

Abstract
Methylation levels have been shown to change with age at sites across the human genome. Change at some of these sites is so consistent across individuals that it can be used as an ‘epigenetic clock’ to predict an individual's chronological age to within a few years. Here, we examined how the pattern of epigenetic ageing in chimpanzees compares with humans. We profiled genome-wide blood methylation levels by microarray for 113 samples from 83 chimpanzees aged 1–58 years (26 chimpanzees were sampled at multiple ages during their lifespan). Many sites (greater than 65 000) showed significant change in methylation with age and around one-third (32%) of these overlap with sites showing significant age-related change in humans. At over 80% of sites showing age-related change in both species, chimpanzees displayed a significantly faster rate of age-related change in methylation than humans. We also built a chimpanzee-specific epigenetic clock that predicted age in our test dataset with a median absolute deviation from known age of only 2.4 years. However, our chimpanzee clock showed little overlap with previously constructed human clocks. Methylation at CpGs comprising our chimpanzee clock showed moderate heritability. Although the use of a human microarray for profiling chimpanzees biases our results towards regions with shared genomic sequence between the species, nevertheless, our results indicate that there is considerable conservation in epigenetic ageing between chimpanzees and humans, but also substantial divergence in both rate and genomic distribution of ageing-associated sites.
This article is part of the theme issue ‘Evolution of the primate ageing process'.
Keywords: senescence, ageing, life history, Pan troglodytes, methylation, longevity
1. Introduction
Widespread epigenetic changes with age have been characterized in humans, potentially implicating epigenetic mechanisms in senescence [1]. In particular, levels of CpG methylation, an epigenetic modification involving the addition of a methyl group to the DNA base cytosine (C) when it occurs next to the base guanine (G), show pervasive age-related change [1,2]. At a subset of these CpG sites, change is so predictable across individuals that their methylation levels can be used to estimate chronological age using ‘epigenetic clock’ models (for a brief introduction, see [3]). The degree to which these changes contribute to, or are the result of, other aspects of ageing remains unclear [4–6], but the rate of change at age-predictive sites varies slightly among individuals of the same chronological age [4,7,8] and these deviations reflect individual acceleration or deceleration in biological ageing [4,6,9]. Several recent studies in multiple large cohorts have found that accelerated methylation age is associated with phenotypic correlates of ageing, including increased frailty [10], decreased grip strength and lung function [11], diminished cognitive performance [11] and all-cause mortality risk [12–15].
Studies of age-related epigenetic change in other mammals, including mice, whales and canids, show that some, but not all, of the same loci as in humans undergo age-associated methylation changes [16–18]. Additional comparative work suggests that when only CpGs that show age-related change across mammals are studied, the rate of methylation change is negatively correlated with species' maximum lifespan [19].
An in-depth comparison of chimpanzees with humans is of interest because the two species are genetically similar but differ in lifespan. The maximum recorded human lifespan is 122 [20], whereas the oldest known chimpanzees have lived into their 60s in the wild and in captivity at least one individual is estimated to have lived into her 70s [21–25]. Although human life expectancy has increased dramatically over the last 200 years in many industrialized societies, it is estimated to have been twice as high in preindustrial times as that observed in living non-human apes [26,27]. These differences suggest that a long human lifespan emerged sometime over the past approximately 7–8 Myr since our lineage split with Pan [28].
Genetic regulatory mechanisms have long been thought to underlie phenotypic divergence between humans and chimpanzees [29]. Previous comparisons of genome-wide methylation patterns in the two species have identified numerous differentially methylated regions, including at genes that show perfect sequence identity between the two species [30]. It is thus conceivable that epigenetic differences in gene regulation might play a role in the lifespan differences of these two lineages.
In previous research, a commonly used epigenetic clock was applied to a small sample of adult chimpanzees and found to predict chronological age accurately [4], suggesting overall conservation of epigenetic ageing between the two species. In addition, Ito et al. [31] assayed methylation in chimpanzees at 14 CpG sites showing age-related change in humans and found a strong correlation with age at five of these sites, suggesting some degree of species divergence, and allowing them to predict chimpanzee age with a mean absolute deviation of 5.41 years.
In this study, we investigated patterns of epigenetic change with age in a large sample of chimpanzees using genome-wide methylation profiling. We applied two commonly used human epigenetic age estimators (see Material and methods [4,7]) to the chimpanzee data and also built a chimpanzee-specific epigenetic clock. We compared age-related differences in genome-wide methylation between our chimpanzee dataset and a human dataset compiled from samples generated from previous studies [2,7,32].
2. Material and methods
(a). Methylation dataset
Genomic DNA was isolated from whole blood from chimpanzees at Yerkes National Primate Research Center at Emory University and Keeling Center for Comparative Medicine and Research at the University of Texas MD Anderson Cancer Center. Blood was collected at annual physical exams following IACUC protocols approved at each home institution and used as part of other, ongoing research projects [33,34]. The 113 samples included those from 83 individuals spanning ages 1–58.9 years (electronic supplementary material, figure S1). Twenty-six individuals were sampled at two or three ages across the lifespan. Seven individuals were wild-born, whereas all other individuals were captive-born. For the wild-born and two captive-born individuals, only birth year, not exact birth date, was known, in which cases 1 January was used. DNA was extracted as described in Ely et al. [35] or using the QIAGEN DNeasy Blood and Tissue kit following the manufacturer's instructions for automation on the QIACUBE. DNA samples were brought to a concentration of approximately 70 ng µl−1, bisulfite converted and run on the Illumina Infinium Methylation EPIC array (‘EPIC array’ hereafter), which assays methylation levels at greater than 850 000 CpG sites across the genome. To avoid batch effects, we took care to combine a mix of ages and sexes on each array. We restricted the dataset to CpG sites falling on probes previously identified [36] as mapping to the chimpanzee genome with one or zero mismatches.
