two weeks post-insertion across the entire frequency spectrum (Fig. 7c). Furthermore, while power did not change in a depth-dependent manner between cuprizone-treated and control mice over time, similar fluctuations in evoked power over the first two weeks were observed in cuprizone-treated mice. Conversely, evoked power in control mice remained relatively stable except for slight increases in power at 1 and 6 weeks post-insertion (Fig. 7d). These fluctuations in LFP activity were further reflected by averaging evoked power over the entire frequency range between cuprizone-treated and control mice (Fig. 7e). Additionally, cuprizone-treated mice demonstrated a reduced peak in evoked power within the first week of insertion compared to control mice before gradually increasing and stabilizing over time (Fig. 7f). Finally, evoked power was evaluated at distinct frequency ranges between cuprizone-treated and control mice over time (Fig. 7g). Cuprizone-treated mice demonstrated reduced power within delta (24 Hz), theta (5–7 Hz), and alpha (8–14 Hz) frequency bands compared to control animals. Alternatively, cuprizone-treated mice exhibited an increase in power in gamma (30–90 Hz) and high frequency bands (>90 Hz). At all frequency bands, cuprizone-treated mice demonstrated high fluctuations in evoked power compared to control mice, which remained relatively stable over time. Together, these results indicate that cuprizone-induced oligodendrocyte loss and demyelination alters evoked oscillatory activity in a frequency-dependent manner within the mouse visual cortex.
Figure 7. Cuprizone-induced oligodendrocyte loss and demyelination reduces evoked power in a frequency-dependent manner.

(a) Representative power spectra of one mouse from each group (cuprizone-treated, blue; control, black) taken at the same electrode site in layer IV demonstrating evoked (solid) and spontaneous (dashed) power over frequency. (b) Normalized power spectrum taken by subtracting the spontaneous power from evoked power. (c) Heat maps of average evoked power as a function of frequency and time between cuprizone-treated and control animals. (d) Heat maps of average evoked power as a function of depth along microelectrode array and time between cuprizone-treated and control animals. (e) Cuprizone-administration induces oscillations in mean power over entire frequency spectrum for first two weeks compared to control mice. (f) Cuprizone-administration reduces peaks power compared to control mice acutely during insertion. (g) Relative power within different frequency bands demonstrate differential outcomes following cuprizone administration. PEVO: evoked power; PSPON, spontaneous power.
4.7. Cuprizone-induced oligodendrocyte loss and demyelination alters evoked laminar communication within the visual cortex
Since the evoked oscillatory activity differed between wildtype and oligodendrocyte depleted mice, we next investigated the influence of oligodendrocytes and myelin on functional communication within and between cortical laminar circuits through coherence analysis. It is expected that the loss of myelin, a critical substance considered to facilitate information transmission of far-reaching projections, would have significant impacts on interlaminar, if not intralaminar, communication following cuprizone administration. Coherence, reported as a value between 0 and 1, was first normalized by subtracting spontaneous coherence from evoked coherence and then calculated for all 16 pairs of electrode channels, resulting in heat maps of evoked coherence values in a 16 × 16 site diagram (Fig. 8a–b). The greatest difference in coherence between cuprizone-treated and control mice was observed at 8 days post-insertion. Therefore, this time point was compared alongside the experimental endpoint of 49 days post-insertion, which has demonstrated comparable performance between cuprizone-treated and control mice for all electrophysiological metrics observed so far. At 8 days post-insertion, cuprizone-treated mice demonstrated increases in evoked coherence within superficial (layer II/III) and deep layers (layer V/VI) as well as between granular-supragranular (G-SG: layer IV to layer II/III), granular-infragranular (G-IG: layer IV to layer V/VI), and supragranular-infragranular (SG-IG: layer II/III to layer V/VI) regions (Fig. 8a). These responses in cuprizone-treated mice differed from control mice most within higher frequency beta (7–30 Hz) and gamma (30–90 Hz) bands. By day 49 post-insertion, previously coherent regions in cuprizone-treated mice were reduced, demonstrating similar or reduced coherence heat maps compared to control mice (Fig. 8b). These temporal trends in evoked coherence between cuprizone-treated and control mice are further reflected by observing interlaminar connectivity as a function of frequency. Cuprizone-treated mice demonstrated the greatest difference in coherence compared to control mice in G-IG and G-SG regions at 8 days post-insertion (Fig. 8c). Due to a decrease in evoked coherence in cuprizone-treated mice by 49 days post-insertion, coherence between cuprizone-treated and control mice were virtually the same (Fig. 8d). Finally, evaluating the coherence averaged between 7 and 90 Hz between laminar regions demonstrated a significant increase in coherence within G-SG regions over the first 4 weeks post-insertion between cuprizone-treated and control mice (Fig. 8e). Furthermore, synchronicity between neuronal populations is an important property of functional resting-state activity. Therefore, we measured coherence between and within different cortical layers and demonstrated similar trends in coherence during resting-state in cuprizone-treated mice (Supplementary Fig. 1). This analysis indicates that the loss in function of oligodendrocyte and myelin compromises functional communication during resting-state and evoked activity within and between laminar structures of the visual cortex.
Figure 8. Oligodendrocyte depletion and demyelination increases visually-evoked coherence within and between different cortical layers.

Normalized pairwise coherence, taken as the difference in coherence during visually-evoked and spontaneous activity, between cuprizone-treated and control animals at day 8 (a) and day 49 (b) post-insertion. Missing coherence values in (a) are an indication of lost information following electrode depth adjustment to layer IV. Evoked coherence between supragranular-infragranular (SG-IG), granular-infragranular (G-IG), and granular-supragranular (G-SG) regions over frequency between cuprizone-treated and control mice at day 8 (c) and day 49 (d) post-insertion. (e) Mean coherence between 7 and 90 Hz (shaded region of c and d), averaged across animals in each group. * indicates non-overlapping 95% confidence intervals at each time point as determined by likelihood ratio test applied to a linear mixed effects model for cuprizone-treated and control mice. Data presented as mean ± SEM.
4.8. Histological analysis of microelectrode explants reveals sustained oligodendrocyte loss around chronically implanted microelectrode arrays
Following 7 weeks of implantation in control and 12 weeks of cuprizone administration, cortical tissue around microelectrode explants were analyzed for changes in cellular and subcellular dynamics resulting from device-induced inflammation and cuprizone-mediated toxicity. A chronic loss of oligodendrocytes and myelin coupled with damage incurred during chronic device implantation was expected to impair neurons around the device-tissue interface and account for the late-stage decline in recording performance of mice fed control diet. Surprisingly, staining for NeuN+ neurons showed no device-dependent changes in neuronal density between cuprizone-treated or control mice (Fig. 9a–b). However, staining for NF-200+ axons demonstrated increased, yet not significant, levels in neurofilament immunoreactivity in cuprizone-treated mice around the microelectrode implant, suggesting a cuprizone-induced impact on axonal pathology following injury (Supplementary Fig. 2). Further staining for SMI-31+ phosphorylated NF200 axons demonstrated increased immunoreactivity as well as increased neurofilament accumulations in cuprizone-treated mice around an implanted microelectrode array (Supplementary Fig. 2 and 3). Additionally, cuprizone-treated mice maintained low oligodendrocyte density following 7 weeks post-implantation (Fig. 9c–d), consistent with previous cuprizone studies reporting a chronic depletion of oligodendrocytes by 12 weeks of cuprizone administration. Cuprizone-treated mice demonstrated markedly reduced oligodendrocyte density compared to control animals. However, there was no significant differences in oligodendrocyte density between cuprizone-treated animals and controls, on account of high variability in CC1+ cell counts in control mice. This variability may be explained by the heterogeneous presence of oligodendrocytes throughout the cortex, which may be differentially susceptible to device insertion injury [26]. Staining for MBP+ myelin revealed comparable fluorescence intensities in control mice at 7 weeks post-insertion (Fig. 9e–f) relative to pre-insertion results (Fig. 1c). Analysis of contralateral tissue demonstrated significantly reduced MBP+ fluorescence intensities in cuprizone-treated mice compared to controls, confirming that 12 weeks of cuprizone administration induced a chronic state of demyelination within the visual cortex. (Supplementary Fig. 4). Interestingly, cuprizone-treated mice demonstrated an increase in myelin intensity around the site of microelectrode implantation relative to baseline (Fig. 9e–f). As a result, no significant differences in MBP+ myelin intensity compared to control mice were reported at 7 weeks post-insertion, similar to observed oligodendrocyte density. Together, the results suggest that impaired recording performances over time could be explained by the chronic depletion of oligodendrocytes and myelin following long-term cuprizone administration.
Figure 9. Explant histology 7 weeks post-insertion reveals sustained depletion of cortical oligodendrocytes alongside comparable neuronal densities.