For the genome-wide comparative analysis, we downloaded human blood methylation data generated on the Illumina Infinium Methylation 450 K array (‘450 K array’ hereafter) for 274 individuals aged 10–84 years [2,7,32] from the Gene Expression Omnibus online database (accessions: GSE40279, GSE87571, GSE56105). Only probes on both the EPIC and 450 K arrays were retained in this analysis. As the human dataset only included adolescents and adults, we removed juveniles [37] from the chimpanzee dataset, resulting in a sample size of 87 (73 individuals). Wild-born individuals were not included in this analysis.
For all analyses, we pre-processed data using the ChAMP v. 2.12.4 R package [38]. We first filtered the raw intensity data to remove probes (i) with spectral intensities not above background levels, (ii) that do not target CpG dinucleotides, (iii) that contain known SNPs, (iv) that are on the sex chromosomes, and (v) for which fewer than three beads were counted for 5% or more of the samples (electronic supplementary material, figure S1). We then normalized the data using Beta Mixture Quantile dilation (BMIQ), which accounts for the two different probe types on the 450 K and EPIC arrays [39]. Singular value decomposition analysis [40] was performed to identify significant batch effects and we used ComBat (Combining Batches) normalization [41] to counteract variation due to array in the chimpanzee data and due to dataset in the human data.
(b). Identification of differentially methylated sites with age
We used linear models to identify age-associated differentially methylated probes (aDMPs; electronic supplementary material, figure S1). For the chimpanzee data, we used a mixed-effects model implemented with the lmekin function of the R package coxme [42] using a relatedness matrix estimated from a known pedigree and individual ID to account for repeat sampling of the same individuals at different points in their lives as random effects and parallelized using GNU parallel [43]. For the human data, we used the dmpFinder function, which is an extension of the lmFit linear regression function in the limma R package [44], in the ChAMP package [38]. We corrected for the number of tests by estimating q-values at a 5% false discovery rate [45]. We filtered out CpG sites for which the mean methylation was greater than 0.9 or less than 0.1 or for which variation in methylation among samples was less than 0.2, following previous studies [46,47].
For aDMPs shared by both species, we compared rates of change with age in the two species through analysis of covariance (ANCOVA) in R [48]. For this analysis, we only included unrelated chimpanzee individuals to avoid confounds related to heritability of methylation. Because methylation change with age often decreases with advancing age [49], we sought to ensure that differences in rate between species are not solely due to an attenuating rate of change over the longer lifespan in humans. To this end, we performed an additional ANCOVA including only data for human individuals under 59, matching the oldest chronological age in our chimpanzee dataset. We finally determined which genes showed enrichment for either shared, chimpanzee-specific or human-specific aDMPs. Association of CpGs with genes was based on the IlluminaHumanMethylation450kmanifest Bioconductor package v. 0.4.0 [50] in ChAMP [38].
(c). Age prediction analysis
We assessed the accuracy of chimpanzee age prediction using two human epigenetic age estimators: (i) the ‘Hannum Clock’ [7], a clock trained on human blood methylation data generated using the 450 K array; and (ii) the ‘Horvath Clock’ [4], a clock trained on methylation data from many different tissues generated using the 450 K array and the Illumina Infinium Methylation 27 K array. Next, to build a chimpanzee-specific age predictor clock model, we split the dataset into sex- and age-balanced training (N = 58) and test (N = 55) data (electronic supplementary material, figure S1), with all individuals with uncertain birth dates, including wild-born individuals, and repeat samples for the same individual assigned to the test dataset. We restricted our data to probes on the 450 K array to make the results more directly comparable to the Hannum Clock for humans. We used elastic net regression—a form of penalized linear regression appropriate for large datasets with many more predictors than samples—to build the age prediction model [51]. We set the tuning parameter, α, to 0.5, which is considered a reasonable intermediate between regularization of ridge regression and reduced predictor selection in LASSO regression for building interpretable models with many intercorrelated predictors while avoiding overparametrization [51]. We set the shrinkage parameter, λ, to 0.617, the value that was found to minimize the error in the model using 10-fold cross-validation with the function cv.fit in glmnet. We performed bootstrapping of the model building procedure, sampling the training dataset with replacement 100 times, to assess repeatability in CpGs selected.
We estimated the heritability of methylation at CpG sites comprising our chimpanzee clock model, as well as an equal number of non-clock CpGs. For each CpG, we estimated the genetic component of variance in methylation by fitting an animal model using the known population pedigree through Bayesian inference via a Markov chain Monte Carlo (MCMC) approach in the R package MCMCglmm [52]. Models were run for 1 000 000 iterations with a burn-in period of 100 000 iterations and a thinning interval of 500 iterations. Age and sex were included as fixed effects and individual ID as a random effect. We assessed MCMC convergence and mixing visually by plotting the traces and densities of sampled values across iterations and testing for Heidelberg stationarity for all fixed and random effects. For models that did not show MCMC convergence, we re-ran the MCMC algorithm for at least 10 million iterations with a burn-in period of at least 500 000 iterations. We did not further consider CpGs for which models failed to converge by 100 million iterations. Because the inclusion of fixed effects (in this case, age and sex) in animal models can lead to inflated estimates of h2 due to a decrease in residual variance when these effects are included [53], we followed de Villemereuil et al. [54] and included variance attributable to fixed effects in the total phenotypic variance.
Finally, we investigated the possibility of accurately predicting chimpanzee age from a much smaller number of CpG sites, which might be useful in the development of a more widely applicable, simplified assay. To this end, we fitted a linear model for the top 10 CpGs in our full predictive model showing the strongest correlation with age using the same training dataset. We then fitted linear models of (i) just the CpGs found to be significant predictors of age in our 10-CpG model and (ii) the single CpG showing the strongest correlation with age. We calculated the median absolute deviation (MAD) between known age and age predicted by these simplified models on our test dataset.
3. Results
We identified 65 829 significant age-associated differentially methylated probes (aDMPs) in chimpanzees and 53 224 aDMPs in humans at a 5% false discovery rate. Of these, 20 792 CpGs showed age-associated change in both species (figure 1a). We found that 87% (18 142) of these shared aDMPs showed a significant difference in rate of change with age between species, with 98% (17 792) of these showing faster change in chimpanzees (figure 1b). When only humans within the same chronological age range as chimpanzees (under the age of 59) were included in the comparison of rate, the results were broadly similar (electronic supplementary material, figure S2). Twenty-one genes were significantly enriched for aDMPs shared by chimpanzees and humans (electronic supplementary material, table S1), five genes were significantly enriched for chimpanzee-specific aDMPs (electronic supplementary material, table S2) and a single gene was enriched for human-specific aDMPs (electronic supplementary material, table S3). One gene, PCDHGA4 (protocadherin gamma subfamily A, 4) was enriched for both shared and chimpanzee-specific aDMPs.
Figure 1.
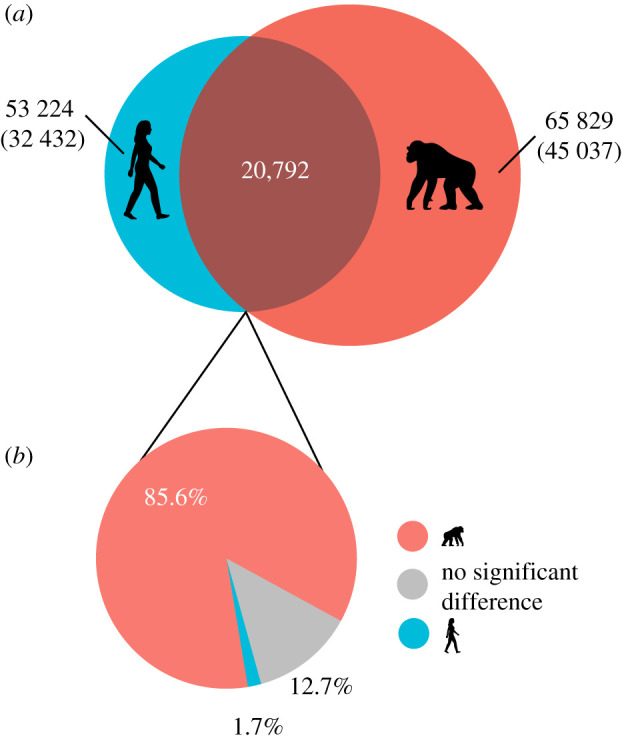
Results of the global comparative epigenetic ageing analysis. (a) Overlap between humans and chimpanzees in aDMPs at a 5% false discovery rate. (b) Relative rate of change in methylation with age of aDMPs shared between humans and chimpanzees. The numbers in parentheses are the numbers of aDMPs unique to each species. (Online version in colour.)
The Hannum Clock predicted the ages of the chimpanzees in our sample with a MAD of 5.2 years (figure 2a). The ages of nearly 80% of individuals (89) were overestimated. The Horvath Clock predicted chimpanzee age with a MAD of only 2.8 years (figure 2b).
Figure 2.

Results of the epigenetic clock analysis. Correlations are Pearson's product-moment correlations, p-values and slopes of predicted age regressed on known age. (a) Predictive accuracy of the Hannum Clock, (b) predictive accuracy of the Horvath Clock, (c) predictive accuracy of the new chimpanzee clock, (d) individual epigenetic ageing trajectories for chimpanzees with repeat samples using the chimpanzee clock. Lines are coloured by individual ID and connect dots from the same individual. (Online version in colour.)