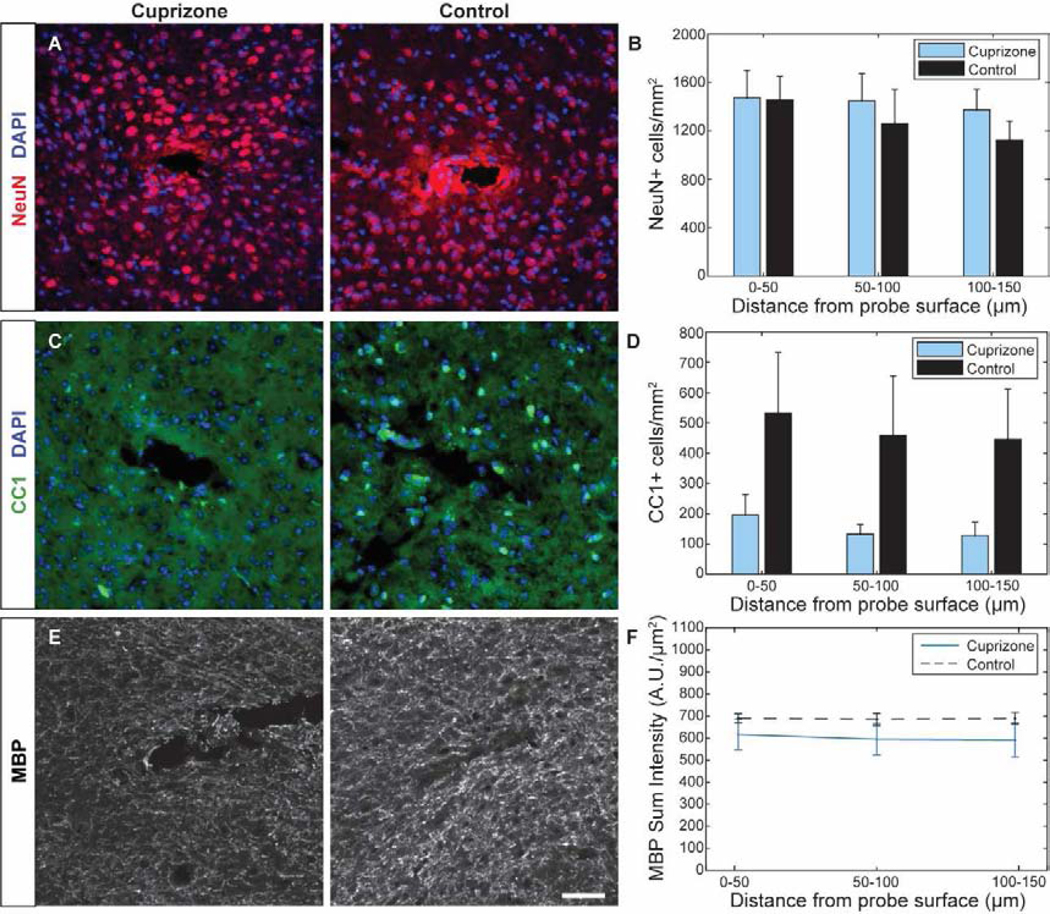
(a) Representative histological stain for NeuN+ neurons around a microelectrode probe hole in cuprizone-treated and control animals. (b) Cell counts demonstrate comparable neuronal populations between the two groups within 150 μm from the probe surface. (c) Representative stain for CC1+ oligodendrocytes around a microelectrode probe hole in cuprizone-treated and control animals. (d) Cell counts demonstrate reduced oligodendrocyte density within 150 μm from the probe surface in cuprizone-treated mice. (e) Representative stain for MBP+ myelin around a microelectrode probe hole in cuprizone-treated and control animals. (f) Sum fluorescence intensity demonstrates a slight reduction in MBP intensity in cuprizone-treated animals compared to controls. Scale bar = 50 μm.
Since glial cell activation is commonly reported in both models of cuprizone administration and intracortical electrode implantation, we wanted to determine the extent of glial activation by experimental endpoint. Chronic implantation of a microelectrode array induced glial reactivity with spatial preference around the device interface (Fig. 10). However, only certain glial cells responded to chronic cuprizone administration. The number of NG2+ oligodendrocyte precursor cells within 150 μm radius from site of device insertion was lower in cuprizone-treated mice compared to mice on control diet, demonstrating significant reduction within 50 μm from the site of insertion (Fig. 10a–b). While oligodendrocyte precursor cells in cuprizone-treated mice maintained similar densities within each bin, control mice demonstrated greater NG2+ cell density with increasing proximity to the site of probe implantation and compared to pre-implantation cell counts, indicating specific glial reactivity in response to device implantation injury. Chronic implantation of a microelectrode array induced similar distance-dependent activation of Iba-1+ microglia and GFAP+ astrocytes (Fig. 10c–f). However, the average Iba-1+ fluorescence intensity was virtually the same between cuprizone-treated and control mice indicating no cuprizone-specific influences on microglia activation post-injury (Fig. 10c–d). On the other hand, cuprizone demonstrated specific astrocyte reactivity, with increased, and more highly varied, GFAP+ signal intensity in cuprizone-treated animals compared to controls (Fig. 10e–f). These results indicate that long-term cuprizone toxicity induces a reduction in oligodendrocyte precursor density as well as mild activation of astrocyte populations around chronically implanted microelectrode arrays.
Figure 10. Cuprizone-induced glial reactivity around implanted microelectrode arrays at 7 weeks post-insertion.
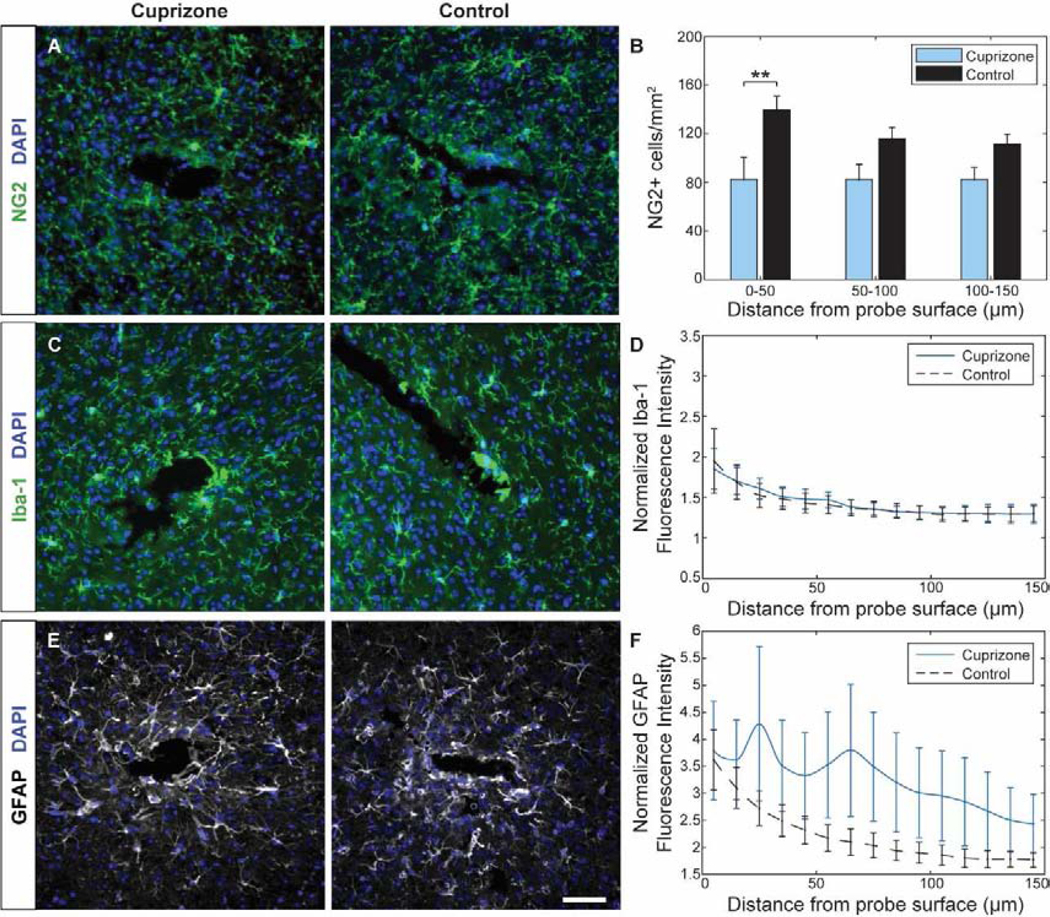
(a) Representative histological stain for NG2+ glial cells around a microelectrode probe hole in cuprizone-treated and control animals. (b) Cell counts demonstrated reduced NG2+ precursor population within 150 μm from the probe surface in cuprizone-treated mice. (c) Representative stain for Iba-1+ microglia in cuprizone-treated and control animals around a microelectrode probe hole. (d) Normalized intensity demonstrated comparable Iba-1+ intensity between control and cuprizone-treated mice. (e) Representative stain for GFAP+ astrocytes in cuprizone-treated and control animals around a microelectrode probe hole. (f) Normalized intensity demonstrated elevated GFAP+ intensity within 150 μm from the probe surface in cuprizone-treated mice. Fluorescence intensities were normalized to contralateral hemisphere. Scale bar = 50 μm. ** indicates p < 0.01.
5.0. Discussion
The contribution of oligodendrocytes and myelin on electrophysiological activity within the mouse visual cortex was observed in a cuprizone-inducible model of demyelination. Sixteen channel, single-shank microelectrode arrays were used to record neuronal data over a 7 week period following 5 weeks of cuprizone administration in oligodendrocyte- and myelin-depleted mice compared to control mice fed with normal diets. Changes in single- and multi-unit activity as well as changes in local field potentials were analyzed to reveal the influence of oligodendrocyte loss and demyelination on the electrophysiological properties of individual neurons, activity of local neuronal populations, neuronal oscillatory activity, and laminar connectivity of cortical circuits. Overall, cuprizone administration induced significant reductions in the evoked firing properties of recordable neurons, decreased firing rates of resting-state and evoked neuronal activity, decreased multi-unit activity in a latency-dependent manner, disrupted the evoked frequency of neuronal oscillations, and altered interlaminar communication within the cortex. Furthermore, post-mortem histology revealed chronic depletion of oligodendrocytes alongside quantitatively matched neuronal densities near the site of microelectrode insertion in cuprizone-treated mice compared to control mice, promoting the idea that loss or dysfunction of oligodendrocytes is a possible source of recording performance failure around intracortical neural interfaces.
5.1. Targeted depletion of cortical oligodendrocytes following 5 weeks of cuprizone administration
After 5 weeks of cuprizone exposure, oligodendrocytes and myelin were significantly (p<0.001) depleted within the visual cortex (Fig. 1). It is important to note that cuprizone depletion does not lead to complete elimination of oligodendrocytes, since a complete loss of oligodendrocyte would likely be fatal. Even though cuprizone does not target myelin directly, loss of oligodendrocytes and inability for oligodendrocytes to regenerate and re-myelinate during cuprizone administration results in demyelinating injury [76]. In vitro analysis determined that cuprizone exposure does not affect the viability of cultured neurons, microglia, or astrocytes, nor does it affect the proliferation or survival of oligodendrocyte progenitor cells [77]. Furthermore, cuprizone treatment does not induce a copper deficiency or toxicity in cultured neurons, demonstrating that cuprizone does not readily pass through neuronal cell membranes [64]. Thus, cuprizone acts with high specificity on oligodendrocyte cells through cellular dysfunctions in a copper-dependent and independent manner [78].While the underlying mechanisms of cuprizone-induced oligodendrocyte toxicity are unclear, the most common theories are disturbances in mitochondrial function important for energy metabolism [59, 78, 79]. Oligodendrocytes require high amounts of generated ATP to sustain their metabolic output due to continuous synthesis of lipid myelin membranes, which heightens their vulnerability to mitochondrial injury above other metabolically active cells within the CNS [46]. Cuprizone treatment reportedly alters mitochondrial enzymes dependent on copper as a co-factor and can disrupt electron transport function of complexes I-III within the respiratory chain [80–82]. As a result, reductions in intracellular energy, accumulation of reactive oxidative species, and inherently low antioxidant levels in oligodendrocytes can lead to cellular apoptosis following cuprizone administration [30, 59]. Alternatively, cuprizone-induced copper deficiency has the potential to directly affect the production or deposition of myelin within oligodendrocytes, which requires the translation, transcription, and translocation of myelin-related proteins within endosomes that fuse with an expanding myelin membrane [83]. Endosome fusion to the oligodendrocyte cellular membrane is the principle method for depositing myelin proteins and lipids necessary to generate and maintain myelination. Copper has been implicated in the regulation of intrinsic cellular mechanisms which control fatty acid synthesis within the brain as well as mRNA gene expression of myelin-related proteins during cuprizone-induced toxicity [84–86]. Furthermore, as a transitional metal ion whose abundance can lead to cellular toxicity, copper can induce vesicular fusion with the plasma membrane (exocytosis) in order to maintain homeostatic levels, and in the process promotes the repair or replacement of damaged cellular membranes [87]. As a result, a lack of copper due to cuprizone administration could possibly inhibit this myelin forming process within oligodendrocytes, resulting in CNS demyelination.
In response to oligodendrocyte loss, oligodendrocyte precursor cells migrate, proliferate, and differentiate into myelinating oligodendrocytes in order to restore homeostatic density [40]. As a result, an increase in oligodendrocyte precursors within the cortex is expected following acute cuprizone toxicity (Fig. 1d). Existing myelin is degraded following cuprizone-induced oligodendrocyte cell death and the remaining debris is cleared away by microglia (Fig. 1c). Additionally, protection of mature oligodendrocytes during cuprizone administration results in attenuation of myelin loss and reduced microglia or astrocyte reactivity in white matter tracts, suggesting that debris left from oligodendrocyte and myelin degeneration induces glial cell activation [74]. Therefore, glial cell activation is commonly reported in white matter tracts of the cuprizone model [88, 89]. However, we report no significant differences in activation of microglia or astrocytic markers in cortex by 5 weeks of cuprizone administration (Fig. 1e–f). This could be accounted for by a different temporal course of cortical glial activation and extent of glial response due to lower amount of myelin and, consequently, accumulation of myelin debris in the cortex compared to the heavily myelinated white matter tracts. Furthermore, this phenomenon is known to be strain-dependent, which would explain why we demonstrate little to no activation of microglia or astrocytes within the cortex of C57BL/6J mice following 5 weeks of cuprizone toxicity [58].
5.2. Loss of cortical oligodendrocytes and myelin on electrophysiological recording properties of individual neurons
Neural electrophysiology was evaluated in the mouse visual cortex due to the fact that visual stimulation can elicit robust neural responses and is a more reliable and reproducible method of stimulation compared to somatosensory or motor cortex stimulation. Single-shank Michigan-style microelectrode arrays were implanted to a depth of 800 μm within the cortex (layer V/VI) in order to avoid insertion into white matter areas beneath the cortex which consist of a different distribution of oligodendrocyte cells and myelin fibers. Mice were recorded while head-fixed on a movable platform for linear ambulation and while awake to avoid unwanted influences in neural responses seen with anesthetized recording [70]. Motion artifacts that appeared during recording were minimized by repeating trials as needed to collect continuous, undisrupted data [90]. Current source density analysis showed that the electrode did not drift more than the distance occupied by one electrode site in either direction (<50 μm) within the first two weeks of insertion before stabilizing, indicating electrode position was relatively stable throughout the course of implantation (Fig. 2). Control mice demonstrated a slight increase in layer IV position along the implanted microelectrode array, which is a result of tissue swelling that can occur following brain injury [91]. Interestingly, cuprizone mice experienced, on average, a decrease in layer IV position along the implanted microelectrode array indicative of tissue shrinkage, which may be a result of a bulk loss and clearance of oligodendrocyte and myelin following cuprizone administration (Fig. 2j). The insulating properties of myelin decrease the capacitance of axonal membranes [32]. Therefore, the reduction of myelin via cuprizone toxicity would effectively increase axonal membrane capacitance and may reduce impedances around an implanted microelectrode. Here, we show that electrode impedances were significantly reduced in cuprizone-treated animals for the first week after insertion (Fig. 3f). Abnormally high or low impedances, which would be indicative of mechanical or material failure, were not reported in either cuprizone-treated or control mice, demonstrating electrode stability and functionality throughout the course of implantation [23, 92]. This may be due to revised fabrication methods or subtle design differences compared to previously reported work using 100 μm site spacing probes [93]. Regardless, hardware issues should equally affect both experimental group and control, which should not diminish the significant differences of the electrophysiological recording properties reported here.
Recording yield, which is a measure of device recording efficacy, was significantly impaired at the onset of implantation in cuprizone-treated mice (Fig. 3a). Given that other established modes of recording performance failure, such as glial scar formation and neurodegeneration, are identified as late-stage events that occur following chronic device implantation (>2–4 weeks post-implantation), this decrease in yield can likely be attributed to the absence of cortical oligodendrocytes and myelin membranes following the cuprizone pretreatment [94]. Reduced single-unit yields can suggest a failure in signal transduction of demyelinated axons, resulting in decreased signal amplitudes of more distal neurons, which would not be threshold detected by the recording device. Indeed, the amplitude of evoked neuronal responses in cuprizone-treated mice were decreased compared to controls (Fig. 3d). This is consistent with an observed decrease in signal-to-noise ratios (SNRs), a metric that measures the strength of detected signals against the recorded noise (Fig. 3b). SNRs are dependent on the amplitude or strength of neuronal action potentials as well as electrophysiological noise (i.e. neuronal noise, glial reactivity) for each contact site, which also appeared reduced in cuprizone-treated mice. Furthermore, oligodendrocytes and myelin populate the gray matter cortex in a layer-specific manner, with oligodendrocyte and myelin densities the highest in deeper laminar structures [33]. This spatial distribution is consistent with a recorded loss in yield, SNR, and amplitudes in layer IV and V of cuprizone-treated mice, indicating that recording performance is altered as a function of depth along the implanted microelectrode array due to spatial specificity of oligodendrocytes and myelin within the cortex (Fig. 4). This may also explain the plateau in recording yield between 20–40% in cuprizone-treated animals (Fig. 3a), which equates to ~3–6 recording sites (~150–300 μm of cortical depth). Considering that cuprizone toxicity significantly depletes most oligodendrocytes (Fig. 1b), but not all oligodendrocytes within the CNS, as well as the fact that not all neurons within the gray matter cortex are myelinated (and therefore unaffected from cuprizone demyelination) suggests that this is the maximum possible deficiency in recording performance we can observe given the limitations of the model and the myeloarchitecture of the brain.
Trends in single-unit activity were relatively stable throughout the 7 week implantation period in cuprizone-treated mice (Fig. 3). Recording performance in the oligodendrocyte-depleted group began at a low performance state on day 1 and remained plateaued at that low performance state over time. In contrast, recordings in control mice, which began with healthy oligodendrocytes were significantly higher (p<0.05) relative to cuprizone-treated mice before experiencing a gradual decline over time, which is consistent with previously published data [21, 95, 96]. By 7 weeks post-insertion, electrophysiological performance in control mice was indistinguishable from that of cuprizone-treated mice. These results were contradictory to our initial hypothesis, that recording performance in cuprizone-treated mice would decline more rapidly than control animals given a lack of oligodendrocyte- and myelin-mediated support for neurons and axons. In fact, by 7 weeks post-insertion, neuron density was similar between the two groups (Fig. 9B). However, it is important to note that previous studies have demonstrated that the presence of NeuN staining does not necessarily indicate that the neuron is functional as opposed to being silenced or otherwise dysfunctional [97]. Indeed, previous evaluation of cortical neurogenesis following injury or via induced differentiation has demonstrated the inability for new neurons to integrate functionally due to an unfavorable microenvironment [98, 99]. Instead, an alternative hypothesis is that the initial pre-insertion depletion of oligodendrocytes (Fig. 1) leads to an environment where neurons are more susceptible to becoming dysfunctional even without (or following) a microelectrode implantation. This could explain why recording performance starts low and remains plateaued over the course of cuprizone treatment (Fig. 3–8). Taken together, significant decreases in oligodendrocytes and myelin with cuprizone-treatment prior to array implantation (Fig. 1b–c) leads to significantly lower initial recording performance (Fig. 3–8) without significantly impacting neuronal density (Fig. 9b) compared to controls. However, over the course of 7 weeks, there are complex cell dynamics and the significant differences between the two groups are lost for oligodendrocyte density (Fig. 9d; cuprizone 0–50 μm: 168.6 ± 55.5 cells/mm2, control 0–50 μm: 449.3 ± 124.2 cells/mm2), which is accounted for in large part by the drop in CC1+ cells in control mice and a small increase of CC1 in the cuprizone group (compare Fig. 1 with Fig. 9), myelin intensity (Fig. 9f; cuprizone 0–50 μm: 617.3 ± 137.1 A.U./μm2, control 0–50 μm: 690.9 ± 45.6 A.U./μm2), which is largely accounted for by the increase in MBP staining in the cuprizone group (compare control in Fig. 1 and Fig. 9), and recording performances (Fig. 3–8). Interestingly, the number of NG2 cells in control mice is unchanged at 7 weeks versus the time of implantation while it is reduced to half in the cuprizone group. The latter could represent a depletion of NG2 cells that have differentiated into oligodendrocytes, possibly explaining the increase in CC1+ cells in the cuprizone group and leading to increase in myelin density. This supports the notion that over time in wildtype controls (that recapitulates normal implantation injury), the microenvironment around the electrode within the recording radius becomes depleted of oligodendrocytes and myelin, which contribute to recording performance degradation. While the current study does not establish causation, this work motivates future mechanistic studies that link oligodendrocyte health and their metabolic and/or neurotrophic function to recording performance.