The chimpanzee clock that we built from our training dataset comprised 80 CpG sites, 61% of which showed an increase in methylation over the lifespan and 39% of which showed a decrease (electronic supplementary material, table S4). They were distributed across coding, regulatory and intergenic regions; none of these types of genomic regions was enriched among clock CpGs (electronic supplementary material, figure S3). The chimpanzee clock model predicted age in our test dataset with a MAD of 2.4 years (figure 2c; individual ageing trajectories are shown in figure 2d). About half (55%) of the age estimates were overestimates and about half (45%) were underestimates. Six of the CpGs in the chimpanzee clock were also in the Hannum Clock, which reflects significant enrichment (Fisher's exact test: p-value < 0.001), while one additional CpG (cg01459453) in the chimpanzee clock was part of the Horvath Clock, which is not more than would be expected by chance (Fisher's exact test, p = 0.09). There was no significant difference between males and females in age acceleration using either our chimpanzee clock (F1,53 = 0.003, p = 0.957; electronic supplementary material, figure S4A) or the Horvath Clock (F1,111 = 0.486, p = 0.487; electronic supplementary material, figure S4B). Males showed a significantly greater epigenetic age acceleration using the Hannum Clock (F1,111 = 18.58, p < 0.001; electronic supplementary material, figure S4C).
A total of 1134 CpGs comprised the 100 bootstrap clock models of chimpanzee epigenetic ageing, which had a mean size of 67 CpGs. Only one CpG (cg16867657) was present in all 100 models and only 23 CpGs were present in over half of the models. Fifteen of the CpGs selected across all bootstrap models were also part of the human Hannum Clock and 13 were part of the Horvath Clock. The chimpanzee clock did not accurately predict human age, with a MAD of greater than 20 years (electronic supplementary material, figure S5). Our simplified predictive model consisted of three CpG sites and had a MAD of 2.6 years (electronic supplementary material, figure S6A; table S5). The model fitted with only a single CpG had a MAD of 3.1 years (electronic supplementary material, figure S6B; table S5).
The heritability of methylation at CpG sites in the chimpanzee clock ranged from 0.02 to 0.73, with an average heritability of 0.20 (electronic supplementary material, figure S7A). The non-clock CpGs showed an average heritability of 0.30 (range = 0.13–0.89), which is significantly higher than that of clock CpGs (Kruskal–Wallis, X2 = 50.771, d.f. = 1, p < 0.001; electronic supplementary material, figure S7B). Models for five CpGs failed to converge by 100 million iterations.
4. Discussion
We observed pervasive and significant change in methylation with age across the chimpanzee genome, as has been observed in humans [2,7]. In fact, we identified more aDMPs in chimpanzees than in humans, despite a larger sample size in humans, which may reflect relative homogeneity in our chimpanzee sample and/or greater variability in human ageing with respect to methylation patterns. The chimpanzees in this study shared a similar, fairly controlled environment. By contrast, humans are characterized by considerable variation in lifestyle and environment. Our compiled human dataset consisted of data derived from US Caucasian and Hispanic populations [7], individuals of European descent from the Brisbane Systems Genetics Study [32] and participants in the Northern Swedish Population Health Study, who live in rural communities and have either traditional or more typical modern European lifestyles [2]. Characterization of other populations of chimpanzees, in particular wild chimpanzee populations, would significantly aid interpretation of the differences between species that we found and the degree to which they are due to greater variability in human ageing. The contribution of methodological differences (i.e. the use of the 450 K v. EPIC array) may also be a factor in the differences we observed.
Many of the genes enriched for CpGs found to be aDMPs in both chimpanzees and humans are involved in development (e.g. HOXC4/homeobox C4, IRX2/Iroquois homeobox 2, LHX8/LIM homeobox 8, MSX1/Msh homeobox 1, PITX2/paired like homeodomain 2, TFAP2A/Transcription factor AP-2-alpha) and, in particular, neurodevelopment (e.g. DCC/netrin 1 receptor, FOXG1/Forkhead box G1, PAX3/Paired box 3, PCDHGA4/Protocadherin gamma subfamily A 4, SIM1/Single-minded homolog 1, ZIC1/Zinc finger protein ZIC 1, and ZIC4/Zinc finger protein ZIC 4), which is consistent with the results of previous studies in humans [2,4]. This finding further lends support to the idea that some portion of patterns of epigenetic ageing reflect the unfurling of developmental processes and potential eventual loss of developmentally established regulatory conditions [55,56]. Given that in chimpanzees, as well as humans, neurodevelopment is markedly protracted [57], it makes sense that signatures of neurodevelopmental processes specifically would be found among age-related epigenetic changes. The dearth of genes showing species-specific enrichment of aDMPs suggests that although chimpanzees and humans may differ in their pattern of ageing at many individual CpG sites, these may not translate to gene-level and/or regulatory differences.
Our finding of a faster rate of age-related methylation change in chimpanzees is consistent with the results of previous studies comparing the rate of epigenetic change among species at conserved sites [19] and a scenario in which faster epigenetic ageing in chimpanzees could reflect faster biological ageing, which in turn may contribute to shorter lifespan. This possibility also agrees with our finding that the human Hannum Clock tended to overestimate chimpanzee epigenetic age, indicating that chimpanzees show accelerated epigenetic ageing at these human age-predictive sites. By contrast, the human Horvath Clock did not tend to overestimate chimpanzee age and, in fact, predicted chimpanzee age about as well as it predicts human age [4]. The Horvath Clock was developed from methylation levels measured in many different tissue types and, therefore, likely reflects an overall more conserved component of epigenetic ageing; this conservation may thus extend to chimpanzees as well. Our results further indicate that the Horvath Clock can be used to predict chimpanzee age across the lifespan. It is notable that we did not see acceleration of epigenetic ageing in males relative to females for either our chimpanzee clock or the Horvath Clock, as has been found in humans [58] and would be predicted, given apparent sex differences in lifespan ([59], though see [60]). This finding may partially reflect our relatively small sample size and the paucity of males of older ages in our sample. However, the fact that male age acceleration is observed using the Hannum Clock suggests that although this clock predicts chronological age less accurately, it might be better at capturing biological age.