While recording performances trend slightly higher in the cuprizone treated group at later time points, these changes are non-significant (Fig. 3). In contrast to NeuN, neurofilament expression was actually increased in cuprizone-treated mice (Supplementary Fig. 2–3). However, SMI-31 labeling was also substantially elevated around the implant, which is associated with dystrophic neurite formation and abnormal axonal transport [100]. Therefore, it is important to keep in mind that the presence of antibody labeling (NF-200) does not necessarily indicate that those cells or cellular elements are functional as previously shown with NeuN staining [97]. Given the limitation in endpoint analyses with the cuprizone model, it is uncertain whether recording performance in control mice would have plateaued at this level by 7 weeks post-insertion or would have continued declining over time. However, these findings suggest that the microenvironment around chronically implanted electrodes in healthy mice gradually becomes increasingly similar to cuprizone-treated cortical tissue. Furthermore, the observed decrease in electrophysiological properties of control mice to a level that is comparable to recordings in cuprizone-treated mice suggest that loss or dysfunction in oligodendrocytes and myelin could potentially act as a source of biological failure for chronically implanted intracortical devices. These findings support the premise that preserving the function of oligodendrocytes and myelin can help improve neural recording performances following injury induced by chronically implanted microelectrode devices.
5.3. Cuprizone-induced oligodendrocyte loss and demyelination impairs multi-unit activity within the cortex
Analysis of multi-unit recordings provided insight on how oligodendrocytes and myelin modulate the functional activity of local neuronal populations within the visual cortical circuit. Cuprizone-induced oligodendrocyte loss and demyelination reduced neuronal firing rates compared to control mice at the onset of device insertion and remained reduced throughout the course of implantation (Fig. 5). This was observed in a depth-dependent manner, with cuprizone-treated mice experiencing the most reduction in multi-unit firing rate within layers II/III. Beyond two weeks post-insertion, firing rate appeared to non-significantly increase in cuprizone-treated mice, which may be due to the unexpected increase in oligodendrocyte density and myelination observed during explant histology. Myelin sheaths cluster voltage-gated Na+ sodium channels into nodes along axons, providing saltatory propagation of neuronal signals [32]. The loss or addition of myelin can result in a redistribution of those sodium channels, weakening or strengthening the propagative effect of action potentials down the axon [101]. Therefore, weakened axonal conduction velocities due to cuprizone-induced demyelination could diminish signals before they are detected by the microelectrode, reducing recorded firing rates. One study investigating the effects of increases in myelin sheath thicknesses of ERK1/2-mutant mice demonstrated a decrease in latency-to-peak of evoked amplitudes within the auditory cortex compared to wild-type mice, suggesting that myelin provides direct functional consequences of neuronal activity by regulating conduction velocity [102]. However, significant differences were not detected during the 1st action potential peak, which would be indicative of a conduction velocity issue during myelin sheathing, but during the 2nd, 3rd, and 5th peak after stimulus presentation, suggesting perhaps a deficiency in metabolic or trophic support from damaged or dysfunctional oligodendrocytes, which would be necessary for repetitive neuronal firing [103]. Demyelinated axons require more energy to support repolarization of active neurons as well as match the levels of metabolic transport within previously myelinated axons [104]. Indeed, we show comparable recording outcomes within the first 100 ms after stimulation onset in multi-unit yield and SNFRR between cuprizone-treated and control mice, which is within the range of normal physiological latencies reported for visual information to reach the cortex (Fig. 6). Furthermore, we report that significant differences in multi-unit dynamics, where recording of multi-unit activity was increased (i.e. repetitive neuronal firing) in control mice over cuprizone-treated mice (Fig. 6), were more apparent following sustained evoked visual stimulation. These results are more indicative of a loss in energetic support from oligodendrocytes impairing recurrent neuronal firing activity than a reduction in myelin-mediated conduction velocities. In either case, these results establish that loss of oligodendrocytes and myelin can reduce the firing properties of local neuronal populations.
5.4. Oligodendrocyte depletion and demyelination alters oscillatory activity and functional laminar connectivity within the cortex
Cognition and other brain functions are highly dependent on synchronized neural circuits to achieve highly coordinated levels of rhythmic activity. Alterations in myelin are known to have direct influences on neuronal oscillatory dynamics given their regulation on axonal conduction velocities important for fast information transmission over far-reaching brain regions [105–107]. Here, we demonstrated that peak LFP power was visibly reduced in cuprizone-treated mice compared to control mice and that cuprizone administration induced large fluctuations in average power within the first two weeks post-insertion (Fig. 7). Changes in power were not reflected in a depth-dependent manner, which is expected given that neuronal oscillations occur between large neuronal networks in the brain. However, relative changes in power (evoked power in relation to spontaneous power) were observed to be altered within distinct frequency bands following cuprizone administration. At lower frequency ranges, such as the theta (2–4 Hz), delta (5–7 Hz), and alpha (8–15 Hz) frequency range, relative power was reduced in cuprizone-treated mice compared to controls. Oscillations in neuronal activity over larger distances are often governed by these lower frequency ranges given that small changes in neuronal conduction velocities can have profound impacts on network synchronization [105]. This would explain the reduced power observed within lower frequency bands, given demyelination of far-reaching neuronal projections following cuprizone administration. Alternatively, power at higher frequency bands, such as gamma (30–90 Hz) bands and beyond (high-frequency oscillations, >90 Hz), were increased in cuprizone-treated mice relative to controls. Satellite oligodendrocytes have been shown to be entrained to high-frequency activity of pyramidal neurons [36], which could influence the high-frequency oscillations within these local circuits depending on the cuprizone-induced depletion of satellite versus non-satellite oligodendrocyte cells. One study reported higher survivability in satellite oligodendrocytes above other oligodendrocytes following cuprizone administration [108]. Perineuronal oligodendrocytes also demonstrated active re-myelination following demyelinating injury and could explain the observed increase in power within higher frequency bands observed in cuprizone-treated mice. In all, these alterations in spectral power demonstrate that cuprizone-induced oligodendrocyte loss and demyelination alter the synchronicity of neuronal circuits within the cortex.
Brain connectivity can also be evaluated in terms of coherence, which is a statistical metric measuring how populations of neurons within different regions are activated in a similar fashion [109, 110]. Changes in resting-state coherence between brain regions have been observed to occur naturally throughout development and adulthood [111]. During development, increases in coherence between hemispheric brain regions are hypothesized due to increased myelination by enhancing neural transmission and reducing phase lag [112]. However, we report that coherence actually increases following cuprizone-induced oligodendrocyte depletion and demyelination (Fig. 8). We demonstrate that cuprizone-treated mice exhibit the greatest difference in resting-state and evoked G-SG coherence and G-IG coherence, with relatively little change between SG-IG regions. This disruption in layer IV communication is reflected by reductions in evoked single- and multi-unit activity of cuprizone-treated mice compared to controls. Furthermore, cuprizone administration altered communication within cortical layers, particularly superficial and deeper layers of the cortex. Despite the fact that myelin is traditionally viewed as insulating far-projecting axons, mainly the excitatory pyramidal neurons that connect both cortical hemispheres and other distal brain regions, recent advances in electron microscopy (EM) techniques have revealed that a significant portion of cortical interneurons, which are important for local circuit function, are also myelinated [113]. Specifically, EM revealed that parvalbumin-positive (PV+) interneurons possess about half of all myelinated axons in layer II/III and a quarter of myelinated axons in layer IV. Since inhibitory interneurons are important for regulating the excitability of different regions of the cortex, it is possible that a significant demyelinating insult to cortical interneurons following cuprizone administration can reduce the inhibitory influence of local circuits, altering the intricate balance in neuronal excitation/inhibition and, by extension, the functional coherence within and between different cortical layers. Indeed, vulnerability of inhibitory interneurons has been observed previously in experimental demyelinating models of MS [114, 115]. Furthermore, abnormal increases in cortical coherence are commonly observed in different brain disorders. For example, EEG studies track increases in coherence between different brain regions to predict areas of seizure onset in epilepsy patients [116, 117]. Fluctuations in coherence are also observed in ADHD patients in an age-dependent manner [118]. The sensitive balance in neuronal excitation and inhibition is necessary for different brain regions to function coherently. In all, we show that myelinating oligodendrocytes are important regulators of communication between different cortical circuits and that cuprizone-induced depletion of oligodendrocytes and myelin alters normal intra- and interlaminar coherence within the cortex.
5.5. Histological state of explanted cortical tissue following chronic cuprizone administration
Explant histology provided insight into cellular and subcellular tissue changes following 7 weeks of device implantation and 12 weeks of cuprizone administration that led to functional neural activity (Fig. 9–10). Labeling for neurons showed that 12 weeks of cuprizone administration did not have an effect on neuronal distribution around chronically implanted microelectrode arrays (Fig. 9ab), which is consistent with previously reported findings that cuprizone itself does not induce neuronal damage or cell death within young mice [59]. However, NF-200 fluorescence demonstrated increased immunoreactivity of axonal neurofilaments in cuprizone-treated mice compared to controls (Sup. Fig. 2). Indeed, histological labeling demonstrated altered neurofilament distribution around the implanted probe hole for cuprizone-treated mice, indicating axonal re-organization in response to combined influence of implantation injury and cuprizone administration. This result stands contrary to our hypothesis that a lack of trophic support from oligodendrocytes and myelin due to cuprizone administration would reduce the number of neurons and axons during device implantation injury, particularly given the limited capacity for the CNS to regenerate. One possible explanation is the lack of inhibitory chondroitin sulfate proteoglycans (CSPGs) secreted by astrocytes and NG2 glia in cuprizone-treated animals, which can prevent axons from extending through the lesion to re-establish functional neural circuits following injury [119–121]. Furthermore, the presence of NG2 glial cells themselves, which can form specialized synapses with neurons, are hypothesized to be axon inhibitory by acting as regulators of neurite outgrowth [122]. Of note is that the elevation of NG2 cells reverses between cuprizone-treated and control groups before device implantation (Fig. 1d, cuprizone 0–50 μm: 169.4 ± 31.2 cells/mm2, control 0–50 μm: 83.1 ± 29.2 cells/mm2) and 7 weeks after implantation (Fig. 10b; cuprizone 0–50 μm: 82.2 ± 18.3 cells/mm2, control 0–50 μm: 139.5 ± 11.2 cells/mm2). These observations, together with changes in MBP, NF200, and SMI31 may provide an avenue for future investigations. This hinderance to axonal regeneration coupled with an exhausted population of oligodendrocyte precursor cells (and therefore fewer deposited CSPGs) following chronic cuprizone administration may explain the increase in neurofilament staining observed around the site of injury. However, it is again important to note that the presence of antibody labeling does not necessarily indicate functionality [97].