The size of our chimpanzee clock model and its relative composition of CpG sites that increase or decrease with age is similar to the Hannum Clock. The finding that our bootstrap models collectively represented over 1000 CpG sites indicates that many sites in the genome can be used in combination to predict chimpanzee age. Only 15 of these, however, overlapped with the Hannum Clock. Nevertheless, the three CpG sites retained in our simplified clock model are all among the sites in the Hannum Clock—cg16867657 (associated with ELOVL2), cg25478614 (associated with SST) and cg16054275 (associated with F5)—indicating conservation in the most predictive sites between the two species. The single-CpG model was built using cg16867657, which was also the only CpG selected across model building bootstrap replicates and was also the aDMP most significantly associated with age. Methylation at this and other sites associated with the gene ELOVL2 has been correlated with age in many studies in humans [61–68], as well as in the previous study of epigenetic age estimation in chimpanzees [31], and this specific CpG has been included in multiple simplified human epigenetic age estimators [62,63], though the biological significance of age-related changes in ELOVL2 methylation remains uncertain. SST encodes somatostatin, a peptide hormone with important endocrine function that has been found to show decreases in expression with age in the brain in humans [69] and non-human primates (Japanese macaques [70]) and has been implicated in Alzheimer's disease [71]. The significance of age-related change in blood, however, is unclear. F5 encodes coagulation factor V, a protein involved in blood coagulation [72].
The comparable performance of our simplified chimpanzee epigenetic clock to our full clock model is encouraging of the possibility of developing a lower cost, simplified assay to accurately estimate age. This prospect holds promise for studies of wild populations, though it will be necessary to test this model in additional chimpanzee individuals and sample types.
Relatedly, it is interesting that six out of the seven wild-born individuals' ages were underestimated by our full chimpanzee clock model. The underestimation was in all cases greater than 1 year (mean deviation: 4.8 years), suggesting that this observation is not simply the result of overestimating chronological age slightly by using a birthday of 1 January. This likely reflects the fact that wild-born individuals were all in the oldest age class (greater than 40) and we observe a moderate increase in underestimation of age at the oldest ages (Pearson's correlation between age acceleration and chronological age = −0.56, p < 0.001).
Our mean heritability estimate of h2 = 0.20 is within the range of estimates of the mean heritability of methylation across the genome in humans (15–34% [73–76]). That it is significantly lower than the mean of non-clock CpGs examined (0.30) makes sense, given the expected high proportion of variance accounted for by age for clock CpGs.
One limitation of this study is that by measuring methylation using a microarray designed for humans, we are biasing our study towards conserved genomic regions. We chose to use a microarray in order to cost-effectively generate high coverage data that are consistent across samples and can be compared relatively straightforwardly to human methylation ageing datasets, which have been generated by methylation microarray. However, previous studies have found that differential methylation among species tends to be greater in regions that also show greater sequence divergence [77]. We thus may be missing significant regions of age-associated divergence.
In conclusion, we found that humans and chimpanzees show considerable overlap but also substantial differences in patterns of epigenetic ageing. The component of shared epigenetic ageing between species is largely characterized by a faster rate of epigenetic ageing in chimpanzees that may reflect accelerated biological ageing relative to humans. There are also many CpG sites that show epigenetic ageing unique to each species, which may hold promise for future study. Finally, we were able to predict chimpanzee chronological age with high accuracy from methylation levels, including from just a few sites. Although our models require validation in independent chimpanzee samples/populations, this bodes well for efforts to implement simplified age estimation assays that might be of broad utility.
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
{kind=link}
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank colleagues from the Yerkes National Primate Research Center and the Michale E. Keeling Center for Comparative Medicine and Research, Gary Aronson, Cheryl Stimpson, D. Rick Lee, D.V.M. and Evelyn Ng for providing laboratory assistance; the Yale Center for Research Computing for use of the research computing infrastructure (supported by NIH grants no.s RR19895 and RR029676-01) and support; Yuan Tian for assistance using the R package ChAMP; Steve Horvath, Matteo Pelligrini, Jenny Tung, Noah Snyder-Mackler, Amanda Lea and Tauras Vilgalys for help with study development and discussion; David Watts, Alison Richard, Eric Sargis, two anonymous reviewers and the members of the GW Primate Genomics Laboratory and Laboratory for Evolutionary Neuroscience for helpful discussion and/or comments on previous drafts. We are grateful to Melissa Emery-Thompson for inviting us to submit this manuscript as part of this Special Issue.
Ethics
All aspects of this research adhered to the American Psychological Associations guidelines for the ethical treatment of animals in research. All blood samples were collected prior to changes in US Federal guidelines concerning endangered species in 2015.
Data accessibility
Raw data are available on the National Center forBiotechnology Information's Gene Expression Omnibus (Accession GSE136296). Code is available on the National Chimpanzee Brain Resource website Data Repository page (https://www.chimpanzeebrain.org/data-repository).
Authors' contributions
E.E.G., R.R.L., B.J.B. and C.C.S. conceived of the study. W.D.H., J.J.E. and N.S. contributed to experimental design and data acquisition. N.S., E.E.G. and C.M.W. conducted the laboratory work. E.E.G. conducted the data analysis and drafted the manuscript with input from all coauthors.
Competing interests
We declare we have no competing interests.
Funding
Funding for this work was provided by the Leakey Foundation, National Science Foundation (grant no. BCS-1733896), Yale MacMillan Center for International Studies, Yale Institute for Biospheric Studies, The George Washington University and Yale University.
References
- 1.Pal S, Tyler JK. 2016. Epigenetics and aging. Sci. Adv. 2, e1600584 ( 10.1126/sciadv.1600584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johansson Å, Enroth S, Gyllensten U. 2013. Continuous aging of the human DNA methylome throughout the human lifespan. PLoS ONE 8, e67378 ( 10.1371/journal.pone.0067378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guevara EE, Lawler RR. 2018. Epigenetic clocks. Evol. Anthropol. 27, 256–260. ( 10.1002/evan.21745) [DOI] [PubMed] [Google Scholar]
- 4.Horvath S. 2013. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 ( 10.1186/gb-2013-14-10-r115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Aquila P, Rose G, Bellizzi D, Passarino G. 2013. Epigenetics and aging. Maturitas 74, 130–136. ( 10.1016/j.maturitas.2012.11.005) [DOI] [PubMed] [Google Scholar]
- 6.Weidner CI, Wagner W. 2014. The epigenetic tracks of aging. Biol. Chem. 395, 1307–1314. ( 10.1515/hsz-2014-0180) [DOI] [PubMed] [Google Scholar]
- 7.Hannum G, et al. 2013. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 49, 359–367. ( 10.1016/j.molcel.2012.10.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weidner C, et al. 2014. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 15, R24 ( 10.1186/gb-2014-15-2-r24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benayoun BA, Pollina EA, Brunet A. 2015. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 16, 593–610. ( 10.1038/nrm4048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breitling LP, Saum K-U, Perna L, Schöttker B, Holleczek B, Brenner H. 2016. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin. Epigenet. 8, 21 ( 10.1186/s13148-016-0186-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marioni RE, et al. 2015. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 44, 1388–1396. ( 10.1093/ije/dyu277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marioni RE, et al. 2015. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 16, 25 ( 10.1186/s13059-015-0584-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen BH, et al. 2016. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging 8, 1844–1859. ( 10.18632/aging.101020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, Christensen K. 2015. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 15, 149–154. ( 10.1111/acel.12421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perna L, Zhang Y, Mons U, Holleczek B, Saum K-U, Brenner H. 2016. Epigenetic age acceleration predicts cancer, cardiovascular, and all-cause mortality in a German case cohort. Clin. Epigenetics 8, 64 ( 10.1186/s13148-016-0228-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polanowski AM, Robbins J, Chandler D, Jarman SN. 2014. Epigenetic estimation of age in humpback whales. Mol. Ecol. Resour. 14, 5 ( 10.1111/1755-0998.12247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spiers H, Hannon E, Wells S, Williams B, Fernandes C, Mill J. 2016. Age-associated changes in DNA methylation across multiple tissues in an inbred mouse model. Mech. Ageing 154, 20–23. ( 10.1016/j.mad.2016.02.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson MJ, vonHoldt B, Horvath S, Pellegrini M. 2017. An epigenetic aging clock for dogs and wolves. Aging 9, 1055–1068. ( 10.18632/aging.101211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowe R, et al. 2018. Ageing-associated DNA methylation dynamics are a molecular readout of lifespan variation among mammalian species. Genome Biol. 19, 22 ( 10.1186/s13059-018-1397-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ice GH. 2005. Biological anthropology and aging. J. Cross-Cult. Gerontol. 20, 87–90. ( 10.1007/s10823-005-9084-6) [DOI] [PubMed] [Google Scholar]
- 21.Hill K, Boesch C, Goodall J, Pusey A, Williams J, Wrangham R. 2001. Mortality rates among wild chimpanzees. J. Hum. Evol. 40, 437–450. ( 10.1006/jhev.2001.0469) [DOI] [PubMed] [Google Scholar]
- 22.Thompson ME, et al. 2007. Aging and fertility patterns in wild chimpanzees provide insights into the evolution of menopause. Curr. Biol. 17, 2150–2156. ( 10.1016/j.cub.2007.11.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura M, Nishie H. 2012. Death of the oldest female at Mahale and some notes about longevity of wild chimpanzees. Pan Africa News 19, 19–21. ( 10.5134/168179) [DOI] [Google Scholar]
- 24.Levitis DA, Burger O, Lackey LB. 2013. The human post-fertile lifespan in comparative evolutionary context. Evol. Anthropol. 22, 66–79. ( 10.1002/evan.21332) [DOI] [PubMed] [Google Scholar]