By 7 weeks post-insertion and 12 weeks of cuprizone exposure, cuprizone-treated mice demonstrated reduced oligodendrocyte densities around chronically implanted microelectrode arrays compared to control mice. (Fig. 9cd) This level of oligodendrocyte loss is consistent with previous studies employing long-term administration of the cuprizone toxin, which can induce chronic depletion of oligodendrocytes in white and gray matter tissue. However, cuprizone-treated mice remarkably demonstrated an increase in oligodendrocyte density following 7 weeks of insertion and 12 weeks of cuprizone administration (Fig. 9d; cuprizone 0–50 μm: 168.6 ± 55.5 cells/mm2) compared to pre-insertion at 5 weeks of cuprizone treatment (Fig. 1b; cuprizone 0–50 μm: 69.5 ± 33.9 cells/mm2). Furthermore, while myelin staining demonstrated reduced fluorescence intensity in cuprizone-treated mice compared to control mice at the experimental endpoint, there were no significant differences in myelin fluorescence between the two groups, and myelin intensity of cuprizone-treated mice was in fact upregulated compared to pre-insertion baseline (Fig. 9ef). Furthermore, myelin expression was upregulated near the site of insertion in both cuprizone-treated and control animals compared to myelin intensity on contralateral hemispheres (Sup Fig. 4). Despite this increase in myelination near the electrode, deficits in recording performance persisted, suggesting a lack of functional connectivity with newly generated myelin or newly myelinated axons with the local neuronal circuit. This may be explained by an inability for the MBP+ antibody to differentiate intact versus degenerated myelin in cuprizone-treated (and control) animals, obfuscating the possibility of accumulated or unresolved myelin debris around the microelectrode array. The dual insult of cuprizone toxicity with injury from penetrating cortical insertion and implantation has not been observed before and may indicate that oligodendrocyte and myelin loss experienced during chronic cuprizone exposure is counteracted by tissue healing events that occur following CNS injury. Unlike previous cuprizone studies without a second insult of brain injury [58, 60], these results indicate a potential increased activation of oligodendrocyte regeneration and re-myelination pathways with simultaneous chronic cuprizone exposure and probe implantation injury. Additionally, an important aspect of device implantation injury is the upregulation of oxidative stress events, which can occur from blood-brain barrier disruption, impairment of waste clearance pathways, and activation of inflammatory glial cells [123]. Generation of reactive oxidative species and free radicals can impact tissue health by reducing neuron viability as well as the material properties of the electrode by inducing corrosion and delamination [124–126]. This is relevant due to the fact that cuprizone, a chelator of copper ions important for metabolic redox reactions, can exert its own oxidative stress on CNS tissue. However, as mentioned previously, cuprizone toxicity and copper chelation is a phenomenon specific to the oligodendrocyte population and would not be expected to influence the redox reactions occurring within neurons and other CNS cells. Oligodendrocytes are particularly susceptible to oxidative damage given their intrinsically low supply of antioxidant enzymes, which may contribute to their vulnerability during cuprizone exposure. Previous studies have applied pharmacological or biomaterial approaches to alleviate the build-up of oxidative damage that can occur around implanted devices, such as application of the anti-oxidant resveratrol or surface modification with superoxide dismutase, a free radical scavenger [125, 127–129], which can potentially protect oligodendrocytes and other CNS cells sensitive to harmful oxidative species. These intrinsic mechanisms that can lead to oligodendrocyte dysfunction induced during device implantation and inflammation could provide further insight into the inflammatory biological microenvironment and deteriorating electrophysiological signals detected by microelectrode arrays.
As expected, the number of NG2 glia around the site of device insertion was significantly reduced in cuprizone-treated mice compared to controls following chronic cuprizone toxicity (Fig. 10ab). Oligodendrocyte regeneration and re-myelination typically occurs when mice return to a normal diet after 5–6 weeks of cuprizone treatment due to local progenitor cells proliferating and differentiating to restore depleted cells [130]. During chronic cuprizone administration, however, oligodendrocytes are unable to repopulate and re-myelinate the affected tissue due to an exhausted precursor population that cannot support the continual renewal and immediate loss of oligodendrocyte cells. Interestingly, the number of NG2 glia in control mice was increased compared to baseline data, indicating a proliferative response to the insertion of a microelectrode device at 7 weeks post-insertion (Fig. 1,10). This data is in line with previously reported reactivity of the NG2 precursor population following acute electrode implantation as well as increased proliferation up to 4 weeks post-insertion of a microelectrode array [28, 57]. Both cuprizone-treated and control mice demonstrated similar fold changes in microglia fluorescence intensity compared to contralateral control tissue as well as a device-dependent increase in intensity with proximity to the site of insertion, suggesting no cuprizone-specific activation of microglia cells following injury (Fig. 10cd). Interestingly, astrocyte intensities were increased in cuprizone-treated animals compared to control mice 7 weeks post-insertion (Fig. 10ef). The dual incidence of decreased NG2 glia alongside increased astrocytic intensities could be indicative of previously reported phenomenon describing the conversion of oligodendrocyte precursors into reactive astrocytes following CNS injury [131, 132]. These results may suggest that damage and inflammation during device insertion coupled with oligodendrocyte precursor depletion/exhaustion from cuprizone administration preferentially directs differentiation of NG2 glia toward a more astrocytic cell fate. Alternatively, intercellular communication between oligodendrocytes and astrocytes is facilitated via membrane gap junctions linking the cytosol of both cells [133]. This connection is critical for the diffusion of potassium ions and other neurotransmitters released from depolarizing neurons to prevent axonal swelling and is suggested to support myelin integrity following demyelinating injury [134, 135]. A reduction in connexin proteins involved in oligodendrocyte-astrocyte gap junctions reportedly occurs alongside increased astrogliosis within gray matter lesions of MS patients [136]. Furthermore, cuprizone administration and subsequent recovery can alter the distribution of these same connexins within myelinating oligodendrocytes and astrocytes [137]. As a result, loss or dysfunction in intercellular communication between oligodendrocytes and astrocytes due to cuprizone toxicity may explain elevated astrocyte reactivity observed in cuprizone-treated animals.
5.6. Holistic summary of combined electrophysiology and histological outcomes
Summarizing the immunohistochemical results between cuprizone-treated and control mice at 7 weeks post-implantation with respect to their pre-implant histology is necessary to understand how it relates to the measured electrophysiology. Prior to implantation following 5 weeks of cuprizone administration, oligodendrocytes and myelin were significantly reduced (~85% and ~65%, respectively) within the cortex compared to control animals. Due to cuprizone toxicity, the number of oligodendrocyte precursor cells were elevated while astrocyte activation was slightly, yet not significantly, increased in cuprizone-treated mice. At the time of implantation, cuprizone-treated mice, experiencing an oligodendrocyte- and myelin- depleted state, demonstrated significantly reduced recording metrics compared to control animals, which persisted between the two experimental groups until ~1–2 weeks after implantation. Beyond the first two weeks, recording performance in control animals began to decline steadily over time while cuprizone-treated animals remained relatively stable until week 6 and 7 after implantation at which point cuprizone-treated animals experienced a slight increase in recording metrics such that both groups demonstrated similar levels of performance by experimental endpoint. Histological analysis around the microelectrode array at 7 weeks post-implantation determined that neuronal densities between cuprizone-treated and control mice were matched. However, cuprizone-treated animals demonstrated a slight increase in oligodendrocyte density and a relatively large increase in axonal and myelin intensity around the implant compared to baseline. Control animals on the other hand experienced a slight decrease in oligodendrocyte density compared to pre-insertion values. While microglia activation was matched between the two groups, cuprizone-treated mice experienced a large reduction in the number of oligodendrocyte precursor cells around the implant as well as greater and more variable astrocyte activation.
5.7. Limitations of study and future directions
There were a few limitations inherent to the experimental design of the study that should be considered. First is the constraint of chronic cuprizone treatment. Most studies traditionally observe the effects of cuprizone administration acutely at 5–6 weeks or chronically at 12 weeks [138]. Mice treated with cuprizone within 13–16 weeks reportedly experienced clonic-tonic seizures, systemic symptoms, and death, particularly male C57BL/6J mice [62, 65]. This restricted the experimental window of device insertion to 7 weeks of implantation following 5 weeks of cuprizone-induced oligodendrocyte depletion and demyelination. While 7 weeks was enough to observe electrophysiological metrics of control mice gradually decline down to the level of cuprizone-treated mice, recording performances could potentially continue to decline up to 12 weeks post-insertion before plateauing [21]. This suggests that further discrepancies in recording quality beyond 7 weeks of insertion may be observed but would necessitate an alternative method of oligodendrocyte and myelin depletion. One study, however, reported the use of cuprizone for up to 34 weeks in female C57BL/6J mice, which may be explained by sex specificity of the cuprizone model, and would be worth considering in the future [139]. Furthermore, cuprizone administration and neural recording performed in parallel with in vivo imaging would provide insight into the loss or regeneration of oligodendrocytes around the microelectrode implant over time, allowing for spatial and temporal correlations between tissue responses and device performance. The second limitation is the unknown contribution of non-myelinating satellite oligodendrocyte cells, which maintain close contact with neuronal soma instead of only extending cellular projections for myelin ensheathment, to neuronal health and function [108, 140]. It is known that these satellite cells are electrically-coupled within a glial syncytium and can modulate the excitability and oscillations of local circuits [36]. However, to what extent cuprizone exposure impacts the function of these perineuronal oligodendrocytes is unknown and was not specifically examined in this study. The third limitation considers the fact that cuprizone-induced oligodendrocyte cell death and demyelination induces activation of phagocytic glial cells within the brain, such as reactive microglia and astrocytes [60]. Still, more recent work has suggested that this glia activation (microglia and astrocytes) is in response to rapid oligodendrocyte loss and myelin debris accumulation as opposed to direct activation of glia by cuprizone [141].
While we did not observe increased activation of glial cells at 5 weeks post-cuprizone administration, GFAP+ fluorescence intensities were elevated in cuprizone-treated animals compared to controls at 7 weeks post-implantation of a microelectrode array, suggesting a long-term effect of cuprizone-induced astrogliosis. It is worth noting that neurons cannot store glucose as glycogen and do not generally engage glycogenolysis for energy production [142, 143]. However, over short periods, hypoxic neurons are able to rely on lactate from nearby astrocytes and oligodendrocytes (that are connected to astrocytes via gap junctions) to support neuronal oxidative metabolism [142, 143]. Therefore, one future direction may be elucidating if metabolic support mechanisms of nearby astrocytes play a crucial role in neuronal dysfunction and silencing around chronically implanted microelectrode arrays [97]. Since glial activation and scar formation can alter device impedances and attenuate recorded neural signals, an alternative method of oligodendrocyte and myelin knockout without a glial-activating component should be considered to delineate the effects of oligodendrocyte and myelin loss on neuronal electrophysiology and electrode performance in the absence of cuprizone-induced glial cell reactivity. In addition, further investigation of whether NG2 glia proliferation, oligodendrocyte differentiation and regeneration, or remyelination events have greater influences in maintaining or improving long-term neural interface performance would facilitate focused development of biomaterial design and intervention strategies.
6.0. Conclusion
Oligodendrocyte depletion and demyelination via cuprizone administration resulted in deficits in neuronal activity and recording performance around chronically implanted microelectrode arrays. Electrophysiological analysis revealed reduced single-unit and multi-unit recording metrics at the onset of device insertion in oligodendrocyte-depleted cortical tissue compared to control animals and remained reduced throughout the duration of device implantation. Furthermore, cuprizone changed global connectivity of neural circuits within the cortex, altering oscillatory activity and laminar communication. Recordings in control mice gradually declined to the level of performance seen in cuprizone-treated animals over time, suggesting oligodendrocyte loss as a critical regulator of neuronal physiology and a potential biological failure mode of neural electrode arrays. This study highlights the importance of preserving oligodendrocyte and myelin function to improve the electrophysiological properties of neurons around chronic intracortical microelectrode devices and reveals a potentially novel target for therapeutic intervention to enhance functional tissue recovery following CNS injury and increase the longevity and stability of neural interfacing technology.
Supplementary Material
9.0. Acknowledgements
The authors would like to thank Jazmyn Charles for assistance with bin application of histological images.
8.0. Funding
This work was supported by NIH NINDS R01NS094396 and a diversity supplement to this parent grant as well as NIH NINDS R21NS108098, NIH NINDS R01NS089688, NIH NINDS R01NS062019, and VA BLRD I21BX003237.
Footnotes
Declaration of interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
☐The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hatsopoulos NG and Donoghue JP, The science of neural interface systems. Annual review of neuroscience, 2009. 32: p. 249–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz AB, et al. , Brain-controlled interfaces: movement restoration with neural prosthetics. Neuron, 2006. 52(1): p. 205–220. [DOI] [PubMed] [Google Scholar]
- 3.Buzsáki G, et al. , Tools for probing local circuits: high-density silicon probes combined with optogenetics. Neuron, 2015. 86(1): p. 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hochberg LR, et al. , Neuronal ensemble control of prosthetic devices by a human with tetraplegia. Nature, 2006. 442(7099): p. 164–71. [DOI] [PubMed] [Google Scholar]
- 5.Collinger JL, et al. , High-performance neuroprosthetic control by an individual with tetraplegia. The Lancet, 2013. 381(9866): p. 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iordanova B, et al. , Optogenetic investigation of the variable neurovascular coupling along the interhemispheric circuits Journal of Cerebral Blood Flow & Metabolism, 2018. 38(4): p. 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michelson NJ, et al. , Calcium activation of cortical neurons by continuous electrical stimulation: Frequency-dependence, temporal fidelity and activation density. Journal of Neuroscience Research, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stocking KC, Vazquez AL, and Kozai TDY, Intracortical neural stimulation with untethered, ultrasmall carbon fiber electrodes mediated by the photoelectric effect. IEEE Transactions on Biomedical Engineering, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Eles JR and Kozai TDY, In vivo imaging of calcium and glutamate responses to intracortical microstimulation reveals distinct temporal responses of the neuropil and somatic compartments in layer II/III neurons. . Biomaterials, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alba NA, et al. , In vivo electrochemical analysis of a PEDOT/MWCNT neural electrode coating. Biosensors, 2015. 5: p. 618–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozai TD, et al. , Comprehensive chronic laminar single-unit, multi-unit, and local field potential recording performance with planar single shank electrode arrays. Journal of neuroscience methods, 2015. 242: p. 15–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrese JC, et al. , Failure mode analysis of silicon-based intracortical microelectrode arrays in non-human primates. Journal of neural engineering, 2013. 10(6): p. 066014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chestek CA, et al. , Long-term stability of neural prosthetic control signals from silicon cortical arrays in rhesus macaque motor cortex. Journal of neural engineering, 2011. 8(4): p. 045005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams JC, Rennaker RL, and Kipke DR, Long-term neural recording characteristics of wire microelectrode arrays implanted in cerebral cortex. Brain Research Protocols, 1999. 4(3): p. 303–313. [DOI] [PubMed] [Google Scholar]
- 15.Salatino JW, et al. , Glial responses to implanted electrodes in the brain. Nature BME, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rousche PJ and Normann RA, Chronic recording capability of the Utah Intracortical Electrode Array in cat sensory cortex. Journal of Neuroscience Methods, 1998. 82(1): p. 1–15. [DOI] [PubMed] [Google Scholar]
- 17.Cody PA, et al. , Unique electrophysiological and impedance signatures between encapsulation types: An analysis of biological Utah array failure and benefit of a biomimetic coating in a rat model. Biomaterials, 2018. 161: p. 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eles J, et al. , Meningeal inflammatory response and fibrous tissue remodeling around intracortical implants: An in vivo two-photon imaging study. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozai TDY, et al. , Two-photon imaging of chronically implanted neural electrodes: Sealing methods and new insights. Journal of Neuroscience Methods, 2016. 256: p. 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozai TDY, et al. , Reduction of neurovascular damage resulting from microelectrode insertion into the cerebral cortex using in vivo two-photon mapping. J Neural Eng, 2010. 7(4): p. 046011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozai TD, et al. , Effects of caspase-1 knockout on chronic neural recording quality and longevity: insight into cellular and molecular mechanisms of the reactive tissue response. Biomaterials, 2014. 35(36): p. 9620–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasad A, et al. Coupling biotic and abiotic metrics to create a testbed for predicting neural electrode performance. in 2011 Annual International Conference of the IEEE Engineering in Medicine and Biology Society 2011. [DOI] [PubMed] [Google Scholar]
- 23.Wellman SM, et al. , A Materials Roadmap to Functional Neural Interface Design. Advanced Functional Materials, 2018. 28(12): p. 201701269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michelson NJ, et al. , Multi-scale, multi-modal analysis uncovers complex relationship at the brain tissueimplant neural interface: new emphasis on the biological interface. Journal of neural engineering, 2018. 15(3): p. 033001–033001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wellman SM and Kozai TD, Understanding the inflammatory tissue reaction to brain implants to improve neurochemical sensing performance. 2017, ACS Publications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wellman SM, Cambi F, and Kozai TDY, The role of oligodendrocytes and their progenitors on neural interface technology: A novel perspective on tissue regeneration and repair. Biomaterials, 2018. 183: p. 200–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eles J, et al. , In vivo imaging of neuronal calcium during electrode implantation: Spatial and temporal mapping of damage and recovery. Biomaterials, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wellman SM, et al. , Revealing spatial and temporal patterns of cell death, glial proliferation, and blood-brain barrier dysfunction around implanted intracortical neural interfaces. 2019. 13: p. 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baumann N. and Pham-Dinh D, Biology of Oligodendrocyte and Myelin in the Mammalian Central Nervous System. Physiological Reviews, 2001. 81(2): p. 871–927. [DOI] [PubMed] [Google Scholar]
- 30.Bradl M. and Lassmann H, Oligodendrocytes: biology and pathology. Acta neuropathologica, 2010. 119(1): p. 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dougherty KD, Dreyfus CF, and Black IB, Brain-Derived Neurotrophic Factor in Astrocytes, Oligodendrocytes, and Microglia/Macrophages after Spinal Cord Injury. Neurobiology of Disease, 2000. 7(6): p. 574–585. [DOI] [PubMed] [Google Scholar]
- 32.Hartline DK, What is myelin? Neuron Glia Biology, 2009. 4(2): p. 153–163. [DOI] [PubMed] [Google Scholar]
- 33.Tomassy GS, et al. , Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science (New York, N.Y.), 2014. 344(6181): p. 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valério-Gomes B, et al. , The Absolute Number of Oligodendrocytes in the Adult Mouse Brain. Frontiers in Neuroanatomy, 2018. 12(90). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takasaki C, et al. , Cytochemical and cytological properties of perineuronal oligodendrocytes in the mouse cortex. European Journal of Neuroscience, 2010. 32(8): p. 1326–1336. [DOI] [PubMed] [Google Scholar]