- 25.Wood BM, Watts DP, Mitani JC, Langergraber KE. 2017. Favorable ecological circumstances promote life expectancy in chimpanzees similar to that of human hunter-gatherers. J. Hum. Evol. 105, 41–56. ( 10.1016/j.jhevol.2017.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirkwood TBL. 1997. The origins of human ageing. Phil. Trans. R. Soc. Lond. B 352, 1765–1772. ( 10.1098/rstb.1997.0160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finch CE. 2009. Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc. Natl Acad. Sci. USA 107, 1718–1724. ( 10.1073/pnas.0909606106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.dos Reis M, Gunnell GF, Barba-Montoya J, Wilkins A, Yang Z, Yoder AD. 2018. Using phylogenomic data to explore the effects of relaxed clocks and calibration strategies on divergence time estimation: primates as a test case. Syst. Biol. 67, 594–615. ( 10.1093/sysbio/syy001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King M, Wilson A. 1975. Evolution at two levels in humans and chimpanzees. Science 188, 107–116. ( 10.1126/science.1090005) [DOI] [PubMed] [Google Scholar]
- 30.Hernando-Herraez I, et al. 2013. Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet. 9, e1003763 ( 10.1371/journal.pgen.1003763) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito H, Udono T, Hirata S, Inoue-Murayama M. 2018. Estimation of chimpanzee age based on DNA methylation. Sci. Rep. 8, 9998 ( 10.1038/s41598-018-28318-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McRae AF, et al. 2018. Identification of 55,000 replicated DNA methylation QTL. Sci. Rep. 8, 17605 ( 10.1038/s41598-018-35871-w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ely JJ, Zavaskis T, Lammey ML. 2012. Hypertension increases with aging and obesity in chimpanzees (Pan troglodytes). Zoo Biol. 32, 79–87. ( 10.1002/zoo.21044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staes N, et al. 2017. FOXP2 variation in great ape populations offers insight into the evolution of communication skills. Sci. Rep. 7, 16866 ( 10.1038/s41598-017-16844-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ely JJ, Gonzalez DL, Reeves-Daniel A, Stone WH. 1998. Individual identification and paternity determination in chimpanzees (Pan troglodytes) using human short tandem repeat (STR) markers. Int. J. Primatol. 19, 255–271. [Google Scholar]
- 36.Needhamsen M, Ewing E, Lund H, Gomez-Cabrero D, Harris RA, Kular L, Jagodic M. 2017. Usability of human infinium methylationEPIC BeadChip for mouse DNA methylation studies. BMC Bioinf. 18, 486 ( 10.1186/s12859-017-1870-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamada Y, Udono T, Teramoto M, Sugawara T. 1996. The growth pattern of chimpanzees: somatic growth and reproductive maturation in Pan troglodytes. Primates 37, 279–295. ( 10.1007/bf02381860) [DOI] [Google Scholar]
- 38.Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, Teschendorff AE. 2017. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984. ( 10.1093/bioinformatics/btx513) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teschendorff AE, West J, Beck S. 2013. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum. Mol. Genet. 22, R7–R15. ( 10.1093/hmg/ddt375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teschendorff AE, et al. 2009. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS ONE 4, e8274 ( 10.1371/journal.pone.0008274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson WE, Li C, Rabinovic A. 2006. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. ( 10.1093/biostatistics/kxj037) [DOI] [PubMed] [Google Scholar]
- 42.Therneau TM. 2015. Coxme: mixed effects Cox models. R package version 2.2-5.
- 43.Tange O. 2015. Gnu parallel: the command-line power tool. The USENIX Magazine 2015, 42–47. ( 10.5281/zenodo.16303) [DOI]
- 44.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 ( 10.1093/nar/gkv007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Storey J, Bass AJ, Dabney A, Robinson D. 2018. qvalue: Q-value estimation for false discovery rate control. R package version 2.14.0.
- 46.Emes RD, Farrell WE. 2012. Make way for the ‘next generation’: application and prospects for genome-wide, epigenome-specific technologies in endocrine research. J. Mol. Endocrinol. 49, R19–R27. ( 10.1530/jme-12-0045) [DOI] [PubMed] [Google Scholar]
- 47.Lea AJ, Altmann J, Alberts SC, Tung J. 2016. Resource base influences genome-wide DNA methylation levels in wild baboons (Papio cynocephalus). Mol. Ecol. 25, 1681–1696. ( 10.1111/mec.13436) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.R Core Team. 2019. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See https://www.R-project.org/. [Google Scholar]
- 49.Snir S, Farrell C, Pellegrini M. 2019. Human epigenetic ageing is logarithmic with time across the entire lifespan. Epigenetics 14, 912–926. ( 10.1080/15592294.2019.1623634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bibikova M, et al. 2011. High density DNA methylation array with single CpG site resolution. Genomics 98, 288–295. ( 10.1016/j.ygeno.2011.07.007) [DOI] [PubMed] [Google Scholar]
- 51.Zou H, Hastie T. 2005. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 67, 301–320. ( 10.1111/j.1467-9868.2005.00503.x) [DOI] [Google Scholar]
- 52.Hadfield JD. 2010. MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. J. Stat. Softw. 33, 1–22.20808728 [Google Scholar]
- 53.Wilson DS. 2008. Why h2 does not always equal VA/VP? J. Evol. Biol. 21, 648–650. ( 10.1111/j.1420-9101.2008.01500.x) [DOI] [PubMed] [Google Scholar]