- 36.Battefeld A, Klooster J, and Kole MHP, Myelinating satellite oligodendrocytes are integrated in a glial syncytium constraining neuronal high-frequency activity. Nature Communications, 2016. 7: p. 11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.LeVine SM and Torres MV, Satellite oligodendrocytes and myelin are displaced in the cortex of the reeler mouse. Developmental Brain Research, 1993. 75(2): p. 279–284. [DOI] [PubMed] [Google Scholar]
- 38.Lee Y, et al. , Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature, 2012. 487(7408): p. 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fünfschilling U, et al. , Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature, 2012. 485(7399): p. 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hughes EG, et al. , Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nature neuroscience, 2013. 16(6): p. 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tripathi RB, et al. , NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2010. 30(48): p. 16383–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levine JM and Reynolds R, Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Experimental neurology, 1999. 160(2): p. 333–347. [DOI] [PubMed] [Google Scholar]
- 43.Kuhlmann T, et al. , Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain, 2008. 131(7): p. 1749–1758. [DOI] [PubMed] [Google Scholar]
- 44.Ettle B, Schlachetzki JCM, and Winkler J, Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Molecular neurobiology, 2016. 53(5): p. 3046–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith KJ and McDonald WI, The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Philosophical transactions of the Royal Society of London. Series B, Biological sciences, 1999. 354(1390): p. 1649–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosko L, et al. , Oligodendrocyte Bioenergetics in Health and Disease. The Neuroscientist, 2018: p. 1073858418793077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roth AD and Núñez MT, Oligodendrocytes: functioning in a delicate balance between high metabolic requirements and oxidative damage, in Glial Cells in Health and Disease of the CNS. 2016, Springer; p. 167–181. [DOI] [PubMed] [Google Scholar]
- 48.Jana A, Hogan EL, and Pahan K, Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. Journal of the Neurological Sciences, 2009. 278(1): p. 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.JUURLINK BH, Response of glial cells to ischemia: roles of reactive oxygen species and glutathione. Neuroscience & Biobehavioral Reviews, 1997. 21(2): p. 151–166. [DOI] [PubMed] [Google Scholar]
- 50.Thorburne SK and Juurlink BH, Low glutathione and high iron govern the susceptibility of oligodendroglial precursors to oxidative stress. Journal of neurochemistry, 1996. 67(3): p. 1014–1022. [DOI] [PubMed] [Google Scholar]
- 51.Dewar D, Underhill SM, and Goldberg MP, Oligodendrocytes and Ischemic Brain Injury. Journal of Cerebral Blood Flow & Metabolism, 2003. 23(3): p. 263–274. [DOI] [PubMed] [Google Scholar]
- 52.Dent KA, et al. , Oligodendrocyte birth and death following traumatic brain injury in adult mice. PloS one, 2015. 10(3): p. e0121541-e0121541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flygt J, et al. , Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. European Journal of Neuroscience, 2013. 38(1): p. 2153–2165. [DOI] [PubMed] [Google Scholar]
- 54.Lotocki G, et al. , Oligodendrocyte vulnerability following traumatic brain injury in rats. Neuroscience Letters, 2011. 499(3): p. 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winslow BD and Tresco PA, Quantitative analysis of the tissue response to chronically implanted microwire electrodes in rat cortex. Biomaterials, 2010. 31(7): p. 1558–1567. [DOI] [PubMed] [Google Scholar]
- 56.Winslow BD, et al. , A comparison of the tissue response to chronically implanted Parylene-C-coated and uncoated planar silicon microelectrode arrays in rat cortex. Biomaterials, 2010. 31(35): p. 9163–9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wellman SM and Kozai TD, In vivo spatiotemporal dynamics of NG2 glia activity caused by neural electrode implantation. Biomaterials, 2018. 164: p. 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Skripuletz T, et al. , Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. The American journal of pathology, 2008. 172(4): p. 1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Praet J, et al. , Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neuroscience & Biobehavioral Reviews, 2014. 47: p. 485–505. [DOI] [PubMed] [Google Scholar]
- 60.Gudi V, et al. , Glial response during cuprizone-induced de-and remyelination in the CNS: lessons learned. Frontiers in cellular neuroscience, 2014. 8: p. 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sen MK, et al. , Behavioural phenotypes in the cuprizone model of central nervous system demyelination. Neuroscience & Biobehavioral Reviews, 2019. 107: p. 23–46. [DOI] [PubMed] [Google Scholar]
- 62.Matsushima GK and Morell P, The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain pathology, 2001. 11(1): p. 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gudi V, et al. , Regional differences between grey and white matter in cuprizone induced demyelination. Brain research, 2009. 1283: p. 127–138. [DOI] [PubMed] [Google Scholar]
- 64.Benetti F, et al. , Cuprizone neurotoxicity, copper deficiency and neurodegeneration. NeuroToxicology, 2010. 31(5): p. 509–517. [DOI] [PubMed] [Google Scholar]
- 65.Hoffmann K, et al. , Epileptic seizures and hippocampal damage after cuprizone-induced demyelination in C57BL/6 mice. Experimental Neurology, 2008. 210(2): p. 308–321. [DOI] [PubMed] [Google Scholar]
- 66.Nicolai E, et al. , Design choices for next-generation neurotechnology can impact motion artifact in electrophysiological and fast-scan cyclic voltammetry measurements. 2018. 9(10): p. 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kozai TDY, et al. , Chronic In Vivo Evaluation of PEDOT/CNT for Stable Neural Recordings. IEEE transactions on bio-medical engineering, 2016. 63: p. 111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kolarcik CL, et al. , Elastomeric and soft conducting microwires for implantable neural interfaces. Soft Matter, 2015. 11(24): p. 4847–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ludwig KA, et al. , Using a common average reference to improve cortical neuron recordings from microelectrode arrays. Journal of neurophysiology, 2009. 101(3): p. 1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Michelson NJ and Kozai TDY, Isoflurane and ketamine differentially influence spontaneous and evoked laminar electrophysiology in mouse V1. Journal of Neurophysiology, 2018. 120(5): p. 2232–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kozai TD, et al. , Chronic tissue response to carboxymethyl cellulose based dissolvable insertion needle for ultrasmall neural probes. Biomaterials, 2014. 35(34): p. 9255–9268. [DOI] [PubMed] [Google Scholar]
- 72.Du ZJ, et al. , Ultrasoft microwire neural electrodes improve chronic tissue integration. Acta Biomaterialia, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eles JR, et al. , Neuroadhesive L1 coating attenuates acute microglial attachment to neural electrodes as revealed by live two-photon microscopy. Biomaterials, 2017. 113: p. 279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xing B, et al. , Conditional depletion of GSK3b protects oligodendrocytes from apoptosis and lessens demyelination in the acute cuprizone model. Glia, 2018. 66(9): p. 1999–2012. [DOI] [PubMed] [Google Scholar]
- 75.Rowley CD, et al. , Assessing intracortical myelin in the living human brain using myelinated cortical thickness. Frontiers in Neuroscience, 2015. 9: p. 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Torkildsen Ø, et al. , The cuprizone model for demyelination. Acta Neurologica Scandinavica, 2008. 117(s188): p. 72–76. [DOI] [PubMed] [Google Scholar]
- 77.Bénardais K, et al. , Cuprizone [bis (cyclohexylidenehydrazide)] is selectively toxic for mature oligodendrocytes. Neurotoxicity research, 2013. 24(2): p. 244–250. [DOI] [PubMed] [Google Scholar]
- 78.Taraboletti A, et al. , Cuprizone Intoxication Induces Cell Intrinsic Alterations in Oligodendrocyte Metabolism Independent of Copper Chelation. Biochemistry, 2017. 56(10): p. 1518–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Werner SR, et al. , Proteomic analysis of demyelinated and remyelinating brain tissue following dietary cuprizone administration. Journal of molecular neuroscience, 2010. 42(2): p. 210–225. [DOI] [PubMed] [Google Scholar]
- 80.Faizi M, et al. , Toxicity of cuprizone a Cu2+ chelating agent on isolated mouse brain mitochondria: a justification for demyelination and subsequent behavioral dysfunction. Toxicology mechanisms and methods, 2016. 26(4): p. 276–283. [DOI] [PubMed] [Google Scholar]
- 81.Pasquini LA, et al. , The Neurotoxic Effect of Cuprizone on Oligodendrocytes Depends on the Presence of Proinflammatory Cytokines Secreted by Microglia. Neurochemical Research, 2007. 32(2): p. 279–292. [DOI] [PubMed] [Google Scholar]
- 82.Acs P, et al. , Distribution of oligodendrocyte loss and mitochondrial toxicity in the cuprizone-induced experimental demyelination model. Journal of Neuroimmunology, 2013. 262(1): p. 128–131. [DOI] [PubMed] [Google Scholar]
- 83.Krämer EM, Schardt A, and Nave KA, Membrane traffic in myelinating oligodendrocytes. Microscopy research and technique, 2001. 52(6): p. 656–671. [DOI] [PubMed] [Google Scholar]
- 84.Gybina AA and Prohaska JR, Copper deficiency results in AMP-activated protein kinase activation and acetylCoA carboxylase phosphorylation in rat cerebellum. Brain Research, 2008. 1204: p. 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Franco-Pons N, et al. , Discoidin Domain Receptor 1, a Tyrosine Kinase Receptor, is Upregulated in an Experimental Model of Remyelination and During Oligodendrocyte Differentiation In Vitro. Journal of Molecular Neuroscience, 2009. 38(1): p. 2–11. [DOI] [PubMed] [Google Scholar]
- 86.Morell P, et al. , Gene Expression in Brain during Cuprizone-Induced Demyelination and Remyelination. Molecular and Cellular Neuroscience, 1998. 12(4): p. 220–227. [DOI] [PubMed] [Google Scholar]
- 87.Peña K, Coblenz J, and Kiselyov K, Brief exposure to copper activates lysosomal exocytosis. Cell calcium, 2015. 57(4): p. 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kotter MR, et al. , Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. Journal of Neuroscience, 2006. 26(1): p. 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Napoli I. and Neumann H, Protective effects of microglia in multiple sclerosis. Experimental neurology, 2010. 225(1): p. 24–28. [DOI] [PubMed] [Google Scholar]
- 90.Nicolai EN, et al. , Design Choices for Next-Generation Neurotechnology Can Impact Motion Artifact in Electrophysiological and Fast-Scan Cyclic Voltammetry Measurements. Micromachines, 2018. 9(10): p. 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Donkin JJ and Vink R, Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol, 2010. 23(3): p. 293–9. [DOI] [PubMed] [Google Scholar]
- 92.Williams JC, et al. , Complex impedance spectroscopy for monitoring tissue responses to inserted neural implants. Journal of neural engineering, 2007. 4(4): p. 410. [DOI] [PubMed] [Google Scholar]
- 93.Kozai TDY, et al. , Mechanical failure modes of chronically implanted planar silicon-based neural probes for laminar recording. Biomaterials, 2015. 37: p. 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Streit WJ, et al. , Electrode Failure: Tissue, Electrical, and Material Responses. IEEE Pulse, 2012. 3(1): p. 30–33. [DOI] [PubMed] [Google Scholar]
- 95.Golabchi A, et al. , Melatonin improves quality and longevity of chronic neural recording. Biomaterials, 2018. 180: p. 225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kozai TDY, et al. , Comprehensive chronic laminar single-unit, multi-unit, and local field potential recording performance with planar single shank electrode arrays. Journal of Neuroscience Methods, 2015. 242: p. 15–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Michelson NJ, et al. , Multi-scale, multi-modal analysis uncovers complex relationship at the brain tissueimplant neural interface: New Emphasis on the Biological Interface. Journal of Neural Engineering, 2018. 15(033001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arvidsson A, et al. , Neuronal replacement from endogenous precursors in the adult brain after stroke. Nature Medicine, 2002. 8(9): p. 963–970. [DOI] [PubMed] [Google Scholar]
- 99.Berninger B, et al. , Functional Properties of Neurons Derived from <em>In Vitro</em> Reprogrammed Postnatal Astroglia. The Journal of Neuroscience, 2007. 27(32): p. 8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vickers JC, et al. , Dystrophic neurite formation associated with age-related β amyloid deposition in the neocortex: clues to the genesis of neurofibrillary pathology. Experimental neurology, 1996. 141(1): p. 1–11. [DOI] [PubMed] [Google Scholar]
- 101.Hamada MS and Kole MHP, Myelin loss and axonal ion channel adaptations associated with gray matter neuronal hyperexcitability. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2015. 35(18): p. 7272–7286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jeffries MA, et al. , ERK1/2 Activation in Preexisting Oligodendrocytes of Adult Mice Drives New Myelin Synthesis and Enhanced CNS Function. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2016. 36(35): p. 9186–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Philips T. and Rothstein JD, Oligodendroglia: metabolic supporters of neurons. The Journal of clinical investigation, 2017. 127(9): p. 3271–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alizadeh A, Dyck SM, and Karimi-Abdolrezaee S, Myelin damage and repair in pathologic CNS: challenges and prospects. Frontiers in Molecular Neuroscience, 2015. 8(35). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pajevic S, Basser PJ, and Fields RD, Role of myelin plasticity in oscillations and synchrony of neuronal activity. Neuroscience, 2014. 276: p. 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Filley CM and Fields RD, White matter and cognition: making the connection. Journal of neurophysiology, 2016. 116(5): p. 2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Almeida RG and Lyons DA, On Myelinated Axon Plasticity and Neuronal Circuit Formation and Function. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2017. 37(42): p. 10023–10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ludwin SK, The perineuronal satellite oligodendrocyte. Acta Neuropathologica, 1979. 47(1): p. 49–53. [DOI] [PubMed] [Google Scholar]
- 109.Bowyer SM, Coherence a measure of the brain networks: past and present. Neuropsychiatric Electrophysiology, 2016. 2(1): p. 1. [Google Scholar]
- 110.Maier A, et al. , Distinct superficial and deep laminar domains of activity in the visual cortex during rest and stimulation. Frontiers in systems neuroscience, 2010. 4: p. 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fleck JI, et al. , The Impact of Age and Cognitive Reserve on Resting-State Brain Connectivity. Frontiers in aging neuroscience, 2017. 9: p. 392–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barry RJ, et al. , Age and gender effects in EEG coherence: I. Developmental trends in normal children. Clinical Neurophysiology, 2004. 115(10): p. 2252–2258. [DOI] [PubMed] [Google Scholar]
- 113.Micheva KD, et al. , A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons. eLife, 2016. 5: p. e15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rossi S, et al. , Inflammation inhibits GABA transmission in multiple sclerosis. Multiple Sclerosis Journal, 2012. 18(11): p. 1633–1635. [DOI] [PubMed] [Google Scholar]
- 115.Falco A, et al. , Reduction in parvalbumin-positive interneurons and inhibitory input in the cortex of mice with experimental autoimmune encephalomyelitis. Experimental brain research, 2014. 232(7): p. 2439–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Song J, et al. , Methods for examining electrophysiological coherence in epileptic networks. Frontiers in neurology, 2013. 4: p. 55–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang G, et al. , Epileptic Seizure Detection Based on Partial Directed Coherence Analysis. IEEE Journal of Biomedical and Health Informatics, 2016. 20(3): p. 873–879. [DOI] [PubMed] [Google Scholar]
- 118.Clarke AR, et al. , EEG coherence in adults with Attention-Deficit/Hyperactivity Disorder. International Journal of Psychophysiology, 2008. 67(1): p. 35–40. [DOI] [PubMed] [Google Scholar]
- 119.Zhong Y. and Bellamkonda RV, Dexamethasone-coated neural probes elicit attenuated inflammatory response and neuronal loss compared to uncoated neural probes. Brain research, 2007. 1148: p. 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Friedlander DR, et al. , The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. The Journal of cell biology, 1994. 125(3): p. 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dou CL and Levine JM, Inhibition of neurite growth by the NG2 chondroitin sulfate proteoglycan. The Journal of neuroscience : the official journal of the Society for Neuroscience, 1994. 14(12): p. 7616–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Filous AR, et al. , Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. The Journal of neuroscience : the official journal of the Society for Neuroscience, 2014. 34(49): p. 16369–16384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kozai TD, et al. , Brain tissue responses to neural implants impact signal sensitivity and intervention strategies. ACS chemical neuroscience, 2015. 6(1): p. 48–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Prasad A, et al. , Comprehensive characterization and failure modes of tungsten microwire arrays in chronic neural implants. Journal of neural engineering, 2012. 9(5): p. 056015. [DOI] [PubMed] [Google Scholar]
- 125.Potter KA, et al. , The effect of resveratrol on neurodegeneration and blood brain barrier stability surrounding intracortical microelectrodes. Biomaterials, 2013. 34(29): p. 7001–7015. [DOI] [PubMed] [Google Scholar]
- 126.Takmakov P, et al. , Rapid evaluation of the durability of cortical neural implants using accelerated aging with reactive oxygen species. Journal of neural engineering, 2015. 12(2): p. 026003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nguyen JK, et al. , Influence of resveratrol release on the tissue response to mechanically adaptive cortical implants. Acta biomaterialia, 2016. 29: p. 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Potter-Baker KA, et al. , Development of superoxide dismutase mimetic surfaces to reduce accumulation of reactive oxygen species for neural interfacing applications. Journal of Materials Chemistry B, 2014. 2(16): p. 2248–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zheng XS, et al. , A superoxide scavenging coating for improving tissue response to neural implants. Acta Biomaterialia, 2019. 99: p. 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baxi EG, et al. , Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone-induced demyelination. Glia, 2017. 65(12): p. 2087–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hackett AR, et al. , STAT3 and SOCS3 regulate NG2 cell proliferation and differentiation after contusive spinal cord injury. Neurobiology of disease, 2016. 89: p. 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dimou L. and Gallo V, NG 2-glia and their functions in the central nervous system. Glia, 2015. 63(8): p. 14291451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nualart-Marti A, Solsona C, and Fields RD, Gap junction communication in myelinating glia. Biochimica et Biophysica Acta (BBA) - Biomembranes, 2013. 1828(1): p. 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rash JE, Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience, 2010. 168(4): p. 982–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kıray H, et al. , The multifaceted role of astrocytes in regulating myelination. Experimental neurology, 2016. 283(Pt B): p. 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Markoullis K, et al. , Oligodendrocyte Gap Junction Loss and Disconnection From Reactive Astrocytes in Multiple Sclerosis Gray Matter. Journal of Neuropathology & Experimental Neurology, 2014. 73(9): p. 865–879. [DOI] [PubMed] [Google Scholar]
- 137.Parenti R, et al. , Dynamic expression of Cx47 in mouse brain development and in the cuprizone model of myelin plasticity. Glia, 2010. 58(13): p. 1594–1609. [DOI] [PubMed] [Google Scholar]
- 138.Kipp M, et al. , The cuprizone animal model: new insights into an old story. Acta neuropathologica, 2009. 118(6): p. 723–736. [DOI] [PubMed] [Google Scholar]
- 139.Nomura T, et al. , Pathological changes in mice with long term cuprizone administration. Neurochemistry International, 2019. 126: p. 229–238. [DOI] [PubMed] [Google Scholar]
- 140.Taniike M, et al. , Perineuronal Oligodendrocytes Protect against Neuronal Apoptosis through the Production of Lipocalin-Type Prostaglandin D Synthase in a Genetic Demyelinating Model. The Journal of Neuroscience, 2002. 22(12): p. 4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xing B, et al. , Conditional depletion of GSK3b protects oligodendrocytes from apoptosis and lessens demyelination GLIA, 2018. [DOI] [PubMed] [Google Scholar]
- 142.Kasischke KA, et al. , Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science, 2004. 305(5680): p. 99–103. [DOI] [PubMed] [Google Scholar]
- 143.Howarth C, Gleeson P, and Attwell D, Updated energy budgets for neural computation in the neocortex and cerebellum. Journal of Cerebral Blood Flow & Metabolism, 2012. 32(7): p. 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.