- 54.De Villemereuil P, Morrissey MB, Nakagawa S, Schielzeth H. 2018. Fixed-effect variance and the estimation of repeatabilities and heritabilities: issues and solutions. J. Evol. Biol. 31, 621–632. ( 10.1111/jeb.13232) [DOI] [PubMed] [Google Scholar]
- 55.Horvath S, Raj K. 2018. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384. ( 10.1038/s41576-018-0004-3) [DOI] [PubMed] [Google Scholar]
- 56.Yang JH, et al. 2019. Erosion of the epigenetic landscape and loss of cellular identity as a cause of aging in mammals. bioRxiv 1, 808642 ( 10.1101/808642) [DOI] [Google Scholar]
- 57.Bianchi S, et al. 2013. Synaptogenesis and development of pyramidal neuron dendritic morphology in the chimpanzee neocortex resembles humans. Proc. Natl Acad. Sci. USA 110, 10 395–10 401. ( 10.1073/pnas.1301224110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horvath S, et al. 2016. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 17, 171 ( 10.1186/s13059-016-1030-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bronikowski AM, et al. 2011. Aging in the natural world: comparative data reveal similar mortality patterns across primates. Science 331, 1325–1328. ( 10.1126/science.1201571) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Havercamp K, Watanuki K, Tomonaga M, Matsuzawa T, Hirata S. 2019. Longevity and mortality of captive chimpanzees in Japan from 1921 to 2018. Primates 60, 525–535. ( 10.1007/s10329-019-00755-8) [DOI] [PubMed] [Google Scholar]
- 61.Jakobsson A, Westerberg R, Jacobsson A. 2006. Fatty acid elongases in mammals: their regulation and roles in metabolism. Prog. Lipid Res. 45, 237–249. ( 10.1016/j.plipres.2006.01.004) [DOI] [PubMed] [Google Scholar]
- 62.Garagnani P, et al. 2012. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 11, 1132–1134. ( 10.1111/acel.12005) [DOI] [PubMed] [Google Scholar]
- 63.Florath I, Butterbach K, Muller H, Bewerunge-Hudler M, Brenner H. 2013. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet. 23, 1186–1201. ( 10.1093/hmg/ddt531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steegenga WT, et al. 2014. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age 36, 1523–1540. ( 10.1007/s11357-014-9648-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marttila S, Kananen L, Häyrynen S, Jylhävä J, Nevalainen T, Hervonen A, Jylhä M, Nykter M, Hurme M. 2015. Ageing-associated changes in the human DNA methylome: genomic locations and effects on gene expression. BMC Genomics 16, 179 ( 10.1186/s12864-015-1381-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rönn T, et al. 2015. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum. Mol. Genet. 24, 3792–3813. ( 10.1093/hmg/ddv124) [DOI] [PubMed] [Google Scholar]
- 67.Kananen L, Marttila S, Nevalainen T, Jylhävä J, Mononen N, Kähönen M, Raitakari OT, Lehtimäki T, Hurme M. 2016. Aging-associated DNA methylation changes in middle-aged individuals: the Young Finns study. BMC Genomics 17, 103 ( 10.1186/s12864-016-2421-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bysani M, Perfilyev A, de Mello VD, Rönn T, Nilsson E, Pihlajamäki J, Ling C. 2017. Epigenetic alterations in blood mirror age-associated DNA methylation and gene expression changes in human liver. Epigenomics 9, 105–122. ( 10.2217/epi-2016-0087) [DOI] [PubMed] [Google Scholar]
- 69.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. 2004. Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. ( 10.1038/nature02661) [DOI] [PubMed] [Google Scholar]
- 70.Hayashi M, Yamashita A, Shimizu K. 1997. Somatostatin and brain-derived neurotrophic factor mRNA expression in the primate brain: decreased levels of mRNAs during aging. Brain Res. 749, 283–289. ( 10.1016/S0006-8993(96)01317-0) [DOI] [PubMed] [Google Scholar]
- 71.Saito T, Iwata N, Tsubuki S, Takaki Y, Takano J, Huang SM, Suemoto T, Higuchi M, Saido TC. 2005. Somatostatin regulates brain amyloid β peptide Aβ 42 through modulation of proteolytic degradation. Nat. Med. 11, 434–439. ( 10.1038/nm1206) [DOI] [PubMed] [Google Scholar]
- 72.Duga S, Asselta R, Tenchini ML. 2004. Coagulation factor V. Int. J. Biochem. Cell Biol. 36, 1393–1399. ( 10.1016/j.biocel.2003.08.002) [DOI] [PubMed] [Google Scholar]
- 73.Grundberg E, et al. 2013. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am. J. Hum. Genet. 93, 876–890. ( 10.1016/j.ajhg.2013.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McClay JL, et al. 2015. High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol. 16, 1–16. ( 10.1186/s13059-015-0842-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taudt A, Colomé-Tatché M, Johannes F. 2016. Genetic sources of population epigenomic variation. Nat. Rev. Genet. 17, 319–332. ( 10.1038/nrg.2016.45) [DOI] [PubMed] [Google Scholar]
- 76.van Dongen J, et al. 2016. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 7, 1 ( 10.1038/ncomms11115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hernando-Herraez I, Heyn H, Fernandez-Callejo M, Vidal E, Fernandez-Bellon H, Prado-Martinez J, Sharp AJ, Esteller M, Marques-Bonet T. 2015. The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res. 43, 8204–8214. ( 10.1093/nar/gkv693) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available on the National Center forBiotechnology Information's Gene Expression Omnibus (Accession GSE136296). Code is available on the National Chimpanzee Brain Resource website Data Repository page (https://www.chimpanzeebrain.org/data-repository).